
Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs



Abstract
:1. Introduction
2. Importance of Solubility on the Bioavailability of Drugs

| Freely Soluble | 100–1000 mg/mL |
|---|---|
| Soluble | 33–100 mg/mL |
| Sparingly soluble | 10–33 mg/mL |
| Slightly soluble | 1–10 mg/mL |
| Very slightly soluble | 0.1–1 mg/mL |
| Practically insoluble | <0.1 mg/mL |
- Class 1: High Solubility–High Permeability
- Class 2: Low Solubility–High Permeability
- Class 3: High Solubility–Low Permeability
- Class 4: Low Solubility–Low Permeability
3. The Polymorphism of Drugs: Anhydrous and Solvated Forms
4. Polymorph Screening
5. Case Studies of Polymorphic Drugs
| Drug Substance | Polymorphism Aspects | Bioavailability Issues |
|---|---|---|
Chloramphenicol palmitate | Chloramphenicol palmitate is a prodrug of chloramphenicol with antibiotic properties [64]. Chloramphenicol palmitate exist in three polymorphic forms: (A, B, C) [65,66], the stable form A (biologically inactive modification), the metastable form B (active modification) and unstable form C [67,68,69]. The three crystalline forms were also called α, β and γ. The α form is unstable at room temperature and gradually transforms to β on storage [70,71]. | Form B (β) dissolves faster than Form A (α), and has a much higher solubility [72,73,74]. Low serum levels for the stable polymorph A were observed [75]. |
Oxytetracycline | Oxytetracycline is a broad spectrum antibiotic. It exists in two different polymorphs [76]. | Oxytetracycline showed differences in patients’ blood levels [77] or differences in in vitro dissolution of tablets [78] because of differences in polymorphic forms. |
Carbamazepine | Carbamazepine is used in the treatment of epilepsy and trigeminal neuralgia. Different polymorphic forms were described [79,80,81,82,83,84,85,86,87,88,89,90,91]. Four anhydrous polymorphs were characterized: I, II, III, and IV, respectively identified as triclinic, trigonal, monoclinic, and monoclinic [77]. | In spite different studies demonstrated similar pharmacokinetics in humans of anhydrous and dihydrate forms of carbamazepine [92] and no differences in bioavailability between a generic carbamazepine product and an innovator product [93], several clinical failures were reported concerning carbamazepine [94,95], in particular with generic carbamazepine tablets [96]. More recently, it was confirmed that the initial dissolution rate of carbamazepine was in the order of form III > form I > dihydrate, while the order of AUC values was form I > form III > dihydrate. This discrepancy may be attributed to the rapid transformation from form III to dihydrate in GI fluids [97]. |
Ritonavir | Ritonavir is an antiretroviral drug belonging to protease inhibitor class and used to treat HIV-1 infection. Ritonavir exhibits conformational polymorphism [98] and a total of five forms were described [60]. The forms I and II were more extensively characterized [98]. | 2 years after the launch of the first ritonavir product, several batches failed dissolution specifications because the presence of a different polymorphic form having ~50% lower intrinsic solubility of reference form [36]. |
Atorvastatin calcium | Atorvastatin calcium is an inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, with strong ability to lowering blood cholesterol. At least 60 polymorphic forms/solvates/hydrates have been patented [99,100,101]. It is not unusual to verify the presence of polymorphic impurities in the marketed atorvastatin calcium (API) with consequences on drug bioavailability and stability [102]. | Atorvastatin is unstable and the hydroxy acid form is converted to lactone form that is 15 times less soluble than the hydroxyl acid form [103,104]. This instability of atorvastatin calcium leading to poor solubility (0.1 mg/mL) is the main cause for low bioavailability of the drug after oral administration as the absolute bioavailability of atorvastatin calcium is only 14% [105]. |
Axitinib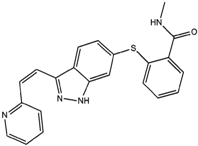 | Axitinib is a tyrosine kinase inhibitor of endothelial growth factor interrupting tumor angiogenesis and thus, preventing the growth of cancer cells. 60 solvates, polymorphs of solvates, and five anhydrous forms were discovered [106,107,108,109]. | The commercial formulation under trade name Inlyta® contains the stable anhydrous form [107]. |
Phanylbutazone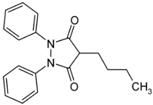 | Phenylbutazone is a potent anti-rheumatic drug existing in different polymorphic and solvated forms [110,111,112,113]. Anhydrous forms I and II were more extensively described and form II resulted more soluble than form I. The Form III is a highly unstable form [110]. | Anhydrous forms I and II polymorphic forms exhibited different solubilities, dissolution rates and oral absorption [110,114]. |
Rifaximin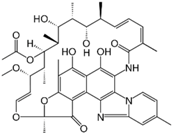 | Rifaximin is a synthetic derivative of rifamycin, with very low gastrointestinal absorption, but still displaying a broad spectrum of antibacterial activity [115,116,117]. Rifaximin shows crystal polymorphism (poolymorphs α, β, γ, δ, ε) [118,119]. The polymorph α is the most thermodynamically stable form and the commercial one. | In vitro studies show different dissolution and solubility rates for these polymorphs, and in vivo investigations in dogs found different pharmacokinetic patterns, with δ and γ polymorphs displaying the highest systemic bioavailability [119]. The most PK parameters were significantly higher after administration of generic rifaximin, because of the presence of both rifaximin-α and amorphous forms [120]. |
5.1. Chloramphenicol Palmitate
5.2. Oxytetracycline
5.3. Carbamazepine
5.4. Ritonavir
5.5. Atorvastatin Calcium
5.6. Axitinib
5.7. Phenylbutazone
5.8. Rifaximin
6. Regulatory Considerations
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Serajuddin, A.T.M. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, N.; Newman, A. Pharmaceutical cocrystals and their physicochemical properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onouea, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Cho, J.M.; Yun, G.; Lee, J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, D.; Chen, M. Drug nanocrystals for the formulation of poorly soluble drugs and its application as a potential drug delivery system. J. Nanopart. Res. 2008, 10, 845–862. [Google Scholar] [CrossRef]
- Chen, H.; Khemtong, C.; Yang, X.; Chang, X.; Gao, J. Nanonization strategies for poorly-soluble drugs. Drug Discov. Today 2011, 16, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Saravana, K.K.; Prasanna, R.Y. Dissolution enhancement of poorly soluble drugs by using complexation technique. A review. J. Pharm. Sci. Res. 2013, 5, 120–124. [Google Scholar]
- Challa, R.; Ahuja, A.; Ali, J.; Khar, R.K. Cyclodextrins in drug delivery: An updated review. AAPS Pharm. Sci. Technol. 2005, 6, E329–E357. [Google Scholar] [CrossRef] [PubMed]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Snider, D.A.; Addicks, W.; Owens, W. Polymorphism in generic drug product development. Adv. Drug Deliv. Rev. 2004, 56, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.F.; Tong, W.Q. Impact of solid state properties on developability assessment of drug candidates. Adv. Drug Deliv. Rev. 2004, 56, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Babu, N.J.; Nangia, A. Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Murdande, S.B.; Pikal, M.J.; Shanker, R.M.; Bogner, R.H. Aqueous solubility of crystalline and amorphous drugs: challenges in measurement. Pharm. Dev. Technol. 2011, 16, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.G.; Law, D.; Schmitt, E.A.; Qiu, Y. Phase transformation considerations during process development and manufacture of solid oral dosage forms. Adv. Drug Deliv. Rev. 2004, 56, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Ku, M.S. Salt and polymorph selection strategy based on the biopharmaceutical classification system for early pharmaceutical Development. Am. Pharm. Rev. 2010, 20, 30. [Google Scholar]
- Lachman, L.; Lieberman, H.; Kanig, J.L. The Theory and Practise of Industrial Pharmacy, 3rd ed.; Lea & Febiger: Philadelphia, PA, USA, 1986. [Google Scholar]
- Merisko, E.; Liversidge, G.G. Nanocrystals: Resolving pharmaceutical formulation issues associated with poorly water-soluble compounds. In Particles; Marty, J.J., Ed.; Marcel Dekker: Orlando, FL, USA, 2002. [Google Scholar]
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Ku, M.S.; Dulin, W. A biopharmaceutical classification-based Right-First-Time formulation approach to reduce human pharmacokinetic variability and project cycle time from First-In-Human to clinical Proof-Of-Concept. Pharm. Dev. Technol. 2012, 17, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Dow Jones Newswires. GlaxoSmithKline on Track to Launch 11 Drugs by Dec. 2003; Dow Jones Newswires: New York, NY, USA, 2003. [Google Scholar]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Guidance for Industry: Waiver of in Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System; Food and Drug Administation: Rockville, MD, USA, 2000.
- Benet, L.Z. The role of BCS (Biopharmaceutics Classification System) and BDDCS (Biopharmaceutics Drug Disposition Classification System) in drug development. J. Pharm. Sci. 2013, 102, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012. [Google Scholar] [CrossRef] [PubMed]
- Vippagunta, S.R.; Brittain, H.G.; Grant, D.J.W. Crystalline solids. Adv. Drug Deliv. Rev. 2001, 48, 3–26. [Google Scholar] [CrossRef]
- Rodriguez-Spong, B.; Price, C.P.; Jayasankar, A.; Matzger, A.J.; Rodriguez-Hornedo, N. General principles of pharmaceutical solid polymorphism: a supramolecular perspective. Adv. Drug Deliv. Rev. 2004, 56, 241–274. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert-Brandstätter, M. Thermomicroscopy in the Analysis of Pharmaceuticals; Pergamon Press: Oxford, UK, 1971. [Google Scholar]
- Borka, L.; Haleblian, J.K. Crystal polymorphism of pharmaceuticals. Acta Pharm. Jugosl. 1990, 40, 71–94. [Google Scholar]
- Borka, L. Review on crystal polymorphism of substances in the European Pharmacopoeia. Pharm. Acta Helv. 1991, 66, 6–22. [Google Scholar]
- Giron, D. Thermal analysis and calorimetric methods in thecharacterization of polymorphs and solvates. Thermochim. Acta 1995, 248, 1–59. [Google Scholar] [CrossRef]
- Hilfiker, R.; Blatter, F.; von Raumer, M. Relevance of solid-state properties for pharmaceutical products polymorphism. In the Pharmaceutical Industry; Hilfiker, R., Ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Pudipeddi, M.; Serajuddin, A.T. Trends in solubility of polymorphs. J. Pharm. Sci. 2005, 94, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Chemburkar, S.R.; Bauer, J.; Deming, K.; Spiwek, H.; Patel, K.; Morris, J.; Henry, R.; Spanton, S.; Dziki, W.; Porter, W.; et al. Dealing with the impact of ritonavir polymorphs on the late stages of bulk drug process development. Org. Process Res. Dev. 2000, 4, 413–417. [Google Scholar] [CrossRef]
- Dubbini, A.; Censi, R.; Martena, V.; Hoti, E.; Ricciutelli, M.; Malaj, L.; di Martino, P. Influence of pH and method of crystallization on the solid physical form of indomethacin. Int. J. Pharm. 2014, 473, 536–544. [Google Scholar] [CrossRef]
- Censi, R.; Rascioni, R.; di Martino, P. Changes in the solid state of anhydrous and hydrated forms of sodium naproxen under different grinding and environmental conditions: Evidence of the formation of new hydrated forms. Eur. J. Pharm. Biopharm. 2015, 92, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Seddon, K.R. Pseudopolymorph: A Polemic. Cryst. Growth Des. 2004, 4. [Google Scholar] [CrossRef]
- Morris, K.R. Structural aspects of hydrates and solvates. In Polymorphism in Pharmaceutical Sciences, Drugs and the Pharmaceutical Sciences; Brittain, H., Ed.; Marcel Dekker: New York, NY, USA, 1999; Volume 95, pp. 125–181. [Google Scholar]
- Khankari, R.J.; Grant, D.J.W. Pharmaceutical hydrates. Thermochim. Acta 1995, 248, 61–79. [Google Scholar] [CrossRef]
- Shefter, E.; Higuchi, T. Dissolution behavior of crystalline solvated and nonsolvated forms of some pharmaceuticals. J. Pharm. Sci. 1963, 52, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Shan, N.; Zaworotko, M.J. Polymorphic Crystal Forms and Cocrystals in Drug Delivery (Crystal Engineering). Drug Dev. 2010. [Google Scholar] [CrossRef]
- Allen, P.V.; Rahn, P.D.; Sarapu, A.C.; Vanderwielen, A.J. Physical characterization of erythromycin: Anhydrate, monohydrate, and dihydrate crystalline solids. J. Pharm. Sci. 1978, 67, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Blagden, N.; de Matas, M.; Gayan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Grant, D.J.W. Crystal structures of drugs: Advances in determination, prediction and engineering. Nat. Rev. Drug Discov. 2004, 3, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, P.; Barthélémy, C.; Palmieri, G.F.; Martelli, S. Physical characterization of naproxen sodium hydrate and anhydrate forms. Eur. J. Pharm. Sci. 2001, 14, 293–300. [Google Scholar] [CrossRef]
- Di Martino, P.; Barthélémy, C.; Joiris, E.; Capsoni, D.; Masic, A.; Massarotti, V.; Gobetto, R.; Bini, M.; Martelli, S. A new tetrahydrated form of sodium naproxen. J. Pharm. Sci. 2007, 96, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Malaj, L.; Censi, R.; di Martino, P. Mechanism for dehydration of three sodium naproxen hydrates. Cryst. Growth Des. 2009, 9, 2128–2136. [Google Scholar] [CrossRef]
- Di Martino, P.; Malaj, L.; Censi, R.; Martelli, S. Physico-chemical and technological properties of sodium naproxen granules prepared in a high-shear mixer-granulator. J. Pharm. Sci. 2008, 97, 5263–5273. [Google Scholar] [CrossRef] [PubMed]
- Stahly, G.P. Diversity in Single- and Multiple-Component Crystals. The Search for and Prevalence of Polymorphs and Cocrystals. Cryst. Growth Des. 2007, 6, 1007–1026. [Google Scholar] [CrossRef]
- Pepinsky, R. Crystal engineering—A new concept in crystallography. Phys. Rev. 1955, 100, 971. [Google Scholar]
- Schmidt, G.M.J. Photodimerization in the solid state. Pure Appl. Chem. 1971, 27, 647–678. [Google Scholar] [CrossRef]
- Desiraju, G.R. Chemistry beyond the molecule. Nature 2001, 412, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. Crystal engineering: A brief overview. J. Chem. Sci. 2010, 122, 667–675. [Google Scholar] [CrossRef]
- Biradha, K.; Su, C.Y.; Vittal, J.J. Recent developments in crystal engineering. Cryst. Growth Des. 2011, 11, 875–886. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Met. 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Caira, M.R. Crystalline Polymorphism of Organic Compounds. Top. Curr. Chem. 1998, 198, 163–208. [Google Scholar]
- Price, S.L. The computational prediction of pharmaceutical crystal structures and polymorphism. Adv. Drug Deliv. Rev. 2004, 56, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Morissette, S.L.; Soukasene, S.; Levinson, D.A.; Cima, M.J.; Almarsson, O. Elucidation of crystal form diversity of the HIV protease inhibitor ritonavir by high-throughput crystallization. Proc. Natl. Acad. Sci. USA 2003, 100, 2180–2184. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.L.; Morissette, S.L.; McNulty, C.; Goldsweig, A.; Shaw, P.; le Quesne, M.; Monagle, J.; Encina, N.; Marchionna, J.; Johnson, A.; et al. Iterative high-throughput polymorphism studies on acetaminophen and an experimentally derived structure for form III. J. Am. Chem. Soc. 2002, 124, 10958–10959. [Google Scholar] [CrossRef] [PubMed]
- Almarsson, O.; Hickey, M.B.; Peterson, M.L.; Morissette, S.L.; Soukasene, S.; McNulty, C.; Tawa, M.; MacPhee, J.M.; Remenar, J.F. High-Throughput surveys of crystal form diversity of highly polymorphic pharmaceutical compounds. Cryst. Growth Des. 2003, 3, 927–933. [Google Scholar] [CrossRef]
- Kojima, T.; Tsutsumi, S.; Yamamoto, K.; Ikeda, Y.; Moriwaki, T. High-throughput cocrystal slurry screening by use of in situ Raman microscopy and multi-well plate. Int. J. Pharm. 2010, 399, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Edgerton, W.H. Chloramphenicol Esters and Method for Obtaining Same. U.S. Patent 2,662,906, 15 December 1953. [Google Scholar]
- Borka, L.; Backe-Hansen, K. IR spectroscopy of chloramphenicol palmitate. Polymorph alteration caused by the KBr disc technique. Acta Pharm. Suec. 1968, 5, 271–278. [Google Scholar] [PubMed]
- Kanenewa, N.; Otsuka, M. Effect of grinding on the transformation of polymorphs of chloramphenicol palmitate. Chem. Pharm. Bull. 1985, 33, 1660–1668. [Google Scholar] [CrossRef]
- Burger, A. Neue untersuchungergebnisse von chloramphenicolpalmitat. Sci. Pharm. 1977, 45, 269–281. [Google Scholar]
- Gamberini, M.C.; Baraldi, C.; Tinti, A.; Rustichelli, C.; Ferioli, V.; Gamberini, G. Solid state characterization of chloramphenicol palmitate. Raman spectroscopy applied to pharmaceutical polymorphs. J. Mol. Struct. 2006, 785, 216–224. [Google Scholar] [CrossRef]
- Mishra, R.; Srivastava, A.; Sherma, A.; Tandon, P.; Baraldi, C.; Gamberini, M.C. Structural, electronic, thermodynamical and charge transfer properties of chloramphenicol palmitate using vibrational spectroscopy and DFT calculations. Spectrochim. Acta Part A Mol. Biomol. Spectr. 2013, 101, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Iitaka, Y. The β-form of chloramphenicol palmitate. Acta Cryst. 1974, B30, 2781–2783. [Google Scholar] [CrossRef]
- Szulzewsky, K.; Kulpe, S.; Schulz, B.; Kunath, D. The structure of the b modification of chloramphenicol palmitate. A redetermination. Acta Cryst. 1981, B37, 1673–1676. [Google Scholar] [CrossRef]
- Aguiar, A.J.; Krc, J.; Kinkel, A.W.; Samyn, J.C. Effect of polymorphism on the absorption of chloramphenicol from chloramphenicol palmitate. J. Pharm. Sci. 1967, 56, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, A.J.; Zelmer, J.E. Dissolution behavior of polymorphs of chloramphenicol palmitate and mefanamic acid. J. Pharm. Sci. 1969, 58, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Glazko, A.J.; Edgerton, W.H.; Dill, W.A.; Lenz, W.R. Chloromycetin palmitate—A synthetic ester of chloromycetin. Antibiot. Chemother. 1952, 2, 234–242. [Google Scholar]
- Maeda, T.; Takenaka, H.; Yamahira, Y.; Noguchi, T. Use of rabbits for absorption studies on polymorphs of chloramphenicol palmitate. Chem. Pharm. Bull. 1980, 28, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Liebenberg, W.; de Villiers, M.; Wurster, D.E.; Swanepoel, E.; Dekker, T.G.; Lotter, A.P. The effect of polymorphism on powder compaction and dissolution properties of chemically equivalent oxytetracycline hydrochloride powders. Drug Dev. Ind. Pharm. 1999, 25, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Brice, G.W.; Hammer, H.F. Therapeutic nonequivalence of oxytetracycline capsules. J. Am. Med. Assoc. 1969, 208, 1189–1190. [Google Scholar] [CrossRef]
- Groves, M.J. Solution tests on generic brands of oxytetracycline tablets. Pharm. J. 1973, 210, 318–319. [Google Scholar]
- Reboul, J.P.; Cristau, B.; Soyfer, J.C.; Astier, J.P. 5H-5-Dibenzyl[b,f]azepinecarboxamide (carbamazepine). Acta Crystallogr. Sect. B Struct. Commun. 1981, 37, 1844–1848. [Google Scholar]
- Himes, V.L.; Mighell, A.D.; Decamp, W.H. Structure of carbamazepine-5H-dibenz[b,f]azepine-5-carboxamide. Acta Crystallogr. Sect. B Struct. Commun. 1981, 37, 2242–2245. [Google Scholar] [CrossRef]
- Chang, C.H.; Yang, D.S.C.; Yoo, C.S.; Wang, B.L.; Pletcher, J. The crystal structures of (S) and (R) baclofen and carbamazepine. Acta Crystallogr. 1981, A37. [Google Scholar] [CrossRef]
- Reck, G.; Dietz, G. The order-disorder structure of carbamazepine dihydrate: 5H-Dibenz[b,f] azepine-5-carboxamide dihydrate, C15H12N2O···2 H2O. Cryst. Res. Technol. 1986, 21, 1463–1468. [Google Scholar] [CrossRef]
- Lowes, M.M.J.; Caira, M.R.; Lotter, A.P.; Vanderwatt, J.G. Physicochemical properties and X-ray structural studies of the trigonal polymorph of carbamazepine. J. Pharm. Sci. 1987, 76, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Lisgarten, J.N.; Palmer, R.A.; Saldanha, J.W.J. Crystal and molecular structure of 5-carbamyl-5H-dibenzo[b,f]azepine. Crystallogr. Spectrosc. Res. 1989, 19, 641–649. [Google Scholar] [CrossRef]
- Ceolin, R.; Toscani, S.; Gardette, M.F.; Dzyabchenko, V.N.; Bachet, B. X-ray characterization of the triclinic polymorph of carbamazepine. J. Pharm. Sci. 1997, 86, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Rustichelli, C.; Gamberini, G.; Ferioli, V.; Gamberini, M.C.; Ficarra, R.; Tommasini, S. Solid-state study of polymorphic drugs: carbamazepine. J. Pharm. Biomed. Anal. 2000, 23, 41–54. [Google Scholar] [CrossRef]
- Lang, M.D.; Kampf, J.W.; Matzger, A.J. Form IV of carbamazepine. J. Pharm. Sci. 2002, 91, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.D.; Grzesiak, A.L.; Matzger, A.J. The use of polymer heteronuclei for crystalline polymorph selection. J. Am. Chem. Soc. 2002, 124, 14834–14835. [Google Scholar] [CrossRef] [PubMed]
- Grzesiak, A.L.; Lang, M.; Kim, K.; Matzger, A.J. Comparison of the four anhydrous polymorphs of carbamazepine and the crystal structure of form I. J. Pharm. Sci. 2003, 92, 2260–2271. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, S.G.; Kuduva, S.S.; McMahon, J.A.; Moulton, B.; Walsh, R.D.B.; Zaworotko, M.J.; Rodríguez-Hornedo, N. Crystal engineering of the composition of pharmaceutical phases: Multiple-component crystalline solids involving carbamazepine. Cryst. Growth Des. 2003, 3, 909–919. [Google Scholar] [CrossRef]
- Young, W.W.L.; Suryanarayanan, R. Kinetics of transition of anhydrous carbamazepine to carbamazepine dihydrate in aqueous suspensions. J. Pharm. Sci. 1991, 80, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Kahela, P.; Aaltonen, R.; Lewing, E.; Anttila, M.; Kristoffersson, E. Pharmacokinetics and dissolution of two crystalline forms of carbamazepine. Int. J. Pharm. 1983, 14, 103–112. [Google Scholar] [CrossRef]
- Jumao-as, A.; Bella, I.; Craig, B.; Lowe, J.; Dasheiff, R.M. Comparison of steady-state blood levels of two carbamazepine formulations. Epilepsia 1989, 30, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Koch, G.; Allan, J. Untoward effects of generic carbamazepine therapy. Arch. Neurol. 1987, 44, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Sachdeo, R.; Chokroverty, S.; Beleldiuk, G. Risk of switching from brand-name to generic drugs in seizure disorder. Epilepsia 1987, 28, 581. [Google Scholar]
- Meyer, M.C.; Straughn, A.B.; Jarvi, E.J.; Wood, G.C.; Pelsor, F.R.; Shah, V.P. The bioinequivalence of carbamazepine tablets with a history of clinical failures. Pharm. Res. 1992, 9, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Ito, S.; Itai, S.; Yamamoto, K. Physicochemical properties and bioavailability of carbamazepine polymorphs and dihydrate. Int. J. Pharm. 2000, 193, 137–146. [Google Scholar] [CrossRef]
- Bauer, J.; Spanton, S.; Henry, R.; Quick, J.; Dziki, W.; Porter, W.; Morris, J. Ritonavir: An extraordinary example of conformational polymorphism. Pharm. Res. 2001, 18, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Mckenzie, A.T. Applicant: Warner-Lambert Company. Form III crystalline (R-(R*,R*)-2-(4-fluorophenyl)-beta, delta-dihyxory-5-(1-methyl-ethyl)-3-phenyl-4-phenylamino)carbonyl)-1H-pyreol-1-heptanoic acid hemi calcium salt (Atorvastatin). WO97/03958, 6 February 1997. [Google Scholar]
- Jin, Y.S.; Ulrich, J. New crystalline solvates of atorvastatin calcium. Chem. Eng. Technol. 2010, 33, 839–844. [Google Scholar] [CrossRef]
- Chadha, R.; Kuhad, A.; Arora, P.; Kishor, S. Characterisation and evaluation of pharmaceutical solvates of atorvastatin calcium by thermoanalytical and spectroscopic studies. Chem. Cent. J. 2012, 6, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Shete, G.; Puri, V.; Kumar, L.; Bansal, A.K. Solid state characterization of commercial crystalline and amorphous atorvastatin calcium samples. AAPS Pharm. Sci. Technol. 2010, 11, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Kerc, J.; Salobir, M.; Bavec, S. Atorvastatin Calcium in a Pharmaceutical form Composition Thereof and Pharmaceutical Formulation Comprising Atorvastatin Calcium. U.S. Patent 7,030,151, 18 April 2006. [Google Scholar]
- Kerc, J. Stable Pharmaceutical Formulation Comprising a HMGCoAreductase Inhibitor. U.S. Patent Application US 2009/0264497 A1, 22 October 2009. [Google Scholar]
- Khan, F.N.; Dehghan, M.H.G. Enhanced bioavailability of atorvastatin calcium from stabilized gastric resident formulation. AAPS Pharm. Sci. Technol. 2011, 12, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Chekal, B.P.; Campeta, A.M.; Abramov, Y.A.; Feeder, N.; Glynn, P.P.; McLaughlin, R.W.; Meenan, P.A.; Singer, R.A. The challenges of developing an API crystallization process for a complex polymorphic and highly solvating system. Part I. Org. Process. Res. Dev. 2009, 13, 1327–1337. [Google Scholar] [CrossRef]
- Campeta, A.M.; Chekal, B.P.; Abramov, Y.A.; Meenan, P.A.; Henson, M.J.; Shi, B.; Singer, R.A.; Horspool, K.R. Development of a targeted polymorph screening approach for a complex polymorphic and highly solvating API. J. Pharm. Sci. 2010, 99, 3874–3886. [Google Scholar] [CrossRef] [PubMed]
- Abramov, Y.A. QTAIM application in drug development: prediction of relative stability of drug polymorphs from experimental crystal structures. J. Phys. Chem. A 2011, 115, 12809–12817. [Google Scholar] [CrossRef] [PubMed]
- Vasileiadis, M.; Pantelides, C.C.; Adjiman, C.S. Prediction of the crystal structures of axitinib, a polymorphic pharmaceutical molecule. Chem. Eng. Sci. 2015, 121, 60–76. [Google Scholar] [CrossRef]
- Matsunaga, J.; Nambu, N.; Nagai, T. Physicochemical approach to biopharmaceutical phenomena. XXX. Polymorphism of phenylbutazone. Chem. Pharm. Bull. 1976, 24, 1169–1172. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.G.; Pisano, F.; Bruno, A. Polymorphism of phenylbutazone: Properties and comparisonal behaviour of crystals. J. Pharm. Sci. 1977, 66, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Kawaguchi, S.; Kobayashi, H.; Nishijo, J. Polymorphism of phenylbutazone by spray dried methods. J. Pharm. Pharmacol. 1980, 32, 579–580. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, T.; Datta, S.; Sheth, A.R.; Grant, D.J.W. Relationships between crystal structures and thermodynamic properties of phenylbutazone solvates. Cryst. Eng. Commun. 2004, 6, 243–249. [Google Scholar] [CrossRef]
- Pandit, J.K.; Gupta, S.K.; Gode, K.D.; Mishra, B. Effect of crystal form on the oral absorption of phenylbutazone. Int. J. Pharm. 1984, 21, 129–132. [Google Scholar] [CrossRef]
- Marchi, E.; Montecchi, L.; Venturini, A.P.; Mascellani, G.; Brufani, M.; Cellai, L. 4-Deoxypyri-do[1),2):1,2]imidazo[5,4-c]rifamycin SV derivatives. A new series of semisynthetic rifamycins with high antibacterial activity and low gastroenteric absorption. J. Med. Chem. 1985, 28, 960–963. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.D.; DuPont, H.L. Rifaximin: In vitro and in vivo antibacterial activity—Review. Chemotherapy 2005, 51, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Adachi, J.A.; DuPont, H.L. Rifaximin: A novel nonabsorbed rifamycin for gastrointestinal disorders. Clin. Infect. Dis. 2006, 42, 541–547. [Google Scholar] [CrossRef] [PubMed]
- European Pharmacopoeia. Rifaximin (Revised Monograph). 2011. Suppl 7.1:2362. Available online: http://www.edqm.eu/en/european-pharmacopoeia-8th-edition-1563.html (accessed on 15 September 2014).
- Viscomi, G.C.; Campana, M.; Barbanti, M.; Grepioni, F.; Polito, M.; Confortini, D.; Rosini, G.; Righi, P.; Cannata, V.; Braga, D. Crystal forms of rifaximin and their effect on pharmaceutical properties. Cryst. Eng. Commun. 2008, 10, 1074–1081. [Google Scholar] [CrossRef]
- Blandizzi, C.; Viscomi, G.C.; Scarpignato, C. Impact of crystal polymorphism on the systemic bioavailability of rifaximin, an antibiotic acting locally in the gastrointestinal tract, in healthy volunteers. Drug Des. Dev. Ther. 2015, 9, 1–11. [Google Scholar]
- Lipitor. Package Insert, Pfizer Ireland Pharmaceuticals, Dublin, Ireland. Parke Davis; Division of Pfizer Inc.: New York, NY, USA, 2009. [Google Scholar]
- Byrn, S.; Pfeiffer, R.; Ganey, M.; Hoiberg, C.; Poochikian, G. Pharmaceutical solids: A strategic approach to regulatory consideration. Pharm. Res. 1995, 12, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Singhal, D.; Curatolo, W. Drug polymorphism and dosage form design: A practical perspective. Adv. Drug Deliv. Rev. 2004, 23, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Evans, J.M.B.; Myerson, A.S. Determination of solubility of polymorphs using Differential Scanning Calorimetry. J. Cryst. Growth Des. 2003, 3, 991–995. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Censi, R.; Di Martino, P. Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules 2015, 20, 18759-18776. https://doi.org/10.3390/molecules201018759
Censi R, Di Martino P. Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules. 2015; 20(10):18759-18776. https://doi.org/10.3390/molecules201018759
Chicago/Turabian StyleCensi, Roberta, and Piera Di Martino. 2015. "Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs" Molecules 20, no. 10: 18759-18776. https://doi.org/10.3390/molecules201018759
APA StyleCensi, R., & Di Martino, P. (2015). Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules, 20(10), 18759-18776. https://doi.org/10.3390/molecules201018759