
Synthesis, Hydrolysis, and Protonation-Promoted Intramolecular Reductive Breakdown of Potential NRTIs: Stavudine α-P-Borano-γ-P-N-l-tryptophanyltriphosphates

Abstract
:
1. Introduction
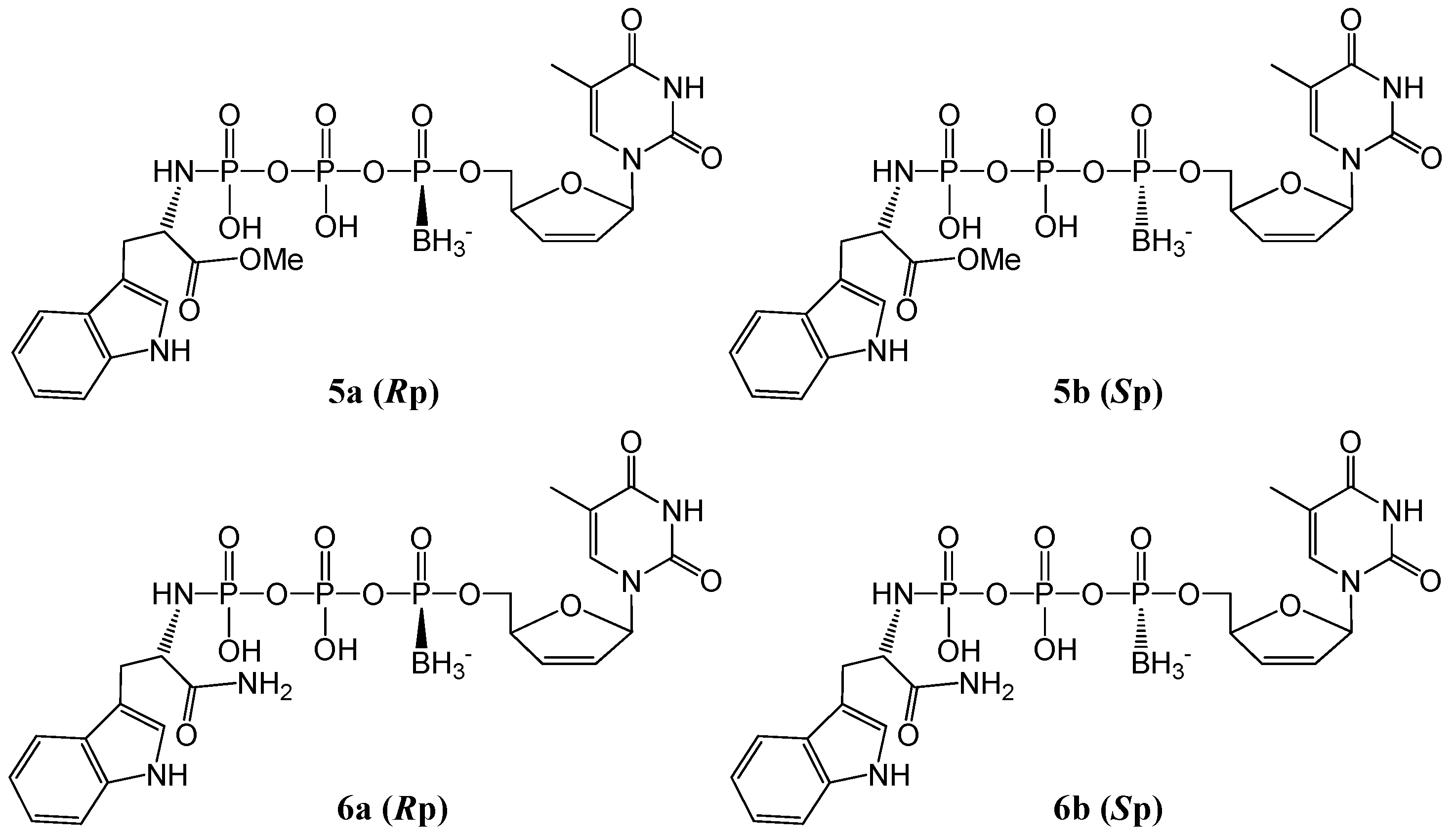
2. Results and Discussion
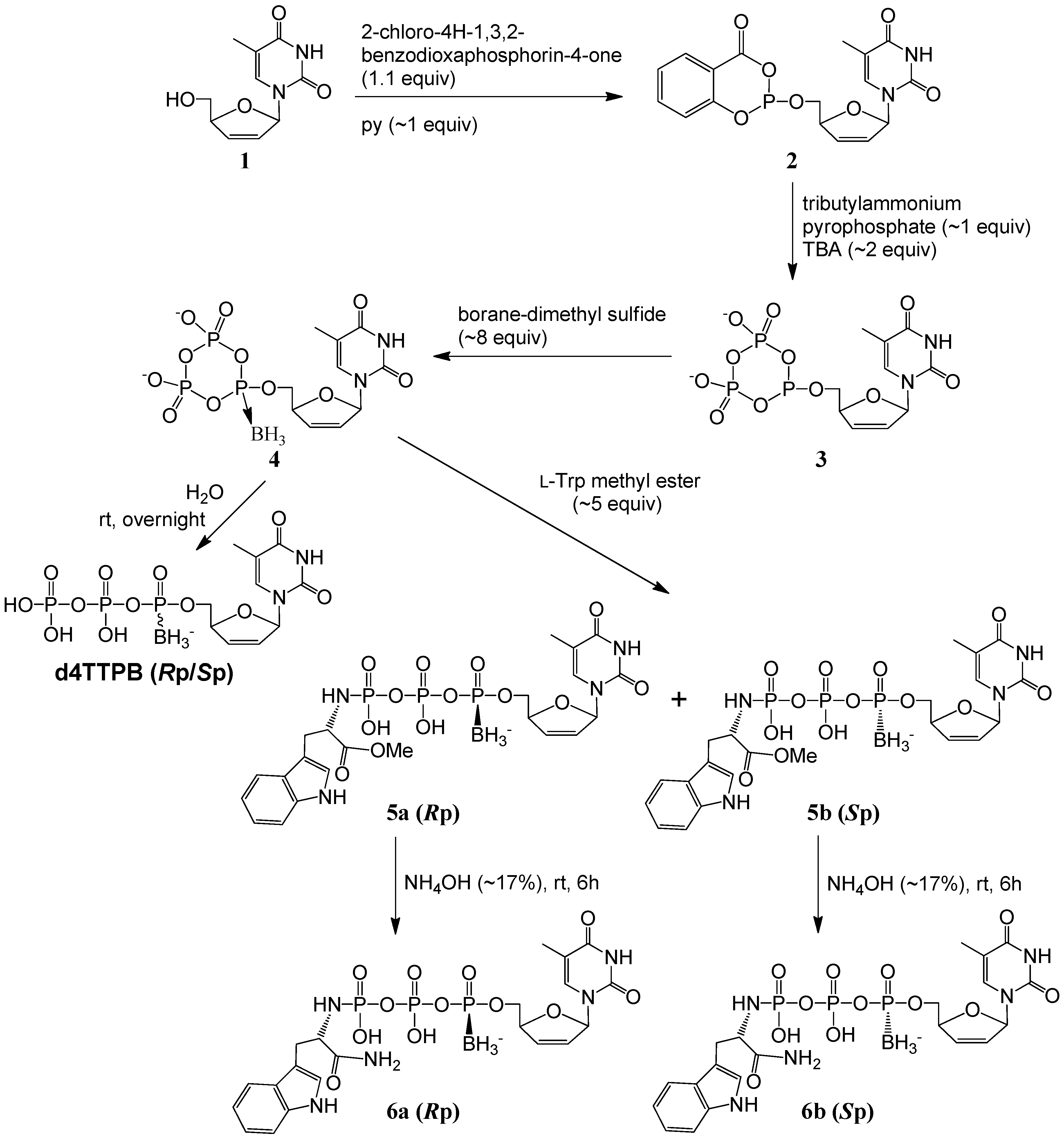
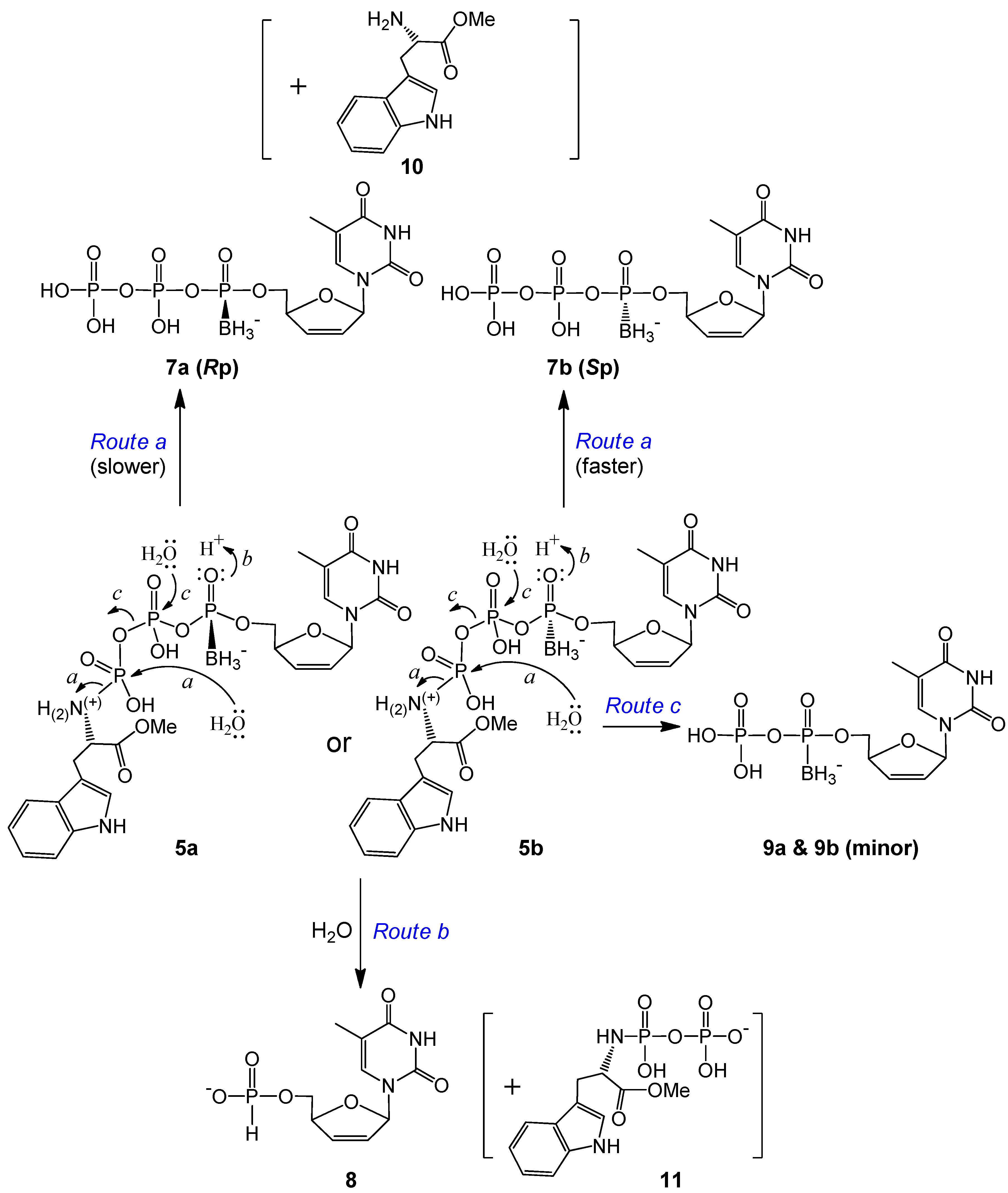
2.1. Synthesis and Isolation
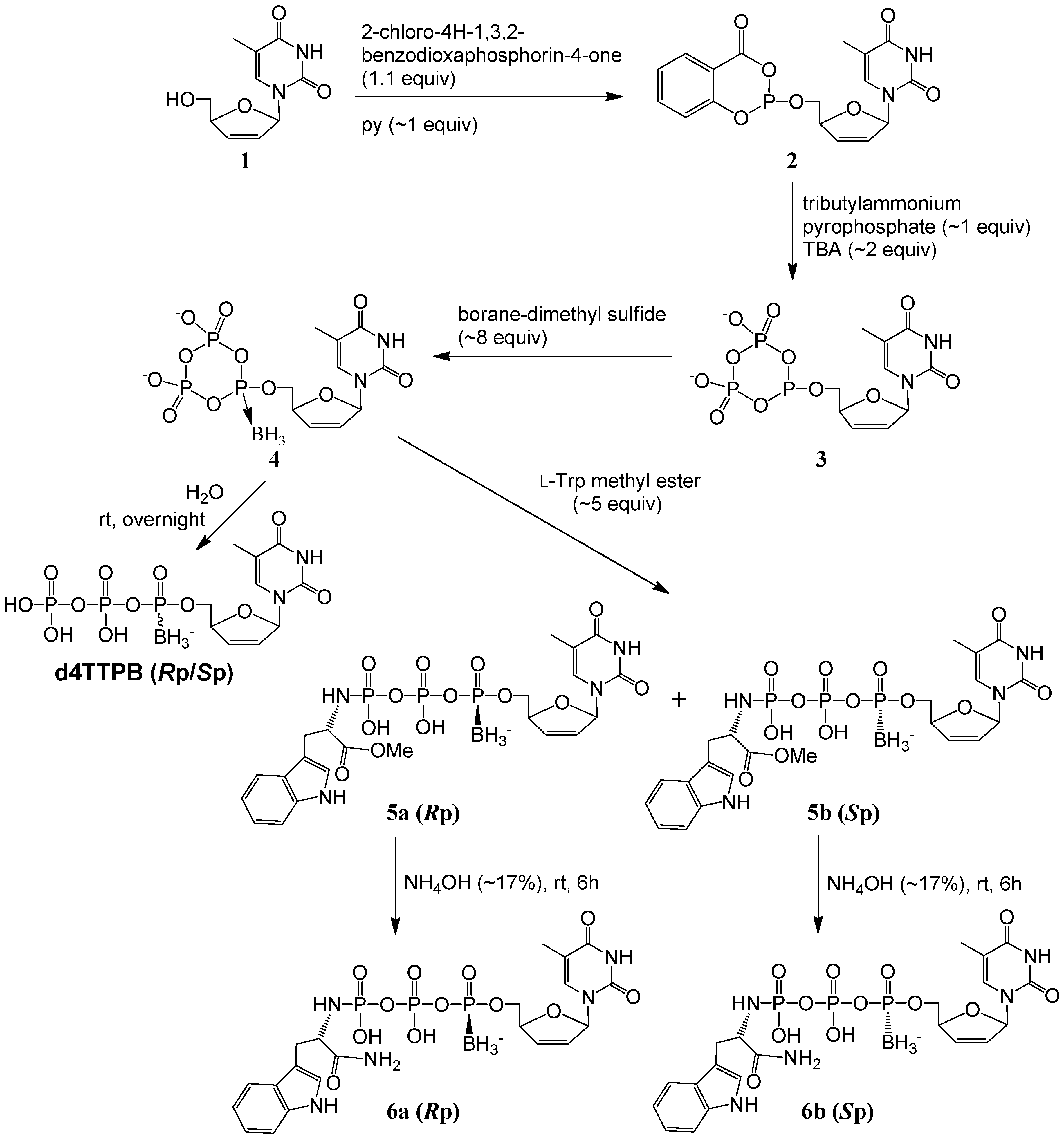

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| d4TTPαB Analog | MW by LC-MS | % Yield a | HPLC b | |
|---|---|---|---|---|
| ACN (%) | tR (min) | |||
| 5a | 661 | ~30 | 15 | 7.9 |
| 5b | 661 | 15 | 11 | |
| 6a | 646 | ~12 | 15 | 4.1 |
| 6b | 646 | 15 | 4.4 | |
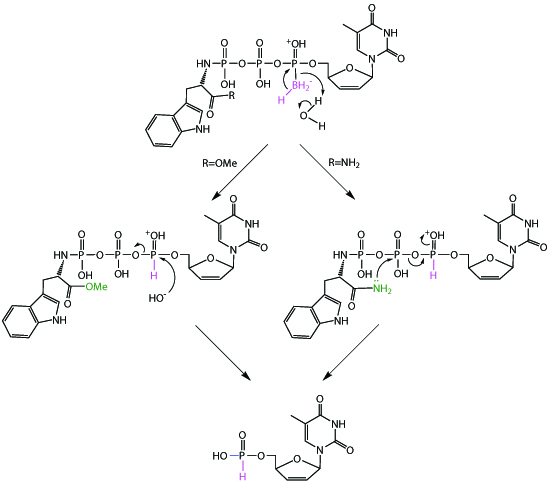
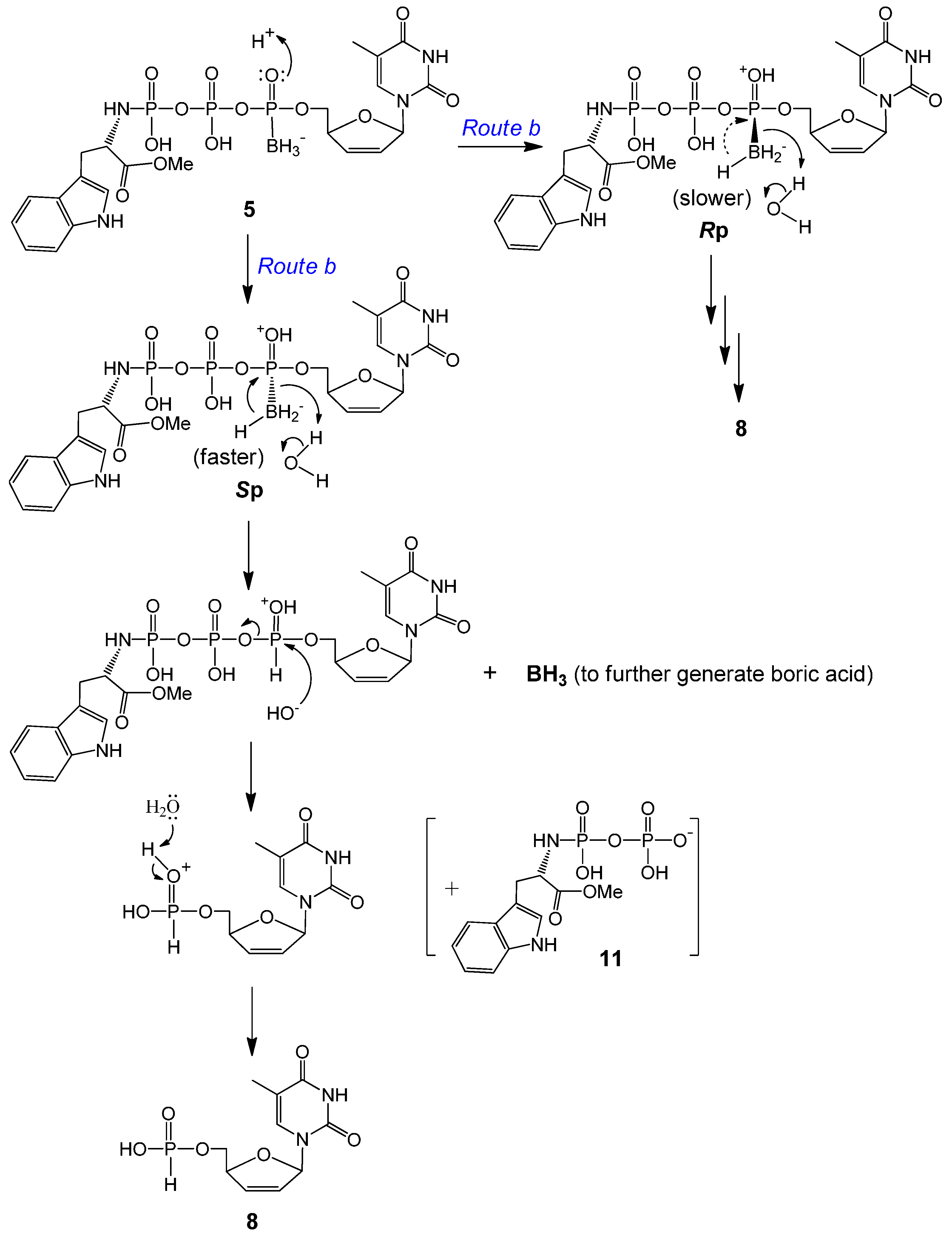
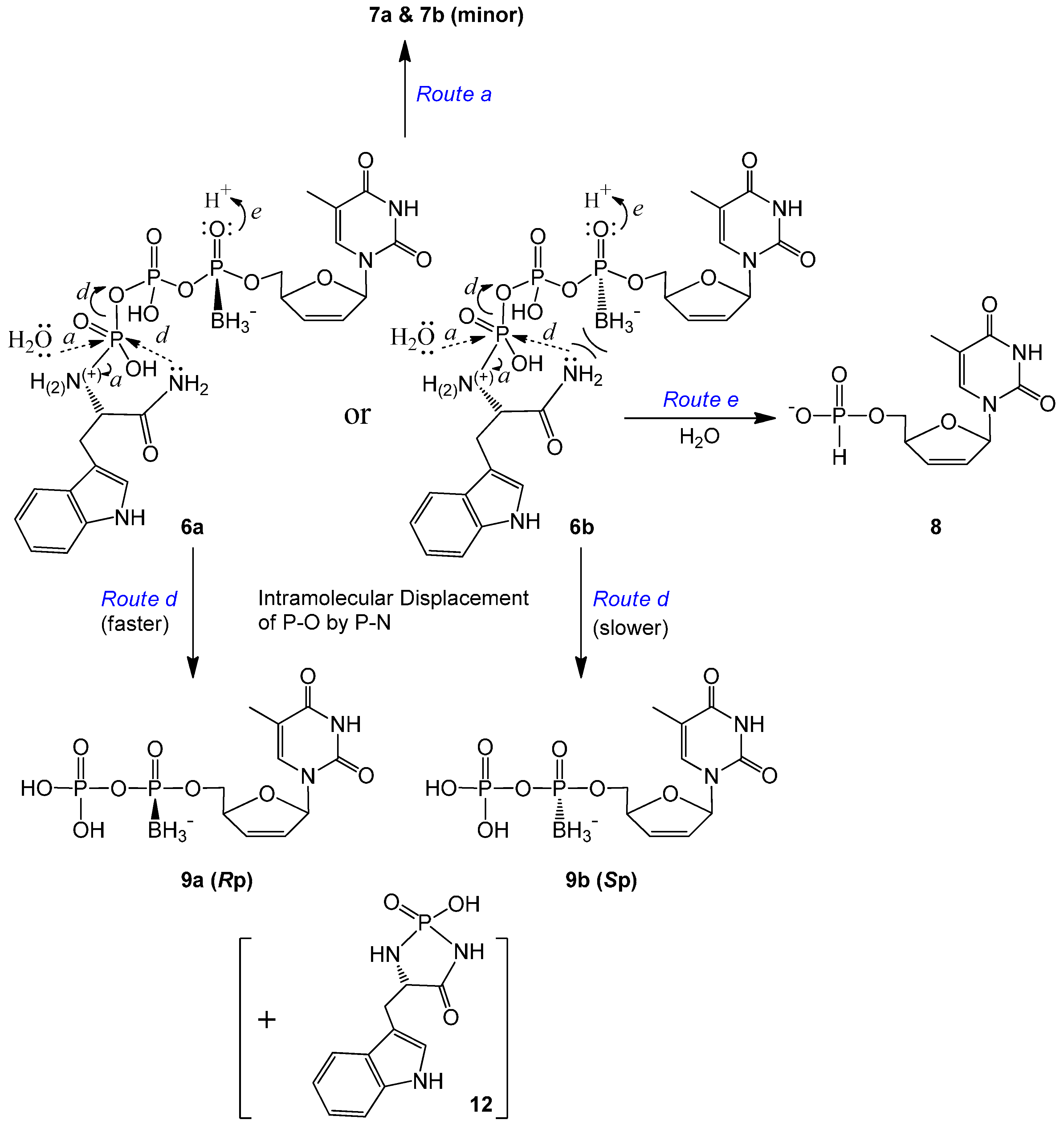
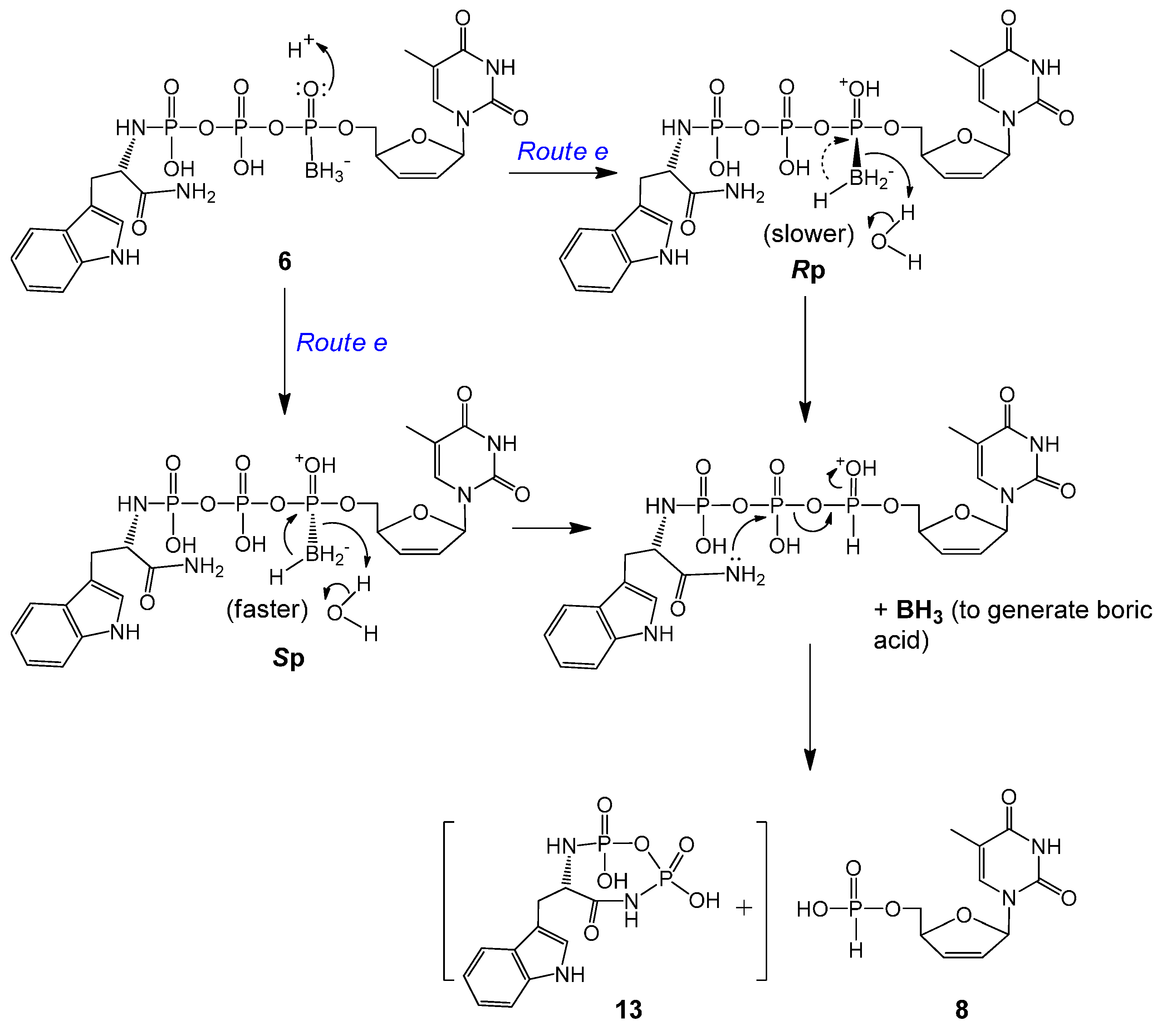
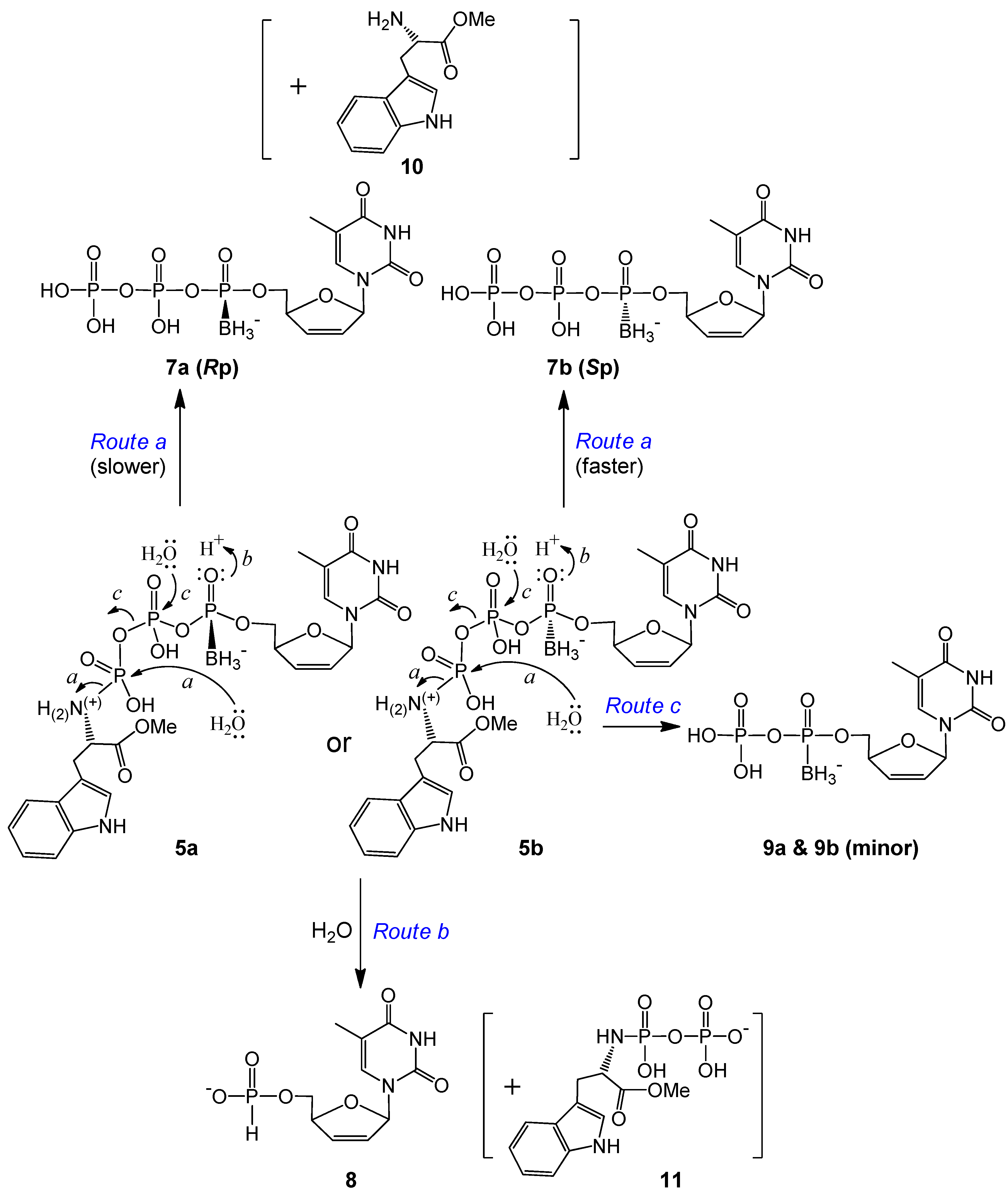
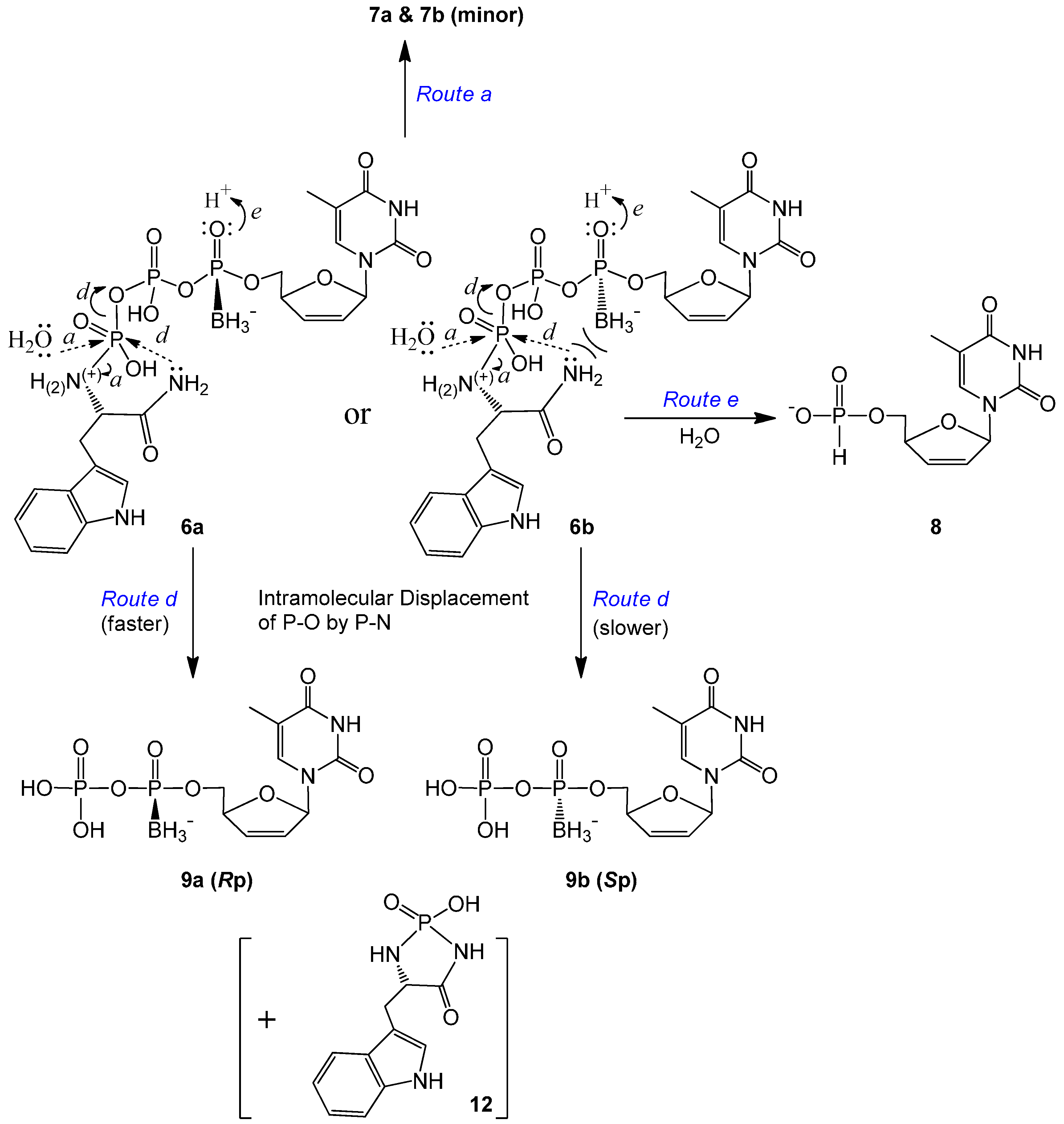
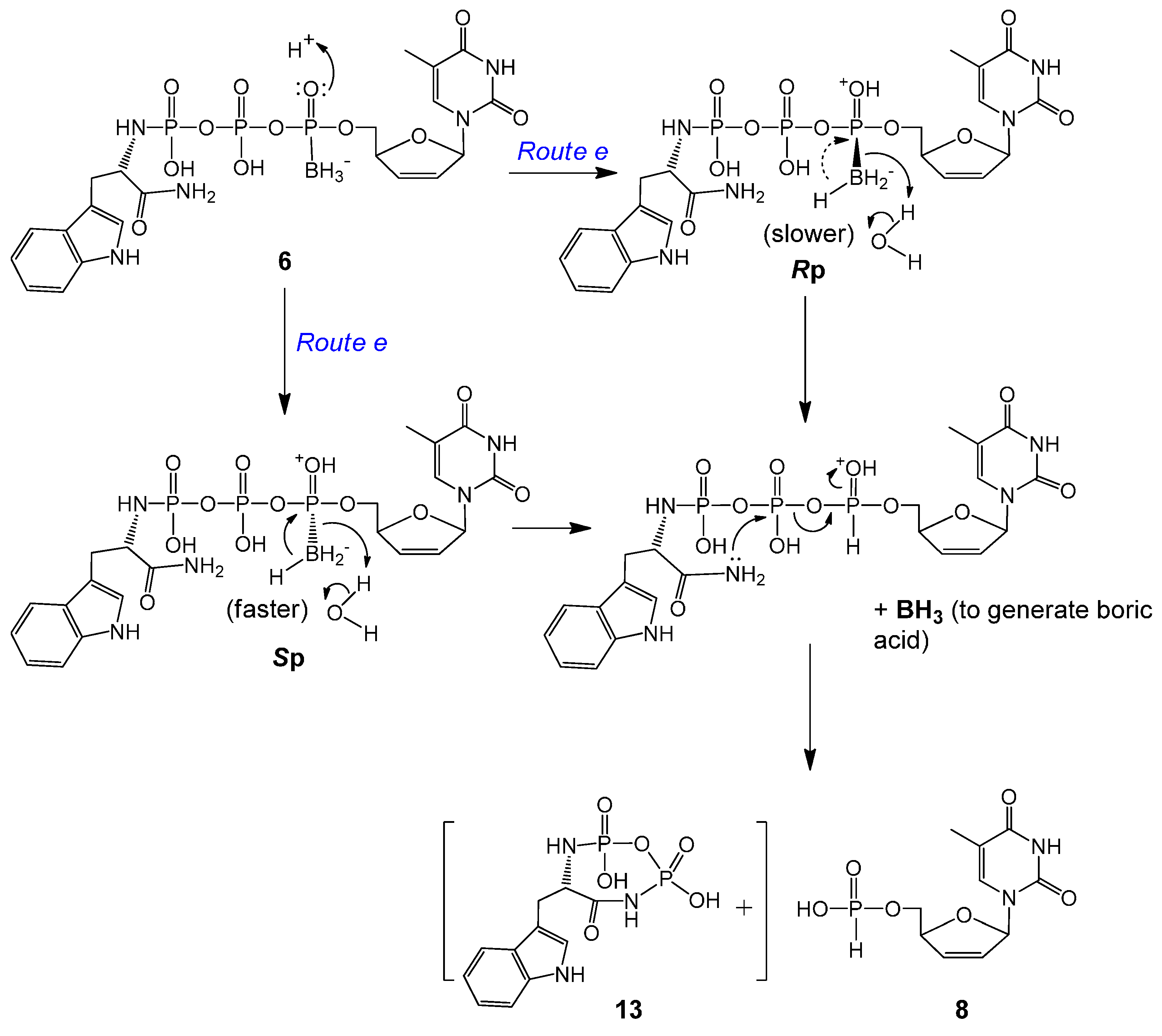
2.2. Stability Studies
| d4TTPαB Analog | t1/2 (d) | Major Degradation Products (a%, b% *) | % of Intact Molecule ǂ |
|---|---|---|---|
| 5a | 35.9 | d4TTPαB Rp 7a (1.8%, 7.1%) | ~81% |
| d4TH-P 8 (2.3%, 2.8%) | |||
| 5b | 26.6 | d4TTPαB Sp 7b (8.8, 16%) | ~68% |
| d4TH-P 8 (6.7%, 7.9%) | |||
| 6a | 5.0 | d4TDPαB Rp 9a (>14%, >39%) | ~24% |
| d4TH-P 8 (2.6%, 4.9%) | |||
| 6b | 6.4 | d4TDPαB Sp 9b (~13%, ~31%) | ~30% |
| d4TH-P 8 (2.6%, ~8%) |

3. Experimental Section
3.1. General Information
3.2. Procedures for the Synthesis and Isolation of 5a, 5b, 6a, 6b, and d4T α-P-Boranotriphosphates
3.3. LC-MS Analysis of Degradation Products from d4T Triphosphate Mimics
3.4. Spectroscopic Data
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wagner, C.R.; Iyer, V.V.; McIntee, E.J. Pronucleotides: Toward the in vivo delivery of antiviral and anticancer nucleotides. Med. Res. Rev. 2000, 20, 417–451. [Google Scholar] [CrossRef]
- Jessen, H.J.; Schulz, T.; Balzarini, J.; Meier, C. Bioreversible protection of nucleoside diphosphates. Angew. Chem. Int. Ed. 2008, 47, 8719–8722. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.S.; Hostetler, K.Y. Application of kinase bypass strategies to nucleoside antivirals. Antivir. Res. 2011, 92, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Sofia, M.J. Nucleotide prodrugs for HCV therapy. Antivir. Chem. Chemother. 2011, 22, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Pradere, U.; Garnier-Amblard, E.C.; Coats, S.J.; Amblard, F.; Schinazi, R.F. Synthesis of nucleoside phosphate and phosphonate prodrugs. Chem. Rev. 2014, 114, 9154–9218. [Google Scholar] [CrossRef] [PubMed]
- Weinschenk, L.; Gollnest, T.; Schols, D.; Balzarini, J.; Meier, C. Bis(benzoyloxybenzyl)-DiPPro nucleoside diphosphates of anti-HIV active nucleoside analogues. ChemMedChem 2015, 10, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z. A review on the chemical synthesis of pyrophosphate bonds in bioactive nucleoside diphosphate analogs. Bioorg. Med. Chem. Lett. 2015, 25, 3777–3783. [Google Scholar] [CrossRef] [PubMed]
- Meier, C. Pro-nucleotides—Recent advances in the design of efficient tools for the delivery of biologically active nucleoside monophosphates. Synlett 1998, 3, 233–242. [Google Scholar] [CrossRef]
- Furman, P.A.; Murakami, E.; Niu, C.; Lam, A.M.; Espiritu, C.; Bansal, S.; Bao, H.; Tolstykh, T.; Steuer, H.M.; Keilman, M.; et al. Activity and the metabolic activation pathway of the potent and selective hepatitis C virus pronucleotide inhibitor PSI-353661. Antivir. Res. 2011, 91, 120–132. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Harris, S.A.; Daluge, S.M.; Gudmundsson, K.S.; McLean, E.W.; Burnette, T.C.; Marr, H.; Hazen, R.; Condreay, L.D.; Johnson, L.; et al. Application of phosphoramidate pronucleotide technology to abacavir leads to a significant enhancement of antiviral potency. J. Med. Chem. 2005, 48, 3504–3515. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Hassan-Abdallah, A.; Srinivasan, S.; Wang, Y.; Siddiqui, A.; Daluge, S.M.; Gudmundsson, K.S.; Zhou, H.; McLean, E.W.; Peckham, J.P.; et al. Application of phosphoramidate protide technology significantly improves antiviral potency of carbocyclic adenosine derivatives. J. Med. Chem. 2006, 49, 7215–7226. [Google Scholar] [CrossRef] [PubMed]
- Cho, A.; Zhang, L.; Xu, J.; Lee, R.; Butler, T.; Metobo, S.; Aktoudianakis, V.; Lew, W.; Ye, H.; Clarke, M.; et al. Discovery of the first C-nucleoside HCV polymerase inhibitor (GS-6620) with demonstrated antiviral response in HCV infected patients. J. Med. Chem. 2014, 57, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Cahard, D.; McGuigan, C.; Balzarini, J. Aryloxy phosphoramidate triesters as protides. Mini-Rev. Med. Chem. 2004, 4, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Drontle, D.P.; Wagner, C.R. Designing a pronucleotide stratagem: Lessons from amino acid phosphoramidates of anticancer and antiviral pyrimidines. Mini-Rev. Med. Chem. 2004, 4, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.; Goerbig, U.; Mueller, C.; Balzarini, J. cycloSal-PMEA and cycloAmb-PMEA: Potentially new phosphonate prodrugs based on the cycloSal-pronucleotide approach. J. Med. Chem. 2005, 48, 8079–8086. [Google Scholar] [CrossRef] [PubMed]
- Vukadinovic, D.; Boege, N.P.H.; Balzarini, J.; Meier, C. “Lock-in” modified cycloSal nucleotides—The second generation of cycloSal prodrugs. Nucleosides Nucleotides Nucleic Acids 2005, 24, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Maiti, M.; Maiti, M.; Rozenski, J.; de Jonghe, S.; Herdewijn, P. Aspartic acid based nucleoside phosphoramidate prodrugs as potent inhibitors of hepatitis C virus replication. Org. Biomol. Chem. 2015, 13, 5158–5174. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.F.; Shaw, B.R.; Spielvogel, B.F. Boron-containing nucleic acids. Syntheysis of oligodeoxynucleoside boranophosphates. J. Am. Chem. Soc. 1990, 112, 9000–9001. [Google Scholar] [CrossRef]
- Summers, J.S.; Roe, D.; Boyle, P.D.; Colvin, M.; Shaw, B.R. Structural studies of a borane-modified phosphate diester linkage: Ab initio calculations on the dimethylboranophosphate anion and the single-crystal X-ray structure of its diisopropylammonium salt. Inorg. Chem. 1998, 37, 4158–4159. [Google Scholar] [CrossRef] [PubMed]
- Shaw, B.R.; Wan, J.; Wang, X.; Dobrikov, M.; He, K.; Porter, K.; Lin, J.L.; Rait, V.; Sergueev, D.; Sergueeva, Z.A. Reading and writing genetic information with boranophosphate (borane phosphate) mimics of nucleotides, DNA, and RNA. Collect. Symp. Ser. 2002, 5, 169–180. [Google Scholar]
- Shaw, B.R.; Dobrikov, M.; Wang, X.; Wan, J.; He, K.; Lin, J.L.; Li, P.; Rait, V.; Sergueeva, Z.A.; Sergueev, D. Reading, writing, and modulating genetic information with boranophosphate mimics of nucleotides, DNA, and RNA. Ann. N. Y. Acad. Sci. 2003, 1002, 12–29. [Google Scholar] [CrossRef] [PubMed]
- Dobrikov, M.I.; Sergueeva, Z.A.; Shaw, B.R. Incorporation of (α-P-borano)-2ʹ,3ʹ-dideoxycytidine 5′-triphosphate into DNA by drug-resistant MMLV reverse transcriptase and Taq DNA polymerase. Nucleosides Nucleotides Nucleic Acids 2003, 22, 1651–1655. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.W.; Briley, J.D.; Shaw, B.R. Direct PCR sequencing with boronated nucleotides. Nucleic Acids Res. 1997, 25, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.; Schneider, B.; Sarfati, S.; Deville-Bonne, D.; Guerreiro, C.; Boretto, J.; Janin, J.; Véron, M.; Canard, B. Structural basis for activation of α-boranophosphate nucleotide analogues targeting drug-resistant reverse transcriptase. EMBO J. 2000, 19, 3520–3529. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Meyer, P.; Sarfati, S.; Mulard, L.; Guerreiro, C.; Boretto, J.; Janin, J.; Veron, M.; Deville-Bonne, D.; Canard, B. Activation of anti-reverse transcriptase nucleotide analogs by nucleoside diphosphate kinase: Improvement by α-boranophosphate substitution. Nucleosides Nucleotides Nucleic Acids 2001, 20, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Sergueeva, Z.A.; Dobrikov, M.; Shaw, B.R. Nucleoside and oligonucleoside boranophosphates: Chemistry and properties. Chem. Rev. 2007, 107, 4746–4796. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Caruthers, M. Synthesis of DNA/RNA and their analogs via phosphoramidite and H-phosphonate chemistries. Molecules 2013, 18, 14268–14284. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Boyle, N.; Chen, F.; Rajappan, V.; Fagan, P.; Brooks, J.L.; Hurd, T.; Leeds, J.M.; Rajwanshi, V.K.; Jin, Y.; et al. Synthesis of AZT 5′-triphosphate mimics and their inhibitory effects on HIV-1 reverse transcriptase. J. Med. Chem. 2004, 47, 6902–6913. [Google Scholar] [CrossRef] [PubMed]
- Hacker, S.M.; Mex, M.; Marx, A. Synthesis and stability of phosphate modified ATP analogues. J. Org. Chem. 2012, 77, 10450–10454. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.; Eckstein, F.J. Rapid and efficient synthesis of nucleoside 5′-O-(1-thiotriphosphates), 5′-triphosphates and 2′,3′-cyclophosphorothioates using 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one. Org. Chem. 1989, 54, 631–635. [Google Scholar] [CrossRef]
- Krzyzanowska, B.K.; He, K.; Hasan, A.; Shaw, B.R. A convenient synthesis of 2′-deoxyribonucleoside 5′-(α-P-borano)triphosphates. Tetrahedron 1998, 54, 5119–5128. [Google Scholar] [CrossRef]
- Boyle, N.A.; Rajwanshi, V.K.; Prhavc, M.; Wang, G.; Fagan, P.; Chen, F.; Ewing, G.J.; Brooks, J.L.; Hurd, T.; Leeds, J.M.; et al. Synthesis of 2′,3′-dideoxynucleoside 5′-α-P-borano-β,γ-(difluoromethylene)triphosphates and their inhibition of HIV-1 reverse transcriptase. J. Med. Chem. 2005, 48, 2695–2700. [Google Scholar] [CrossRef] [PubMed]
- Boyle, N.A.; Fagan, P.; Brooks, J.L.; Prhavc, M.; Lambert, J.; Cook, P.D. 2′,3′-Dideoxynucleoside 5′-β,γ-(difluoromethylene) triphosphates with α-P-thio or α-P-seleno modifications: Synthesis and their inhibition of HIV-1 reverse transcriptase. Nucleosides Nucleotides Nucleic Acids 2005, 24, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Xu, Z.; Liu, H.; Wennefors, C.K.; Dobrikov, M.I.; Ludwig, J.; Shaw, B.R. Synthesis of α-P-modified nucleoside diphosphates with ethylenediamine. J. Am. Chem. Soc. 2005, 127, 16782–16783. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shaw, B.R. Synthesis, chemical and enzymatic studies on α-P-modified nucleoside-γ-P-N-tryptophanyltriphosphates. In Proceedings of the 232nd ACS National Meeting, San Francisco, CA, USA, 10–14 September 2006.
- Han, Q.; Gaffney, B.L.; Jones, R.A. One-flask synthesis of dinucleoside tetra- and pentaphosphates. Org. Lett. 2006, 8, 2075–2077. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Sarafianos, S.G.; Arnold, E.; Parniak, M.A.; Gaffney, B.L.; Jones, R.A. Synthesis of AZTpSpCX2ppSA and AZTpSpCX2ppSAZT: Hydrolysis-resistant potential inhibitors of the AZT excision reaction of HIV-1 RT. Org. Lett. 2007, 9, 5243–5246. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Sarafianos, S.G.; Arnold, E.; Parniak, M.A.; Gaffney, B.L.; Jones, R.A. Synthesis of boranoate, selenoate, and thioate analogs of AZTp4A and Ap4A. Tetrahedron 2009, 65, 7915–7920. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mohamady, S.; Taylor, S.D. Synthesis of nucleoside 5′-tetraphosphates containing terminal fluorescent labels via activated cyclic trimetaphosphate. J. Org. Chem. 2014, 79, 2308–2313. [Google Scholar] [CrossRef] [PubMed]
- Ora, M.; Ojanperä, J.; Lönnberg, H. Hydrolytic reactions of thymidine 5′-O-phenyl-N-alkylphosphoramidates, models of nucleoside 5′-monophosphate prodrugs. Chem. Eur. J. 2007, 13, 8591–8599. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Porter, K.; Huang, F.; Shaw, B.R. Boron-containing oligodeoxyribonucleotide 14mer duplexes: Enzymatic synthesis and melting studies. Nucleic Acids Res. 1995, 23, 4495–4501. [Google Scholar] [CrossRef] [PubMed]
- Major, D.T.; Nahum, V.; Wang, Y.F.; Reiser, G.; Fischer, B. Molecular recognition in purinergic receptors. 2. Diastereoselectivity of the h-P2Y1-receptor. J. Med. Chem. 2004, 47, 4405–4416. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.S.; Shaw, B.R. Boranophosphates as mimics of natural phosphodiesters in DNA. Curr. Med. Chem. 2001, 8, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z.; Shaw, B.R. Synthesis, Hydrolysis, and Protonation-Promoted Intramolecular Reductive Breakdown of Potential NRTIs: Stavudine α-P-Borano-γ-P-N-l-tryptophanyltriphosphates. Molecules 2015, 20, 18808-18826. https://doi.org/10.3390/molecules201018808
Xu Z, Shaw BR. Synthesis, Hydrolysis, and Protonation-Promoted Intramolecular Reductive Breakdown of Potential NRTIs: Stavudine α-P-Borano-γ-P-N-l-tryptophanyltriphosphates. Molecules. 2015; 20(10):18808-18826. https://doi.org/10.3390/molecules201018808
Chicago/Turabian StyleXu, Zhihong, and Barbara Ramsay Shaw. 2015. "Synthesis, Hydrolysis, and Protonation-Promoted Intramolecular Reductive Breakdown of Potential NRTIs: Stavudine α-P-Borano-γ-P-N-l-tryptophanyltriphosphates" Molecules 20, no. 10: 18808-18826. https://doi.org/10.3390/molecules201018808
APA StyleXu, Z., & Shaw, B. R. (2015). Synthesis, Hydrolysis, and Protonation-Promoted Intramolecular Reductive Breakdown of Potential NRTIs: Stavudine α-P-Borano-γ-P-N-l-tryptophanyltriphosphates. Molecules, 20(10), 18808-18826. https://doi.org/10.3390/molecules201018808