
Therapeutic Oligonucleotides Targeting Liver Disease: TTR Amyloidosis

Abstract
:1. Introduction
2. TTR Amyloidosis
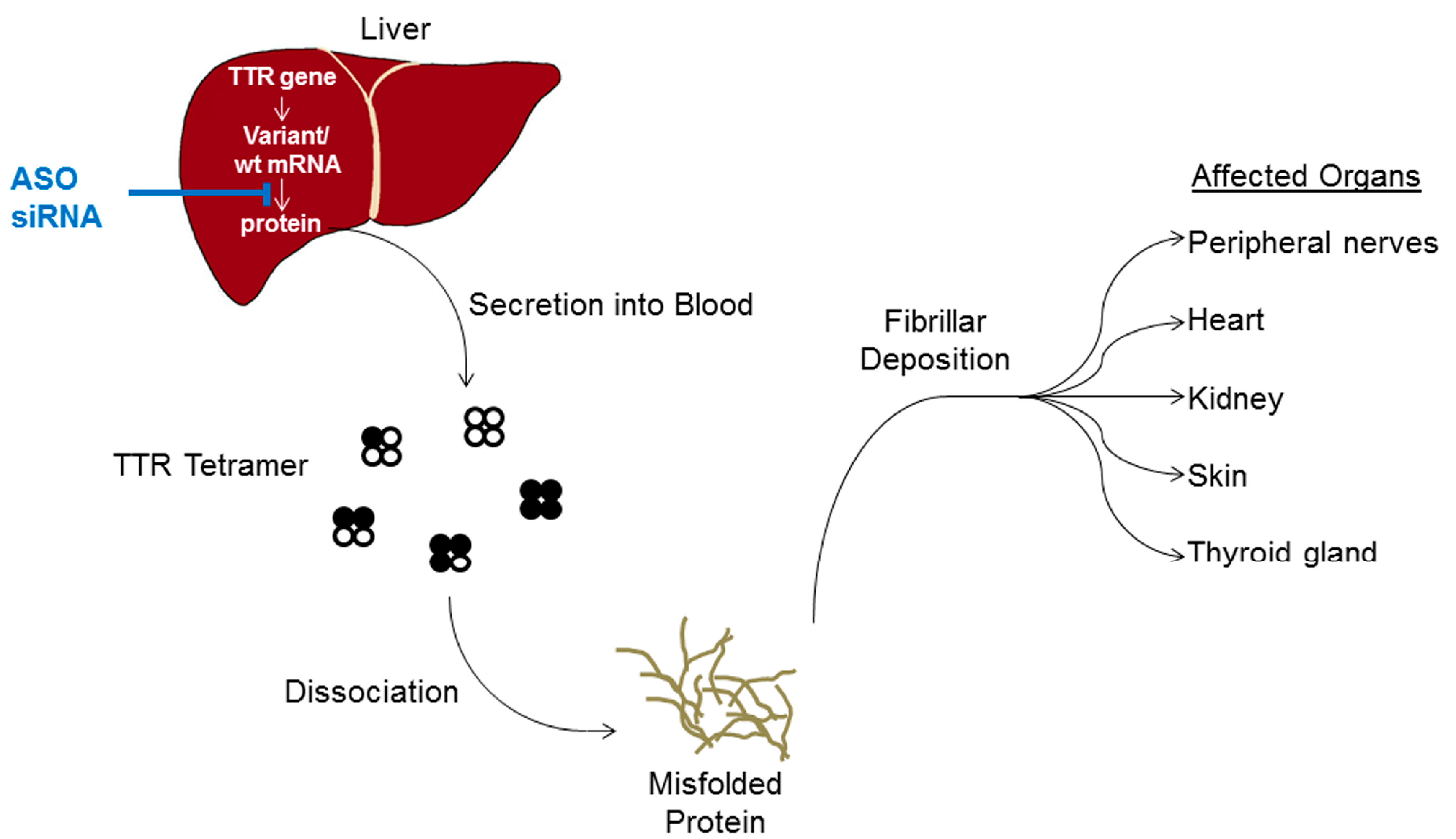
3. Molecular Mechanism of Antisense Oligonucleotides
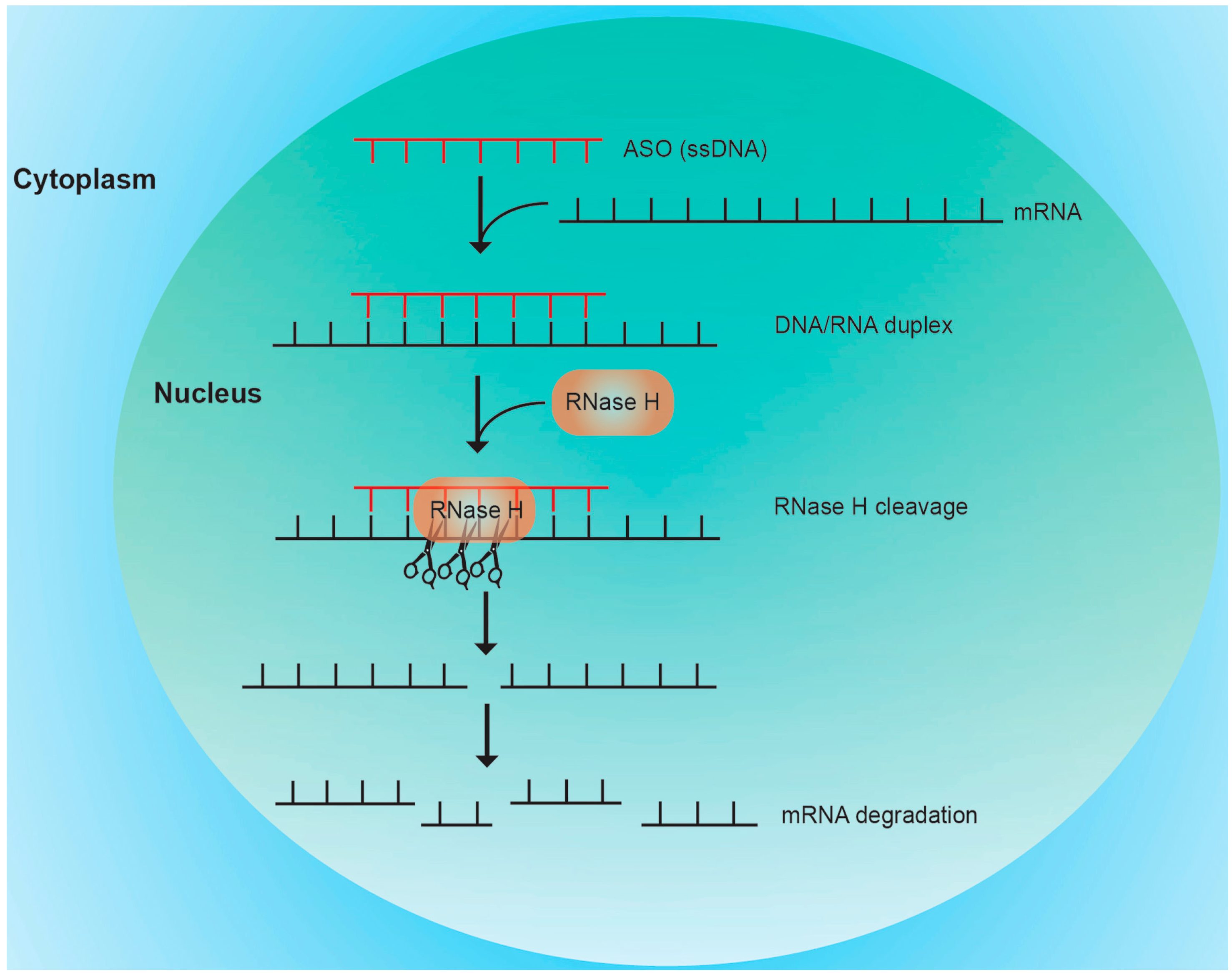
3.1. RNase H Mediated Cleavage
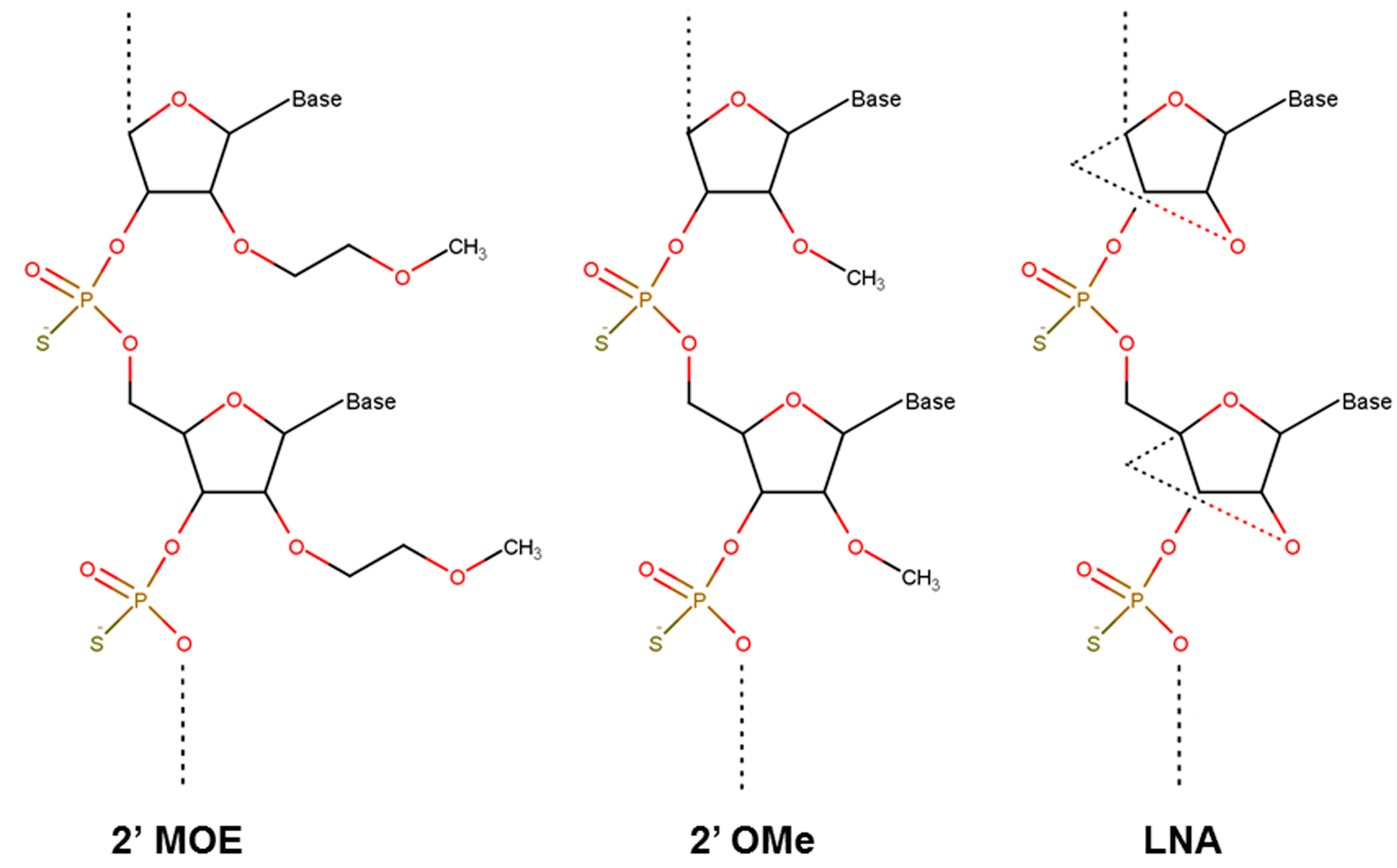
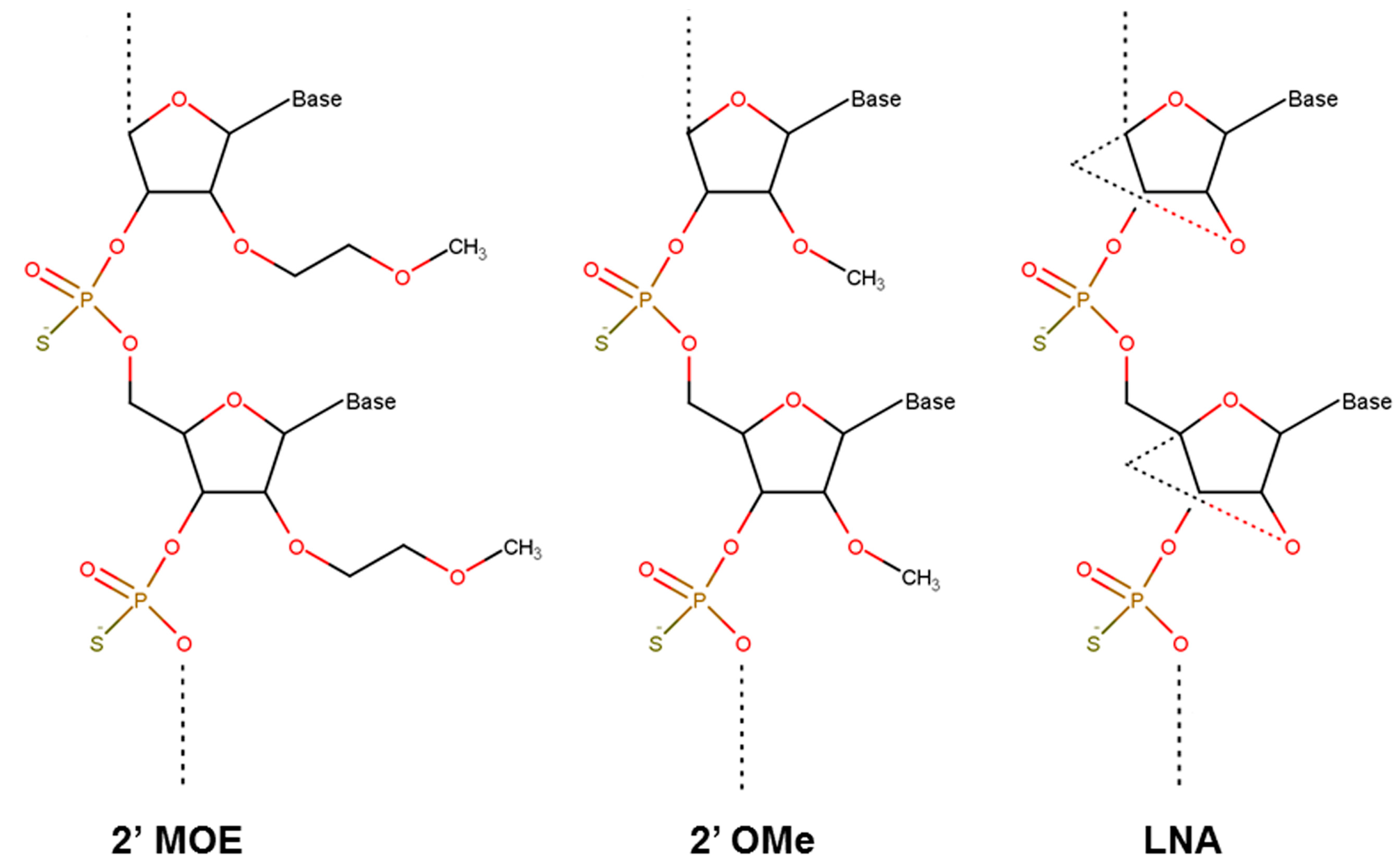
3.2. Nucelotide Modification
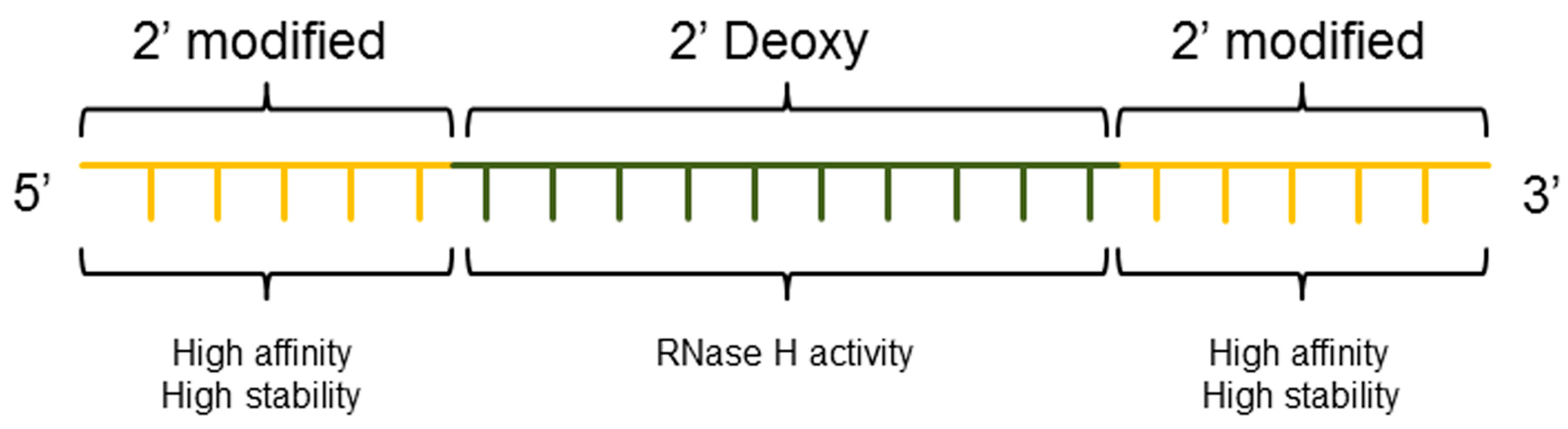
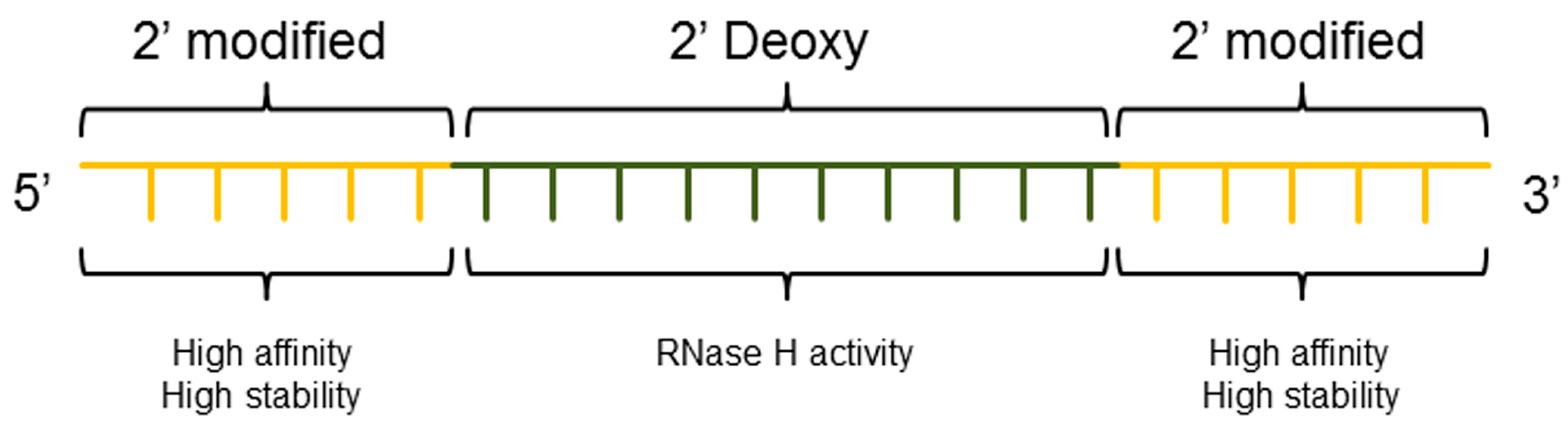
3.3. ASO Gapmer
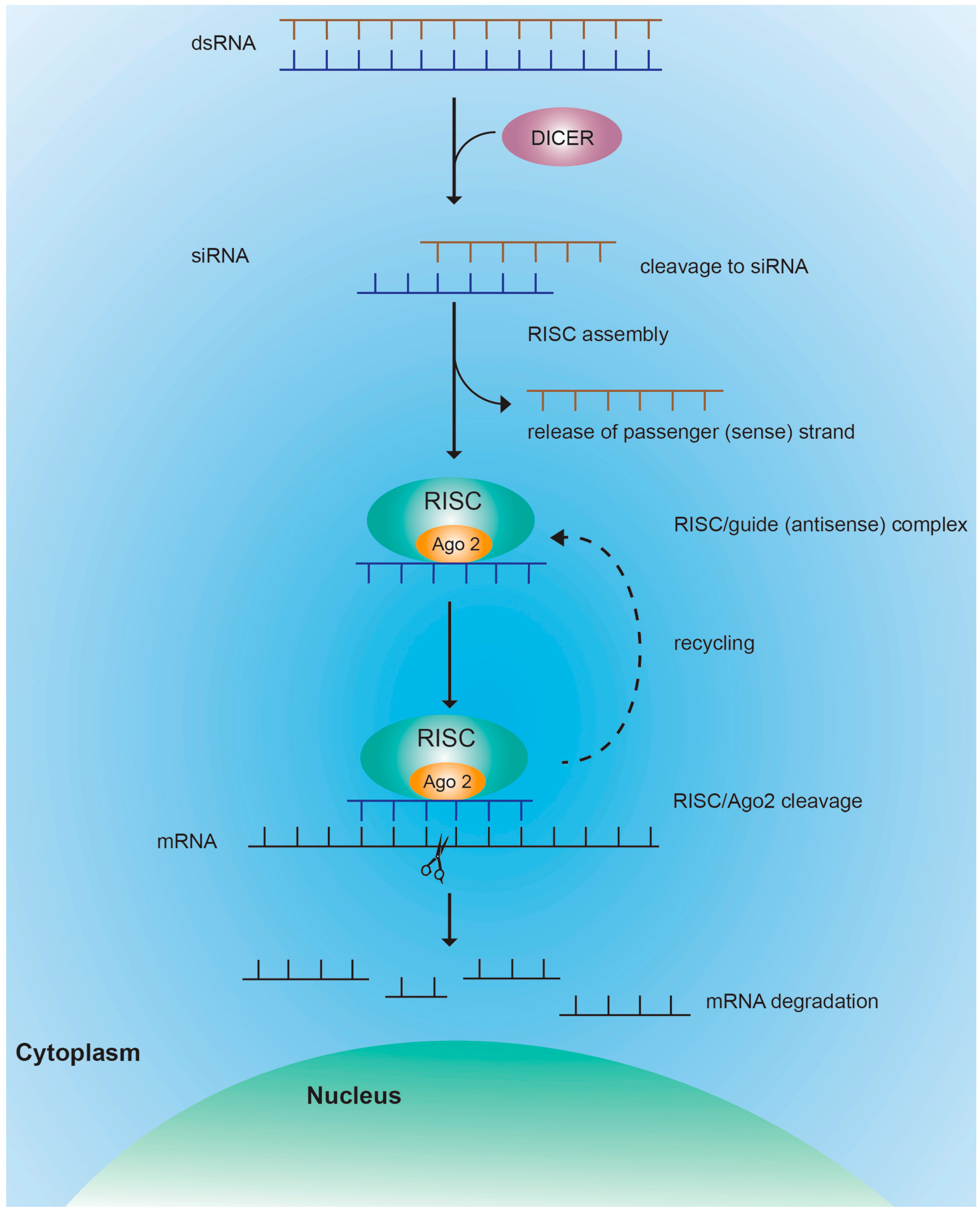
4. RNAi-Mediated Gene Silencing Using siRNAs
4.1. RISC Mediated Cleavage
4.2. Modification of siRNA
5. Delivery of Oligonucleotides to the Liver: An Attractive Target for Therapeutic Oligonucleotides
5.1. Biological Barriers for Liver Targeted Oligonucleotides
5.2. Molecular Strategies for Specific Liver Targeting
6. Clinical Studies Employing ASO Directed against Human TTR
Alternate Clinical Target of TTR ASO
7. Clinical Studies Employing siRNA Directed against Human TTR
GalNac Modification of TTR siRNA
8. Concluding Remarks

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ISIS-TTRRx (ISIS Pharmaceuticals) | ALN-TTR02 (Alnylam Pharmaceuticals) | ALN-TTRSC (Alnylam Pharmaceuticals) | |
|---|---|---|---|
| mRNA target | 3′ UTR | 3′ UTR | 3′ UTR |
| Oligoncleotide | DNA | RNA | RNA |
| Nucleotide modification | PS, 2′-MOE | LNP | GalNAc |
| mRNA degradation | RNase H-dependent | RISC | RISC |
| Primary site of action | nucleus | cytoplasm | cytoplasm |
| Administration | subcutaneous | systemic infusion | subcutaneous |
| Premedication | No | Yes | No |
| Study start-estimated completion | 12/2012–11/2016 a | 11/2013–01/2017 b | 12/2014–12/2018 c |
| Dosing | weekly 300 mg (3 doses first week) | 0.3 mg/kg every 3 weeks | weekly 500 mg (5 doses first week) |
| Serum TTR knockdown | ~80% d | ~80% d | ~80% d |
| Disease | FAP, FAC, SSA | FAP | FAC |
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Ago 2 | Argonaute 2 |
| ALT | Alanine transaminase |
| ASO | Antisense oligonucleotide |
| ASPGR | Asialoglycoprotein receptor |
| CDM | Carboxylated dimethyl maleic acid |
| CPPs | Cell-penetrating peptides |
| ds | Doublestranded |
| FAP; FAC; SSA | Familial amyloid polyneuropathy/cardiomyopathy; senile systemic amyloidosis |
| GalNAc | N-acetylgalactosamine |
| i.p. | Intraperitoneal |
| IRR | infusion-related reactions |
| i.v. | Intravenous |
| kDa | Kilodalton |
| LDL | Low-density lipoprotein |
| LNA | Locked nucleic acid |
| miRNA | Micro RNA |
| NAG | N-acetyl galactosamine |
| ncRNAs | Non-coding RNAs |
| 2′-OMe | 2′-O-methyl |
| 2′-MOE | 2′-O-methoxyethyl |
| PD | Pharmacodynamic |
| PEG | Polyethylene glycol |
| PK | Pharmacokinetic |
| PS | Phosphorothioate |
| RBP | retinol binding protein |
| RES | Reticuloendothelial system |
| RISC | RNA-induced silencing complex |
| RNA | Ribonucleic acid |
| RNAi | RNA interference |
| s.c. | Subcutaneous |
| siRNA | Small interfering RNA |
| SNALP | Stable nucleic-acid-particle |
| TLRs | Toll-like receptors |
| Tm | Melting temperature |
| TTR | Transthyretin |
References
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Goodchild, J. Therapeutic oligonucleotides. Methods Mol. Biol. 2011, 764, 1–15. [Google Scholar] [PubMed]
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.; Mattick, J.S. Non-coding RNAs: Regulators of disease. J. Pathol. 2010, 220, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Hrdlickova, B.; de Almeida, R.C.; Borek, Z.; Withoff, S. Genetic variation in the non-coding genome: Involvement of micro-RNAs and long non-coding RNAs in disease. Biochim. Biophys. Acta 2014, 1842, 1910–1922. [Google Scholar] [CrossRef] [PubMed]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Perez, B.; Vilageliu, L.; Grinberg, D.; Desviat, L.R. Antisense mediated splicing modulation for inherited metabolic diseases: Challenges for delivery. Nucleic Acid Ther. 2014, 24, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, R.P.; Velmeshev, D.; Faghihi, M.A. De-repressing LncRNA-Targeted Genes to Upregulate Gene Expression: Focus on Small Molecule Therapeutics. Mol. Ther. Nucleic Acids 2014, 3, e196. [Google Scholar] [CrossRef] [PubMed]
- Modarresi, F.; Faghihi, M.A.; Lopez-Toledano, M.A.; Fatemi, R.P.; Magistri, M.; Brothers, S.P.; van der Brug, M.P.; Wahlestedt, C. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat. Biotechnol. 2012, 30, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Lorenzer, C.; Dirin, M.; Winkler, A.M.; Baumann, V.; Winkler, J. Going beyond the liver: Progress and challenges of targeted delivery of siRNA therapeutics. J. Control. Release 2015, 203, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, A.; Vaishnaw, A.; Fitzgerald, K. Liver as a target for oligonucleotide therapeutics. J. Hepatol. 2013, 59, 1354–1359. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.L.; McCray, P.B., Jr. Current prospects for RNA interference-based therapies. Nat. Rev. Genet. 2011, 12, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.G.; Crosby, J.; Baker, B.F.; Graham, M.J.; Crooke, R.M. Antisense technology: An emerging platform for cardiovascular disease therapeutics. J. Cardiovasc. Transl. Res. 2013, 6, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. Is RNAi dead? Mol. Ther. 2011, 19, 1001–1002. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.D.; Raal, F.J.; Donovan, J.M.; Cromwell, W.C. Mipomersen preferentially reduces small low-density lipoprotein particle number in patients with hypercholesterolemia. J. Clin. Lipidol. 2015, 9, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, T.H.; Lammert, F.; Thompson, R.J. Genetics of liver disease: From pathophysiology to clinical practice. J. Hepatol. 2015, 62, S6–S14. [Google Scholar] [CrossRef]
- Plante-Bordeneuve, V.; Kerschen, P. Transthyretin familial amyloid polyneuropathy. Handb. Clin. Neurol. 2013, 115, 643–658. [Google Scholar] [PubMed]
- Andrade, C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 1952, 75, 408–427. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.P.; Figueira, A.S.; Bravo, F.R. Amyloid fibril protein related to prealbumin in familial amyloidotic polyneuropathy. Proc. Natl. Acad. Sci. USA 1978, 75, 4499–4503. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.J.; Costa, P.P.; Birken, S.; Goodman, D.S. Presence of an abnormal transthyretin (prealbumin) in Portuguese patients with familial amyloidotic polyneuropathy. Trans. Assoc. Am. Physicians 1983, 96, 261–270. [Google Scholar] [PubMed]
- Holmgren, G.; Costa, P.M.; Andersson, C.; Asplund, K.; Steen, L.; Beckman, L.; Nylander, P.O.; Teixeira, A.; Saraiva, M.J.; Costa, P.P. Geographical distribution of TTR met30 carriers in northern Sweden: Discrepancy between carrier frequency and prevalence rate. J. Med. Genet. 1994, 31, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Araki, S.; Ando, Y. Transthyretin-related familial amyloidotic polyneuropathy-Progress in Kumamoto, Japan (1967–2010). Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.; Andersson, R.; Drugge, U.; Holmgren, G.; Sandgren, O. Familial amyloidotic polyneuropathy in Sweden: Geographical distribution, age of onset, and prevalence. Hum. Hered. 1993, 43, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Kincaid, J.C. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 2007, 36, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Spiekerman, A.M. Nutritional assessment (protein nutriture). Anal. Chem. 1995, 67, 429R–436R. [Google Scholar] [CrossRef] [PubMed]
- Episkopou, V.; Maeda, S.; Nishiguchi, S.; Shimada, K.; Gaitanaris, G.A.; Gottesman, M.E.; Robertson, E.J. Disruption of the transthyretin gene results in mice with depressed levels of plasma retinol and thyroid hormone. Proc. Natl. Acad. Sci. USA 1993, 90, 2375–2379. [Google Scholar] [CrossRef] [PubMed]
- Palha, J.A.; Hays, M.T.; Morreale de Escobar, G.; Episkopou, V.; Gottesman, M.E.; Saraiva, M.J. Transthyretin is not essential for thyroxine to reach the brain and other tissues in transthyretin-null mice. Am. J. Physiol. 1997, 272, E485–E493. [Google Scholar] [PubMed]
- Saraiva, M.J. Transthyretin amyloidosis: A tale of weak interactions. FEBS Lett. 2001, 498, 201–203. [Google Scholar] [CrossRef]
- Merlini, G.; Bellotti, V. Molecular mechanisms of amyloidosis. N. Engl. J. Med. 2003, 349, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Ferrao-Gonzales, A.D.; Souto, S.O.; Silva, J.L.; Foguel, D. The preaggregated state of an amyloidogenic protein: Hydrostatic pressure converts native transthyretin into the amyloidogenic state. Proc. Natl. Acad. Sci. USA 2000, 97, 6445–6550. [Google Scholar] [CrossRef] [PubMed]
- Gorevic, P.D.; Prelli, F.C.; Wright, J.; Pras, M.; Frangione, B. Systemic senile amyloidosis. Identification of a new prealbumin (transthyretin) variant in cardiac tissue: Immunologic and biochemical similarity to one form of familial amyloidotic polyneuropathy. J. Clin. Investig. 1989, 83, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Quarta, C.C.; Guidalotti, P.L.; Longhi, S.; Pettinato, C.; Leone, O.; Ferlini, A.; Biagini, E.; Grigioni, F.; Bacchi-Reggiani, M.L.; Lorenzini, M.; et al. Defining the diagnosis in echocardiographically suspected senile systemic amyloidosis. JACC Cardiovasc. Imaging 2012, 5, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, D.R.; Gorevic, P.D.; Buxbaum, J.N. A homozygous transthyretin variant associated with senile systemic amyloidosis: Evidence for a late-onset disease of genetic etiology. Am. J. Hum. Genet. 1990, 47, 127–136. [Google Scholar] [PubMed]
- Tanskanen, M.; Peuralinna, T.; Polvikoski, T.; Notkola, I.L.; Sulkava, R.; Hardy, J.; Singleton, A.; Kiuru-Enari, S.; Paetau, A.; Tienari, P.J.; et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in α2-macroglobulin and tau: A population-based autopsy study. Ann. Med. 2008, 40, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, D.R.; Pastore, R.; Pool, S.; Malendowicz, S.; Kane, I.; Shivji, A.; Embury, S.H.; Ballas, S.K.; Buxbaum, J.N. Revised transthyretin Ile 122 allele frequency in African-Americans. Hum. Genet. 1996, 98, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Ranlov, I.; Alves, I.L.; Ranlov, P.J.; Husby, G.; Costa, P.P.; Saraiva, M.J. A Danish kindred with familial amyloid cardiomyopathy revisited: Identification of a mutant transthyretin-methionine111 variant in serum from patients and carriers. Am. J. Med. 1992, 93, 3–8. [Google Scholar] [CrossRef]
- Zeldenrust, S.R. Genotype-phenotype correlation in FAP. Amyloid 2012, 19 (Suppl. S1), 22–24. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.G.; Ikeda, S.; Lewis, W.D.; Obici, L.; Plante-Bordeneuve, V.; Rapezzi, C.; et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J. Rare Dis. 2013, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Seca, M.; Ferreira, N.; Coelho, T. Vitreous amyloidosis as the presenting symptom of familial amyloid polyneuropathy TTR Val30Met in a portuguese patient. Case Rep. Ophthalmol. 2014, 5, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Quarta, C.C.; Riva, L.; Longhi, S.; Gallelli, I.; Lorenzini, M.; Ciliberti, P.; Biagini, E.; Salvi, F.; Branzi, A. Transthyretin-related amyloidoses and the heart: A clinical overview. Nat. Rev. Cardiol. 2010, 7, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Nashan, B.; Propsting, M.J.; Nakazato, M.; Flemming, P.; Kubicka, S.; Boker, K.; Pichlmayr, R.; Manns, M.P. Familial Amyloidotic Polyneuropathy: Domino liver transplantation. J. Hepatol. 1999, 30, 293–298. [Google Scholar] [CrossRef]
- Ando, Y.; Tanaka, Y.; Nakazato, M.; Ericzon, B.G.; Yamashita, T.; Tashima, K.; Sakashita, N.; Suga, M.; Uchino, M.; Ando, M. Change in variant transthyretin levels in patients with familial amyloidotic polyneuropathy type I following liver transplantation. Biochem. Biophys. Res. Commun. 1995, 211, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Wixner, J.; Obayashi, K.; Ando, Y.; Ericzon, B.G.; Friman, S.; Uchino, M.; Suhr, O.B. Liver transplantation for familial amyloidotic polyneuropathy: Impact on Swedish patients’ survival. Liver Transpl. 2009, 15, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Terazaki, H.; Ando, Y.; Nakamura, M.; Obayashi, K.; Misumi, S.; Shoji, S.; Yamashita, S.; Nakagawa, K.; Ishizaki, T.; Suhr, O.; et al. Variant transthyretin in blood circulation can transverse the blood-cerebrospinal barrier: Qualitative analyses of transthyretin metabolism in sequential liver transplantation. Transplantation 2001, 72, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Ruberg, F.L.; Berk, J.L. Transthyretin (TTR) cardiac amyloidosis. Circulation 2012, 126, 1286–1300. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, M.; Mitsuhashi, S.; Tokuda, T.; Kametani, F.; Takei, Y.I.; Koyama, J.; Kawamorita, A.; Kanno, H.; Ikeda, S.I. Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. Am. J. Transplant. 2007, 7, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Suhr, O.B.; Friman, S.; Ericzon, B.G. Early liver transplantation improves familial amyloidotic polyneuropathy patients’ survival. Amyloid 2005, 12, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.R.; Macedo, B.; Cardoso, I.; Alves, I.; Valencia, G.; Arsequell, G.; Planas, A.; Saraiva, M.J. Selective binding to transthyretin and tetramer stabilization in serum from patients with familial amyloidotic polyneuropathy by an iodinated diflunisal derivative. Biochem. J. 2004, 381, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Maurer, M.S.; Suhr, O.B. THAOS—The Transthyretin Amyloidosis Outcomes Survey: Initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr. Med. Res. Opin. 2013, 29, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA 2013, 310, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Maia, L.F.; Martins da Silva, A.; Waddington Cruz, M.; Plante-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceicao, I.M.; Schmidt, H.H.; et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 2012, 79, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Plante-Bordeneuve, V.; Judge, D.P.; Schmidt, H.; Obici, L.; Perlini, S.; Packman, J.; Tripp, T.; Grogan, D.R. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J. Cardiovasc. Transl. Res. 2013, 6, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Maia, L.F.; da Silva, A.M.; Cruz, M.W.; Plante-Bordeneuve, V.; Suhr, O.B.; Conceicao, I.; Schmidt, H.H.; Trigo, P.; Kelly, J.W.; et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J. Neurol. 2013, 260, 2802–2814. [Google Scholar] [CrossRef] [PubMed]
- Propsting, M.J.; Blaschke, M.; Haas, R.E.; Genschel, J.; Hedrich, H.J.; Manns, M.P.; Schmidt, H.H. Inosine(15.1) hammerhead ribozymes for targeting the transthyretin-30 mutation. Biochem. Biophys. Res. Commun. 1999, 260, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Propsting, M.J.; Kubicka, S.; Genschel, J.; Manns, M.P.; Lochs, H.; Schmidt, H.H. Inhibition of transthyretin-met30 expression using Inosine(15.1)-Hammerhead ribozymes in cell culture. Biochem. Biophys. Res. Commun. 2000, 279, 970–973. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.F.; Lot, S.S.; Condon, T.P.; Cheng-Flournoy, S.; Lesnik, E.A.; Sasmor, H.M.; Bennett, C.F. 2′-O-(2-Methoxy)ethyl-modified anti-intercellular adhesion molecule 1 (ICAM-1) oligonucleotides selectively increase the ICAM-1 mRNA level and inhibit formation of the ICAM-1 translation initiation complex in human umbilical vein endothelial cells. J. Biol. Chem. 1997, 272, 11994–12000. [Google Scholar] [CrossRef] [PubMed]
- Mercatante, D.R.; Kole, R. Control of alternative splicing by antisense oligonucleotides as a potential chemotherapy: Effects on gene expression. Biochim. Biophys. Acta 2002, 1587, 126–132. [Google Scholar] [CrossRef]
- Cazenave, C.; Frank, P.; Toulme, J.J.; Busen, W. Characterization and subcellular localization of ribonuclease H activities from Xenopus laevis oocytes. J. Biol. Chem. 1994, 269, 25185–25192. [Google Scholar] [PubMed]
- Shoeman, R.L.; Hartig, R.; Huang, Y.; Grub, S.; Traub, P. Fluorescence microscopic comparison of the binding of phosphodiester and phosphorothioate (antisense) oligodeoxyribonucleotides to subcellular structures, including intermediate filaments, the endoplasmic reticulum, and the nuclear interior. Antisense Nucleic Acid Drug Dev. 1997, 7, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Forsha, S.J.; Panyutin, I.V.; Neumann, R.D.; Panyutin, I.G. Intracellular traffic of oligodeoxynucleotides in and out of the nucleus: Effect of exportins and DNA structure. Oligonucleotides 2010, 20, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Bally, M.B.; Madden, T.D. Subcellular trafficking of antisense oligonucleotides and down-regulation of bcl-2 gene expression in human melanoma cells using a fusogenic liposome delivery system. Nucleic Acids Res. 2002, 30, 3632–3641. [Google Scholar] [CrossRef] [PubMed]
- Hartig, R.; Shoeman, R.L.; Janetzko, A.; Grub, S.; Traub, P. Active nuclear import of single-stranded oligonucleotides and their complexes with non-karyophilic macromolecules. Biol. Cell 1998, 90, 407–426. [Google Scholar] [CrossRef] [PubMed]
- Cerritelli, S.M.; Crouch, R.J. Ribonuclease H: The enzymes in eukaryotes. FEBS J. 2009, 276, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Oda, Y.; Iwai, S.; Inoue, H.; Ohtsuka, E.; Kanaya, S.; Kimura, S.; Katsuda, C.; Katayanagi, K.; Morikawa, K.; et al. How does RNase H recognize a DNA.RNA hybrid? Proc. Natl. Acad. Sci. USA 1991, 88, 11535–11539. [Google Scholar] [CrossRef] [PubMed]
- Pallan, P.S.; Egli, M. Insights into RNA/DNA hybrid recognition and processing by RNase H from the crystal structure of a non-specific enzyme-dsDNA complex. Cell Cycle 2008, 7, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lima, W.F.; Crooke, S.T. Properties of cloned and expressed human RNase H1. J. Biol. Chem. 1999, 274, 28270–28278. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Fujii, N.; Yasuhara, H.; Wada, S.; Wada, F.; Shigesada, N.; Harada-Shiba, M.; Obika, S. Evaluation of multiple-turnover capability of locked nucleic acid antisense oligonucleotides in cell-free RNase H-mediated antisense reaction and in mice. Nucleic Acid Ther. 2014, 24, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Mou, T.C.; Gray, D.M. The high binding affinity of phosphorothioate-modified oligomers for Ff gene 5 protein is moderated by the addition of C-5 propyne or 2′-O-methyl modifications. Nucleic Acids Res. 2002, 30, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Kang, S.H.; Gryaznov, S.M.; DeDionisio, L.; Heidenreich, O.; Sullivan, S.; Xu, X.; Nerenberg, M.I. Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. J. Biol. Chem. 1994, 269, 26801–26805. [Google Scholar] [PubMed]
- Lorenz, P.; Baker, B.F.; Bennett, C.F.; Spector, D.L. Phosphorothioate antisense oligonucleotides induce the formation of nuclear bodies. Mol. Biol. Cell 1998, 9, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Yu, R.Z.; Levin, A.A. Pharmacokinetics of phosphorothioate antisense oligodeoxynucleotides. Curr. Opin. Investig. Drugs 2001, 2, 562–573. [Google Scholar] [PubMed]
- Clark, C.L.; Cecil, P.K.; Singh, D.; Gray, D.M. CD, absorption and thermodynamic analysis of repeating dinucleotide DNA, RNA and hybrid duplexes [d/r(AC)]12∙[d/r(GT/U)]12 and the influence of phosphorothioate substitution. Nucleic Acids Res. 1997, 25, 4098–4105. [Google Scholar] [PubMed]
- Warfield, K.L.; Panchal, R.G.; Aman, M.J.; Bavari, S. Antisense treatments for biothreat agents. Curr. Opin. Mol. Ther. 2006, 8, 93–103. [Google Scholar] [PubMed]
- Kurreck, J. Antisense technologies. Improvement through novel chemical modifications. Eur. J. Biochem. 2003, 270, 1628–1644. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Khatsenko, O.; Bunker, K.; Crooke, R.; Moore, M.; Burckin, T.; Truong, L.; Sasmor, H.; Levin, A.A. Absolute bioavailability of 2′-O-(2-methoxyethyl)-modified antisense oligonucleotides following intraduodenal instillation in rats. J. Pharmacol. Exp. Ther. 2001, 296, 898–904. [Google Scholar] [PubMed]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Furdon, P.J.; Dominski, Z.; Kole, R. RNase H cleavage of RNA hybridized to oligonucleotides containing methylphosphonate, phosphorothioate and phosphodiester bonds. Nucleic Acids Res. 1989, 17, 9193–9204. [Google Scholar] [CrossRef] [PubMed]
- Berezney, J.P.; Saleh, O.A. Locked nucleic acid oligomers as handles for single molecule manipulation. Nucleic Acids Res. 2014, 42, e150. [Google Scholar] [CrossRef] [PubMed]
- Suresh, G.; Priyakumar, U.D. Atomistic investigation of the effect of incremental modification of deoxyribose sugars by locked nucleic acid (β-d-LNA and α-l-LNA) moieties on the structures and thermodynamics of DNA-RNA hybrid duplexes. J. Phys. Chem. B 2014, 118, 5853–5863. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Gunic, E.; Girardet, J.L.; Stoisavljevic, V. Conformationally locked nucleosides. Synthesis and hybridization properties of oligodeoxynucleotides containing 2′,4′-C-bridged 2′-deoxynucleosides. Bioorg. Med. Chem. Lett. 1999, 9, 1147–1150. [Google Scholar] [CrossRef]
- Kurreck, J.; Wyszko, E.; Gillen, C.; Erdmann, V.A. Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res. 2002, 30, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Leech, S.H.; Olie, R.A.; Gautschi, O.; Simoes-Wust, A.P.; Tschopp, S.; Haner, R.; Hall, J.; Stahel, R.A.; Zangemeister-Wittke, U. Induction of apoptosis in lung-cancer cells following bcl-xL anti-sense treatment. Int. J. Cancer 2000, 86, 570–576. [Google Scholar] [CrossRef]
- Astriab-Fisher, A.; Fisher, M.H.; Juliano, R.; Herdewijn, P. Increased uptake of antisense oligonucleotides by delivery as double stranded complexes. Biochem. Pharmacol. 2004, 68, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Frazier, K.S. Antisense oligonucleotide therapies: The promise and the challenges from a toxicologic pathologist’s perspective. Toxicol. Pathol. 2015, 43, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Hair, P.; Cameron, F.; McKeage, K. Mipomersen sodium: First global approval. Drugs 2013, 73, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, J.J.; Wedel, M.K.; Baker, B.F.; Su, J.; Bradley, J.D.; Yu, R.Z.; Chuang, E.; Graham, M.J.; Crooke, R.M. Potent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation 2006, 114, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Santos, R.D.; Blom, D.J.; Marais, A.D.; Charng, M.J.; Cromwell, W.C.; Lachmann, R.H.; Gaudet, D.; Tan, J.L.; Chasan-Taber, S.; et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: A randomised, double-blind, placebo-controlled trial. Lancet 2010, 375, 998–1006. [Google Scholar] [CrossRef]
- McGowan, M.P.; Tardif, J.C.; Ceska, R.; Burgess, L.J.; Soran, H.; Gouni-Berthold, I.; Wagener, G.; Chasan-Taber, S. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLoS ONE 2012, 7, e49006. [Google Scholar] [CrossRef] [PubMed]
- Gelsinger, C.; Steinhagen-Thiessen, E.; Kassner, U. Therapeutic potential of mipomersen in the management of familial hypercholesterolaemia. Drugs 2012, 72, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.D.; Duell, P.B.; East, C.; Guyton, J.R.; Moriarty, P.M.; Chin, W.; Mittleman, R.S. Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension. Eur. Heart J. 2015, 36, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Sjouke, B.; Balak, D.M.; Beuers, U.; Ratziu, V.; Stroes, E.S. Is mipomersen ready for clinical implementation? A transatlantic dilemma. Curr. Opin. Lipidol. 2013, 24, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Flaim, J.D.; Grundy, J.S.; Baker, B.F.; McGowan, M.P.; Kastelein, J.J. Changes in mipomersen dosing regimen provide similar exposure with improved tolerability in randomized placebo-controlled study of healthy volunteers. J. Am. Heart Assoc. 2014, 3, e000560. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Lee, S.K.; Wang, J.; Ince, N.; Ouyang, N.; Min, J.; Chen, J.; Shankar, P.; Lieberman, J. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat. Med. 2003, 9, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Rossi, J.J. Strategies for silencing human disease using RNA interference. Nat. Rev. Genet. 2007, 8, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Haynes, M.; Huang, L. Hepatic RNA interference: Delivery by synthetic vectors. Drug Deliv. Transl. Res. 2014, 4, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C.; Doudna, J.A. Molecular mechanisms of RNA interference. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.W.; Bass, B.L. A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science 2001, 293, 2269–2271. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Lendeckel, W.; Tuschl, T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001, 15, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Csorba, T.; Lozsa, R.; Hutvagner, G.; Burgyan, J. Polerovirus protein P0 prevents the assembly of small RNA-containing RISC complexes and leads to degradation of ARGONAUTE1. Plant J. 2010, 62, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef]
- Song, J.J.; Smith, S.K.; Hannon, G.J.; Joshua-Tor, L. Crystal structure of Argonaute and its implications for RISC slicer activity. Science 2004, 305, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Ameres, S.L.; Martinez, J.; Schroeder, R. Molecular basis for target RNA recognition and cleavage by human RISC. Cell 2007, 130, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Cuccato, G.; Polynikis, A.; Siciliano, V.; Graziano, M.; di Bernardo, M.; di Bernardo, D. Modeling RNA interference in mammalian cells. BMC Syst. Biol. 2011, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Hutvagner, G.; Zamore, P.D. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002, 297, 2056–2060. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.W.; Davis, M.E. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Res. 2006, 34, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Ramakrishnaiah, V.; Henry, S.; Fouraschen, S.; de Ruiter, P.E.; Kwekkeboom, J.; Tilanus, H.W.; Janssen, H.L.; van der Laan, L.J. Hepatic cell-to-cell transmission of small silencing RNA can extend the therapeutic reach of RNA interference (RNAi). Gut 2012, 61, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Cernilogar, F.M.; Onorati, M.C.; Kothe, G.O.; Burroughs, A.M.; Parsi, K.M.; Breiling, A.; lo Sardo, F.; Saxena, A.; Miyoshi, K.; Siomi, H.; et al. Chromatin-associated RNA interference components contribute to transcriptional regulation in Drosophila. Nature 2011, 480, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.; Bauman, J.; Kang, H.; Ming, X. Biological barriers to therapy with antisense and siRNA oligonucleotides. Mol. Pharm. 2009, 6, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.L.; Rana, T.M. siRNA function in RNAi: A chemical modification analysis. RNA 2003, 9, 1034–1048. [Google Scholar] [CrossRef] [PubMed]
- Guga, P.; Koziolkiewicz, M. Phosphorothioate nucleotides and oligonucleotides—Recent progress in synthesis and application. Chem. Biodivers. 2011, 8, 1642–1681. [Google Scholar] [CrossRef] [PubMed]
- Bramsen, J.B.; Laursen, M.B.; Nielsen, A.F.; Hansen, T.B.; Bus, C.; Langkjaer, N.; Babu, B.R.; Hojland, T.; Abramov, M.; van Aerschot, A.; et al. A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Res. 2009, 37, 2867–2881. [Google Scholar] [CrossRef] [PubMed]
- Amarzguioui, M.; Holen, T.; Babaie, E.; Prydz, H. Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res. 2003, 31, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Bartz, S.R.; Schelter, J.; Kobayashi, S.V.; Burchard, J.; Mao, M.; Li, B.; Cavet, G.; Linsley, P.S. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 2003, 21, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, Y.; Anderson, E.M.; Birmingham, A.; Reynolds, A.; Karpilow, J.; Robinson, K.; Leake, D.; Marshall, W.S.; Khvorova, A. Off-target effects by siRNA can induce toxic phenotype. RNA 2006, 12, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Burchard, J.; Leake, D.; Reynolds, A.; Schelter, J.; Guo, J.; Johnson, J.M.; Lim, L.; Karpilow, J.; Nichols, K.; et al. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA 2006, 12, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Sledz, C.A.; Holko, M.; de Veer, M.J.; Silverman, R.H.; Williams, B.R. Activation of the interferon system by short-interfering RNAs. Nat. Cell Biol. 2003, 5, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.T.; Williams, B.R. Activation of the mammalian immune system by siRNAs. Nat. Biotechnol. 2005, 23, 1399–1405. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Guenthner-Biller, M.; Bourquin, C.; Ablasser, A.; Schlee, M.; Uematsu, S.; Noronha, A.; Manoharan, M.; Akira, S.; de Fougerolles, A.; et al. Sequence-specific potent induction of IFN-α by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 2005, 11, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.; Frank-Kamenetsky, M.; Shulga-Morskaya, S.; Liebow, A.; Bettencourt, B.R.; Sutherland, J.E.; Hutabarat, R.M.; Clausen, V.A.; Karsten, V.; Cehelsky, J.; et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: A randomised, single-blind, placebo-controlled, phase 1 trial. Lancet 2014, 383, 60–68. [Google Scholar] [CrossRef]
- Crooke, S.T.; Graham, M.J.; Zuckerman, J.E.; Brooks, D.; Conklin, B.S.; Cummins, L.L.; Greig, M.J.; Guinosso, C.J.; Kornbrust, D.; Manoharan, M.; et al. Pharmacokinetic properties of several novel oligonucleotide analogs in mice. J. Pharmacol. Exp. Ther. 1996, 277, 923–937. [Google Scholar] [PubMed]
- Zimmermann, T.S.; Lee, A.C.; Akinc, A.; Bramlage, B.; Bumcrot, D.; Fedoruk, M.N.; Harborth, J.; Heyes, J.A.; Jeffs, L.B.; John, M.; et al. RNAi-mediated gene silencing in non-human primates. Nature 2006, 441, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.J.; Crooke, S.T.; Monteith, D.K.; Cooper, S.R.; Lemonidis, K.M.; Stecker, K.K.; Martin, M.J.; Crooke, R.M. In vivo distribution and metabolism of a phosphorothioate oligonucleotide within rat liver after intravenous administration. J. Pharmacol. Exp. Ther. 1998, 286, 447–458. [Google Scholar] [PubMed]
- Geary, R.S.; Wancewicz, E.; Matson, J.; Pearce, M.; Siwkowski, A.; Swayze, E.; Bennett, F. Effect of dose and plasma concentration on liver uptake and pharmacologic activity of a 2′-methoxyethyl modified chimeric antisense oligonucleotide targeting PTEN. Biochem. Pharmacol. 2009, 78, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, H.L.; Hall, J. Target mRNA inhibition by oligonucleotide drugs in man. Nucleic Acids Res. 2012, 40, 10585–10595. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, H.; de Winter, C.; van Kuik, P.; Heuvelmans, N.; Sabatelli, P.; Rimessi, P.; Braghetta, P.; van Ommen, G.J.; de Kimpe, S.; Ferlini, A.; et al. Preclinical PK and PD studies on 2′-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol. Ther. 2010, 18, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.A.; Geary, R.S.; Levin, A.A. Plasma protein binding of an antisense oligonucleotide targeting human ICAM-1 (ISIS 2302). Oligonucleotides 2006, 16, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhao, J.; Han, G.; Zhang, Y.; Dong, X.; Cao, L.; Wang, Q.; Moulton, H.M.; Yin, H. Effective dystrophin restoration by a novel muscle-homing peptide-morpholino conjugate in dystrophin-deficient mdx mice. Mol. Ther. 2014, 22, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin. Drug Metab. Toxicol. 2009, 5, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Castagner, B.; Leroux, J.C. Is there a future for cell-penetrating peptides in oligonucleotide delivery? Eur. J. Pharm. Biopharm. 2013, 85, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Ming, X. Cellular delivery of siRNA and antisense oligonucleotides via receptor-mediated endocytosis. Expert Opin. Drug Deliv. 2011, 8, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Damke, H. Dynamin and receptor-mediated endocytosis. FEBS Lett. 1996, 389, 48–51. [Google Scholar] [CrossRef]
- Overhoff, M.; Sczakiel, G. Phosphorothioate-stimulated uptake of short interfering RNA by human cells. EMBO Rep. 2005, 6, 1176–1181. [Google Scholar] [CrossRef] [PubMed]
- Rydstrom, A.; Deshayes, S.; Konate, K.; Crombez, L.; Padari, K.; Boukhaddaoui, H.; Aldrian, G.; Pooga, M.; Divita, G. Direct translocation as major cellular uptake for CADY self-assembling peptide-based nanoparticles. PLoS ONE 2011, 6, e25924. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.J.; Jones, S.; Fabani, M.M.; Ivanova, G.; Arzumanov, A.A.; Gait, M.J. RNA targeting with peptide conjugates of oligonucleotides, siRNA and PNA. Blood Cells Mol. Dis. 2007, 38, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, S.; Morris, M.; Heitz, F.; Divita, G. Delivery of proteins and nucleic acids using a non-covalent peptide-based strategy. Adv. Drug Deliv. Rev. 2008, 60, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, D.R.; Leirdal, M.; Sioud, M. Gene silencing by systemic delivery of synthetic siRNAs in adult mice. J. Mol. Biol. 2003, 327, 761–766. [Google Scholar] [CrossRef]
- Song, E.; Zhu, P.; Lee, S.K.; Chowdhury, D.; Kussman, S.; Dykxhoorn, D.M.; Feng, Y.; Palliser, D.; Weiner, D.B.; Shankar, P.; et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005, 23, 709–717. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.O., 2nd.; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Koller, E.; Vincent, T.M.; Chappell, A.; De, S.; Manoharan, M.; Bennett, C.F. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011, 39, 4795–4807. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J. Oligonucleotide conjugates for therapeutic applications. Ther. Deliv. 2013, 4, 791–809. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, P.; Misteli, T.; Baker, B.F.; Bennett, C.F.; Spector, D.L. Nucleocytoplasmic shuttling: A novel in vivo property of antisense phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 2000, 28, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Welz, C.; Neuhuber, W.; Schreier, H.; Repp, R.; Rascher, W.; Fahr, A. Nuclear gene targeting using negatively charged liposomes. Int. J. Pharm. 2000, 196, 251–252. [Google Scholar] [CrossRef]
- Detzer, A.; Overhoff, M.; Wunsche, W.; Rompf, M.; Turner, J.J.; Ivanova, G.D.; Gait, M.J.; Sczakiel, G. Increased RNAi is related to intracellular release of siRNA via a covalently attached signal peptide. RNA 2009, 15, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Franzen, S. Factors determining the efficacy of nuclear delivery of antisense oligonucleotides by gold nanoparticles. Bioconjugate Chem. 2008, 19, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Rozema, D.B.; Lewis, D.L.; Wakefield, D.H.; Wong, S.C.; Klein, J.J.; Roesch, P.L.; Bertin, S.L.; Reppen, T.W.; Chu, Q.; Blokhin, A.V.; et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 12982–12987. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Lima, W.F.; Murray, H.M.; Elbashir, S.; Cantley, W.; Foster, D.; Jayaraman, M.; Chappell, A.E.; Manoharan, M.; Swayze, E.E.; et al. Lipid nanoparticles improve activity of single-stranded siRNA and gapmer antisense oligonucleotides in animals. ACS Chem. Biol. 2013, 8, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Dominska, M.; Dykxhoorn, D.M. Breaking down the barriers: SiRNA delivery and endosome escape. J. Cell Sci. 2010, 123, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.R.; Ming, X.; Nakagawa, O.; Jin, J.; Juliano, R.L. Covalent conjugation of oligonucleotides with cell-targeting ligands. Bioorg. Med. Chem. 2013, 21, 6217–6223. [Google Scholar] [CrossRef] [PubMed]
- Oehlke, J.; Birth, P.; Klauschenz, E.; Wiesner, B.; Beyermann, M.; Oksche, A.; Bienert, M. Cellular uptake of antisense oligonucleotides after complexing or conjugation with cell-penetrating model peptides. Eur. J. Biochem. 2002, 269, 4025–4032. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. Intracellular delivery of oligonucleotide conjugates and dendrimer complexes. Ann. N. Y. Acad. Sci. 2006, 1082, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Szebeni, J. Stealth liposomes and long circulating nanoparticles: Critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog. Lipid Res. 2003, 42, 463–478. [Google Scholar] [CrossRef]
- Kumar, V.; Qin, J.; Jiang, Y.; Duncan, R.G.; Brigham, B.; Fishman, S.; Nair, J.K.; Akinc, A.; Barros, S.A.; Kasperkovitz, P.V. Shielding of Lipid Nanoparticles for siRNA Delivery: Impact on Physicochemical Properties, Cytokine Induction, and Efficacy. Mol. Ther. Nucleic Acids 2014, 3, e210. [Google Scholar] [CrossRef] [PubMed]
- Wooddell, C.I.; Rozema, D.B.; Hossbach, M.; John, M.; Hamilton, H.L.; Chu, Q.; Hegge, J.O.; Klein, J.J.; Wakefield, D.H.; Oropeza, C.E.; et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol. Ther. 2013, 21, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Allen, T.M.; Cullis, P.R. Lipid nanoparticle delivery systems for siRNA-based therapeutics. Drug Deliv. Transl. Res. 2014, 4, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Avino, A.; Ocampo, S.M.; Lucas, R.; Reina, J.J.; Morales, J.C.; Perales, J.C.; Eritja, R. Synthesis and in vitro inhibition properties of siRNA conjugates carrying glucose and galactose with different presentations. Mol. Divers. 2011, 15, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef]
- Oishi, M.; Nagasaki, Y.; Itaka, K.; Nishiyama, N.; Kataoka, K. Lactosylated poly(ethylene glycol)-siRNA conjugate through acid-labile β-thiopropionate linkage to construct pH-sensitive polyion complex micelles achieving enhanced gene silencing in hepatoma cells. J. Am. Chem. Soc. 2005, 127, 1624–1625. [Google Scholar]
- Weigel, P.H.; Oka, J.A. The surface content of asialoglycoprotein receptors on isolated hepatocytes is reversibly modulated by changes in temperature. J. Biol. Chem. 1983, 258, 5089–5094. [Google Scholar] [PubMed]
- Stockert, R.J. The asialoglycoprotein receptor: Relationships between structure, function, and expression. Physiol. Rev. 1995, 75, 591–609. [Google Scholar] [PubMed]
- Chen, S.; Tam, Y.Y.; Lin, P.J.; Leung, A.K.; Tam, Y.K.; Cullis, P.R. Development of lipid nanoparticle formulations of siRNA for hepatocyte gene silencing following subcutaneous administration. J. Control. Release 2014, 196, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.C.; Klein, J.J.; Hamilton, H.L.; Chu, Q.; Frey, C.L.; Trubetskoy, V.S.; Hegge, J.; Wakefield, D.; Rozema, D.B.; Lewis, D.L. Co-injection of a targeted, reversibly masked endosomolytic polymer dramatically improves the efficacy of cholesterol-conjugated small interfering RNAs in vivo. Nucleic Acid Ther. 2012, 22, 380–390. [Google Scholar] [PubMed]
- Nishina, K.; Unno, T.; Uno, Y.; Kubodera, T.; Kanouchi, T.; Mizusawa, H.; Yokota, T. Efficient in vivo delivery of siRNA to the liver by conjugation of α-tocopherol. Mol. Ther. 2008, 16, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Nishina, T.; Numata, J.; Nishina, K.; Yoshida-Tanaka, K.; Nitta, K.; Piao, W.; Iwata, R.; Ito, S.; Kuwahara, H.; Wada, T.; et al. Chimeric Antisense Oligonucleotide Conjugated to α-Tocopherol. Mol. Ther. Nucleic Acids 2015, 4, e220. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Pandey, S.; Witchell, D.; Jazayeri, A.; Siwkowski, A.; Monia, B.; Kluve-Beckerman, B. Antisense oligonucleotide therapy for TTR amyloidosis. Amyloid 2011, 18 (Suppl. S1), 60. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Kluve-Beckerman, B.; Zeldenrust, S.R.; Siesky, A.M.; Bodenmiller, D.M.; Showalter, A.D.; Sloop, K.W. Targeted suppression of an amyloidogenic transthyretin with antisense oligonucleotides. Muscle Nerve 2006, 33, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Smith, R.A.; Hung, G.; Kluve-Beckerman, B.; Showalter, A.D.; Sloop, K.W.; Monia, B.P. Suppression of choroid plexus transthyretin levels by antisense oligonucleotide treatment. Amyloid 2010, 17, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.; Kincaid, J.; Ackermann, E.; Monia, B. A phase 3 study to evaluate ISIS-TTRRx in patients with transthyrethin famial amyloid polyneuropathy (TTR-FAP): Study design and baseline demographics. Neurology 2015, 84 14 Supplement S50.006. [Google Scholar]
- Prakash, T.P.; Graham, M.J.; Yu, J.; Carty, R.; Low, A.; Chappell, A.; Schmidt, K.; Zhao, C.; Aghajan, M.; Murray, H.F.; et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014, 42, 8796–8807. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, E.J.; Guo, S.; Booten, S.; Alvarado, L.; Benson, M.; Hughes, S.; Monia, B.P. Clinical development of an antisense therapy for the treatment of transthyretin-associated polyneuropathy. Amyloid 2012, 19 (Suppl. S1), 43–44. [Google Scholar] [CrossRef] [PubMed]
- Zemany, L.; Bhanot, S.; Peroni, O.D.; Murray, S.F.; Moraes-Vieira, P.M.; Castoldi, A.; Manchem, P.; Guo, S.; Monia, B.P.; Kahn, B.B. Transthyretin Antisense Oligonucleotides Lower Circulating RBP4 Levels and Improve Insulin Sensitivity in Obese Mice. Diabetes 2015, 64, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Suhr, O.B.; Coelho, T.; Buades, J.; Pouget, J.; Conceicao, I.; Berk, J.; Schmidt, H.; Waddington-Cruz, M.; Campistol, J.M.; Bettencourt, B.R.; et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: A phase II multi-dose study. Orphanet J. Rare Dis. 2015, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Sohlenius-Sternbeck, A.K. Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements. Toxicol. In Vitro 2006, 20, 1582–1586. [Google Scholar] [CrossRef] [PubMed]
- Forbes, D.C.; Peppas, N.A. Oral delivery of small RNA and DNA. J. Control. Release 2012, 162, 438–445. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niemietz, C.; Chandhok, G.; Schmidt, H. Therapeutic Oligonucleotides Targeting Liver Disease: TTR Amyloidosis. Molecules 2015, 20, 17944-17975. https://doi.org/10.3390/molecules201017944
Niemietz C, Chandhok G, Schmidt H. Therapeutic Oligonucleotides Targeting Liver Disease: TTR Amyloidosis. Molecules. 2015; 20(10):17944-17975. https://doi.org/10.3390/molecules201017944
Chicago/Turabian StyleNiemietz, Christoph, Gursimran Chandhok, and Hartmut Schmidt. 2015. "Therapeutic Oligonucleotides Targeting Liver Disease: TTR Amyloidosis" Molecules 20, no. 10: 17944-17975. https://doi.org/10.3390/molecules201017944
APA StyleNiemietz, C., Chandhok, G., & Schmidt, H. (2015). Therapeutic Oligonucleotides Targeting Liver Disease: TTR Amyloidosis. Molecules, 20(10), 17944-17975. https://doi.org/10.3390/molecules201017944