
Elevated Expression and Pro-Inflammatory Activity of IL-36 in Patients with Systemic Lupus Erythematosus

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Demographical and Clinical Characteristics of SLE Patients and NC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SLE Patients (n = 43) | Normal Control (n = 16) | ||
|---|---|---|---|
| Inactive SLE (n = 22) | Active SLE (n = 21) | ||
| Demographic Characteristics | |||
| Female sex (n) (%) | 20 (90.9) | 13 (95.2) | 14 (87.5) |
| Age at study (year) | |||
| Mean(SD) | 45.2 (9.5) | 48.2 (10.5) | 41.4 (12.5) |
| Range | 24–59 | 23–60 | 23–57 |
| Clinical Features | |||
| SLE duration (year) | |||
| Mean(SD) | 14.8 (5.9) | 16.6 (7.6) | N/A |
| Range | 1–28 | 10–26 | N/A |
| SLICC median(IQR) | 0 (0–1) | 0 (0–3) | N/A |
| SLEDAI median(IQR) | 2 (2–4) | 8 (6–16) | N/A |
| Flare (n) (%) | 5 (22.7) | 16 (76.2) | N/A |
| Serological Features | |||
| Serum complement C3 (g/L) mean (SD) | 0.80 (0.24) | 0.62 (0.19) | N/A |
| Serum complement C4 (g/L) mean (SD) | 0.15 (0.09) | 0.08 (0.05) | N/A |
| Anti-dsDNA >1000 IU/mL (n) (%) | 2 (9.1) | 3 (14.2) | N/A |
| Anti-dsDNA <60 IU/mL (n) (%) | 3 (13.6) | 1 (4.8) | N/A |
| Anti-dsDNA titer (IU/mL) mean (SD) | 268.4 (185.1) | 409.6 (201.7) | N/A |
| Plasma urea (mmol/L) mean (SD) | 5.8 (1.7) * | 6.3 (1.5) ** | 4.7 (1.2) |
| Plasma creatinine (μmol/L) mean (SD) | 70.2 (28.0) | 79.5 (30.3) * | 61.7 (12.9) |
| Plasma total protein (g/L) mean (SD) | 70.3 (10.9) ** | 66.1 (8.7) *** | 77.6 (3.3) |
| Plasma albumin (g/L) mean (SD) | 38.2 (7.2) ** | 31.9 (5.2) *** | 45.6 (2.3) |
| Major Organ System Involvement (n) (%) | |||
| 0 | 5 (22.7) | 0 (0) | N/A |
| 1 | 10 (45.4) | 7 (33.3) | N/A |
| ≥2 | 7 (32.9) | 14 (66.7) | N/A |
| Clinical Manifestation (n) (%) | |||
| Nephritis | 13 (59.1) | 21 (100.0) | N/A |
| Serositis | 5 (22.7) | 7 (33.3) | N/A |
| Hematologic | 5 (22.7) | 6 (28.6) | N/A |
| Arthritis | 8 (38.1) | 9 (42.8) | N/A |
| Current Immunosuppressive Therapy | |||
| Treatment with prednisolone | |||
| Patients (n) (%) | 17 (77.3) | 19 (90.5) | N/A |
| Daily dose (mg) mean (SD) | 7.5 (3.5) | 9.2 (5.7) | N/A |
| Treatment with hydroxychloroquine | |||
| Patients (n) (%) | 9 (40.9) | 10 (47.6) | N/A |
| Daily dose (mg) mean (SD) | 236.7 (95.1) | 300.0 (115.5) | N/A |
| Treatment with mycophenolatemofetil | |||
| Patients (n) (%) | 10 (45.4) | 10 (47.6) | N/A |
| Daily dose (mg) mean (SD) | 883.3 (458.1) | 1000.0 (250.0) | N/A |
| Treatment with lisinopril | |||
| Patients (n) (%) | 11 (50.0) | 13 (61.9) | N/A |
| Daily dose (mg) mean (SD) | 9.6 (7.4) | 14.4 (5.3) | N/A |
| Treatment with azathioprine | |||
| Patients (n) (%) | 4 (18.2.7) | 7 (33.3) | N/A |
| Daily dose (mg) mean(SD) | 58.3 (38.2) | 100.0 (50.0) | N/A |
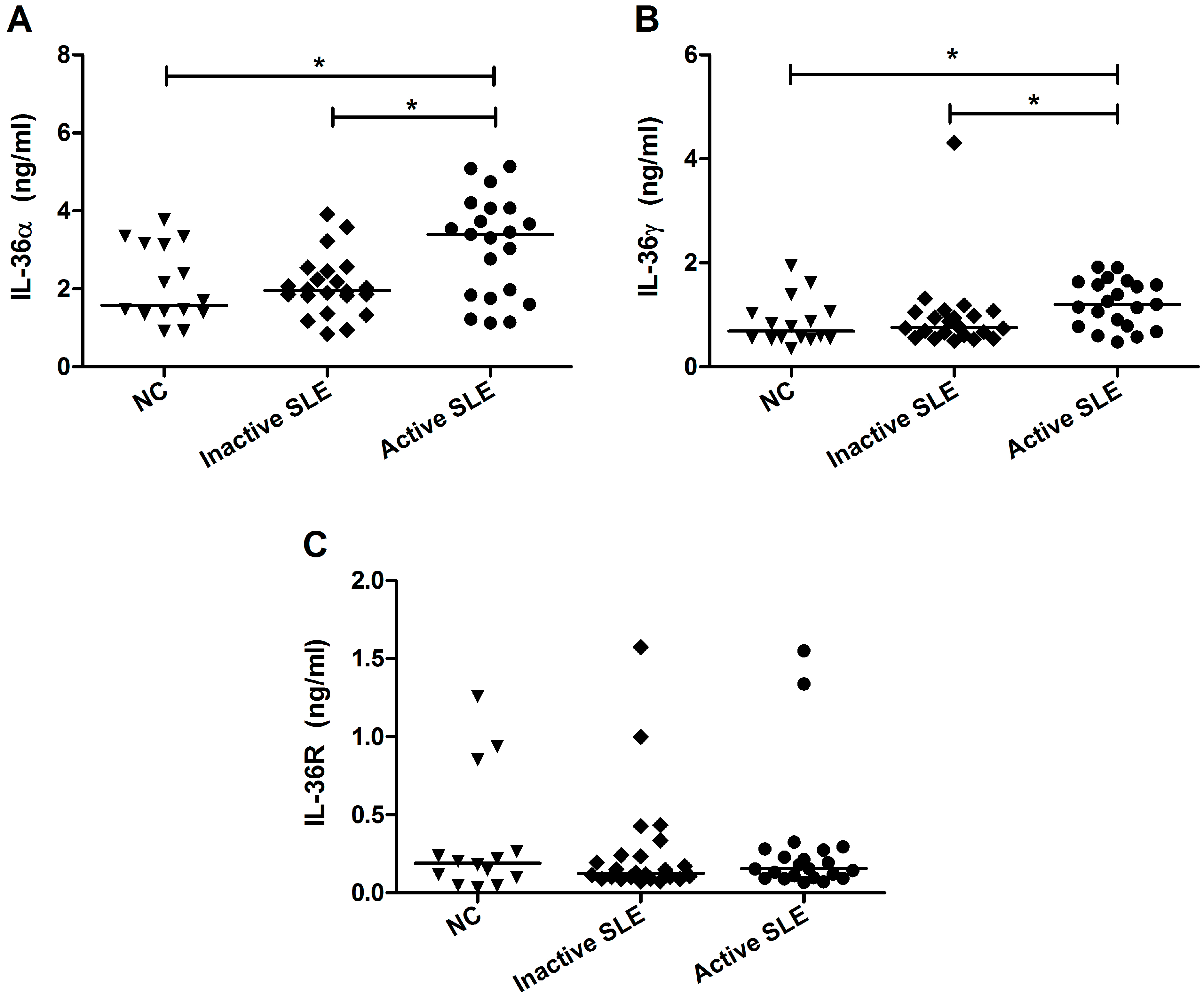
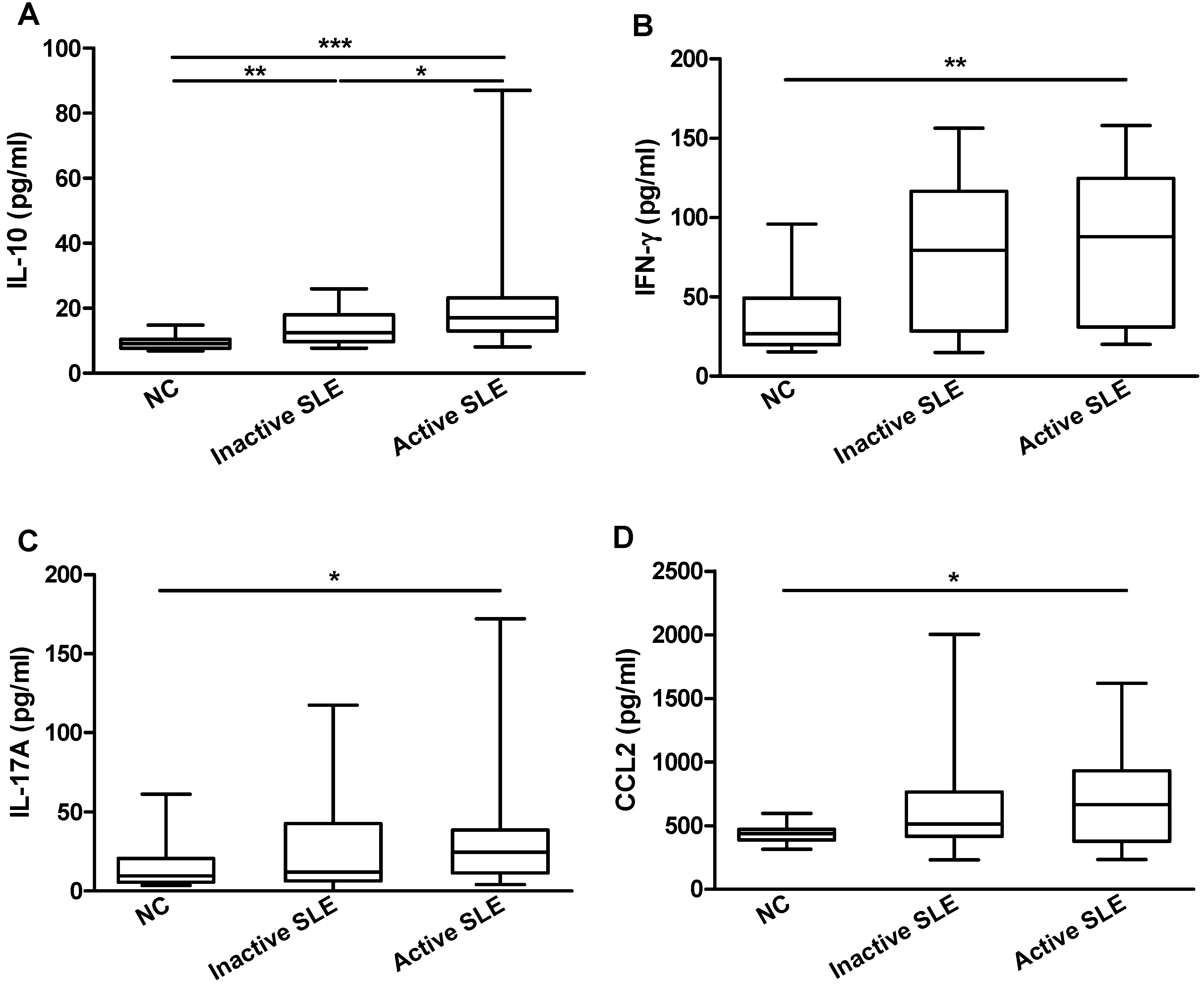
2.2. Elevated Plasma IL-36 Correlated Positively with SLE Disease Activity and Plasma IL-10

| IL-36α (ng/mL) | IL-36γ (ng/mL) | |||
|---|---|---|---|---|
| r Value | p Value | r Value | p Value | |
| SLEDAI | 0.382 | 0.011 * | 0.327 | 0.025 * |
| C3 (g/L) | −0.147 | 0.342 | −0.211 | 0.154 |
| C4 (g/L) | −0.138 | 0.371 | −0.339 | 0.019 * |
| IL-10 (pg/mL) | 0.306 | 0.046 * | 0.338 | 0.023 * |
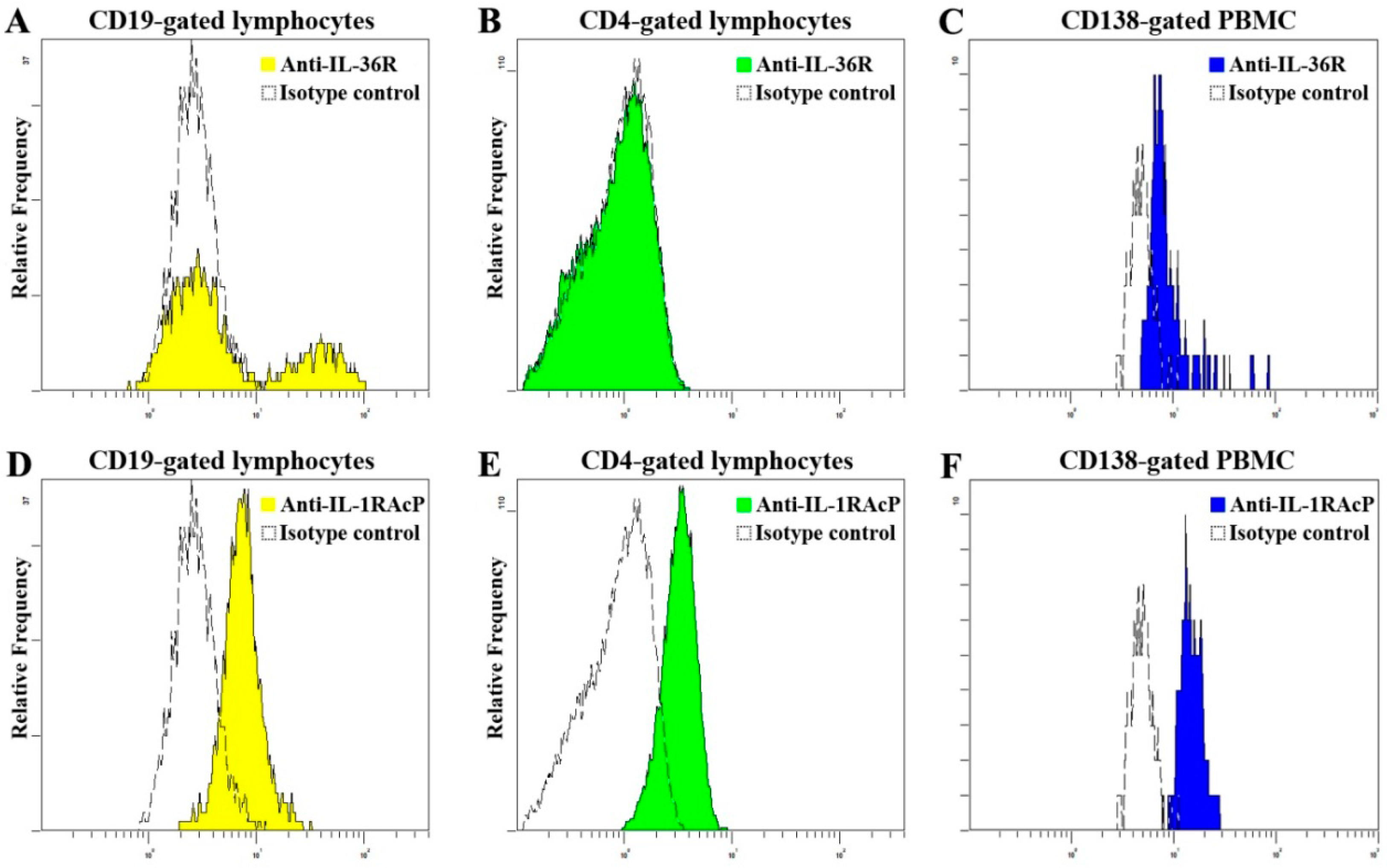
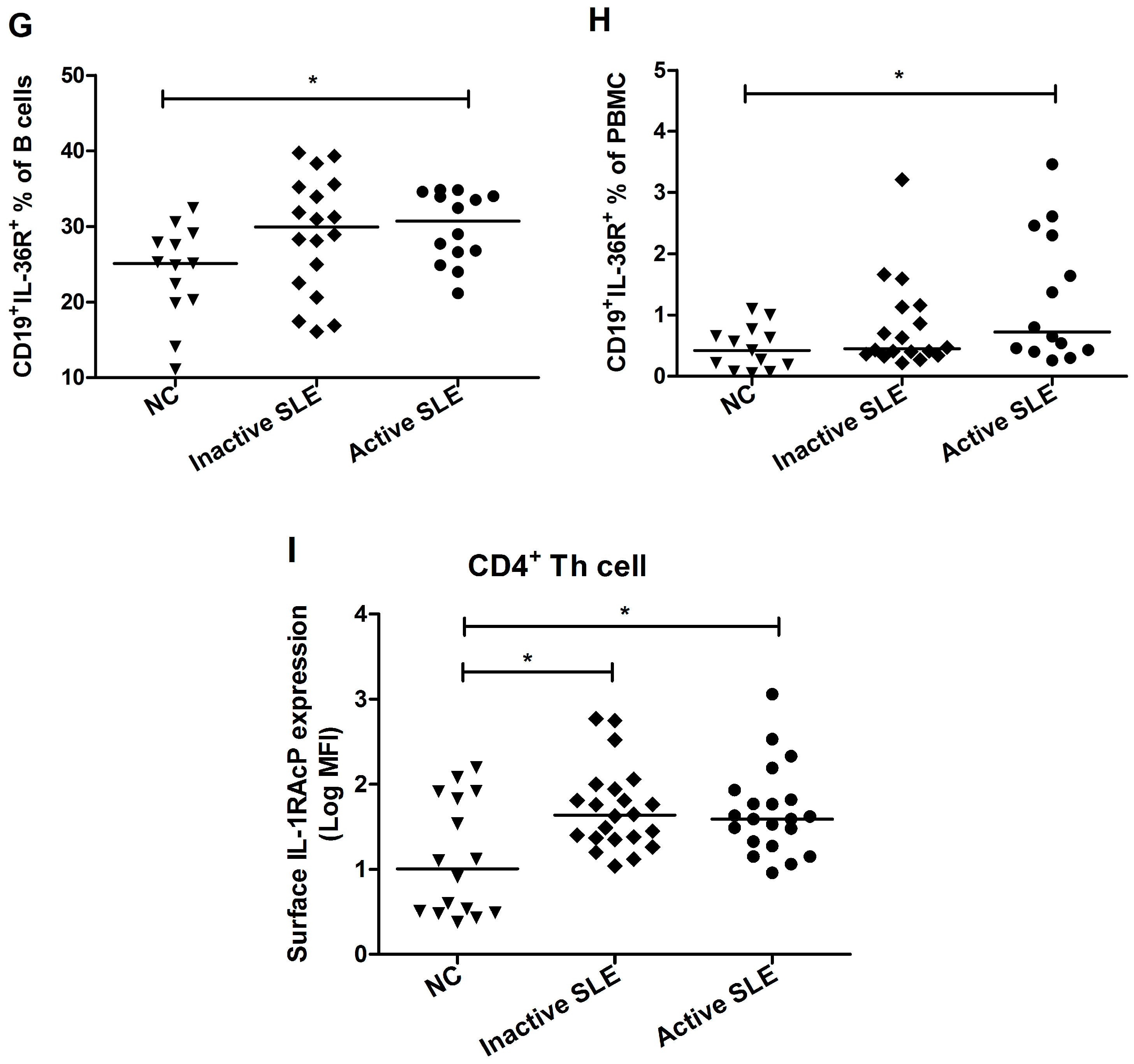
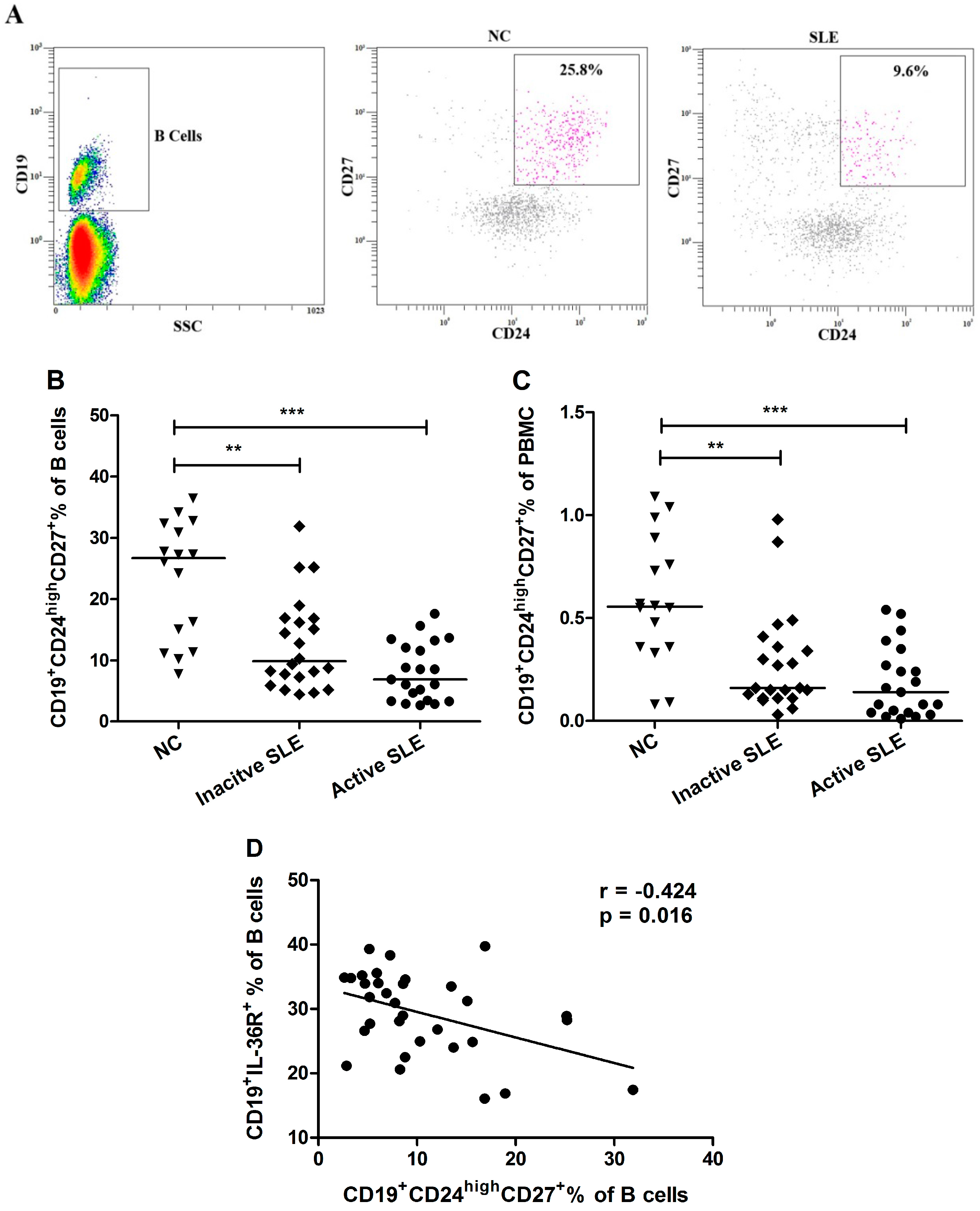
2.3. Elevated Proportion of CD19+ B Cells Expressed with IL-36R in SLE Patients
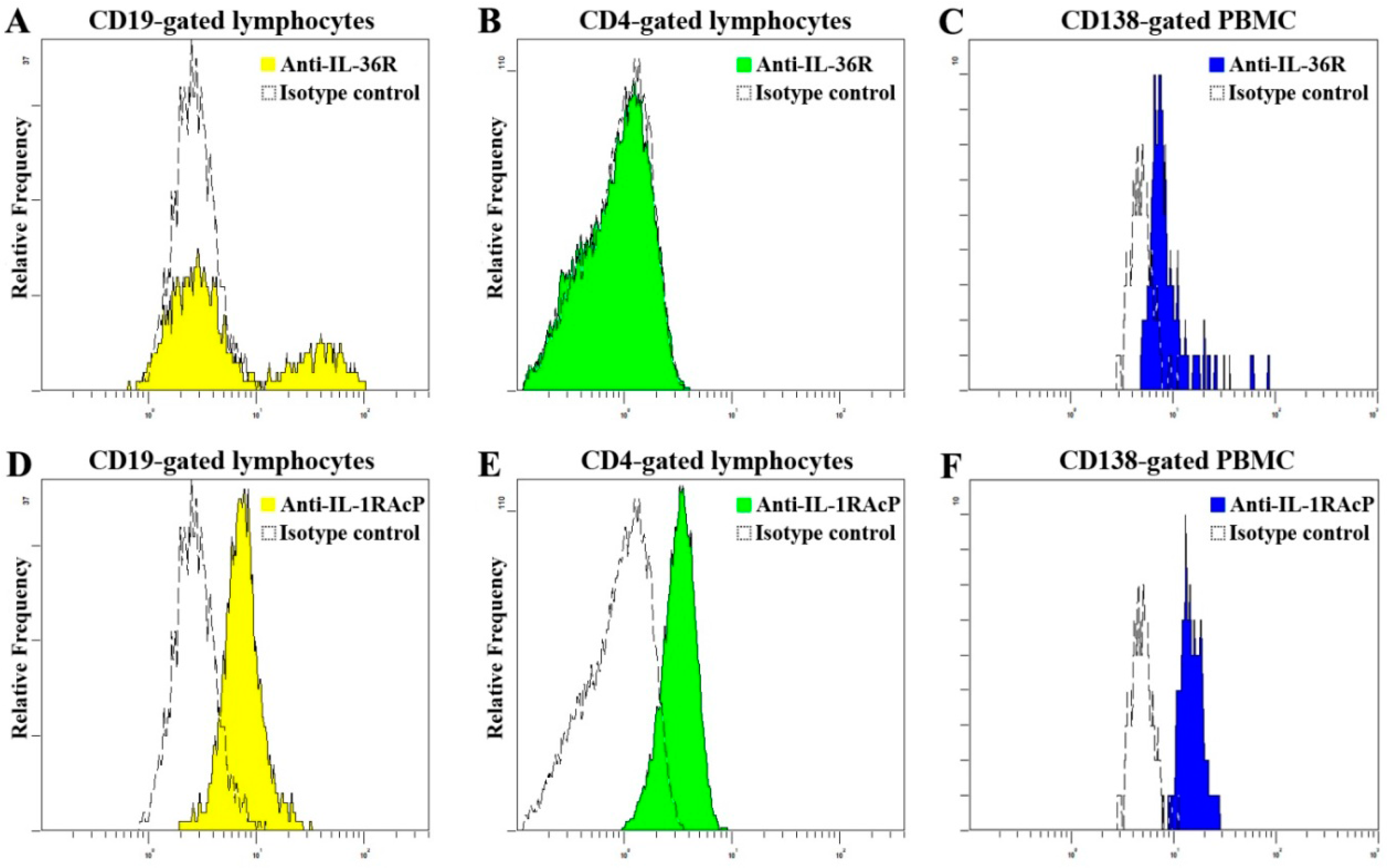
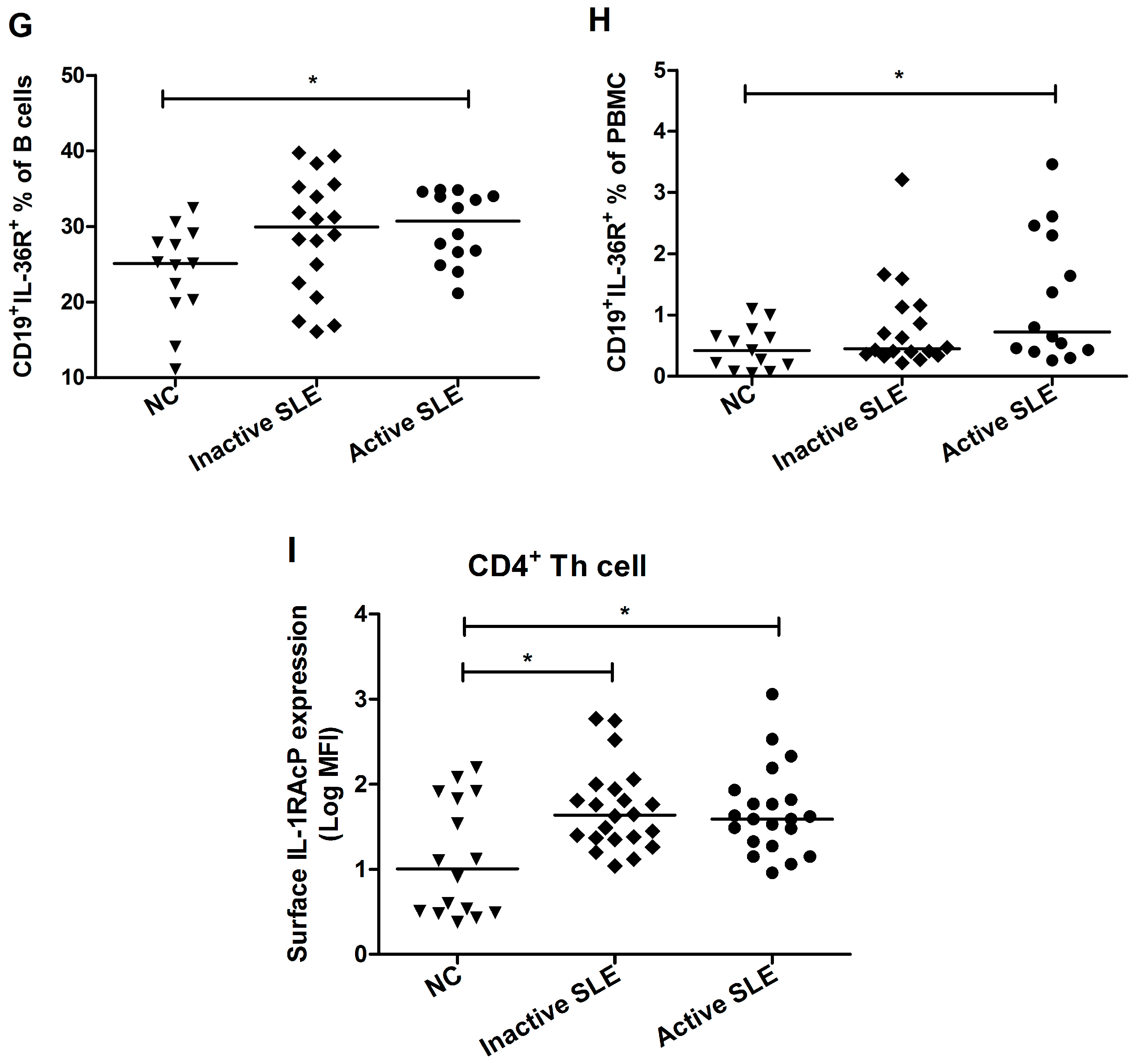
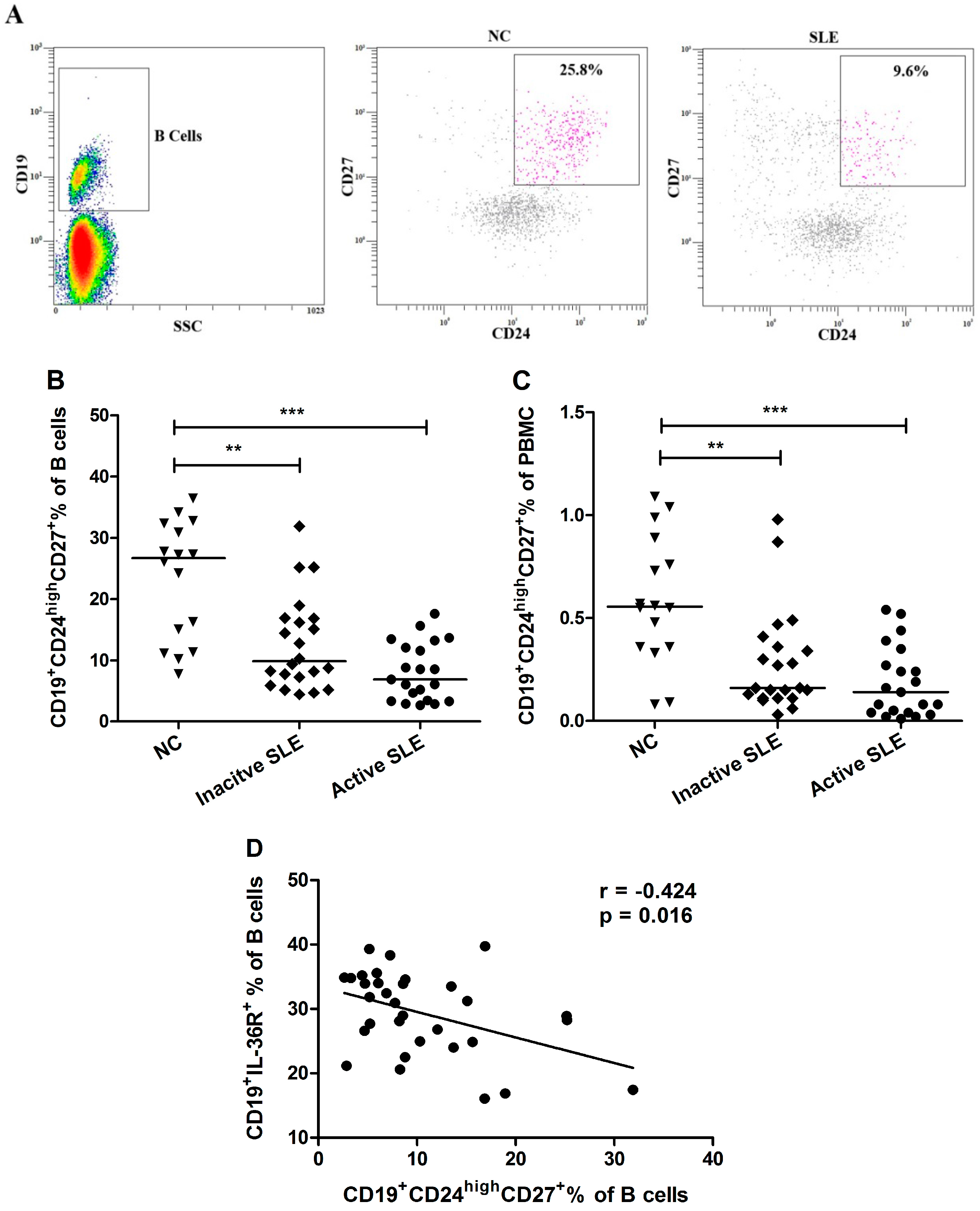
2.4. Decrement of CD19+CD24highCD27+ Breg Lymphocytes in SLE Patients
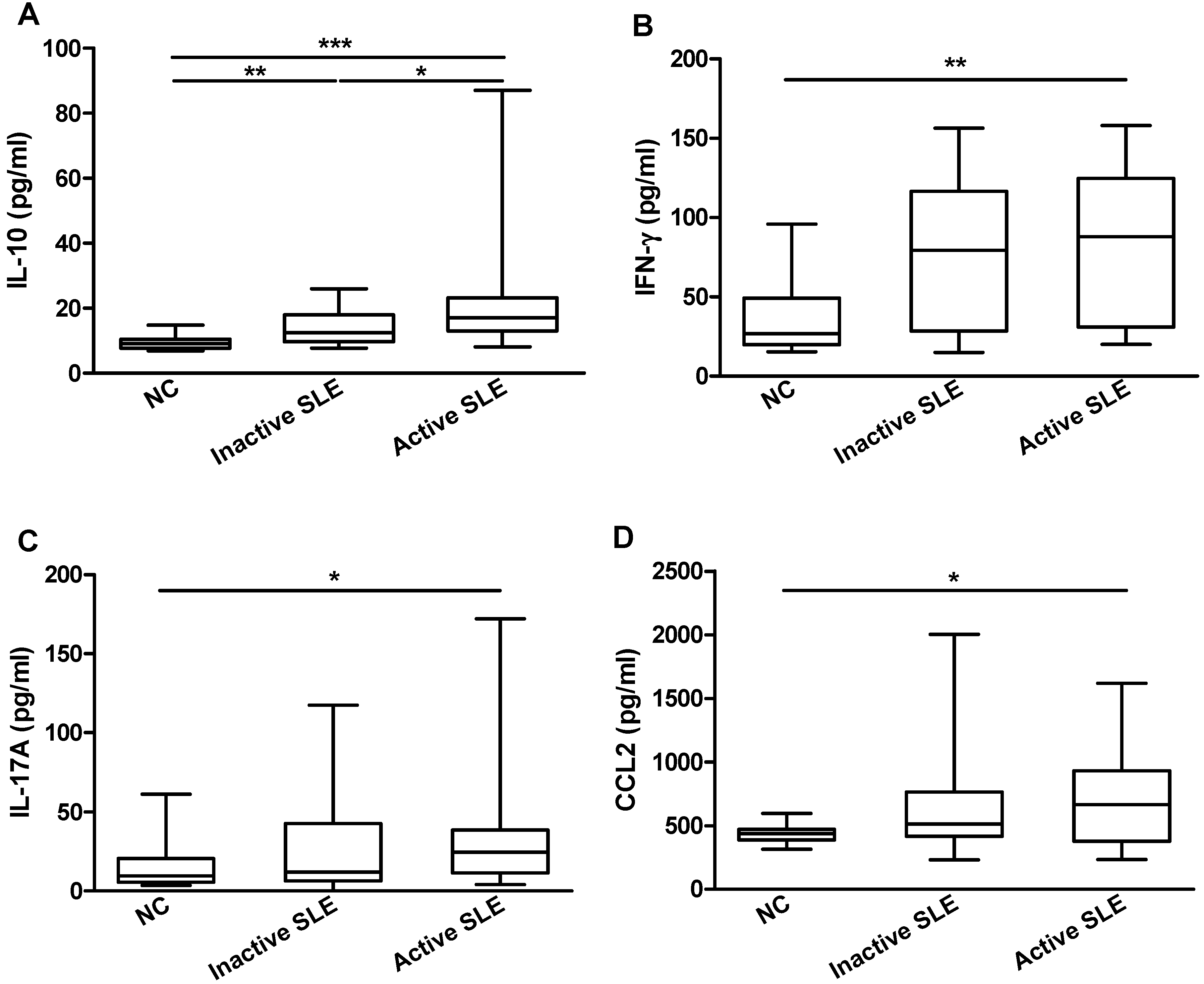
2.5. IL-36 Promotes the Production of Cytokine/Chemokine in PBMC
| Cytokine/Chemokine | Group | Basal Median (IQR) (pg/mL) | Post stimulation with IL-36α Median (IQR) (pg/mL) | Post stimulation with IL-36γ Median (IQR) (pg/mL) |
|---|---|---|---|---|
| IL-6 | NC | 2.3 (0–3.9) | 5.6 (1.9–10.4) # | 3.6 (1.5–6.6) |
| SLE | 26.5 (3.6–43.4) *** | 44.4 (9.9–90.96) ***,& | 35.1 (7.3–65.4) *** | |
| CXCL8 | NC | 548.3 (399.1–676.1) | 1347.0 (785.6–2586.0) ### | 808.6 (505.2–1300.0) # |
| SLE | 1288.0 (1145.0–4487.0) ** | 2661.0 (1145.0–4487.0) *,&& | 1510.0 (678.1–3673) * | |
| IFN-γ | NC | 11.9 (10.4–19.4) | 16.1 (11.9–27.2) | 14.3 (6.6–23.6) |
| SLE | 14.5 (9.3–20.2) | 17.4 (11.3–21.6) | 17.4 (11.9–23.0) | |
| IL-17A | NC | UD | UD | UD |
| SLE | UD | UD | UD |
3. Experimental Section
3.1. Ethics Statement
3.2. SLE Patients and Normal Control (NC) Subjects
3.3. Clinical and Laboratory Parameters
3.4. Assays for the Expression Level of Plasma IL-36α, IL-36γ and IL-36R
3.5. Assays for the Expression Level of Plasma Cytokines/Chemokines
3.6. Flow Cytometry
3.7. Ex vivo Induction of Cytokine/Chemokine from PBMC by Recombinant IL-36
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mok, C.; Lau, C. Pathogenesis of systemic lupus erythematosus. J. Clin. Pathol. 2003, 56, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Ho, C.Y.; Li, E.K.; Lam, C.W. Elevation of proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2 cytokine (IL-4) concentrations in patients with systemic lupus erythematosus. Lupus 2000, 9, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Li, E.K.; Ho, C.Y.; Lam, C.W. Elevation of plasma interleukin-18 concentration is correlated with disease activity in systemic lupus erythematosus. Rheumatology 2000, 39, 1078–1081. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Lit, L.C.; Tam, L.S.; Li, E.K.; Wong, P.T.; Lam, C.W. Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: Implications for Th17-mediated inflammation in auto-immunity. Clin. Immunol. 2008, 127, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Lit, L.C.; Wong, C.K.; Li, E.K.; Tam, L.S.; Lam, C.W. Elevated plasma concentration and ex vivo production of inflammatory chemokines in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2006, 65, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Rönnblom, L.; Elkon, K.B. Cytokines as therapeutic targets in SLE. Nat. Rev. Rheumatol. 2010, 6, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.B.; Lee, S.K.; Kim, D.S.; Lee, J.; Lee, C.H.; Song, C.H. Elevated interleukin-10 levels correlated with disease activity in systemic lupus erythematosus. Clin. Exp. Rheumatol. 1998, 16, 283–288. [Google Scholar] [PubMed]
- Dunn, E.; Sims, J.E.; Nicklin, M.J.; O’Neill, L.A. Annotating genes with potential roles in the immune system: Six new members of the IL-1 family. Trends Immunol. 2001, 22, 533–536. [Google Scholar] [CrossRef]
- Smith, D.E.; Renshaw, B.R.; Ketchem, R.R.; Kubin, M.; Garka, K.E.; Sims, J.E. Four new members expand the interleukin-1 superfamily. J. Biol. Chem. 2000, 275, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Towne, J.E.; Garka, K.E.; Renshaw, B.R.; Virca, G.D.; Sims, J.E. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J. Biol. Chem. 2004, 279, 13677–13688. [Google Scholar] [CrossRef] [PubMed]
- Gresnigt, M.S.; van de Veerdonk, F.L. Biology of IL-36 cytokines and their role in disease. Semin. Immunol. 2013, 25, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Tortola, L.; Rosenwald, E.; Abel, B.; Blumberg, H.; Schafer, M.; Coyle, A.J.; Renauld, J.C.; Werner, S.; Kisielow, J.; Kopf, M. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J. Clin. Investig. 2012, 122, 3965–3976. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.; Xing, X.; Guzman, A.M.; Riblett, M.; Loyd, C.M.; Ward, N.L.; Wohn, C.; Prens, E.P.; Wang, F.; Maier, L.E.; et al. IL-1F5, -F6, -F8, and -F9: A novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J. Immunol. 2011, 186, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Carrier, Y.; Ma, H.L.; Ramon, H.E.; Napierata, L.; Small, C.; O’Toole, M.; Young, D.A.; Fouser, L.A.; Nickerson-Nutter, C.; Collins, M.; et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: Implications in psoriasis pathogenesis. J. Investig. Dermatol. 2011, 131, 2428–2437. [Google Scholar] [CrossRef] [PubMed]
- Vigne, S.; Palmer, G.; Martin, P.; Lamacchia, C.; Strebel, D.; Rodriguez, E.; Olleros, M.L.; Vesin, D.; Garcia, I.; Ronchi, F.; et al. IL-36 signaling amplifies Th1 responses by enhancing proliferation and Th1 polarization of naive CD4+ T cells. Blood 2012, 120, 3478–3487. [Google Scholar] [CrossRef] [PubMed]
- Vigne, S.; Palmer, G.; Lamacchia, C.; Martin, P.; Talabot-Ayer, D.; Rodriguez, E.; Ronchi, F.; Sallusto, F.; Dinh, H.; Sims, J.E.; et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood 2011, 118, 5813–5823. [Google Scholar] [CrossRef] [PubMed]
- Kalampokis, I.; Yoshizaki, A.; Tedder, T.F. IL-10-producing regulatory B cells (B10 cells) in autoimmune disease. Arthritis Res. Ther. 2013, 15, S1. [Google Scholar] [CrossRef] [PubMed]
- Mauri, C.; Blair, P.A. Regulatory B cells in autoimmunity: Developments and controversies. Nat. Rev. Rheumatol. 2010, 6, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Blair, P.A.; Norena, L.Y.; Flores-Borja, F.; Rawlings, D.J.; Isenberg, D.A.; Ehrenstein, M.R.; Mauri, C. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity 2010, 32, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Zha, B.; Wang, L.; Liu, X.; Liu, J.; Chen, Z.; Xu, J.; Sheng, L.; Li, Y.; Chu, Y. Decrease in proportion of CD19+ CD24(hi) CD27+ B cells and impairment of their suppressive function in Graves’ disease. PLoS ONE 2012, 7, e49835. [Google Scholar] [CrossRef] [PubMed]
- de Masson, A.; Bouaziz, J.D.; le Buanec, H.; Robin, M.; O’Meara, A.; Parquet, N.; Rybojad, M.; Hau, E.; Monfort, J.B.; Branchtein, M.; et al. CD24(hi)CD27(+) and plasmablast-like regulatory B cells in human chronic graft-versus-host disease. Blood 2015, 125, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.M.; Baliwag, J.; Chen, C.S.; Guzman, A.M.; Stoll, S.W.; Gudjonsson, J.E.; Ward, N.L.; Johnston, A. IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J. Immunol. 2014, 192, 6053–6061. [Google Scholar] [CrossRef] [PubMed]
- Towne, J.E.; Sims, J.E. IL-36 in psoriasis. Curr. Opin. Pharmacol. 2012, 12, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, H.; Dinh, H.; Dean, C., Jr.; Trueblood, E.S.; Bailey, K.; Shows, D.; Bhagavathula, N.; Aslam, M.N.; Varani, J.; Towne, J.E.; et al. IL-1RL2 and its ligands contribute to the cytokine network in psoriasis. J. Immunol. 2010, 185, 4354–4362. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Accardo-Palumbo, A.; Alessandro, R.; Alessandri, C.; Priori, R.; Guggino, G.; Raimondo, S.; Carubbi, F.; Valesini, G.; Giacomelli, R.; et al. Interleukin-36alpha axis is modulated in patients with primary Sjogren’s syndrome. Clin. Exp. Immunol. 2015, 181, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Lamacchia, C.; Palmer, G.; Rodriguez, E.; Martin, P.; Vigne, S.; Seemayer, C.A.; Talabot-Ayer, D.; Towne, J.E.; Gabay, C. The severity of experimental arthritis is independent of IL-36 receptor signaling. Arthritis. Res. Ther. 2013, 15, R38. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, D.; Gabay, C. Inflammation: IL-36 has proinflammatory effects in skin but not in joints. Nat. Rev. Rheumatol. 2014, 10, 639–640. [Google Scholar] [CrossRef] [PubMed]
- Frey, S.; Derer, A.; Messbacher, M.E.; Baeten, D.L.; Bugatti, S.; Montecucco, C.; Schett, G.; Hueber, A.J. The novel cytokine interleukin-36alpha is expressed in psoriatic and rheumatoid arthritis synovium. Ann. Rheum. Dis. 2013, 72, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xu, W.D.; Zhu, Y.; Wen, P.F.; Leng, R.X.; Pan, H.F.; Ye, D.Q. Serum levels of cytokines in systemic lupus erythematosus. Z. Rheumatol. 2014, 73, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Niyonsaba, F.; Ushio, H.; Akiyama, T.; Kiatsurayanon, C.; Smithrithee, R.; Ikeda, S.; Okumura, K.; Ogawa, H. Interleukin-36 cytokines enhance the production of host defense peptides psoriasin and LL-37 by human keratinocytes through activation of MAPKs and NF-κB. J. Dermatol. Sci. 2012, 68, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Magne, D.; Palmer, G.; Barton, J.L.; Mezin, F.; Talabot-Ayer, D.; Bas, S.; Duffy, T.; Noger, M.; Guerne, P.A.; Nicklin, M.J.; et al. The new IL-1 family member IL-1F8 stimulates production of inflammatory mediators by synovial fibroblasts and articular chondrocytes. Arthritis Res. Ther. 2006, 8, R80. [Google Scholar] [CrossRef] [PubMed]
- Towne, J.E.; Renshaw, B.R.; Douangpanya, J.; Lipsky, B.P.; Shen, M.; Gabel, C.A.; Sims, J.E. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36alpha, IL-36beta, and IL-36gamma) or antagonist (IL-36Ra) activity. J. Biol. Chem. 2011, 286, 42594–42602. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.M.; Cohen, A.S.; Fries, J.F.; Masi, A.T.; Mcshane, D.J.; Rothfield, N.F.; Schaller, J.G.; Talal, N.; Winchester, R.J. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982, 25, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the recombinant IL-36 is available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, M.; Wong, C.K.; Cai, Z.; Dong, J.; Jiao, D.; Kam, N.W.; Lam, C.W.K.; Tam, L.S. Elevated Expression and Pro-Inflammatory Activity of IL-36 in Patients with Systemic Lupus Erythematosus. Molecules 2015, 20, 19588-19604. https://doi.org/10.3390/molecules201019588
Chu M, Wong CK, Cai Z, Dong J, Jiao D, Kam NW, Lam CWK, Tam LS. Elevated Expression and Pro-Inflammatory Activity of IL-36 in Patients with Systemic Lupus Erythematosus. Molecules. 2015; 20(10):19588-19604. https://doi.org/10.3390/molecules201019588
Chicago/Turabian StyleChu, Man, Chun Kwok Wong, Zhe Cai, Jie Dong, Delong Jiao, Ngar Woon Kam, Christopher Wai Kei Lam, and Lai Shan Tam. 2015. "Elevated Expression and Pro-Inflammatory Activity of IL-36 in Patients with Systemic Lupus Erythematosus" Molecules 20, no. 10: 19588-19604. https://doi.org/10.3390/molecules201019588
APA StyleChu, M., Wong, C. K., Cai, Z., Dong, J., Jiao, D., Kam, N. W., Lam, C. W. K., & Tam, L. S. (2015). Elevated Expression and Pro-Inflammatory Activity of IL-36 in Patients with Systemic Lupus Erythematosus. Molecules, 20(10), 19588-19604. https://doi.org/10.3390/molecules201019588