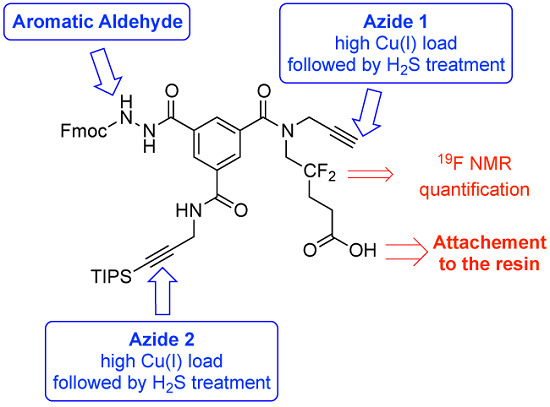
3.2. Syntheses
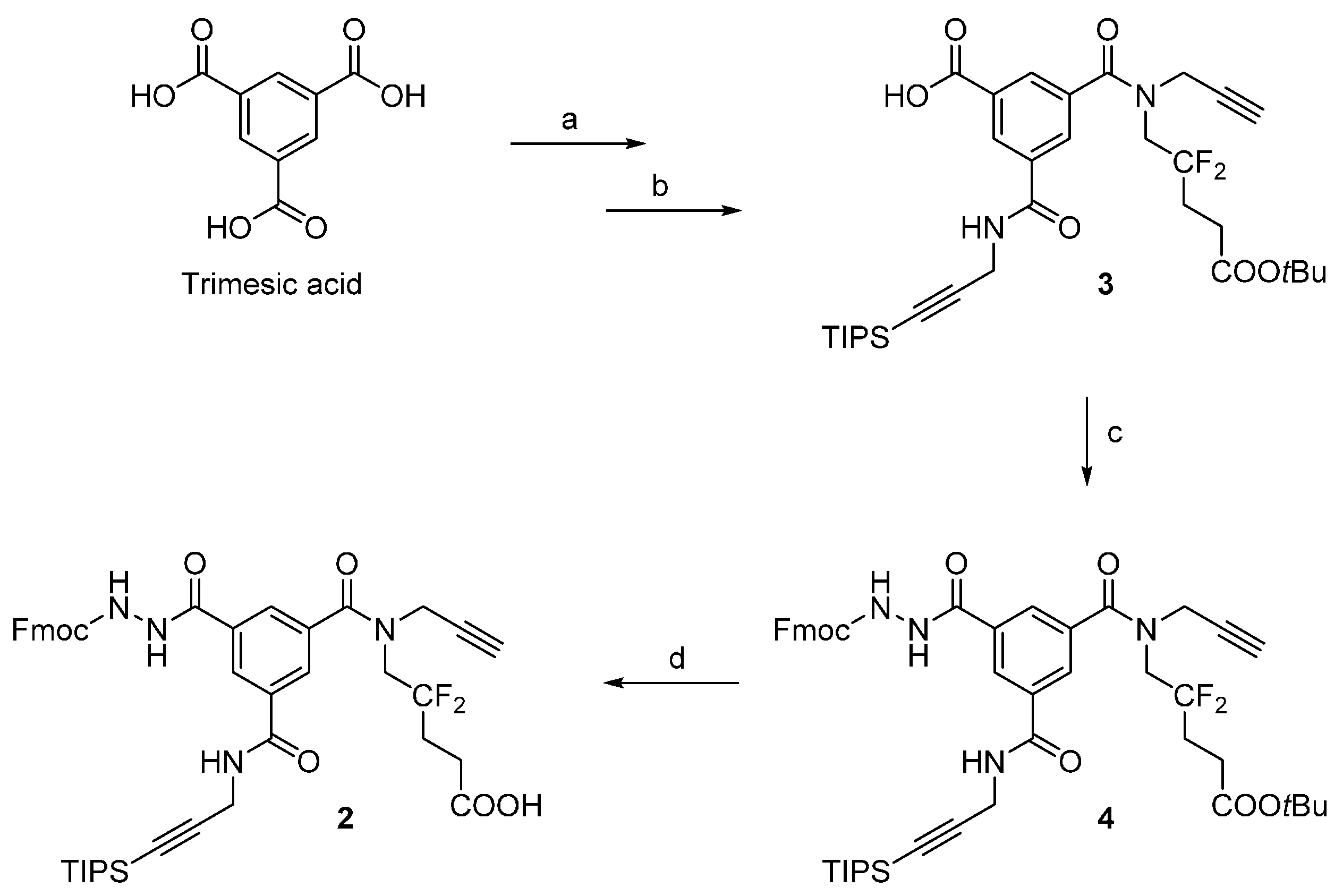
3.2.1. Compound 4
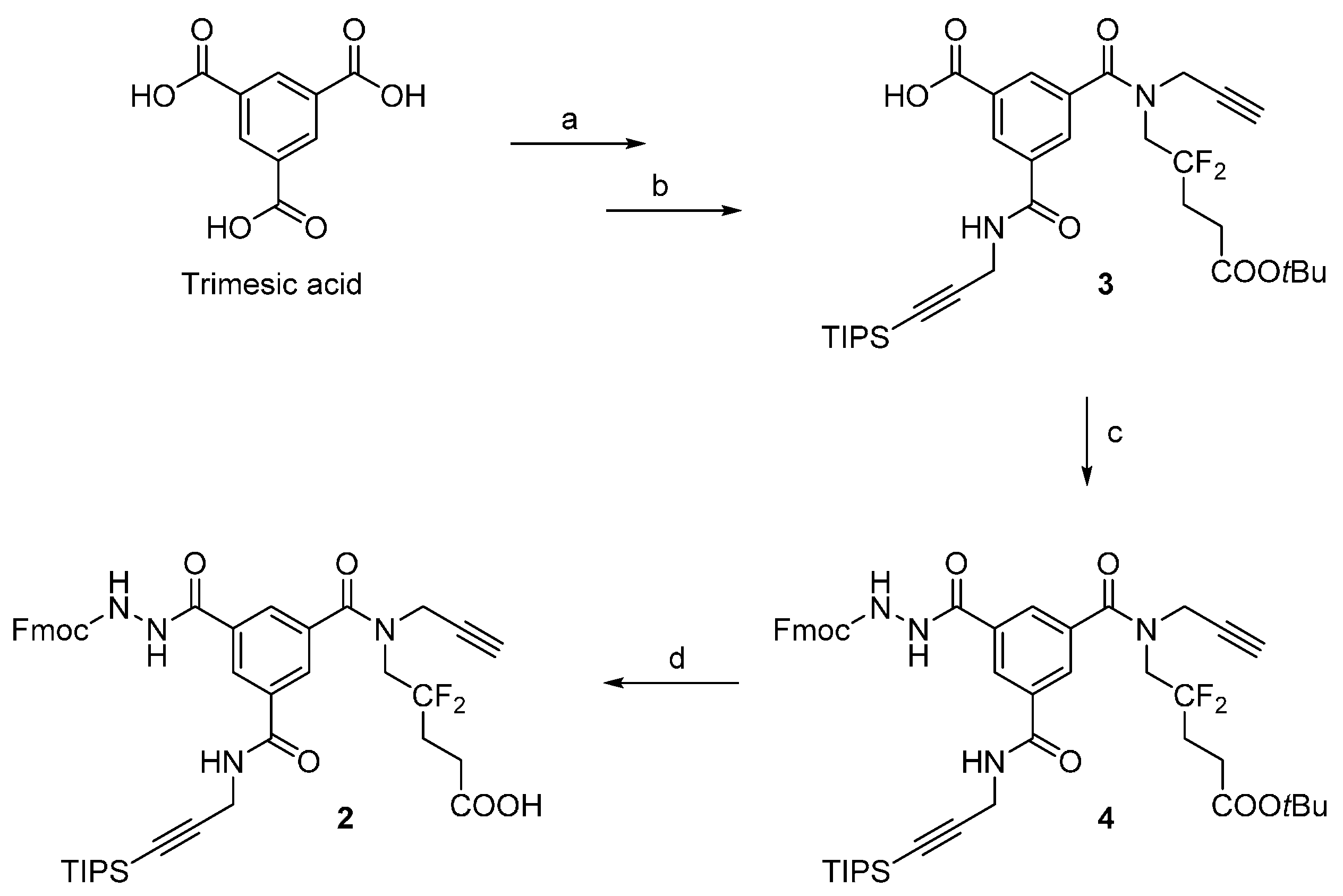
A suspension of compound
3 [
4] (5.6 g, 8.8 mmol), PyBrop (6.2 g, 13.3 mmol, 1.5 eq.), Fmoc-hydrazine [
7] (6.7 g, 26.3 mmol, 3.0 eq.) and DIPEA (3.08 mL, 17.7 mmol, 2.0 eq.) in ACN (150 mL) was stirred at 60 °C. After 3 h, the reaction was cooled to r.t. and then filtered to remove excess Fmoc-hydrazine. The filtrate was evaporated, suspended in EtOAc (60 mL), and filtered again to remove the remaining Fmoc-hydrazine. The EtOAc filtrate was poured into a separatory funnel, washed a saturated NH
4Cl solution (2 × 30 mL) and brine (50 mL), dried over anhydrous sodium sulfate, filtered, and evaporated to give 8.5 g of brown oil. The crude product was purified by flash chromatography on silica (200 g of silica, elution with a linear gradient—5% to 65%—of EtOAc in toluene), resulting in compound
4 as a pale yellow solid (4.5 g, 58%, 57%–67% in repeated runs; R
f = 0.3 in 7:3 toluene/EtOAc).
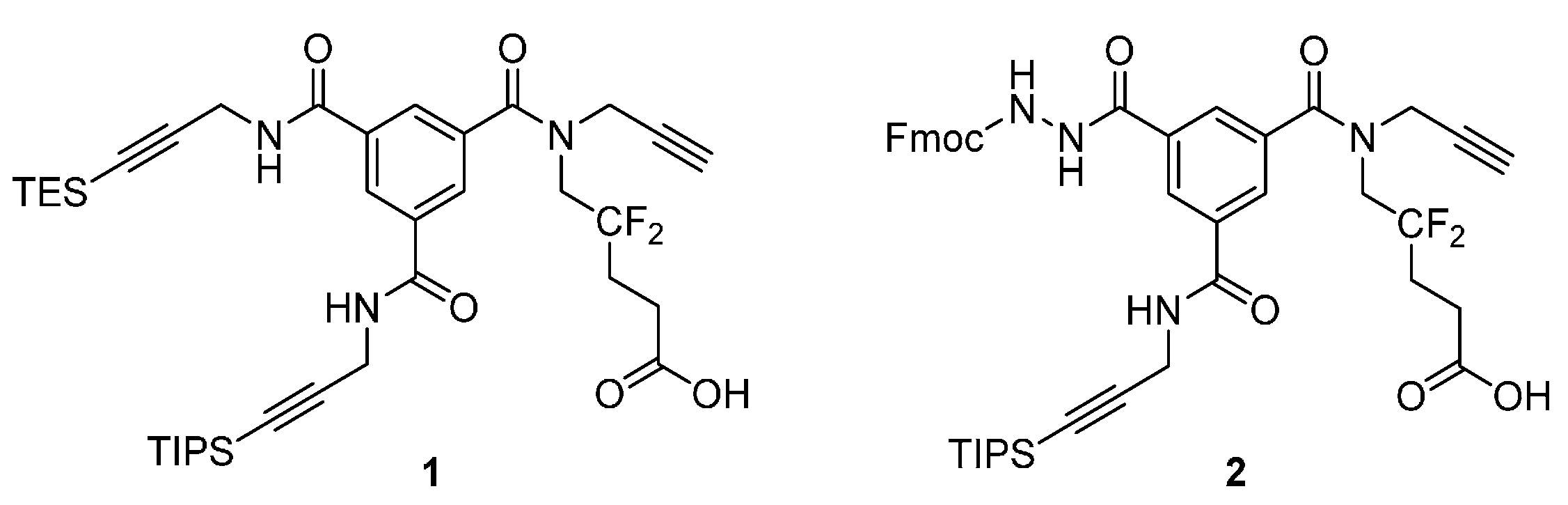
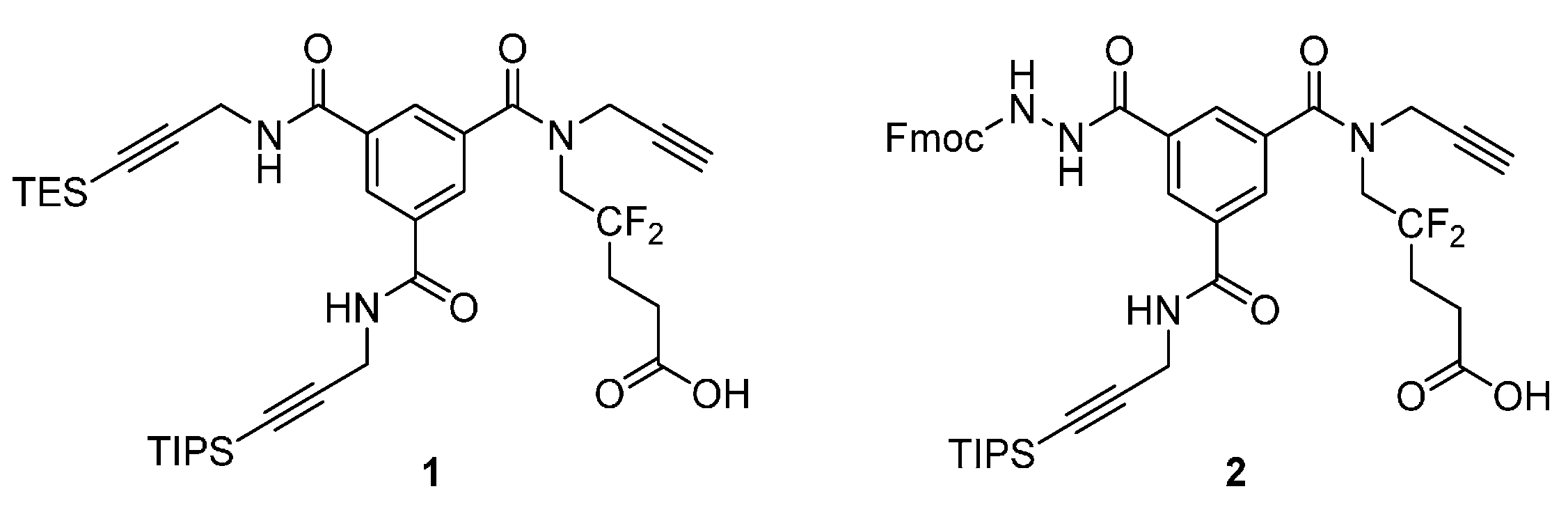
Analytical HPLC: (Method A) tR = 24.5 min. 1H-NMR (500 MHz; DMSO-d6; T = 100 °C): δH 1.06 (21H, m, Si(CH(CH3)2)3), 1.41 (9H, s, C(CH3)3), 2.22 (2H, m, –CH2–), 2.38 (2H, t, 3JHH = 7.5 Hz, –CH2–CO), 3.12 (1H, t, 4JHH = 2.4 Hz, –C≡CH), 4.03 (2H, t, 3JHF = 14.2 Hz, –CH2–N), 4.18 (2H, d, 3JHH = 5.6 Hz, –CH2–N), 4.25 (2H, d, 4JHH = 2.4 Hz, –CH2–N), 4.27 (1H, t, 3JHH = 7.0 Hz, >CH– Fmoc), 4.40 (2H, d, 3JHH = 7.0 Hz, –CH2–O Fmoc), 7.30 (2H, t, 3JHH = 7.5 Hz, 2 × CHarom), 7.40 (2H, t, 3JHH = 7.5 Hz, 2 × CHarom), 7.70 (2H, d, 3JHH = 7.5 Hz, 2 × CHarom), 7.84 (2H, d, 3JHH = 7.5 Hz, 2 × CHarom), 8.04 (1H, t, 3JHH = 1.5 Hz, CHarom), 8.05 (1H, t, 3JHH = 1.5 Hz, CHarom), 8.45 (1H, t, 3JHH = 1.5 Hz, CHarom), 8.87 (1H, t, 3JHH = 5.6 Hz, >NH). 13C-NMR (125.8 MHz; DMSO-d6; T = 100 °C): δC 11.04 (3C, Si(CH(CH3)2)3)), 18.56 (6C, Si(CH(CH3)2)3), 27.99 (3C, –C(CH3)3), 28.22 (1C, –CH2–), 29.94 (1C, –CH2–N), 30.26 (1C, t, 2JCF = 24.4 Hz, –CH2–), 39.40 (1C, –CH2–N), 47.05 (1C, >CH– Fmoc), 49.45 (1C, –CH2–N), 66.75 (1C, –CH2–O Fmoc), 75.72 (1C, ≡C–H), 78.60 (1C, ≡C–), 80.40 (1C, C(CH3)3), 82.36 (1C, ≡C–), 105.64 (1C, ≡C–), 120.13 (2C, 2 × CHarom), 123.47 (1C, t, 1JCF = 244.5 Hz, >CF2), 125.34 (2C, 2 × CHarom), 127.20 (2C, 2 × CHarom), 127.79 (2C, 2 × CHarom), 128.19 (1C, CHarom), 128.21 (1C, CHarom), 128.56 (1C, CHarom), 133.68 (1C, Carom), 135.38 (1C, Carom), 135.88 (1C, Carom), 141.02 (2C, Carom), 143.94 (2C, Carom), 156.32 (1C, –CO–NH), 164.96 (1C, –CO–NH), 170.39 (1C, –CO–NH), 170.74 (1C, –CO–O) (–CO–N< missing). IR (KBr) νmax = 3420 (m, vbr, NH), 3306 (m, C≡C-H), 2944 (s, CH3 TIPS), 2177 (w, C≡C), 1731, 1657, 1526 (s, br, C=O). HRMS (m/z): calcd for C48H58F2N4NaO7Si+ [M + Na]+ 891.39350, found 891.39383.
3.2.2. Scaffold 2
TFA (10 mL) was added to a solution of 4 (4.4 g, 5.1 mmol) in DCM (10 mL) at 0 °C. The reaction mixture was allowed to reach r.t. and was stirred for 1 h. The reaction mixture was co-evaporated with toluene to afford 5.6 g of brownish foam. The crude product was purified by flash chromatography on silica (200 g of silica, elution with a linear gradient—0% to 60% over 40 min—of 1% AcOH/EtOAc (v/v) in toluene), resulting in compound 2 as a beige solid (3.6 g, 87%, 87%–91% in repeated runs; Rf = 0.3 in 1% AcOH/EtOAc (v/v) in toluene 1:1).
Analytical HPLC: (Method A) tR = 17.9 min. 1H-NMR (500 MHz; DMSO-d6; T = 100 °C): δH 1.06 (21H, m, Si(CH(CH3)2)3), 2.24 (2H, m, –CH2–), 2.42 (2H, t, 3JHH = 7.5 Hz, –CH2–CO), 3.12 (1H, t, 4JHH = 2.4 Hz, –C≡CH), 4.04 (2H, t, 3JHF = 14.5 Hz, –CH2–N), 4.18 (2H, d, 3JHH = 5.6 Hz, –CH2–N), 4.25 (2H, d, 4JHH = 2.4 Hz, –CH2–N), 4.27 (1H, t, 3JHH = 7.0 Hz, >CH– Fmoc), 4.40 (2H, d, 3JHH = 7.0 Hz, –CH2–O Fmoc), 7.30 (2H, t, 3JHH = 7.5 Hz, 2 × CHarom), 7.40 (2H, t, 3JHH = 7.5 Hz, 2 × CHarom), 7.71 (2H, d, 3JHH = 7.5 Hz, 2 × CHarom), 7.84 (2H, d, 3JHH = 7.5 Hz, 2 × CHarom), 8.04 (1H, t, 3JHH = 1.5 Hz, CHarom), 8.06 (1H, t, 3JHH = 1.5 Hz, CHarom), 8.44 (1H, t, 3JHH = 1.5 Hz, CHarom), 8.88 (1H, t, 3JHH = 5.6 Hz, >NH). 13C-NMR (125.8 MHz; DMSO-d6; T = 100 °C): δC 10.50 (3C, Si(CH(CH3)2)3)), 18.03 (6C, Si(CH(CH3)2)3), 26.21 (1C, t, 3JCF = 5.2 Hz, –CH2–), 29.40 (1C, –CH2–N), 29.74 (1C, t, 2JCF = 24.2 Hz, –CH2–), 38.81 (1C, –CH2–N), 46.52 (1C, >CH– Fmoc), 66.21 (1C, –CH2–O Fmoc), 75.21 (1C, ≡C–H), 78.10 (1C, ≡C–), 81.83 (1C, ≡C–), 105.12 (1C, ≡C–), 119.60 (2C, 2 × CHarom), 124.00 (1C, t, 1JCF = 244.5 Hz, >CF2), 124.81 (2C, 2 × CHarom), 126.66 (2C, 2 × CHarom), 127.25 (2C, 2x CHarom), 127.64 (1C, CHarom), 127.68 (1C, CHarom), 128.02 (1C, CHarom), 133.14 (1C, Carom), 134.84 (1C, Carom), 135.35 (1C, Carom), 140.49 (2C, 2 × Carom), 143.41 (2C, 2 × Carom), 155.79 (1C, –CO–NH), 164.43 (1C, –CO–NH), 169.86 (1C, –CO–NH), 172.19 (1C, –COOH) (–CO–N< and one –CH2–N missing). IR (KBr) νmax = 3294 (m, br, C≡C-H and NH), 2944 (s, CH3 TIPS), 2178 (w, C≡C), 1723, 1657, 1528 (s, br, C=O). HRMS (m/z): calcd for C44H49F2N4O7Si− [M – H]− 811.33441, found 811.33420.
3.2.3. Compound 5
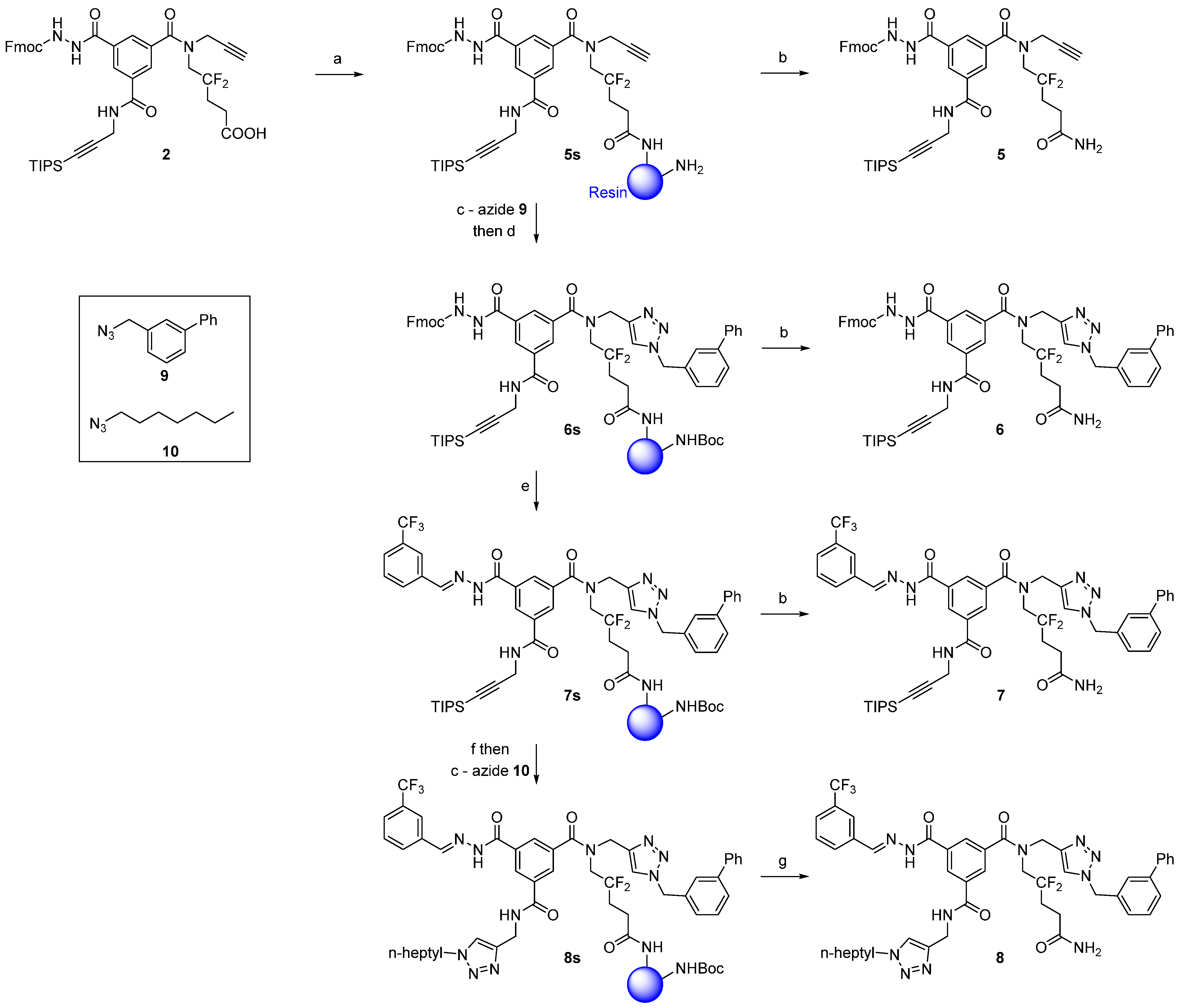
Ramage ChemMatrix® resin (667 mg, 200 μmol, 0.3 mmol/g, Aldrich 727792) with free amino groups was swelled in MeOH, DCM, and ACN (10 minutes each, using 10 mL of solvent/g of resin) in a fritted polypropylene syringe. The resin was then washed with 3 × 5 mL ACN (1 min each). A solution of scaffold 2 (82 mg, 100 μmol), PyBroP (93 mg, 200 μmol, 2.0 eq.), and DIPEA (26 μL, 150 μmol, 1.5 eq.) in ACN (2.5 mL) was added to the resin. The resulting mixture was stirred at r.t. for 6 h. The resin was washed as described for the determination of the loading of compound 2 (vide-infra) and with 3 × 5 mL of MeOH and DCM (1 min each) to afford resin-bound compound 5s.
A small sample of resin-bound compound 5s (ca. 10% of the total amount) was transferred to a fritted glass micro-reactor and dried under reduced pressure. The product was cleaved from the resin with a 5% TFA/DCM (v/v) solution (5 mL) for 30 minutes. The resin was subsequently slowly washed with 3 × 5 mL of 5% TFA/DCM. All solutions were combined and evaporated to dryness. It is recommended that the cleavage procedure be repeated until a constant weight of the dried compound is reached. Typically, two TFA cleavage procedures were sufficient to recover all of the material from the resin. Compound 5 (12 mg) was purified by RP-HPLC (Method A) and lyophilized (5.1 mg, white powder).
Analytical HPLC: (Method A) tR = 14.1 min. 1H-NMR (500 MHz; DMSO-d6; T = 100 °C): δH 1.05 (21H, m, Si(CH(CH3)2)3), 2.19 (2H, m, –CH2–), 2.28 (2H, br t, 3JHH = 7.5 Hz, –CH2–CO), 3.11 (1H, t, 4JHH = 2.4 Hz, –C≡CH), 4.02 (2H, t, 3JHF = 14.5 Hz, –CH2–N), 4.18 (2H, d, 3JHH = 5.5 Hz, –CH2–N), 4.24 (2H, br d, 4JHH = 2.4 Hz, –CH2–N), 4.26 (1H, t, 3JHH = 6.8 Hz, >CH– Fmoc), 4.40 (2H, d, 3JHH = 6.8 Hz, –CH2–O Fmoc), 7.30 (2H, t, 3JHH = 7.5 Hz, 2 × CHarom), 7.40 (2H, t, 3JHH = 7.5 Hz, 2 × CHarom), 7.70 (2H, d, 3JHH = 7.5 Hz, 2 × CHarom), 7.84 (2H, d, 3JHH = 7.5 Hz, 2 × CHarom), 8.02 (1H, t, 3JHH = 1.5 Hz, CHarom), 8.04 (1H, t, 3JHH = 1.5 Hz, CHarom), 8.42 (1H, t, 3JHH = 1.5 Hz, CHarom), 8.88 (1H, br t, 3JHH = 5.6 Hz, >NH), 9.08 (1H, br, >NH), 10.33 (1H, br, >NH). 13C-NMR (125.8 MHz; DMSO-d6; T = 100 °C): δC 10.55 (3C, Si(CH(CH3)2)3)), 18.09 (6C, Si(CH(CH3)2)3), 27.14 (1C, –CH2–), 29.47 (1C, –CH2–N), 30.04 (1C, t, 2JCF = 24.6 Hz, –CH2–), 38.75 (1C, –CH2–N), 46.56 (1C, >CH– Fmoc), 48.81 (1C, –CH2–N), 66.28 (1C, –CH2–O Fmoc), 75.32 (1C, ≡C–H), 78.15 (1C, ≡C–), 81.91 (1C, ≡C–), 105.15 (1C, ≡C–), 119.67 (2C, 2 × CHarom), 123.30 (1C, t, 1JCF = 244.5 Hz, >CF2), 124.87 (2C, 2 × CHarom), 126.74 (2C, 2 × CHarom), 127.34 (2C, 2 × CHarom), 127.72 (2C, 2 × CHarom), 128.09 (1C, CHarom), 133.19 (1C, Carom), 134.90 (1C, Carom), 135.46 (1C, Carom), 140.54 (2C, 2 × Carom), 143.45 (2C, 2 × Carom), 155.87 (1C, –CO–NH), 164.54 (1C, –CO–NH), 169.90 (1C, –CO–NH), 172.26 (1C, –CO–NH2) (–CO–N< missing). HRMS (m/z): calcd for C44H51F2N5NaO6Si+ [M + Na]+ 834.34689, found 834.34718.
3.2.4. Compound 6
The resin
5s was washed with 3 × 5 mL of MeOH and
tBuOH/water (1:1,
v/
v) (1 min each). The following solutions were sequentially added to the resin: (i) azide
9 (105 mg, 500 μmol, 5.0 eq., refer to the
Supporting Information for the synthesis) in 1 mL of
tBuOH/water (1:1,
v/
v); (ii) sodium ascorbate (100 μL of a freshly prepared 0.5 M aqueous solution, 50 μmol, 0.5 eq.) in 1 mL of
tBuOH/water (1:1,
v/
v); and (iii) copper(II) sulfate pentahydrate (100 μL of a freshly prepared 0.1 M aqueous solution, 10 μmol, 0.1 eq.) in 1 mL of
t-BuOH/water (1:1,
v/
v). After stirring for 16 h at r.t., the coupling was complete. The resin was washed with 3 × 5 mL of
tBuOH/water (1:1,
v/
v), DMF, DCM, MeOH and DCM (1 min each) to afford the resin-bound compound
6s.
A small sample of resin-bound compound 6s (ca. 10% of the total amount) was transferred to a fritted glass micro-reactor and dried under reduced pressure. The product was cleaved using the procedure described for compound 5 to give crude compound 6 (ca. 18 mg). Compound 6 was purified by RP-HPLC (Method A) and lyophilized (7.3 mg, white powder).
Analytical HPLC: (Method A) tR = 17.4 min. 1H-NMR (500 MHz; DMSO-d6; T = 100 °C): δH 1.05 (21H, m, Si(CH(CH3)2)3), 2.18 (2H, m, –CH2–), 2.26 (2H, m, –CH2–CO), 3.93 (2H, t, JHF = 14.3 Hz, –CH2–N), 4.17 (2H, d, 3JHH = 5.6 Hz, –CH2–N), 4.26 (1H, t, 3JHH = 7.0 Hz, >CH– Fmoc), 4.39 (2H, d, 3JHH = 7.0 Hz, –CH2–O Fmoc), 4.70 (2H, s, –CH2–N), 5.62 (2H, s, –CH2–N), 7.24 (1H, d, 3JHH = 7.5 Hz, CHarom), 7.29 (2H, t, 3JHH = 7.5 Hz, 2 × CHarom), 7.35 (1H, tt, 3JHH = 7.5 Hz, 4JHH = 1.2 Hz, CHarom), 7.39 (2H, t, 3JHH = 7.5 Hz, 2 × CHarom), 7.44 (3H, t, 3JHH = 7.5 Hz, 3 × CHarom), 7.59 (4H, m, 4 × CHarom), 7.695 (2H, d, 3JHH = 7.5 Hz, 2 × CHarom), 7.84 (2H, d, 3JHH = 7.5 Hz, 2 × CHarom), 8.00 (1H, br s, CHarom), 8.04 (1H, br s, CHarom), 8.05 (1H, br s, CHarom), 8.40 (1H, br s, CHarom), 8.86 (1H, t, 3JHH = 5.6 Hz, >NH), 9.10 (1H, br, >NH), 10.32 (1H, br, >NH). 13C-NMR (125.8 MHz; DMSO-d6; T = 100 °C): δC 10.50 (3C, Si(CH(CH3)2)3)), 18.04 (6C, Si(CH(CH3)2)3), 27.22 (1C, –CH2–), 29.41 (1C, –CH2–N), 30.09 (1C, –CH2–), 43.89 (1C, –CH2–N), 45.51 (1C, >CH– Fmoc), 48.56 (1C, –CH2–N), 52.75 (1C, –CH2–N), 66.22 (1C, –CH2–O Fmoc), 81.83 (1C, ≡C–), 105.12 (1C, ≡C–), 119.61 (2C, CHarom), 123.42 (1C, CHarom), 124.82 (2C, 2 × CHarom), 126.01 (1C, CHarom), 126.10 (1C, CHarom), 126.34 (2C, 2 × CHarom), 126.46 (1C, CHarom), 126.67 (2C, 2 × CHarom), 127.20 (1C, CHarom), 127.27 (2C, 2 × CHarom), 127.38 (1C, CHarom), 127.82 (1C, CHarom), 128.26 (1C, CHarom), 128.48 (2C, 2 × CHarom), 128.97 (1C, CHarom), 134.76 (1C, Carom), 136.01 (1C, Carom), 136.10 (1C, Carom), 139.52 (1C, Carom), 140.49 (2C, 2 × Carom), 140.55 (1C, Carom), 143.40 (2C, 2 × Carom), 155.82 (1C, –CO–NH), 164.55 (1C, –CO–NH), 170.18 (1C, –CO–NH), 172.01 (1C, –CO–NH2) (–CO–N< and two Carom missing). HRMS (m/z): calcd for C57H62F2N8NaO6Si+ [M + Na]+ 1043.44219, found 1043.44263.
3.2.5. Compound 7
- (1)
Capping: The resin 6s was washed with 3 × 5 mL of DCM (1 min each). A cooled solution (0 °C) of Boc2O (ca. 220 mg, ca. 1 mmol, ca. 10 eq.) and DIPEA (35 μL, 200 μmol, 2.0 eq.) in DCM (4 mL) was added to the resin. The resin was left upside down (to avoid projection) at r.t. for 3 h. The resin was washed with 3 × 5 mL of DCM, DMF, DCM and MeOH (1 min each). A Kaiser’s test performed on a few grains of resin confirmed complete capping (negative test).
- (2)
One-step Fmoc-deprotection/hydrazone formation: The Boc-capped resin was washed with 3 × 5 mL of DCM and ACN (1 min each). A solution of 3-(trifluoromethyl)benzaldehyde (67 μL, 500 μmol, 5.0 eq.) in 10% TEA/ACN (v/v, 2.5 mL) was added to the resin. The mixture was stirred at r.t. for 16 h. The resin was then washed with 3 × 5 mL of ACN, DMF, DCM, MeOH, and DCM (1 min each) to afford resin-bound compound 7s.
A small sample of resin-bound compound
7s (
ca. 30% of the total amount) was transferred to a fritted glass micro-reactor and dried under reduced pressure. The product was cleaved using the procedure described for compound
5 to give 24 mg of yellow oil. Compound
7 partially hydrolyzed in the HPLC buffer (0.1% TFA, pH ≈ 2) and was thus purified as follows. First, the low-polarity impurities were extracted from the crude compound
7 with 2 min of sonication in 10 mL of Et
2O. After centrifugation (7200 rpm, 5 min at 20 °C), the supernatant was discarded. The procedure was repeated once, and the final residue was dried to give 16 mg of a pale-yellow solid. Then, the crude product was passed through a C-4 cartridge to remove salts (particularly any remaining copper). Briefly, the crude compound was dissolved in a 30% aqueous AcOH solution (2 mg/mL solution) and loaded onto an activated C-4 cartridge (Chromabond C-4, 3 mL/500 mg). The cartridge was then washed with a 10% aqueous AcOH solution (3 × 2 mL) and water (3 × 2 mL). The product was then eluted with ACN (5 × 2 mL) and lyophilized to give
7 (10 mg, white powder). The purity of crude compound
7 was verified by RP-HPLC (Method A,
Supplementary Figure S5). This crude compound was directly used for NMR and MS analyses.
Analytical HPLC: (Method A) tR = 17.7 min. 1H-NMR (500 MHz; DMSO-d6; T = 100 °C): δH 1.05 (21H, m, Si(CH(CH3)2)3), 2.18 (2H, m, –CH2–), 2.26 (2H, m, –CH2–CO), 3.94 (2H, t, JHF = 14.2 Hz, –CH2–N), 4.18 (2H, d, 3JHH = 5.6 Hz, –CH2–N), 4.72 (2H, s, –CH2–N), 5.64 (2H, s, –CH2–N), 7.25 (1H, d, 3JHH = 7.5 Hz, CHarom), 7.35 (1H, tt, 3JHH = 7.5 Hz, 4JHH = 1.2 Hz, CHarom), 7.44 (3H, t, 3JHH = 7.5 Hz, 3 × CHarom), 7.59 (4H, m, 4 × CHarom), 7.68 (1H, t, 3JHH = 7.6 Hz, CHarom), 7.76 (1H, d, 3JHH = 7.6 Hz, CHarom), 7.99 (1H, d, 3JHH = 7.6 Hz, CHarom), 8.02 (2H, br s, 2 × CHarom), 8.07 (1H, br s, CHarom), 8.11 (1H, br s, CHarom), 8.45 (1H, t, 3JHH = 1.6 Hz, CHarom), 8.56 (1H, s, –N=CH), 8.91 (1H, t, 3JHH = 5.6 Hz, >NH), 11.89 (1H, br s, >NH) (one CH2–N missing). 13C-NMR (125.8 MHz; DMSO-d6; T = 100 °C): δC 10.49 (3C, Si(CH(CH3)2)3)), 18.03 (6C, Si(CH(CH3)2)3), 27.10 (1C, –CH2–), 29.40 (1C, –CH2–N), 30.08 (1C, –CH2–), 44.00 (1C, –CH2–N), 52.76 (1C, –CH2–N), 81.82 (1C, ≡C–), 105.11 (1C, ≡C–), 122.82 (1C, CHarom), 123.46 (1C, CHarom), 126.01 (1C, CHarom), 126.09 (1C, CHarom), 126.32 (3C, 3 × CHarom), 126.43 (1C, CHarom), 127.18 (1C, CHarom), 127.74 (1C, CHarom), 128.11 (1C, CHarom), 128.47 (2C, 2 × CHarom), 128.96 (1C, CHarom), 129.57 (1C, CHarom), 130.56 (1C, CHarom), 139.50 (2C, 2 × Carom), 140.55 (1C, Carom), 164.56 (1C, –CO–NH), 170.20 (1C, –CO–NH) (–CO–N<, –CO–NH and eight Carom missing). HRMS (m/z): calcd for C50H56F5N8O4Si+ [M + H]+ 955.41085, found 955.41159.
3.2.6 Compound 8 (Low Copper Load)
- (1)
TIPS deprotection: The resin 7s was washed with 3 × 5 mL of DMF (1 min each). A solution of TBAF (500 μL of a 1 M solution in DMF, 500 μmol, 5.0 eq.) in DMF (2 mL) was then added to the resin. Three treatments at r.t. for 1.5 h each were necessary to completely remove the TIPS group. Between each treatment, the resin was washed with 3 × 5 mL of DMF (1 min each). The resin was finally washed with 3 × 5 mL of DMF, glacial AcOH, water, DMF, DCM, MeOH, and DCM (1 min each).
- (2)
Final CuAAC: The resin (terminal-alkyne-bound compound) was washed with 3×5 mL of MeOH and
tBuOH/water (1:1,
v/
v) (1 min each). The following solutions were sequentially added to the resin: (i) 1-azidoheptane
10 (71 mg, 500 μmol, 5.0 eq., refer to
Supplementary Materials for the synthesis) in 1 mL of
tBuOH/water (1:1,
v/
v); (ii) sodium ascorbate (100 μL of a freshly prepared 0.5 M aqueous solution, 50 μmol, 0.5 eq.) in 1 mL of
tBuOH/water (1:1,
v/
v); and (iii) copper(II) sulfate pentahydrate (100 μL of a freshly prepared 0.1 M aqueous solution, 10 μmol, 0.1 eq.) in 1 mL of
t-BuOH/water (1:1,
v/
v). Two couplings of 16 h and 4 h at r.t. were necessary for the reaction to reach completion. The resin was washed with 3 × 5 mL of
tBuOH/water (1:1,
v/
v), DMF, DCM, MeOH and DCM (1 min each) to afford resin-bound compound
8s.
Compound
8 was also prepared as described above without partial TFA cleavages to determine the yield of the entire synthesis. The synthesis was performed on a 100 μmol scale (using 100 μmol of scaffold
2 and 200 μmol of the Ramage ChemMatrix resin). After the final TFA cleavage, 57 mg of crude compound
8 were obtained. The crude compound was extracted twice with Et
2O and passed through a C-4 cartridge, as explained for compound
7, to afford compound
8 (23 mg, pale yellow solid, overall yield from compound
2 of 24%). The purity of compound
8 was verified by RP-HPLC (Method B,
Supplementary Figure S6). Crude compound
8 was also analyzed by elemental analysis to check for the presence of residual copper, and
19F-NMR was used to quantify the amount of compounds containing the CF
2 arm present in the crude product (
vide infra).
Analytical HPLC: (Method B) tR = 22.4 min, purity = 71%. 1H-NMR (500 MHz; DMSO-d6; T = 100 °C): δH 0.85 (3H, t, 3JHH = 7.0 Hz, –CH3), 1.25–1.29 (8H, m, 4 × –CH2–), 1.82 (2H, m, –CH2–), 2.18 (2H, m, –CH2–), 2.26 (2H, m, –CH2–CO), 3.94 (2H, t, JHF = 14.2 Hz, –CH2–N), 4.30 (2H, t, 3JHH = 7.2, –CH2–N), 4.57 (2H, d, 3JHH = 5.6 Hz, –CH2–N), 4.71 (2H, s, –CH2–N), 5.64 (2H, s, –CH2–N), 7.25 (1H, d, 3JHH = 7.5 Hz, CHarom), 7.35 (1H, tt, 3JHH = 7.4 Hz, 4JHH = 1.2 Hz, CHarom), 7.44 (3H, m, 3 × CHarom), 7.60 (4H, m, 4 × CHarom), 7.68 (1H, t, 3JHH = 7.5 Hz, CHarom), 7.76 (1H, d, 3JHH = 7.5 Hz, CHarom), 7.895 (1H, s, CHarom), 7.99 (1H, d, 3JHH = 7.5 Hz, CHarom), 8.025 (1H, s, CHarom), 8.03 (1H, s, CHarom), 8.10 (1H, br s, CHarom), 8.11 (1H, br s, CHarom), 8.48 (1H, t, 4JHH = 1.6 Hz, CHarom), 8.57 (1H, s, –N=CH), 8.94 (1H, t, 3JHH = 5.6 Hz, >NH), 11.88 (1H, br s, >NH). 13C-NMR (125.8 MHz; DMSO-d6; T = 100 °C): δC 13.23 (1C, –CH3), 21.42 (1C, –CH2–), 25.50 (1C, –CH2–), 27.10 (1C, –CH2–), 27.55 (1C, –CH2–), 29.24 (2C, 2 × –CH2–), 30.08 (1C, –CH2–), 30.59 (1C, –CH2–), 34.90 (1C, –CH2–N), 43.90 (1C, –CH2–N), 48.73 (1C, –CH2–N), 49.06 (1C, –CH2–N), 52.75 (1C, –CH2–N), 122.32 (1C, CHarom), 122.84 (1C, CHarom), 123.50 (1C, CHarom), 126.00 (1C, CHarom), 126.08 (2C, 2 × CHarom), 126.32 (2C, 2 × CHarom), 126.43 (1C, CHarom), 127.18 (1C, CHarom), 128.14 (1C, CHarom), 128.47 (3C, 3 × CHarom), 128.96 (1C, CHarom), 129.57 (1C, CHarom), 130.57 (1C, CHarom), 133.75 (1C, Carom), 135.31 (1C, Carom), 135.85 (1C, Carom), 136.09 (1C, Carom), 139.51 (1C, Carom), 140.55 (1C, Carom), 142.31 (1C, Carom), 144.25 (1C, Carom), 164.77 (1C, –CO–NH), 165.79 (1C, –CO–NH), 170.24 (1C, –CO–NH) (–CO–N< and four Carom missing). IR (KBr) νmax = 3433 (s, vbr, NH + H2O), 1664 (s, br, C=O). HRMS (m/z): calcd for C48H50F5N11NaO4+ (M + Na)+ 962.38596, found 962.38613. Elemental analysis (Cu): 97–103 ppm.
In a parallel experiment, we prepared 8 by the same methodology as described above, but, before the final cleavage, the resin was treated with gaseous H2S as described below. We obtained 39 mg of crude compound 8 (41 μmol, 41%, if pure) starting from 82 mg of scaffold 2 (100 μmol).
3.2.7. Compound 8 (High Copper Load)
The solid-phase synthesis of compound
8 was repeated as described above but used a higher load of copper (II) sulfate as well as sodium ascorbate. Thus, for the two CuAAC reactions (to produce resin-bound compounds
6s and
8s), the following protocol was applied. The resin was washed with 3 × 5 mL of MeOH and
tBuOH/water (1:1,
v/
v; 1 min each). The following solutions were sequentially added to the resin: (i) azide
9 or
10 (500 μmol, 5.0 eq.) in 1 mL of
tBuOH/water (1:1,
v/
v); (ii) a solution of sodium ascorbate (99 mg, 500 μmol, 5 eq.) in 1 mL of
tBuOH/water (1:1,
v/
v); and (iii) copper(II) sulfate pentahydrate (25 mg, 100 μmol, 1 eq.) in 1 mL of
t-BuOH/water (1:1,
v/
v). After stirring for 16 h at r.t., the coupling was complete. The resin was washed with 3 × 5 mL of
tBuOH/water (1:1,
v/
v), H
2O, AcOH (to solubilize copper(I) salts), H
2O, DMF, DCM, MeOH, and DCM (1 min each). The resin was then treated with gaseous hydrogen sulfide (
vide infra). Before the final cleavage, the resin was washed with 3 × 5 mL DCM and dried over NaOH pellets at 10–100 Pa. The cleavage mixture (75 mg) was extracted twice with Et
2O, passed through a C-4 cartridge as explained for compound
7, and lyophilized to afford final compound
8 (44 mg, pale yellow solid, overall yield from compound
2 of 47%). The purity of compound
8 was verified by RP-HPLC (Method B,
Figure S7B).
Analytical HPLC: (Method B) purity = 64%. Elemental analysis (Cu): 15 ppm.
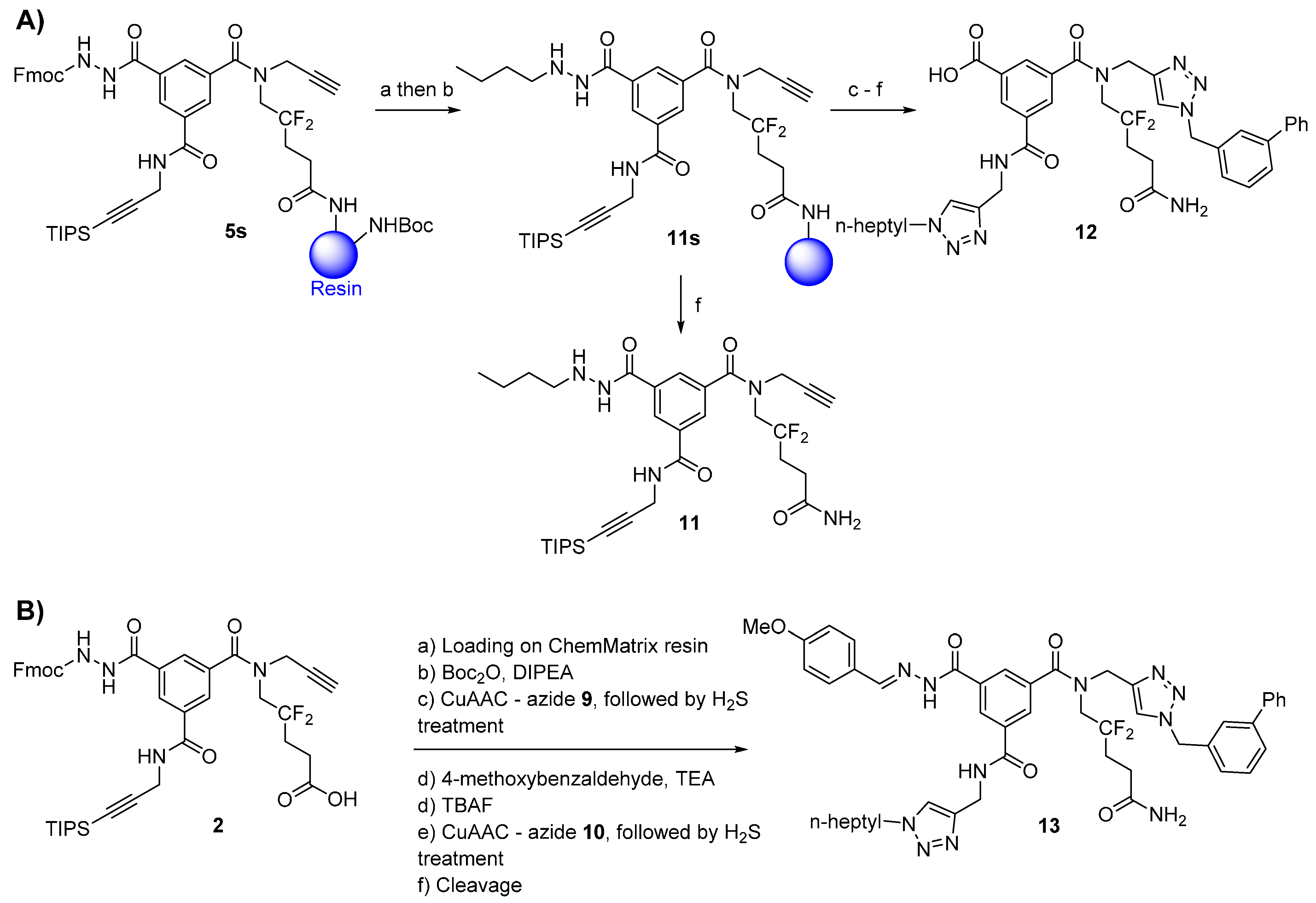
3.2.8. Compound 11
- (1)
Acylhydrazone formation: The resin-bound compound 5s (100 μmol of scaffold) was capped with Boc groups as described for compound 7. The resin was washed with 3 × 5 mL of DCM, DMF, DCM, MeOH, and ACN (1 min each). A solution of 1-butyraldehyde (45 μL, 500 μmol, 5.0 eq.) in 10% TEA/ACN (v/v, 2.5 mL) was added to the resin. The mixture was stirred at r.t. for 16 h. The resin was then washed with 3 × 5 mL of ACN (1 min each).
- (2)
Acylhydrazone reduction: The resin containing the acylhydrazone derivative was washed with 5 × 5 mL of dry EtOH. A cooled solution (0 °C) of sodium borohydride (38 mg, 1000 μmol, 10 eq.) in dry EtOH (3 mL) was added to the resin. The syringe was left upside down (to avoid projection) at 5 °C for 30 min and at r.t. for 3 h. The resin was then washed with 3 × 5 mL of MeOH, H2O, DMF, DCM, MeOH, and DCM (1 min each) to give resin-bound compound 11s.
A sample of resin-bound compound
11s (
ca. 50% of the total amount) was transferred to a fritted glass micro-reactor and dried under reduced pressure. The product was cleaved using the procedure described for compound
5. The purity of crude compound
11 (23 mg) was verified by RP-HPLC (Method B,
Supplementary Figure S8). Compound
11 was purified using RP-HPLC (Method A) and lyophilized (8.9 mg, yield from scaffold
2 ca. 30%, white powder).
Analytical HPLC: (Method B) tR = 19.8 min, crude purity = 70%. 1H-NMR (500 MHz; DMSO-d6; T = 100 °C): δH 0.92 (3H, t, 3JHH = 7.5 Hz, CH3), 1.06 (21H, m, Si(CH(CH3)2)3), 1.40 (2H, m, –CH2–), 1.51 (2H, m, –CH2–), 2.19 (2H, m, –CH2–), 2.28 (2H, m, –CH2–CO), 3.15 (1H, t, 4JHH = 2.4 Hz, –C≡CH), 4.01 (2H, t, 3JHF = 14.5 Hz, –CH2–N), 4.17 (2H, d, 3JHH = 5.6 Hz, –CH2–N), 4.23 (2H, br d, 4JHH = 2.4 Hz, –CH2–N), 7.98 (1H, t, 3JHH = 1.5 Hz, CHarom), 8.00 (1H, t, 3JHH = 1.5 Hz, CHarom), 8.38 (1H, t, 3JHH = 1.5 Hz, CHarom), 8.87 (1H, br t, 3JHH = 5.6 Hz, >NH) (one –CH2–N missing). 13C-NMR (125.8 MHz; DMSO-d6; T = 100 °C): δC 10.50 (3C, Si(CH(CH3)2)3)), 13.22 (1C, –CH3), 18.04 (6C, Si(CH(CH3)2)3), 19.31 (1C, –CH2–), 27.08 (1C, –CH2–), 29.06 (1C, –CH2–), 29.39 (1C, –CH2–N), 29.98 (1C, t, 2JCF = 24.5 Hz, –CH2–), 38.50 (1C, –CH2–N), 48.80 (1C, –CH2–N), 50.35 (1C, –CH2–N), 75.14 (1C, ≡C–H), 78.15 (1C, ≡C–), 81.80 (1C, ≡C–), 105.15 (1C, ≡C–), 123.26 (1C, t, 1JCF = 243.8 Hz, >CF2), 127.33 (1C, CHarom), 127.43 (1C, CHarom), 127.63 (1C, CHarom), 133.60 (1C, Carom), 134.71 (1C, Carom), 135.29 (1C, Carom), 164.54 (1C, –CO–NH), 165.62 (1C, –CO–NH), 172.00 (1C, –CO-NH2) (–CO–N< missing). HRMS (m/z): calcd for C33H50F2N5O4Si+ [M + H]+ 646.35946, found 646.35970.
3.2.9. Compound 12
From resin-bound compound
11s, compound
12 was obtained following the procedures described for compounds
6 and
8 (low copper load). After the final TFA cleavage, 20 mg of crude compound
12 were obtained. The crude compound was extracted twice with Et
2O and passed through a C-4 cartridge, as explained for compound
7. The purity of the crude compound
12 was verified by RP-HPLC (Method B,
Supplementary Figure S9). Crude compound
12 was then purified using RP-HPLC (Method B) and lyophilized to give pure compound
12 (3.1 mg, white powder).
Analytical HPLC: (Method B) tR = 17.9 min. 1H-NMR (500 MHz; DMSO-d6; T = 100 °C): δH 0.86 (3H, t, 3JHH = 7.0 Hz, –CH3), 1.26 (8H, m, 4 × –CH2–), 1.81 (2H, m, –CH2–), 2.19 (2H, m, –CH2–), 2.27 (2H, m, –CH2–CO), 3.91 (2H, t, JHF = 14.4 Hz, –CH2–N), 4.29 (2H, t, 3JHH = 7.1, –CH2–N), 4.56 (2H, d, 3JHH = 5.5 Hz, –CH2–N), 4.65 (2H, s, –CH2–N), 5.64 (2H, s, –CH2–N), 7.30 (1H, dm, 3JHH = 7.5 Hz, CHarom), 7.36 (1H, tt, 3JHH = 7.5 Hz, 4JHH = 1.2 Hz, CHarom), 7.44 (3H, m, 3 × CHarom), 7.60 (4H, m, 4 × CHarom), 7.86 (1H, s, CHarom), 8.02 (1H, s, CHarom), 8.05 (1H, t, 4JHH = 1.6 Hz, CHarom), 8.12 (1H, t, 4JHH = 1.6 Hz, CHarom), 8.50 (1H, t, 4JHH = 1.6 Hz, CHarom), 9.02 (1H, t, 3JHH = 5.5 Hz, >NH), 11.89 (1H, br s, >NH). 13C-NMR (125.8 MHz; DMSO-d6; T = 100 °C): δC 13.20 (1C, –CH3), 21.42 (1C, –CH2–), 25.43 (1C, –CH2–), 27.11 (1C, t, 3JCF = 4.1 Hz, –CH2–), 27.52 (1C, –CH2–), 29.10 (1C, –CH2–), 30.06 (1C, t, 2JCF = 24.3 Hz, –CH2–), 30.58 (1C, –CH2–), 34.89 (1C, –CH2–N), 44.00 (1C, –CH2–N), 48.62 (1C, –CH2–N), 49.16 (1C, –CH2–N), 52.63 (1C, –CH2–N), 122.73 (1C, CHarom), 122.80 (1C, CHarom), 126.07 (1C, CHarom), 126.09 (1C, CHarom), 126.43 (2C, 2 × CHarom), 126.58 (1C, CHarom), 127.19 (1C, CHarom), 128.48 (2C, 2 × CHarom), 128.78 (1C, CHarom), 128.90 (1C, CHarom), 129.17 (1C, CHarom), 129.46 (1C, CHarom), 131.33 (1C, Carom), 134.89 (1C, Carom), 136.09 (1C, Carom), 136.34 (1C, Carom), 139.53 (1C, Carom), 140.54 (1C, Carom), 141.84 (1C, Carom), 144.73 (1C, Carom), 164.60 (1C, –CO–), 165.71 (1C, –CO–), 170.16 (1C, –CO–), 172.01 (1C, –CO–NH2). HRMS (m/z): calcd for C40H44F2N9O5− 768.34390 [M − H]−, found 768.34363; calcd for C40H45F2N9NaO5+ 792.34039 [M + Na]+, found 792.34059.
3.2.10. Compound 13
Compound 13 was synthesized following the procedure described for compound 8 (low copper load), replacing the 3-(trifluoromethyl)benzaldehyde in the synthesis of compound 7 with 4-methoxybenzaldehyde (122 μL, 100 μmol, 10 eq.) and treating the resin with gaseous hydrogen sulfide after each CuAAC. After the final cleavage, the crude product (56 mg) was extracted twice with Et2O, passed through a C-4 cartridge as explained for compound 7, and lyophilized to afford 39 mg (43%) of pale-yellow powder. Part of this powder (20 mg) was purified using RP-HPLC (Method A) and lyophilized (5.4 mg, white powder).
Analytical HPLC: (Method B) tR = 20.4 min. 1H-NMR (500 MHz; DMSO-d6; T = 100 °C): δH 0.85 (3H, t, 3JHH = 7.5 Hz, CH3), 1.24–1.29 (8H, m, 4 × –CH2–), 1.82 (2H, m, –CH2–), 2.19 (2H, m, –CH2–), 2.26 (2H, m, –CH2–), 3.83 (3H, s, –OCH3), 3.94 (2H, br t, 3JHF = 14.2 Hz, N–CH2–), 4.30 (2H, t, 3JHH = 7.1 Hz, N–CH2–), 4.57 (2H, d, 3JHH = 5.6 Hz, N–CH2–), 4.70 (2H, br s, N–CH2–), 5.63 (2H, s, N–CH2–), 7.00 (2H, br d, 3JHH = 8.5 Hz, 2 × CHarom), 7.25 (2H, br d, 3JHH = 7.5 Hz, 2 × CHarom), 7.36 (1H, m, CHarom), 7.45 (3H, m, 3 × CHarom), 7.60 (3H, m, 3 × CHarom), 7.65 (2H, br d, 3JHH = 8.5 Hz, 2 × CHarom), 7.89 (1H, s, CHarom), 8.03 (1H, m, CHarom), 8.08 (2H, br, 2 × CHarom), 8.46 (1H, br, CHarom), 8.93 (1H, br d, 3JHH = 5.6 Hz, NH) (–N=CH missing). 13C-NMR (125.8 MHz; DMSO-d6; T = 100 °C): δC 13.25 (1C, CH3), 21.45 (1C, –CH2–), 25.52 (1C, –CH2–), 27.14 (1C, –CH2–), 27.57 (1C, –CH2–), 29.27 (1C, –CH2–), 30.11 (1C, –CH2–), 30.62 (1C, –CH2–), 34.93 (1C, –CH2–N), 43.94 (1C, –CH2–N), 48.66 (1C, –CH2–N), 49.08 (1C, –CH2–N), 52.77 (1C, –CH2–N), 55.14 (1C, OCH3), 114.23 (2C, 2 × CHarom), 122.33 (1C, CHarom), 123.52 (1C, CHarom), 126.03 (1C, CHarom), 126.11 (1C, CHarom), 126.36 (1C, CHarom), 126.47 (1C, CHarom), 127.22 (1C, CHarom), 127.94 (1C, CHarom), 128.42 (2C, 2 × CHarom), 128.51 (2C, 2 × CHarom), 128.99 (3C, 3 × CHarom), 134.13 (1C, Carom), 134.74 (1C, Carom), 135.79 (1C, Carom), 136.14 (1C, Carom), 139.52 (1C, Carom), 140.57 (1C, Carom), 142.26 (1C, Carom), 144.30 (1C, Carom), 160.89 (1C, Carom), 164.86 (1C, –CO–NH) (–CO–N<, –CO–NH2, –CO–NH and three Carom missing). HRMS (m/z): calcd for C48H53F2N11NaO5+ 924.40914 [M + Na]+, found 924.40973.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}