
Design, Synthesis, Activity and Docking Study of Sorafenib Analogs Bearing Sulfonylurea Unit


Abstract
: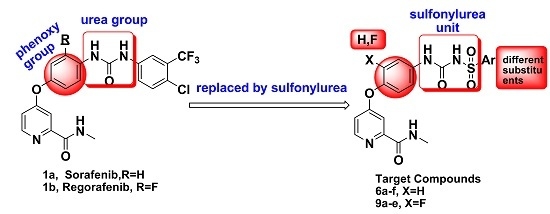
1. Introduction
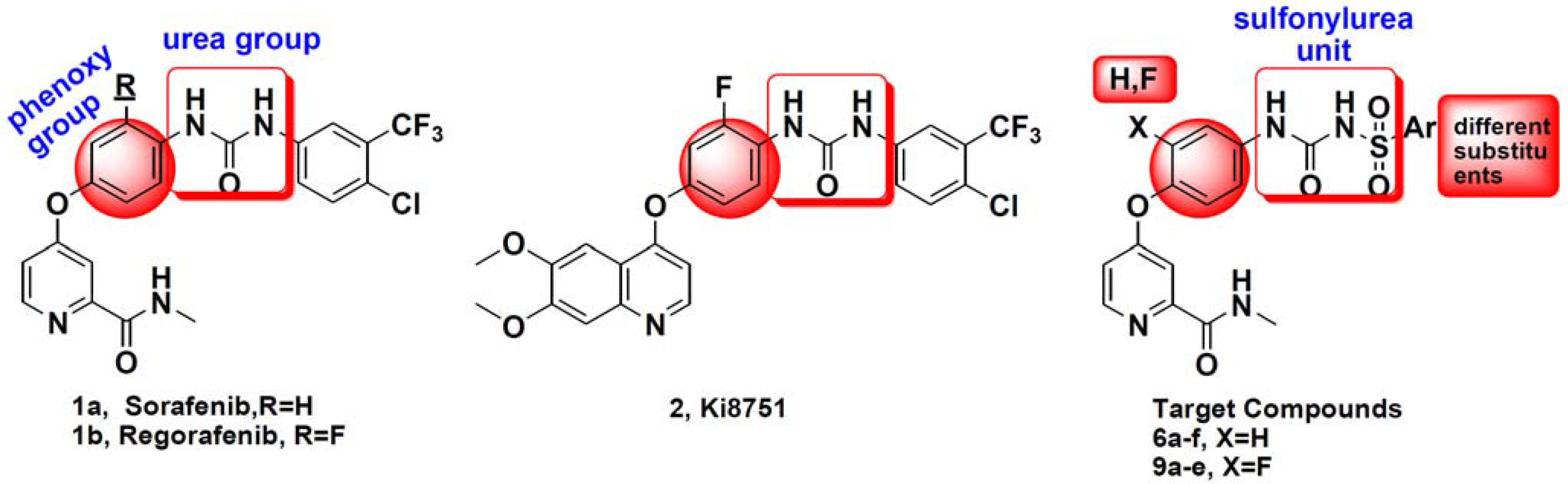
2. Results and Discussion
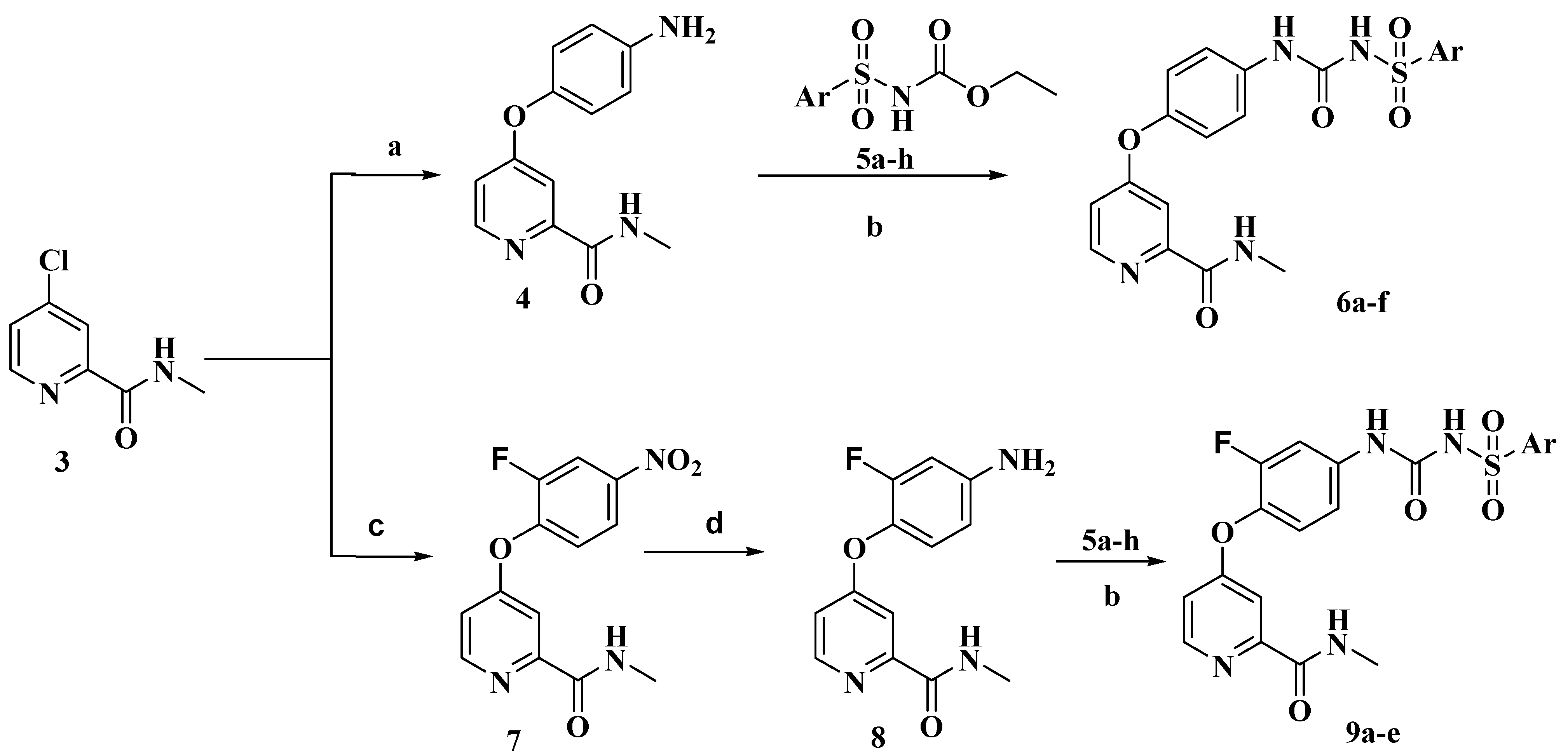
2.1. Biological Evaluation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds No. | Ar | VEGFR2/KDR Inhibitory Rate@10μM (%) | IC50(μM) a | |||
|---|---|---|---|---|---|---|
| A549 | Hela | MCF-7 | PC-3 | |||
| 6a |  | 23.6% ± 12.9% | >100 | >100 | >100 | >100 |
| 6b | 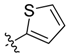 | 54.0% ± 2.7% | 65.86 ± 2.01 | >100 | 72.43 ± 1.96 | >100 |
| 6c |  | 75.8% ± 5.5% | 27.04 ± 1.43 | >100 | >100 | 25.35 ± 1.73 |
| 6d | 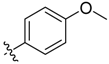 | 61.3% ± 9.6% | 86.91 ± 2.03 | >100 | 80.56 ± 2.04 | >100 |
| 6e | 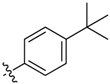 | 31.4% ± 7.2% | >100 | >100 | >100 | 68.87 ± 2.14 |
| 6f | 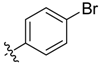 | 46.6% ± 1.8% | 32.59 ± 1.51 | 63.92 ± 1.81 | 16.54 ± 1.22 | 17.97 ± 1.56 |
| 9a |  | <20.0% | >100 | 42.43 ± 1.93 | 17.19 ± 1.54 | ND c |
| 9b |  | <20.0% | >100 | >100 | >100 | ND |
| 9c | 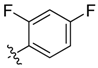 | <20.0% | 57.42 ± 1.89 | >100 | >100 | ND |
| 9d | 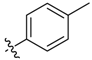 | <20.0% | 33.22 ± 1.82 | 24.65 ± 1.69 | >100 | ND |
| 9e |  | <20.0% | >100 | 44.32 ± 1.75 | 69.25 ± 1.96 | ND |
| Sorafenib b | - | 94.9% ± 1.1% | 6.53 ± 0.82 | 8.08 ± 0.91 | 4.21 ± 0.62 | 11.05 ± 1.07 |
| Staurosporine b | - | 97.3% ± 2.82% | ND | ND | ND | ND |
2.2. Molecular Docking Study
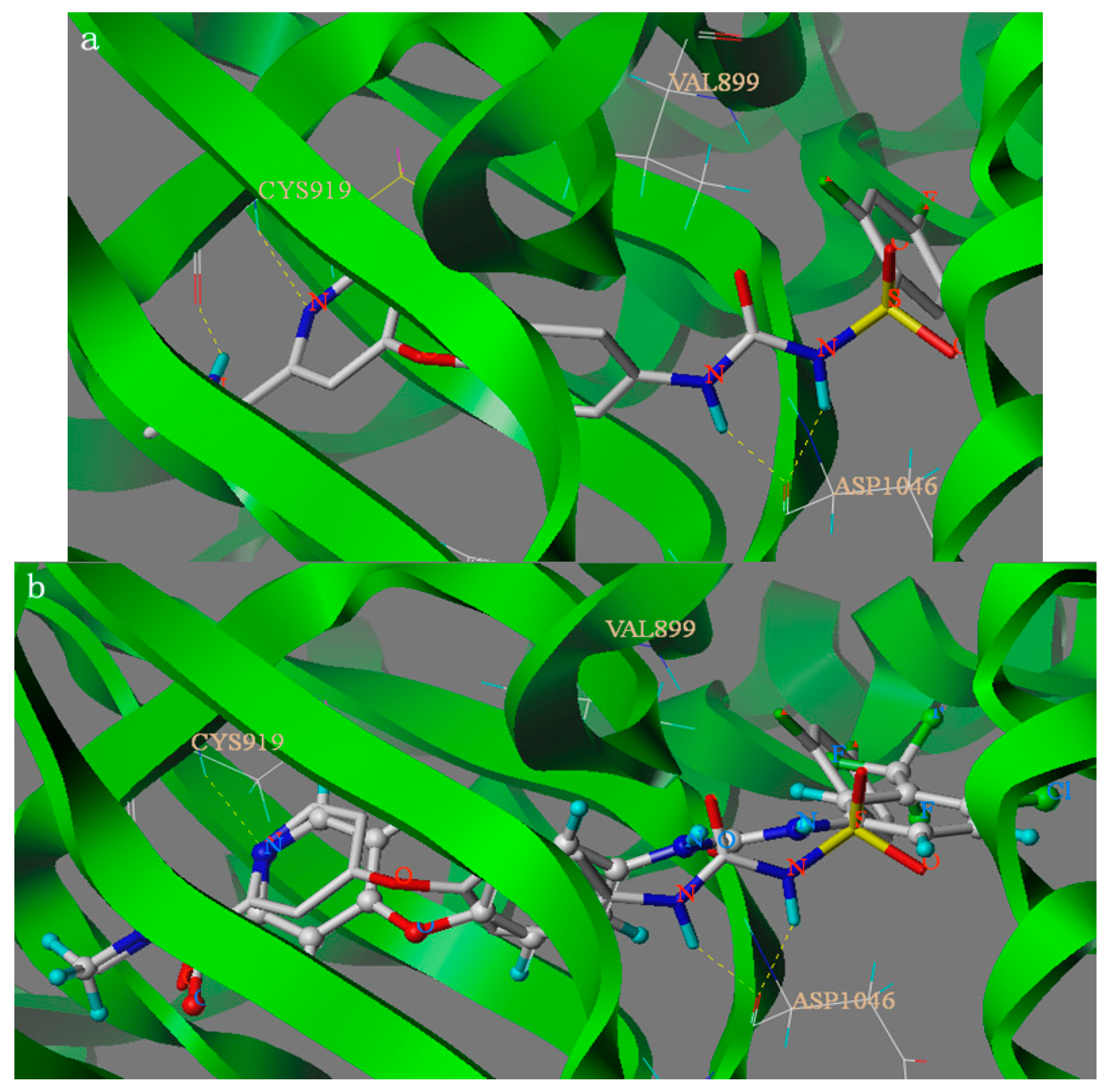
3. Experimental Section
3.1. Chemistry
3.2. VEGFR2/KDR Kinase Assay
3.3. Cytotoxicity Assay in Vitro
3.4. Docking Studies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Word Health Organization. Available online: http://www.who.int/mediacentre/factsheets/fs297/en/ (accessed on 26 March 2015).
- Samadi, A.K.; Bazzill, J.; Zhang, X.; Gallagher, R.; Zhang, H.; Gollapudi, R.; Kindscher, K.; Timmermann, B.; Cohen, M.S. Novel withanolides target medullary thyroid cancer through inhibition of both RET phosphorylation and the mammalian target of rapamycin pathway. Surgery 2012, 6, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- You, W.K.; Sennino, B.; Williamson, C.W.; Falcón, B.; Hashizume, H.; Yao, L.C.; Aftab, D.T.; McDonald, D.M. VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer. Res. 2011, 14, 4758–4768. [Google Scholar] [CrossRef] [PubMed]
- Dayyani, F.; Gallick, G.E.; Logothetis, C.J.; Corn, P.G. Novel therapies for metastatic castrate-resistant prostate cancer. J. Natl. Cancer Inst. 2011, 22, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Hoelder, S.; Clarke, P.A.; Workman, P. Discovery of small molecule cancer drugs: Successes, challenges and opportunities. Mol. Oncol. 2012, 2, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wu, C.; Xu, S.; Li, W.; Fang, H.; Chen, Z.; Tu, X.; Zheng, P. Design, Synthesis, and Biological Evaluation of Sorafenib Analogs Bearing a Sulfonylurea Unit as Novel VEGFR2 Kinase Inhibitors. Adv. Mater. Res. 2014, 1048–1051. [Google Scholar] [CrossRef]
- Tang, Q.; Zhao, Y.; Du, X.; Chong, L.; Gong, P.; Guo, C. Design, synthesis, and structure-activity relationships of novel 6,7-disubstituted-4-phenoxyquinoline derivatives as potential antitumor agents. Eur. J. Med. Chem. 2013, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Zhang, G.; Du, X.; Zhu, W.; Li, R.; Lin, H.; Li, P.; Cheng, M.; Gong, P.; Zhao, Y. Discovery of novel 6,7-disubstituted-4-phenoxyquinoline derivatives bearing 5-(aminomethylene) pyrimidine-2,4,6-trione moiety as c-Met kinase inhibitors. Bioorg. Med. Chem. 2014, 4, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, C.; Sun, C.; Lei, F.; Fang, H.; Zheng, P.; Luo, R.; Zhu, W. Synthesis of Substituted Ethyl Argiosulfonylcarbamates. Adv. Mater. Res. 2014. [Google Scholar] [CrossRef]
- Wang, C.; Gao, H.; Dong, J.; Zhang, Y.; Su, P.; Shi, Y.; Zhang, J. Biphenyl derivatives incorporating urea unit as novel VEGFR–2 inhibitors: Design, synthesis and biological evaluation. Bioorg. Med. Chem. 2014, 1, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, R.M.; Huang, W.; Mehrotra, M.; Zhang, X.; Cannon, H.; Grant, C.M. Preparation of [4-(6-halo-7-substituted-2,4-dioxo-1,4-dihydro-2H-quinazolin-3-yl)-phenyl]-5-chloro-thiophen-2-yl-sulfonylureas as ADP receptor inhibitors for treatment of cardiovascular diseases. WO. 2007056219A2, 18 May 2007. [Google Scholar]
- Tsai, H.J.; Chou, S.Y.; Chuang, S.H.; Chen, C.C.; Hsu, F.L. D-420720, a novel orally active sulfonamide compound dipeptidylpeptidase inhibitor: Structure and activity relationship of arylsulfonamide to dipeptidylpeptidase inhibitor. Drug Dev. Res. 2008, 8, 514–519. [Google Scholar] [CrossRef]
- Roper, J.; Richardson, M.P.; Wang, W.V.; Richard, L.G.; Chen, W.; Coffee, E.M.; Sinnamon, M.J.; Lee, L.; Chen, P.C.; Bronson, R.T.; et al. The dual PI3K/mTOR inhibitor NVP-BEZ235 induces tumor regression in a genetically engineered mouse model of PIK3CA wild-type colorectal cancer. PLoS ONE 2011, 9, e25132. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 7448, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 6a–f and 9a–e are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.; Wang, M.; Tang, Q.; Luo, R.; Chen, L.; Zheng, P.; Zhu, W. Design, Synthesis, Activity and Docking Study of Sorafenib Analogs Bearing Sulfonylurea Unit. Molecules 2015, 20, 19361-19371. https://doi.org/10.3390/molecules201019361
Wu C, Wang M, Tang Q, Luo R, Chen L, Zheng P, Zhu W. Design, Synthesis, Activity and Docking Study of Sorafenib Analogs Bearing Sulfonylurea Unit. Molecules. 2015; 20(10):19361-19371. https://doi.org/10.3390/molecules201019361
Chicago/Turabian StyleWu, Chunjiang, Min Wang, Qidong Tang, Rong Luo, Le Chen, Pengwu Zheng, and Wufu Zhu. 2015. "Design, Synthesis, Activity and Docking Study of Sorafenib Analogs Bearing Sulfonylurea Unit" Molecules 20, no. 10: 19361-19371. https://doi.org/10.3390/molecules201019361
APA StyleWu, C., Wang, M., Tang, Q., Luo, R., Chen, L., Zheng, P., & Zhu, W. (2015). Design, Synthesis, Activity and Docking Study of Sorafenib Analogs Bearing Sulfonylurea Unit. Molecules, 20(10), 19361-19371. https://doi.org/10.3390/molecules201019361