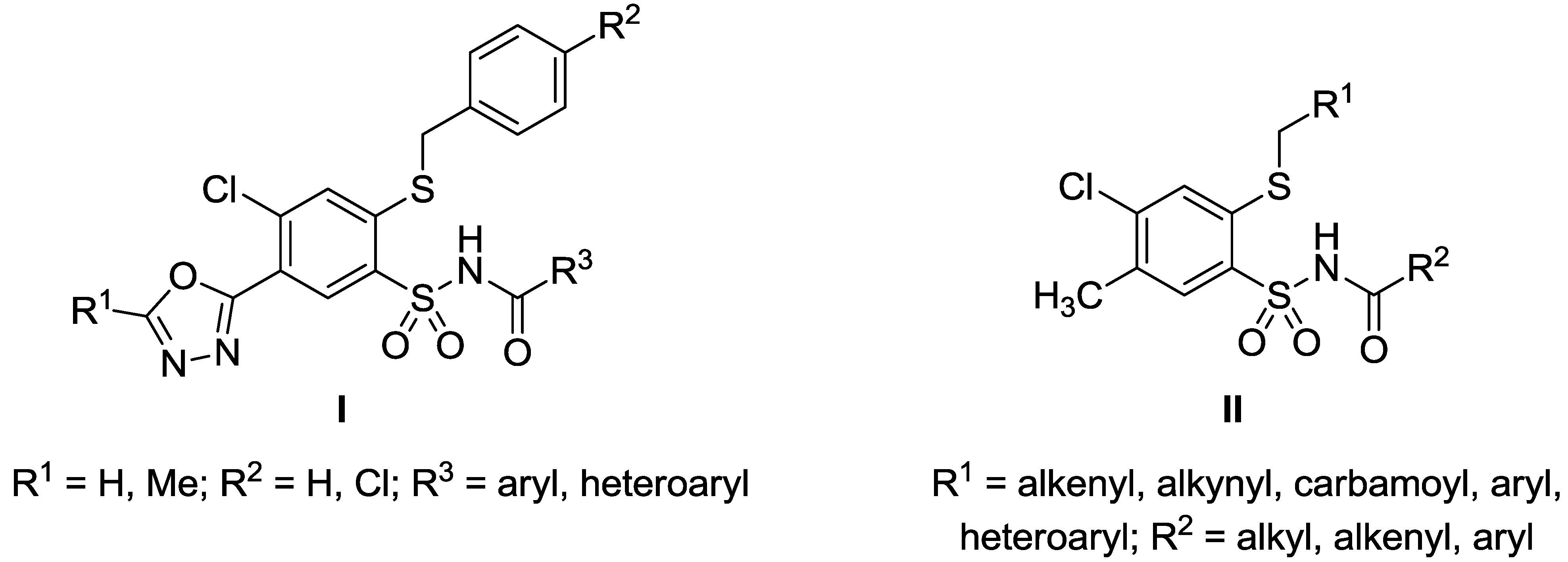
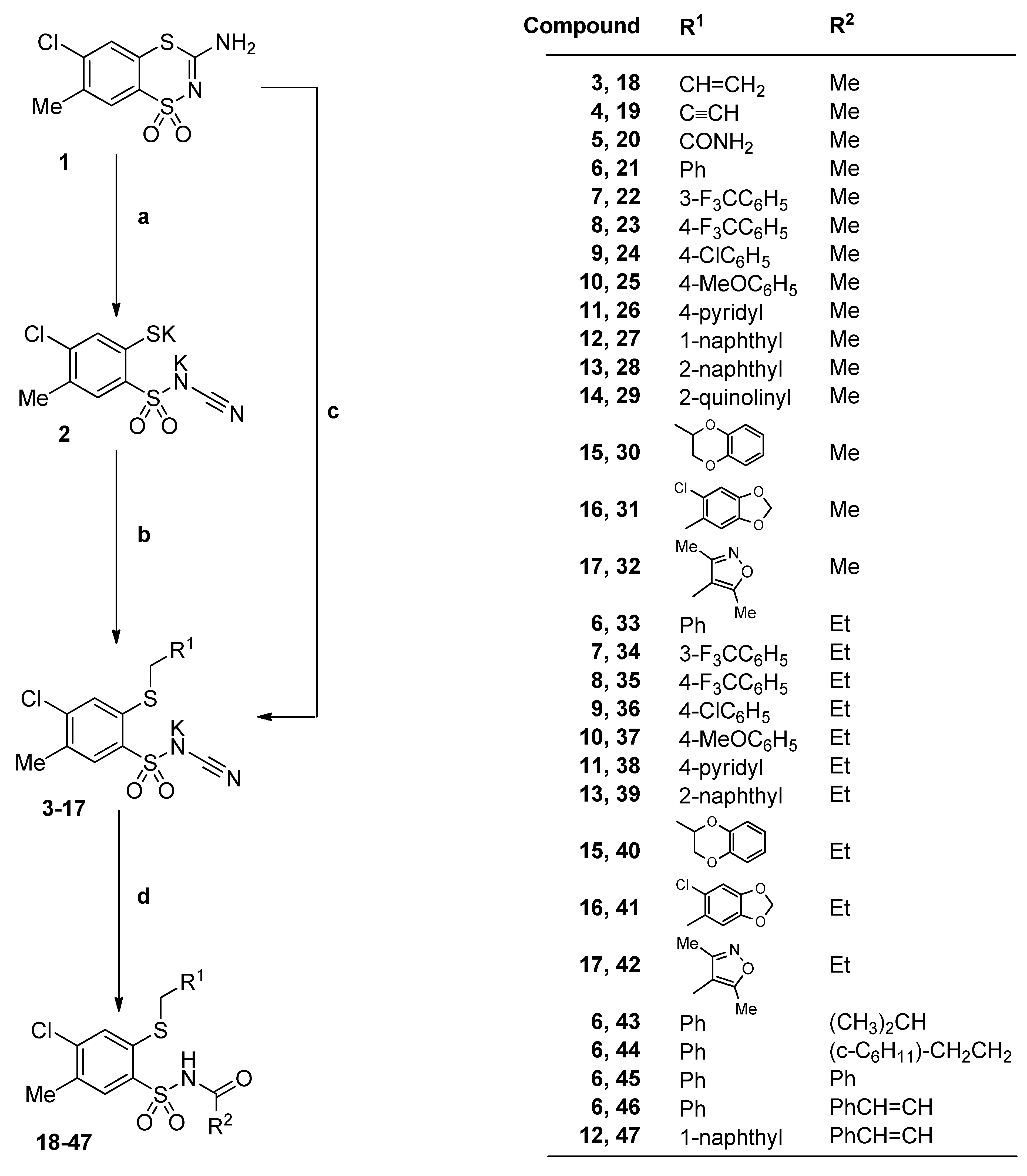
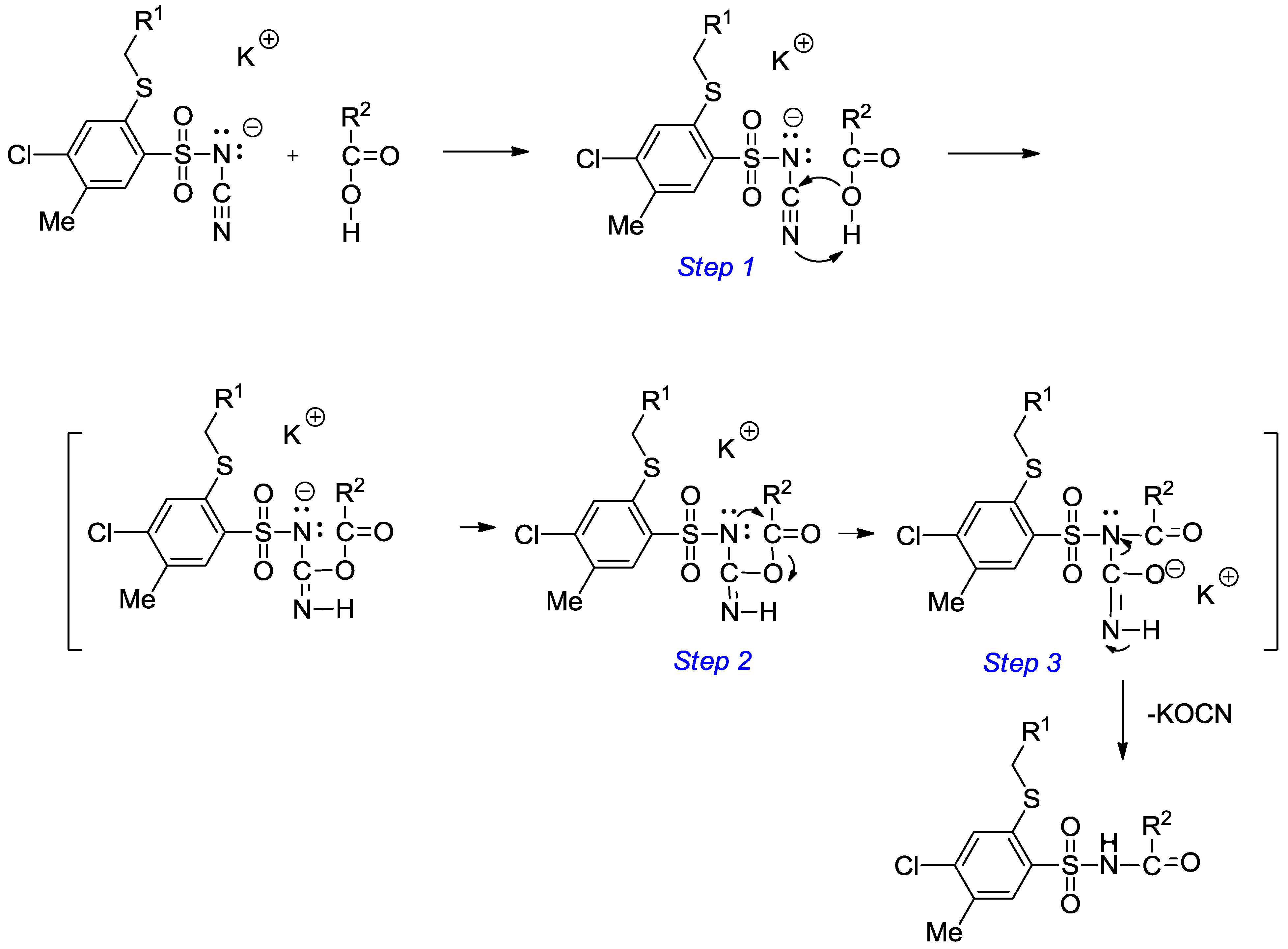
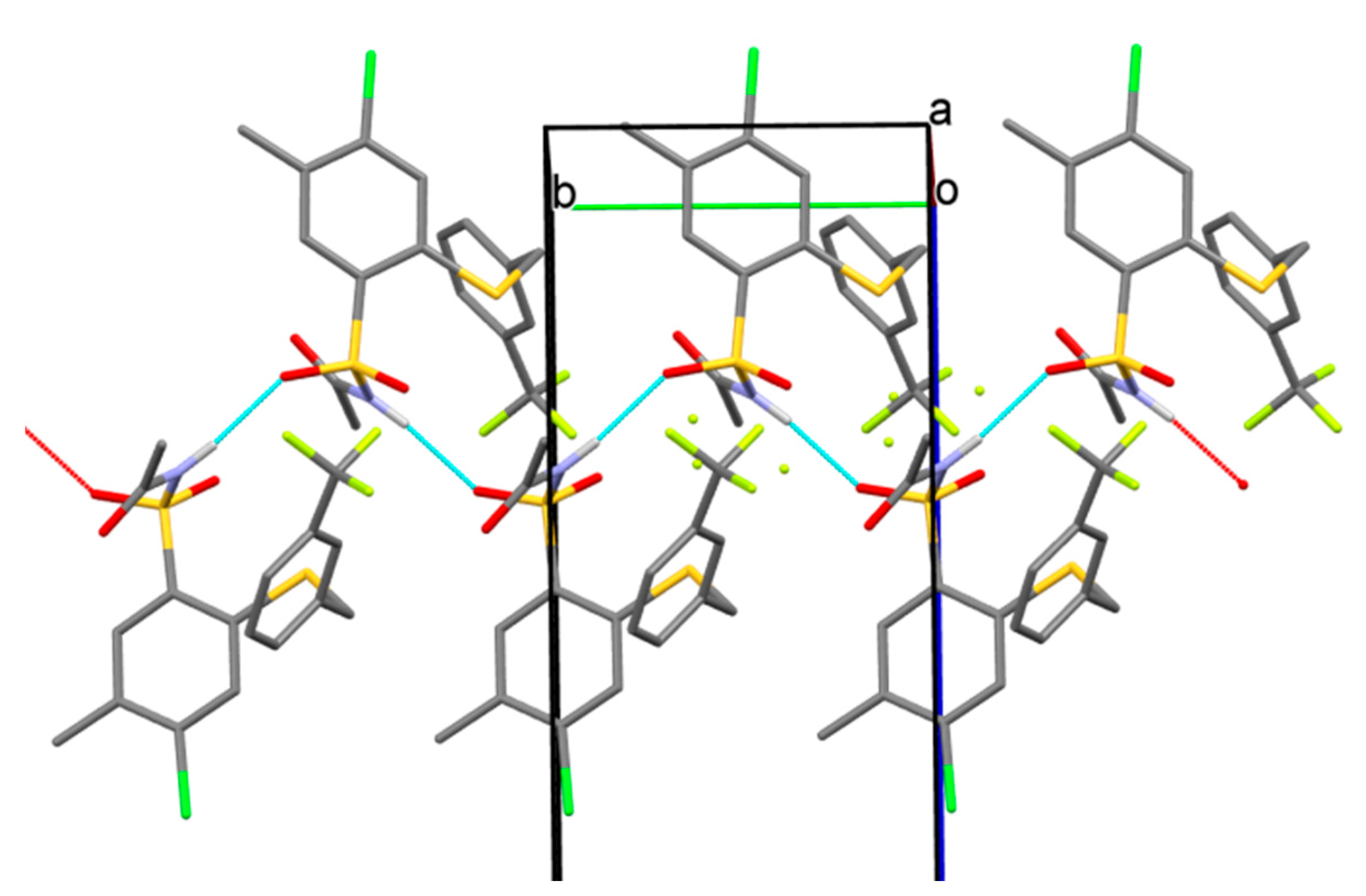
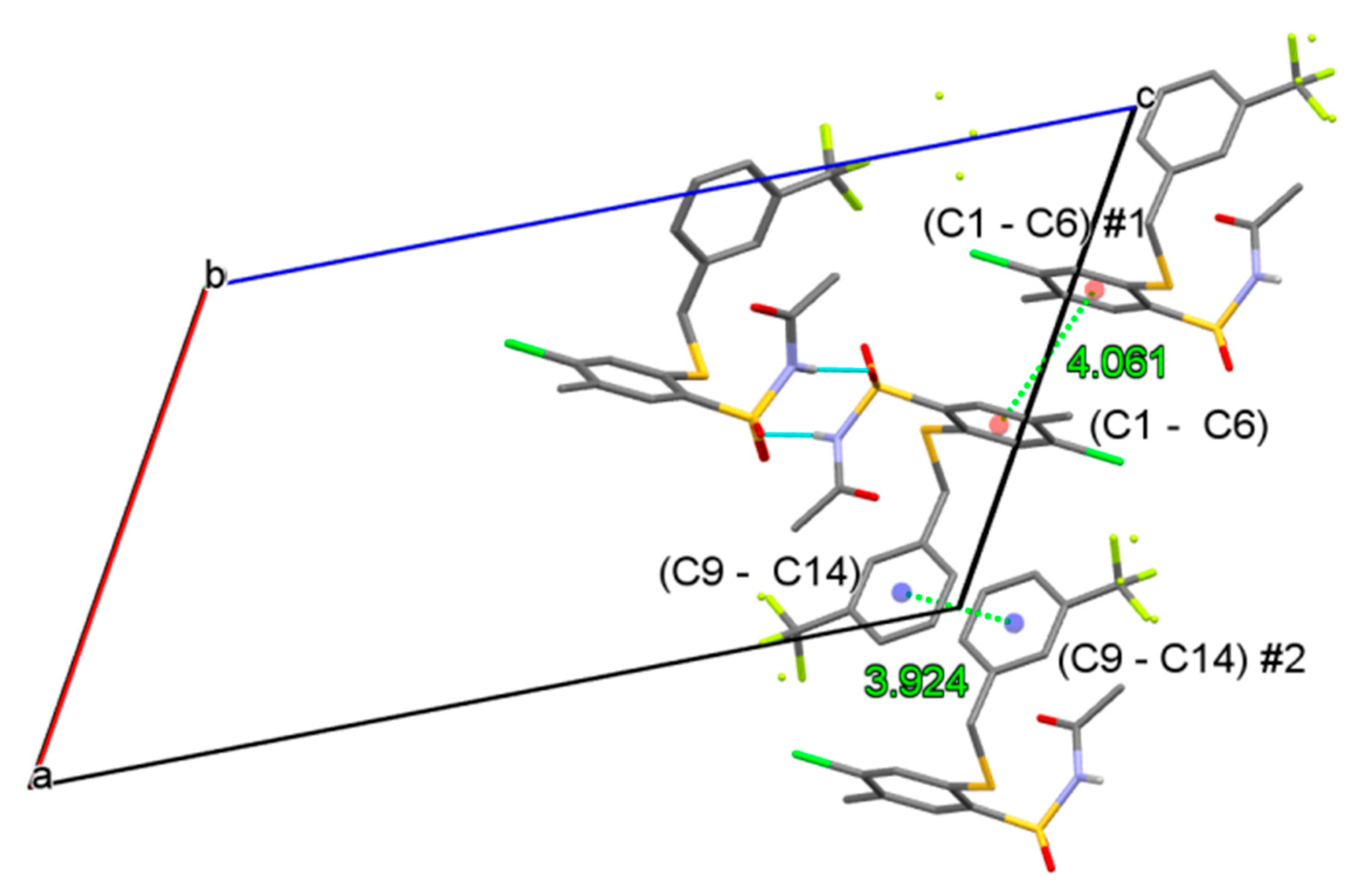
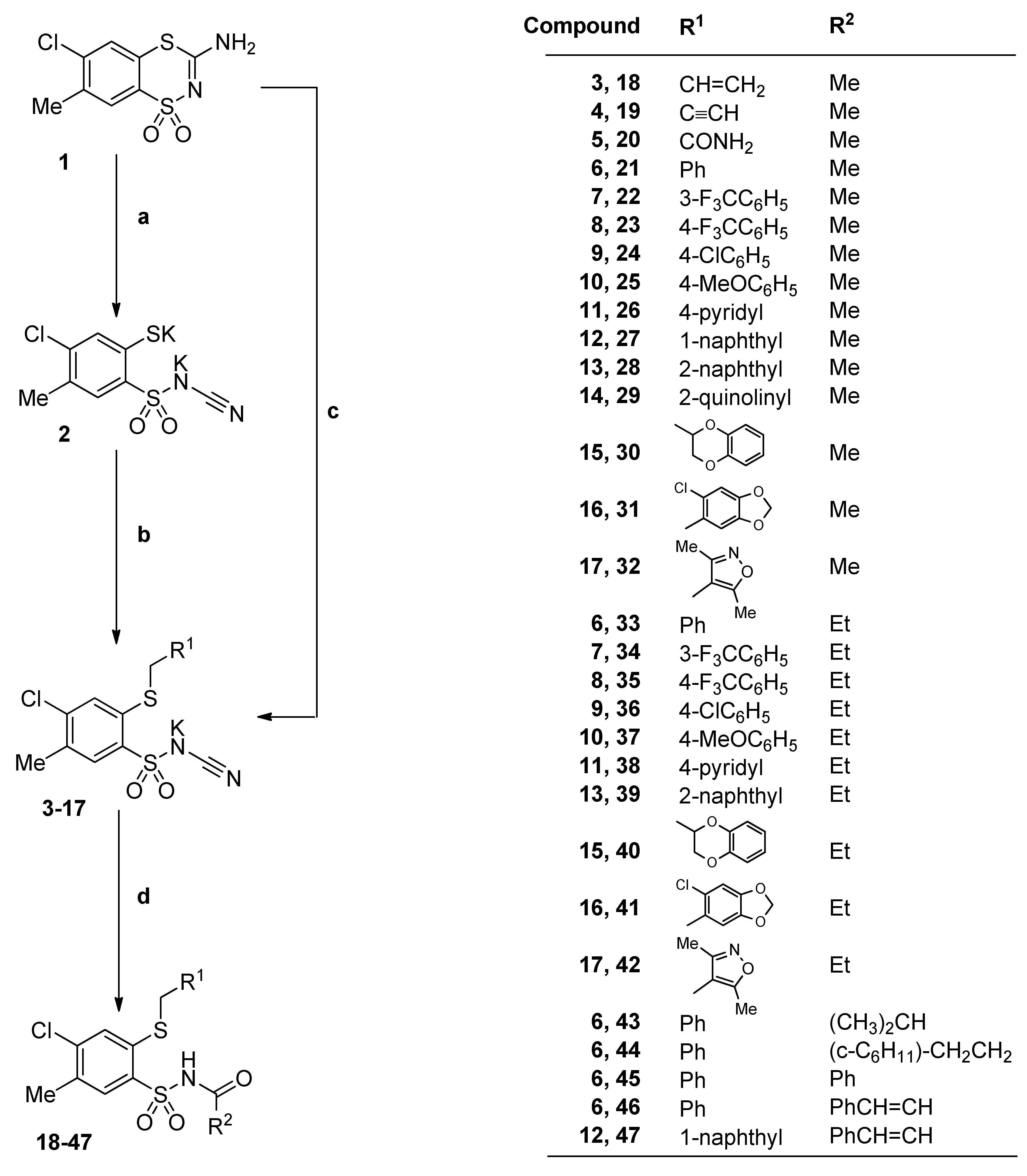
3.2.2. Procedure for the Preparation of N-Acylbenzenesulfonamides 18‒44
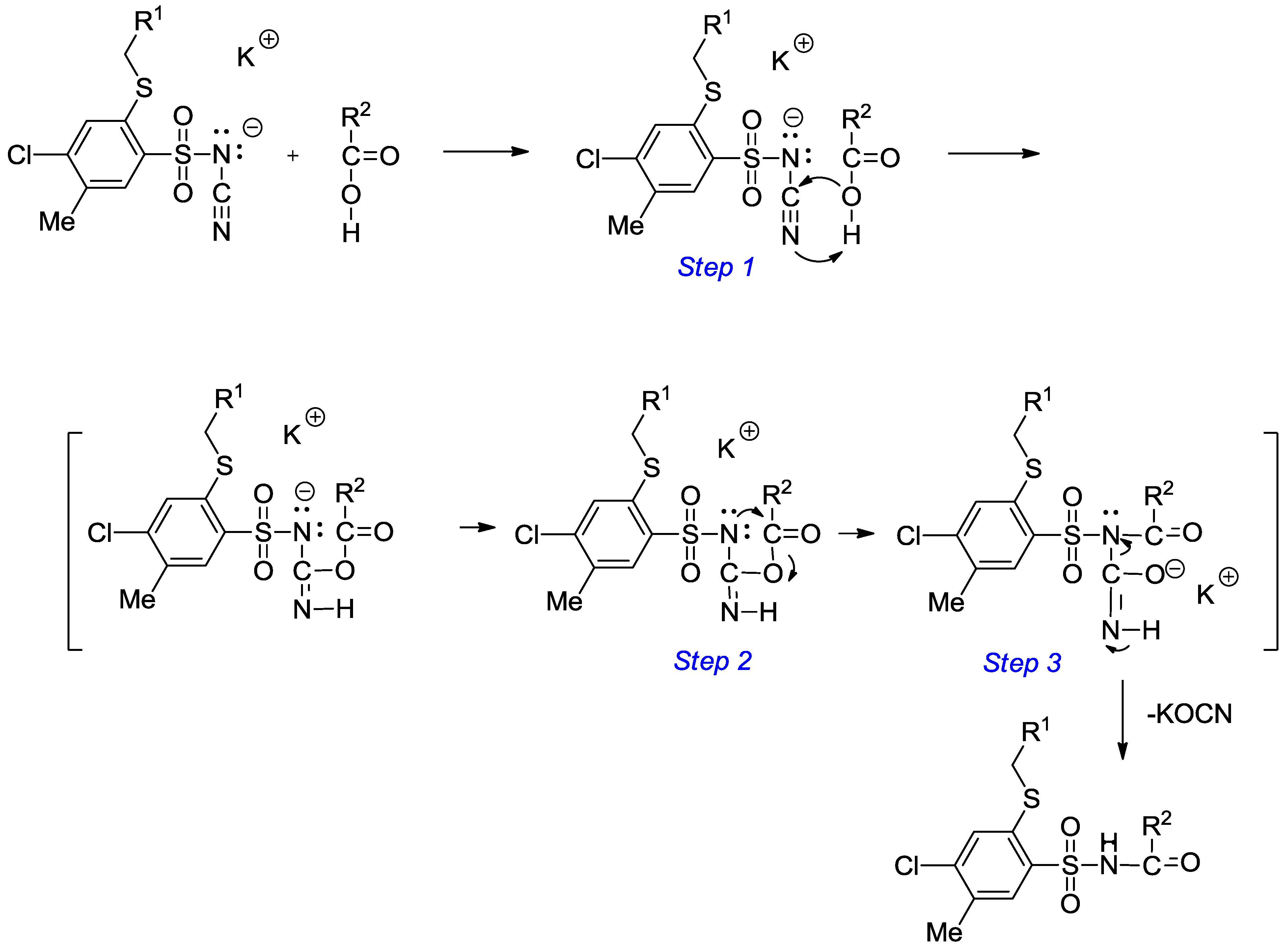
A solution of monopotassium salts 3‒17 (1 mmol) in an appropriate carboxylic acid (40 mmol) was refluxed for 1‒17 h and left overnight at room temperature. The precipitate was filtered off, dried and crystallized from ethanol.
N-(2-Allylthio-4-chloro-5-methylphenylsulfonyl)acetamide (18). Starting from 3 (0.341 g) and acetic acid (2.5 mL) the title compound 18 (0.230 g, 72%) was obtained after 15 h at reflux: mp 138‒140 °C; IR (KBr) 3334, 3241 (NH), 2923 (CH3, CH2), 1686 (C=O), 1352, 1160 (SO2) cm−1; 1H-NMR (500 MHz, DMSO-d6) δ 1.95 (s, 3H, CH3CO), 2.40 (s, 3H, CH3), 3.86 (d, 2H, SCH2), 4.94 (dd, 2H, =CH2), 5.77–5.84 (m, 1H, HC=), 7.62 (s, 1H, H-3), 7.95 (s, 1H, H-6), 12.34 (s, 1H, NH) ppm; anal. C 44.75, H 4.31, N 4.61% calcd for C12H14ClNO3S2, C 45.06, H 4.41, N 4.38%.
N-[4-Chloro-5-methyl-2-(prop-2-ynylthio)phenylsulfonyl]acetamide (19). Starting from 4 (0.339 g) and acetic acid (2.5 mL) the title compound 19 was obtained (0.113 g, 35%) after for 17 h at reflux: mp 148‒150 °C; IR (KBr) 3448, 3298 (NH), 3086 (C≡CH), 2981, 2876 (CH3, CH2), 1692 (C=O), 1472 (N–H), 1353, 1165 (SO2) cm−1; 1H-NMR (500 MHz, DMSO-d6) δ 1.91 (s, 3H, CH3CO), 2.36 (s, 3H, CH3), 3.20 (s, 1H, C≡CH), 4.06 (s, 2H, SCH2), 7.68 (s, 1H, H-3), 7.92 (s, 1H, H-6), 12.34 (s, 1H, NH); 13C-NMR (125 MHz, DMSO-d6) δ 19.69, 21.00, 23.90, 74.95, 80.03, 129.01, 133.66, 134.19, 135.11, 135.82, 139.56, 169.41 ppm; anal. C 44.96, H 3.75, N 4.08% calcd for C12H12ClNO3S2, C 45.35, H 3.81, N 4.41%.
N-(4-Chloro-2-carbamoylmethylthio-5-methylphenylsuflonyl)acetamide (20). Starting from 5 (0.358 g) and acetic acid (2.5 mL) the title compound 20 was obtained (0.101 g, 60%) after 4.5 h at reflux, followed by additional cooling in an ice-water bath for 3 h: mp 220‒225 °C; IR (KBr) 3466, 3355 (NH), 2860, 2701 (CH3, CH2), 1693 (C=O), 1476, 1457 (N–H), 1160, 1119 (SO2) cm−1; 1H-NMR (500 MHz, DMSO-d6) δ 1.93 (s, 3H, CH3CO), 2.34 (s, 3H, CH3), 3.81 (s, 2H, SCH2), 7.27 (s, 1H, CONH2), 7.60 (s, 1H, CONH2), 7.62 (s, 1H, H-3), 7.89 (s, 1H, H-6), 12.30 (s, 1H, NH); 13C-NMR (125 MHz, DMSO-d6) δ 19.63, 23.83, 36.95, 128.79, 133.27, 134.16, 135.21, 136.77, 139.63, 169.42, 169.68 ppm; anal. C 38.84, H 3.78, N 7.95% calcd for C11H13ClN2O4S2, C 39.23, H 3.89, N 8.32%.
N-(2-Benzylthio-4-chloro-5-methylphenylsulfonyl)acetamide (21). Starting from 6 (0.400 g) and acetic acid (2.5 mL) the title compound 21 was obtained (0.200 g, 54%) after 2 h at reflux, followed by additional cooling in an ice-box for 12 h: mp 155‒158 °C; IR (KBr) 3116 (N–H), 2921 (CH3, CH2), 1686 (C=O), 1454 (N–H), 1356, 1165 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 1.92 (s, 3H, CH3CO), 2.35 (s, 3H, CH3), 4.45 (s, 2H, SCH2), 7.26‒7.48 (m, 5H, arom.), 7.67 (s, 1H, H-3), 7.90 (s, 1H, H-6), 12.36 (s, 1H, NH); 13C-NMR (125 MHz, DMSO-d6) δ 19.64, 23.76, 36.77, 128.14, 128.73, 129.18, 129.81, 133.08, 134.16, 135.13, 136.62, 139.59, 169.26 ppm; anal. C 51.89, H 4.35, N 3.77% calcd for C16H16ClNO3S2, C 51.95, H 4.36, N 3.79%.
N-(4-Chloro-5-methyl-2-(3-trifluoromethylbenzylthio)phenylsulfonyl)acetamide (22). Starting from 7 (0.459 g) and acetic acid (2.5 mL) the title compound 22 was obtained (0.367 g, 84%) after 2 h at reflux, followed by additional cooling an ice-box for 12 h: mp 126‒130 °C; IR (KBr) 3221 (N–H), 3065 (C–H arom.), 2986, 2855 (CH3, CH2), 1721 (C=O), 1448 (N–H), 1330, 1164 ( SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 1.92 (s, 3H, CH3CO), 2.35 (s, 3H, CH3), 4.58 (s, 2H, SCH2),7.56‒7.82 (m, 5H, H-3, arom.), 7.91 (s, 1H, H-6), 12.37 (s, 1H, NH) ppm; 13C-NMR (50 MHz, DMSO-d6) δ 19.22, 23.24, 35.66, 124.43, 124.51, 126.06, 126.13, 128.76, 129.10, 129.73, 129.87, 133.12, 133.36, 133.80, 135.11, 135.32, 138.11, 139.21, 168.82 ppm; anal. C 46.49, H 3.42, N 3.13% calcd for C17H15ClF3NO3S2, C 46.63, H 3.45, N 3.20%.
N-(4-Chloro-5-methyl-2-(4-trifluoromethylbenzylthio)phenylsulfonyl)acetamide (23). Starting from 8 (0.459 g) and acetic acid (2.5 mL), the title compound 23 was obtained (0.219 g, 50%) after 1.5 h at reflux, followed by additional cooling in an ice-box for 12 h: mp 180‒181 °C; IR (KBr) 3443 (N–H), 3062 (C–H arom.), 2878, 2710 (CH3, CH2), 1687 (C=O), 1482 (N–H), 1321, 1160 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 1.91 (s, 3H, CH3CO), 2.36 (s, 3H, CH3), 4.56 (s, 2H, SCH2), 7.63‒7.75 (m, 5H, H-3, arom.), 7.91 (s, 1H, H-6), 12.34 (s, 1H, NH) ppm; anal. C 46.66, H 3.45, N 3.21% calcd for C17H15ClF3NO3S2, C 46.63, H 3.45, N 3.20%.
N-[4-Chloro-2-(4-chlorobenzylthio)-5-methylphenylsulfonyl]acetamide (24). Starting from 9 (0.338 g) and acetic acid (2.5 mL), the title compound 24 was obtained (0.219 g, 50%) after 2 h at reflux, followed by additional cooling in an ice-box for 12 h: mp 182‒184 °C; IR (KBr) 3445 (N–H), 3052 (C–H arom.), 2869, 2711 (CH3, CH2), 1683 (C=O), 1490 (N–H), 1350, 1162 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 1.92 (s, 3H, CH3CO), 2.35 (s, 3H, CH3), 4.46 (s, 2H, CH2S), 7.42 (d, 2H, arom.), 7.47 (d, 2H, arom.), 7.68 (s, 1H, H-3), 7.81 (s, 1H, H-6), 12.35 (s, 1H, NH); 13C-NMR (125 MHz, DMSO-d6) δ 19.65, 23.75, 35.97, 128.95, 129.16, 131.57, 132.74, 133.34, 134.18, 135.32, 135.93, 136.12, 139.62, 169.28 ppm; anal. C 47.26, H 3.69, N 3.33% calcd for C16H15ClNO3S2, C 47.53, H 3.74, N 3.46%.
N-[4-Chloro-2-(4-methoxybenzylthio)-5-methylphenylsulfonyl]acetamide (25). Starting from 10 (0.421 g) and acetic acid (2.5 mL) the crude product was obtained after refluxing for 1 h, then evaporating the mixture under reduced pressure. Crystallization from ethanol gave the title compound 25 (0.204 g, 51%): mp 82‒84 °C; IR (KBr) 3427 (N–H), 2924, 2853 (CH3, CH2), 1705 (C=O), 1450 (N–H), 1303, 1032 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 1.79 (s, 3H, CH3CO), 2.32 (s, 3H, CH3), 3.74 (s, 3H, CH3O), 4.25 (s, 2H, CH2S), 6.90 (d, 2H, arom.), 7.35 (d, 2H, arom.), 7.45 (s, 1H, H-3), 7.81 (s, 1H, H-6), 12.20 (br s, 1H, NH) ppm; anal. C 51.20, H 4.57, N 3.58% calcd for C17H18ClNO4S2, C 51.06, H 4.54, N 3.50%.
N-[4-Chloro-2-(4-pyridinylmethylthio)-5-methylphenylsulfonyl]acetamide (26). Starting from 11 (0.392 g) and acetic acid (2.5 mL) the crude product was obtained after 1.5 h at reflux, then evaporating the mixture under reduced pressure. Crystallization from ethanol/chloroform (v/v = 2:1) gave the title compound 26 (0.167 g, 45%): mp 179‒183 °C; IR (KBr) 3413 (N–H), 2923, 2852 (CH3, CH2), 1711 (C=O), 1492 (N–H), 1325, 1153 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 1.92 (s, 3H, CH3CO), 2.34 (s, 3H, CH3), 4.49 (s, 2H, CH2S), 7.44 (d, 2H, pyridine), 7.67 (s, 1H, H-3), 7.90 (s, 1H, H-6), 8.52 (d, 2H, pyridine), 12.40 (s, 1H, NH) ppm; anal. C 48.39, H 4.00, N 7.45% calcd for C15H15ClN2O3S2, C 48.58, H 4.08, N 7.55%.
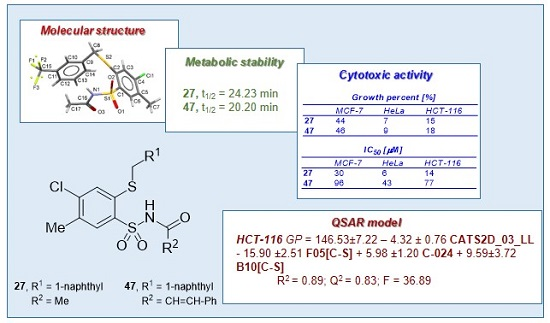
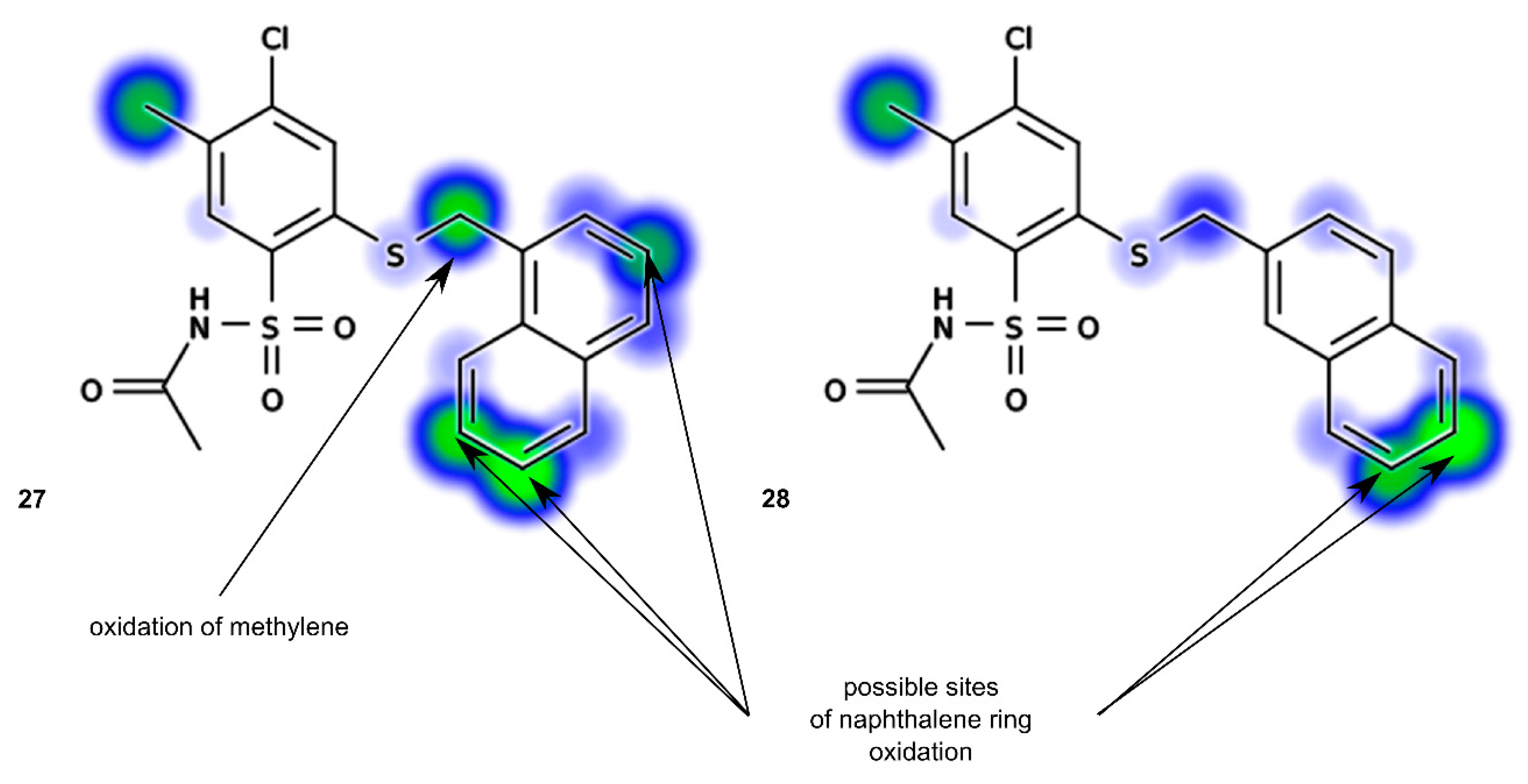
N-[4-Chloro-5-methyl-2-(naphth-1-ylmethyl)phenylsulfonyl]acetamide (27). Starting from 12 (0.442 g) and acetic acid (2.5 mL) a white solid was obtained after refluxing for 1.5 h, followed by additional cooling in an ice-box for 12 h. The product was crystallized from acetonitrile to afford the title compound 27 (0.227 g, 54%): mp 176‒178 °C; IR (KBr) 3438 (N–H), 3061 (C–H arom), 2921, 2852 (CH3, CH2), 1727 (C=O), 1443 (N–H), 1352, 1155 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 1.92 (s, 3H, CH3CO), 2.39 (s, 3H, CH3), 4.91 (s, 2H, CH2S), 7.45‒7.66 (m, 4H, arom.), 7.77 (s, 1H, H-3) 7.89‒8.00 (m, 3H, H-6 and arom.), 8.25 (d, 1H, arom.), 12.20 (s, 1H, NH) ppm; anal. C 57.23, H 4.32, N 3.33% calcd for C20H18ClNO3S2, C 57.20, H 4.32, N 3.34%.
N-[4-Chloro-5-methyl-2-(naphth-2-ylmethyl)phenylsulfonyl]acetamide (28). Starting from 13 (0.442 g) and acetic acid (2.5 mL) the title compound 28 was obtained (0.210 g, 50%) after 2 h at reflux, followed by additional cooling in an ice-box for 12 h: mp 138‒141 °C; IR (KBr) 3423 (N–H); 3066 (C–H arom.); 2919, 2870 (CH3, CH2), 1683 (C=O), 1353, 1161 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 1.93 (s, 3H, CH3CO), 2.33 (s, 3H, CH3), 4.63 (s, 2H, CH2S), 7.40‒7.60 (m, 3H, arom.), 7.75 (s, 1H, H-3), 7.80‒7.96 (m, 5H, H-6 and arom.), 12.36 (s, 1H, NH) ppm; anal. C 57.30, H 4.30, N 3.39% calcd for C20H18ClNO3S2, C 57.20, H 4.32, N 3.34%.
N-[4-Chloro-5-methyl-2-(quinolin-2-yl)phenylsulfonyl]acetamide (29). Starting from 14 (0.421 g) and acetic acid (2.5 mL), the title compound 29 was obtained (0.219 g, 52%) after refluxing with stirring for 2 h, followed by additional cooling in an ice-box for 12 h: mp 153.8‒157 °C; IR (KBr) 3421 (N–H), 3069 (C–H arom.), 2922, 2851 (CH3, CH2), 1719 (C=O), 1457 (N–H), 1351, 1157 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 1.91 (s, 3H, CH3CO), 2.32 (s, 3H, CH3), 3.36 (s, 2H, CH2S), 7.60 (t, 1H, quinol.), 7.68 (s, 1H, H-3), 7.79 (t, 1H, quinol.), 7.88 (s, 1H, H-6), 7.90‒8.00 (m, 3H, quinol.), 8.37 (d, 1H, quinol.), 12.40 (s, 1H, NH) ppm; anal. C 54.60, H 3.92, N 6.37% calcd for C19H17ClN2O3S2, C 54.21, H 4.07, N 6.66%.
N-[4-Chloro-2-(2,3-dihydrobenzo[b][1,4]dioxin-2-ylmethylthio)-5-methylphenylsulfonyl]acetamide (30). Starting from 15 (0.449 g) and acetic acid (2.5 mL), the title compound 30 was obtained (0.235 g, 55%) after refluxing for 1 h 15 min, followed by additional cooling in an ice-box for 12 h: mp 188‒191.5 °C; IR (KBr) 3268 (N–H), 3067 (C–H arom.), 2985, 2883 (CH3, CH2), 1722 (C=O), 1495 (N–H), 1329, 1159 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 1.92 (s, 3H, CH3CO), 2.38 (s, 3H, CH3), 3.50 (d, 2H, CH2O), 4.14 (dd, 1H, CHO), 4.38 (d, 2H, CH2S), 6.66–6.89 (m, 4H, arom.), 7.74 (s, 1H, H-3), 7.94 (s, 1H, H-6), 12.41 (s, 1H, NH) ppm; anal. C 50.50, H 4.24, N 3.28% calcd for C18H18ClNO5S2, C 50.52, H 4.24, N 3.27%.
N-[4-Chloro-2-(6-chlorobenzo[d][1,3]dioxol-5-ylmethylthio)-5-methylphenylsulfonyl]acetamide (31). Starting from 16 (0.469 g) and acetic acid (2.5 mL) the crude product was obtained after 1.5 h at reflux, followed by additional cooling in an ice-box for 12 h,. Crystallization from ethanol/chloroform (v/v = 2.7:1) gave the title compound 31 (0.264 g, 59%): mp 122‒124 °C; IR (KBr) 3424 (N–H), 2922, 2852 (CH3, CH2), 1719 (C=O), 1478 (N–H), 1251, 1159 (SO2) cm−1; 1H NMR (200 MHz, DMSO-d6) δ 1.81 (s, 3H, CH3CO), 2.34 (s, 3H, CH3), 4.30 (s, 2H, CH2S), 6.08 (s, 2H, CH2O), 7,12 (s, 1H, arom.), 7.13 (s, 1H, arom.), 7.44 (s, 1H, H-3), 7.85 (s, 1H, H-6), 12.40 (s, 1H, NH) ppm; anal. C 45.71, H 3.32, N 3.03% calcd for C17H15Cl2NO5S2, C 45.54, H 3.37, N 3.12%.
N-[4-Chloro-2-(3,5-dimethylisoxazol-4-ylmethylthio)-5-methylphenylsulfonyl]acetamide (32). Starting from 17 (0.338 g) and acetic acid (2.5 mL), a product was obtained after 1.5 h at reflux, followed by additional cooling in an ice-box for 12 h. Crystallization from 80% ethanol gave the title compound 32 (0.241 g, 62%): mp 154‒160 °C; IR (KBr) 3421 (N–H), 3086 (C–H arom.), 2922, 2853 (CH3, CH2), 1711 (C=O), 1455 (N–H), 1351, 1161 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 1.92 (s, 3H, CH3CO), 2.26 (s, 3H, CH3), 2.37 (s, 3H, CH3), 2.39 (s, 3H, CH3), 4.28 (s, 2H, CH2S), 7.73 (s, 1H, H-3), 7.93 (s, 1H, H-6), 12.32 (s, 1H, NH) ppm; anal. C 45.95, H 4.25, N 6.88% calcd for C15H17ClN2O4S2, C 46.33, H 4.41, N 7.20%.
N-(2-Benzylthio-4-chloro-5-methylphenylsulfonyl)propionamide (33). Starting from 6 (0.385 g) and propionic acid (3.0 mL) the product was obtained after 1 h at reflux, then evaporating the mixture under reduced pressure and treating the residue with water (10 mL). Crystallization of the crude product from ethyl acetate/petroleum ether gave the title compound 33 (0.330 g, 86%): mp 137‒139 °C; IR (KBr) 3440 (N–H), 3033 (C–H arom.), 2922, 2874 (CH3, CH2), 1673 (C=O), 1469 (N–H), 1355, 1162 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 0.89 (t, 3H, CH3CH2CO), 2.23 (q, 2H, CH2CO), 2,36 (s, 3H, CH3), 4.44 (s, 2H, CH2S), 7.24‒7.45 (m, 5H, arom.), 7.67 (s, 1H, H-3), 7.92 (s, 1H, H-6), 12.31 (s, 1H, NH); 13C-NMR (125 MHz, DMSO-d6) δ 8.94, 19.65, 29.23, 36.73, 128.13, 128.65, 129.17, 129.78, 133.08, 134.19, 135.17, 136.57, 136.62, 139.57, 172.77 ppm; anal. C 53.39, H 4.83, N 3.90% calcd for C17H18ClNO3S2, C 53.18, H 4.73, N 3.65%.
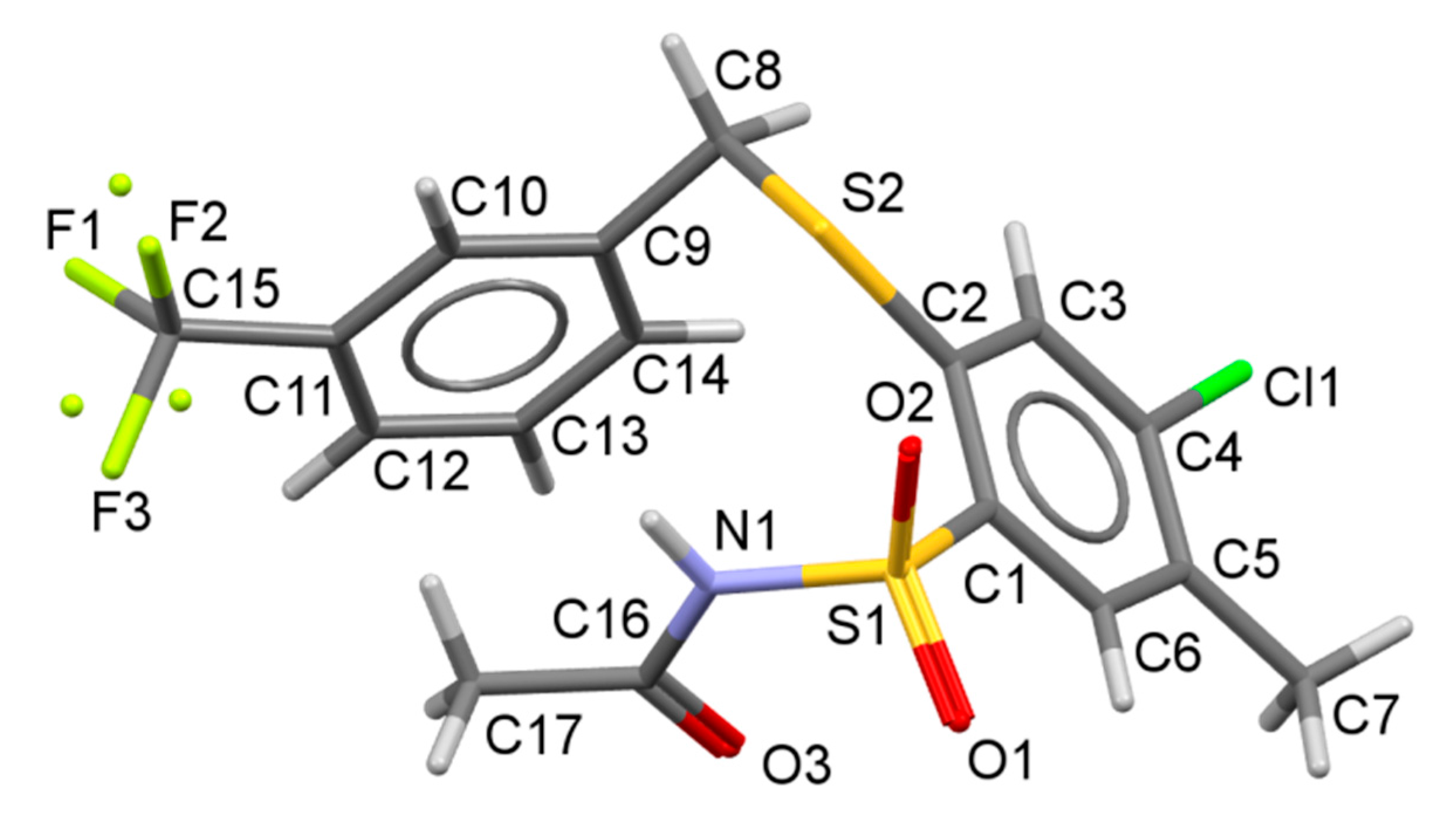
N-[4-Chloro-5-methyl-2-(3-(trifluoromethylbenzylthio)phenylsulfonyl]propionamide (34). Starting from 7 (0.459 g) and propionic acid (3.0 mL) the product was obtained after refluxing for 2 h, then evaporating the mixture under reduced pressure and treating of residue with water (10 mL). Crystallization from 80% ethanol gave the title compound 34 (0.307 g, 68%): mp 123‒127 °C; IR (KBr) 3242 (N–H), 2926, 2853 (CH3, CH2), 1726 (C=O), 1431 (N–H), 1326, 1128 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 0.87 (t, 3H, CH3CH2CO), 2.27 (q, 2H, CH2CO), 2.35 (s, 3H, CH3), 4.56 (s, 2H, CH2S), 7.60‒7.75 (m, 4H, H-3, arom.), 7.80 (m, 1H, arom.), 7.91 (s, 1H, H-6), 12.32 (s, 1H, NH) ppm; anal. C 47.94, H 3.82, N 3.15% calcd for C18H17ClF3NO3S2, C 47.84, H 3.79, N 3.10%.
N-[4-Chloro-5-methyl-2-(4-trifluoromethylbenzylthio)phenylsulfonyl]propionamide (35). Starting from 8 (0.451 g) and propionic acid (3.0 mL) the product was obtained after refluxing for 1 h, then evaporating the mixture under reduced pressure and treating of residue with water (10 mL). Crystallization from ethyl acetate/petroleum ether gave the title compound 35 (0.420 g, 93%): mp 107‒108 °C; IR (KBr) 3218 (N–H), 2988, 2952 (CH3, CH2), 1734 (C=O), 1325, 1122 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 0.86 (t, 3H, CH3CH2CO), 2.19 (q, 2H, CH2CO), 2.36 (s, 3H, CH3), 4.56 (s, 2H, CH2S), 7.66‒7.72 (m, 5H, H-3, arom.), 7.92 (s, 1H, H-6), 12.31 (s, 1H, NH) ppm; anal. C 47.96, H 3.81, N 3.16% calcd for C18H17ClF3NO3S2, C47.84, H 3.79, N 3.10%.
N-[4-Chloro-2-(4-chlorobenzylthio)-5-methylphenylsulfonyl]propionamide (36). Starting from 9 (0.338 g) and propionic acid (3.0 mL) the product was obtained after refluxing for 2 h, then evaporating the mixture under reduced pressure and treating of residue with water (10 mL). Crystallization from 96% ethanol gave the title compound 36 (0.284 g, 68%): mp 87‒91 °C; IR (KBr) 3116 (N–H), 2927, 2865 (CH3, CH2), 1719 (C=O), 1456 (N–H), 1319, 1132 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 0.88 (t, 3H, CH3CH2CO), 2,26 (q, 2H, CH2CO), 2,36 (s, 3H, CH3), 4,44 (s, 2H, CH2S), 7,36 (d, 2H, arom.), 7.44 (d, 2H, arom.), 7.67 (s, 1H, H-3), 7.91 (s, 1H, H-6), 12.27 (s, 1H, NH) ppm; anal. C 48.98, H 4.15, N 3.46% calcd for C17H17Cl2NO3S2, C 48.81, H 4.10, N 3.35%.
N-[4-Chloro-2-(4-methoxybenzylthio)-5-methylphenylsulfonyl]propionamide (37). Starting from 10 (0.421 g) and propionic acid (3.0 mL) the product was obtained after refluxing for 1 h, then evaporating the mixture under reduced pressure and treating of residue with water (10 mL). Crystallization from benzene gave the title compound 37 (0.352 g, 85%): mp 130‒132 °C; IR (KBr) 3268 (N–H), 3032 (C–H arom.), 2935, 2853 (CH3, CH2), 1732 (C=O), 1441 (N–H), 1349, 1175 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 0.89 (t, 3H, CH3CH2CO), 2.22 (q, 2H, CH2CO), 2.36 (s, 3H, CH3), 3.73 (s, 3H, OCH3), 4.37 (s, 2H, CH2S), 6.89 (d, 2H, arom.), 7.34 (d, 2H, arom.), 7.66 (s, 1H, H-3), 7.91 (s, 1H, H-6), 12.27 (s, 1H, NH); 13C-NMR (125 MHz, DMSO-d6) δ 8.92, 19.65, 29.22, 36.29, 55.75, 114.56, 128.20, 128.65, 129.00, 130.99, 132.97, 134.15, 135.13, 136.79, 139.55, 159.27, 172.75 ppm; anal. C 52.11, H 4.85, N 3.32% calcd for C18H20ClNO4S2, C 52.23, H 4.87, N 3.38%.
N-[4-Chloro-5-methyl-2-(pyridin-4-ylmethylthio)phenylsulfonyl]propionamide (38). Starting from 11 (0.392 g) and propionic acid (3.0 mL) the product was obtained after refluxing for 2 h 15 min, then evaporating the mixture under reduced pressure and treating of residue with water (10 mL). Crystallization from ethanol gave the title compound 38 (0.250 g, 65%): mp 151‒154 °C; IR (KBr) 3422 (N–H), 2925, 2852 (CH3, CH2), 1715 (C=O), 1349, 1150 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 0.88 (t, 3H, CH3CH2CO), 2.24 (q, 2H, CH2CO), 2.35 (s, 3H, CH3), 4.50 (s, 2H, CH2S), 7.44 (d, 2H, pyridine), 7.68 (s, 1H, H-3), 7.92 (s, 1H, H-6), 8.51 (d, 2H, pyridine), 12.38 (s, 1H, NH); 13C-NMR (125 MHz, DMSO-d6) δ 8.91, 19.66, 29.25, 35.25, 124.69, 128.87, 133.57, 134.29, 135.46, 135.59, 139.63, 146.34, 150.35, 172.80 ppm; anal. C 50.02, H 4.47, N 7.32% calcd for C16H17ClN2O3S2, C 49.93, H 4.45, N 7.28%.
N-[4-Chloro-5-methyl-2-(naphthalen-2-ylmethylthio)phenylsulfonyl]propionamide (39). Starting from 13 (0.441 g) and propionic acid (3.0 mL) the product was obtained after refluxing for 1.5 h, then evaporating the mixture under reduced pressure and treating of residue with water (10 mL). Crystallization from 80% ethanol gave the title compound 39 (0.373 g, 86%): mp 85‒88 °C; IR (KBr) 3207 (N–H), 3056 (C–H arom.), 2920, 2855 (CH3, CH2), 1731 (C=O), 1446 (N–H), 1321, 1176 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 0.84 (t, 3H, CH3CH2CO), 2.24 (q, 2H, CH2CO), 2.33 (s, 3H, CH3), 4.62 (s, 2H, CH2S), 7.49‒7.60 (m, 4H, arom.), 7.75 (s, 1H, H-3), 7.83‒7.94 (m, 4H, H-6, arom.), 12.30 (s, 1H, NH) ppm; anal. C 58.48, H 4.80, N 3.55% calcd for C21H20ClNO3S2, C 58.12, H 4.65, N 3.23%.
N-[4-Chloro-2-(2,3-dihydrobenzo[b][1,4]dioxin-2-ylmethylthio)-5-mehylphenylsulfonyl]propionamide (40). Starting from 15 (0.449 g) and propionic acid (3.0 mL) the product was obtained after refluxing with stirring for 1 h 45 min, then evaporating the mixture under reduced pressure and treating of residue with water (10 mL). Crystallization from ethanol gave the title compound 40 (0.384 g, 87%): mp 114‒116.2 °C; IR (KBr) 3224 (N–H), 2920, 2852 (CH3, CH2), 1730 (C=O), 1495 (N–H), 1270, 1136 (SO2) cm−1; 1H-NMR (500 MHz, DMSO-d6) δ 0.83 (t, 3H, CH3CH2CO), 2.14 (q, 2H, CH2CO), 2.34 (s, 3H, CH3), 3.36‒3.47 (m, 2H, CH2O), 4.06 (dd, 1H, CHO), 4.32 (d, 2H, CH2S), 6.70‒6.85 (d, 4H, arom.), 7.63 (s, 1H, H-3), 7.88 (s, 1H, H-6), 12.26 (s, 1H, NH) ppm; 13C-NMR (50 MHz, DMSO-d6) δ 9.11, 19.31, 29.95, 33.28, 66.23, 72.08, 117.17, 117.29, 121.66, 121.79, 128.82, 132.73, 133.23, 135.02, 137.93, 142.57, 142.98, 174.53 ppm; anal. C 51.73, H 4.58, N 3.22% calcd for C19H20ClNO5S2, C 51.64, H 4.56, N 3.17%.
N-[4-Chloro-2-(6-chlorobenzo[d][1,3]dioxol-5-ylmethylthio)-5-methylphenylsulfonyl]propionamide (41). Starting from 16 (0.469 g) and propionic acid (3.0 mL) the product was obtained after refluxing for 1 h, then evaporating the mixture under reduced pressure and treating of residue with water (10 mL). Crystallization from ethyl acetate/petroleum ether gave the title compound 41 (0.376 g, 81%): mp 142‒144 °C; IR (KBr) 3205 (N–H), 2922, 2898 (CH3, CH2), 1686 (C=O), 1474 (N–H), 1250, 1116 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 0.88 (t, 3H, CH3CH2CO), 2.23 (q, 2H, CH2CO), 2.38 (s, 3H, CH3), 4.39 (s, 2H, CH2S), 6.08 (s, 2H, CH2), 7.11 (s, 2H, arom.), 7.62 (s, 1H, H-3), 7.93 (s, 1H, H-6), 12.26 (s, 1H, NH); 13C-NMR (125 MHz, DMSO-d6) δ 8.87, 19.68, 29.20, 35.34, 102.90, 110.33, 111.13, 126.15, 126.89, 128.80, 133.38, 134.23, 135.33, 136.46, 139.66, 147.32, 148.48, 172.80 ppm; anal. C 46.70, H 3.70, N 3.01% calcd for C18H17ClNO5S2, C 46.76, H 3.71, N 3.03%.
N-[4-Chloro-2-(3,5-dimethylisoxazol-4-ylmethylthio)-5-methylphenylsulfonyl]propionamide (42). Starting from 17 (0.339 g) and propionic acid (3.0 mL) the product was obtained after refluxing for 1.5 h, then evaporating the mixture under reduced pressure and treating of residue with water (10 mL). Crystallization from ethanol/chloroform (v/v = 2:1) gave the title compound 42 (0.374 g, 68%): mp 160‒163 °C; IR (KBr) 3433 (N–H), 3064 (C–H arom.), 2935, 2859 (CH2, CH2), 1726 (C=O), 1455 (N–H), 1353, 1132 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 0.89 (t, 3H, CH3CH2CO), 2.17‒2.25 (m, 5H, CH2CO and CH3), 2.37 (s, 3H, CH3), 2.39 (s, 3H, CH3), 4.28 (s, 2H, CH2S), 7.71 (s, 1H, H-3), 7.94 (s, 1H, H-6), 12.28 (s, 1H, NH); 13C-NMR (125 MHz, DMSO-d6) δ 8.91, 10.31, 11.29, 19.69, 25.35, 29.15, 109.37, 129.65, 133.61, 134.18, 135.78, 136.14, 139.57, 160.22, 167.67, 172.78 ppm; anal. C 47.75, H 4.75, N 6.97% calcd for C16H19ClN2O4S2 C 47.70, H 4.75, N 6.95%.
N-(2-Benzylthio-4-chloro-5-methylphenylsulfonyl)isobutyramide (43). Starting from 6 (0.391 g) and isobutyric acid (3.7 mL) the product was obtained after refluxing for 2 h, then dropping water (10 mL) into the cooled reaction mixture. Crystallization from ethanol gave the title compound 43 (0.378 g, 95%): mp 122‒125 °C; IR (KBr) 3423 (N–H), 2926, 2868 (CH3, CH2), 1694 (C=O), 1433 (N–H), 1351, 1172 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 0.96 (d, 6H, CH3), 2.36 (s, 3H, CH3), 2.40‒2.60 (m, 1H, CH); 4.43 (s, 2H, CH2S), 7.20‒7.45 (m, 5H, arom.), 7.64 (s, 1H, H-3), 7.92 (s, 1H, H-6), 12.28 (s, 1H, NH) ppm; 13C-NMR (50 MHz, DMSO-d6) δ 24.14, 24.66, 39.81, 41.90, 133.18, 133.45, 134.23, 134.84, 138.00, 139.31, 140.03, 141.41, 141.81, 144.59, 180.90 ppm; anal. C 54.39, H 5.07, N 3.55% calcd for C18H20ClNO3S2, C 54.33, H 5.07, N 3.52%.
N-(2-Benzylthio-4-chloro-5-methylphenylsulfonyl)-3-cyclohexylpropionamide (44). Starting from 6 (0.338 g) and 3-cyclohexenepropionic acid (6.3 mL) the crude product was obtained after refluxing for 1.5 h at 115 °C. Crystallization from acetonitrile gave the title compound 44 (0.378 g, 81%): mp 118‒120 °C; IR (KBr) 3427 (N–H), 3060 (C–H arom.), 2923, 2840 (CH3, CH2), 1678 (C=O), 1448 (N–H), 1359, 1171 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 0.8‒0.98 (m, 2H, cHex.), 1.10‒1.20 (m, 4H, cHex.), 1.40 (q, 2H, CH2), 1.50‒1,80 (m, 5H, cHex.), 2.22 (t, 2H, CH2CO), 2.36 (s, 3H, CH3), 4.43 (s, 2H, CH2S), 7.20‒7.45 (m, 5H, arom.), 7.64 (s, 1H, H-3), 7.92 (s, 1H, H-6), 12.29 (s, 1H, NH) ppm; anal. C 59.19, H 6.04, N 2.97% calcd for C23H28ClNO3S2, C 59.27, H 6.06, N 3.01%.
N-(2-Benzylthio-4-chloro-5-methylphenylsulfonyl)benzamide (45). A mixture of 6 (0.391 g, 1 mmol) and benzoic acid (0.244 g, 2 mmol) in water (4 mL) was stirred in a pressure tube at 100 °C for 144 h. The product was isolated from the hot reaction mixture by filtration. After crystallization from 90% ethanol the title compound 45 was obtained (0.311 g, 72%): mp 168‒170 °C; IR (KBr): 3274 (N–H), 3064 (C–H arom.), 2918 (CH3, CH2), 1693 (C=O), 1453 (N–H), 1332, 1163 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 2.34 (s, 3H, CH3), 4.40 (s, 2H, CH2S), 7.10‒7.20 (m, 3H, arom.), 7.24‒7.36 (m, 2H, arom.), 7.45‒7.58 (m, 2H, arom.), 7.60‒7.72 (m, 2H, H-3 and arom.), 7.88‒7.98 (d, 2H, arom.), 8.02 (s, 1H, H-6), 12.82 (s, 1H, NH); 13C-NMR (125 MHz, DMSO-d6) δ 19.69, 36.65, 128.02, 128.66, 129.03, 129.23, 129.65, 131.98, 133.12, 133.98, 134.44, 135.16, 136.40, 136.66, 139.68, 165.86 ppm; anal. C 58.19, H 4.15, N 3.15% calcd for C21H18ClNO3S2, C 58.39, H 4.20, N 3.24%.
N-(2-Benzylthio-4-chloro-5-methylphenylsulfonyl)-3-phenylacrylamide (46). A mixture of 6 (0.391 g, 1 mmol) and cinnamic acid (0.296 g, 2 mmol) in water (6 mL) was stirred in a pressure tube at 100 °C for 96 h. The product was isolated from the hot reaction mixture by filtration. After crystallization from 80% ethanol the title compound 46 was obtained (0.348 g, 76%): mp 173‒174 °C; IR (KBr) 3199 (N–H), 2919, 2854 (CH3, CH2), 1675 (C=O), 1439 (N–H), 1353, 1131 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 2.37 (s, 3H, CH3), 4.43 (s, 2H, CH2S), 6.74 (d, 1H, CH=), 7.16‒7.24 (m, 3H, arom.), 7.34‒7.68 (m, 9H, arom.), 7.99 (s, 1H, H-6), 12.54 (s, 1H, NH) ppm; 13C-NMR (50 MHz, DMSO-d6) δ 19.25, 36.37, 118.99, 127.59, 128.42, 128.49, 128.62, 129.31, 129.43, 131.04, 132.77, 133.97, 134.13, 134.79, 136.08, 139.26, 144.23, 163.43 ppm; anal. C 60.30, H 4.40, N 3.05% calcd for C23H20ClNO3S2, C 60.32, H 4.40, N 3.06%.
N-[4-Chloro-5-methyl-2-(naphthalen-1-ylmethylthio)phenylsulfonyl]-3-phenylacrylamide (47). A mixture of 12 (0.450 g, 1 mmol) and cinnamic acid (0.148 g, 1.1 mmol) in toluene (5 mL) was stirred in a pressure tube at 110‒120 °C for 120 h. A solvent was evaporated under reduced pressure to dryness and residue was treated with water (150 mL) and stirred using an ultrasonic bath for 15 min. Product was isolated by extraction with chloroform (3 × 50 mL), then the combined organic layers were washed with 1% NaHCO3 (2 × 50 mL), 1 M HCl (50 mL), water (2 × 50 mL) and dried with MgSO4. The solvent was evaporated under reduced pressure and residue was purified by crystallization from ethanol giving the titled compound 47 (0.254 g, 50%): mp 105‒106 °C; IR (KBr) 3192 (N–H), 2923, 2853 (CH3, CH2), 1683 (C=O), 1448 (N–H), 1351, 1178 (SO2) cm−1; 1H-NMR (200 MHz, DMSO-d6) δ 2.42 (s, 3H, CH3), 4.90 (s, 2H, CH2S), 6.58 (d, 1H, CH=), 7.35‒7.64 (m, 10H, CH= and arom.), 7.77 (s, 1H, H-3), 7.82‒7.94 (m, 2H, arom.), 8.03 (s, 1H, H-6), 8.18 (d, 1H, arom.), 12.40 (s, 1H, NH); 13C-NMR (125 MHz, DMSO-d6) δ 19.70, 35.22, 119.30, 124.81, 126.09, 126.57, 127.00, 128.79, 128.90, 129.09, 129.15, 129.79, 131.47, 131.72, 132.01, 133.13, 134.08, 134.32, 134.48, 134.83, 137.35, 139.81, 144.59, 163.87 ppm; anal. C 63.70, H 4.33, N 2.69% calcd for C27H22ClNO3S2, C 63.83, H 4.36, N 2.76%.



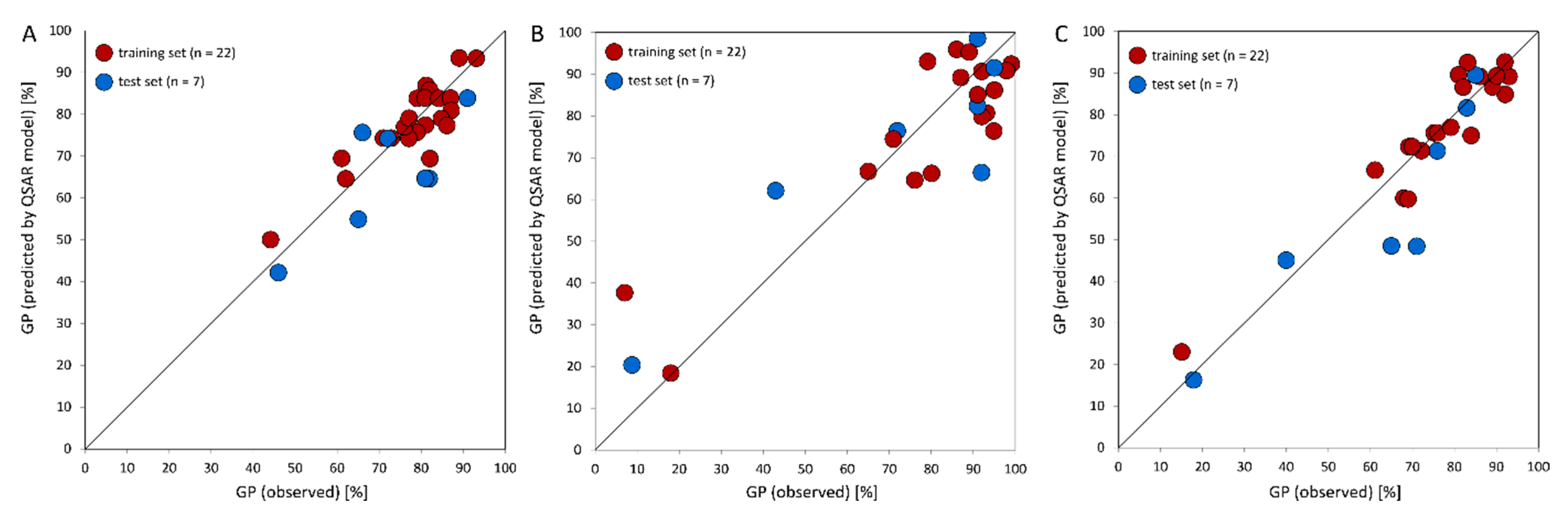
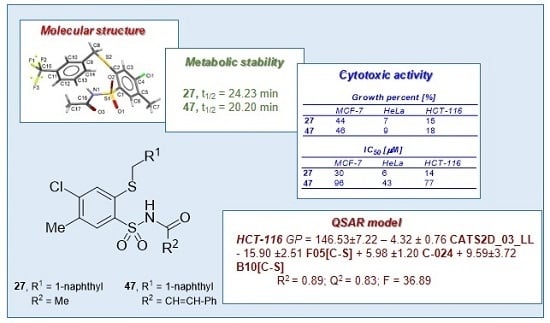
 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}