
Charting a Path to Success in Virtual Screening

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
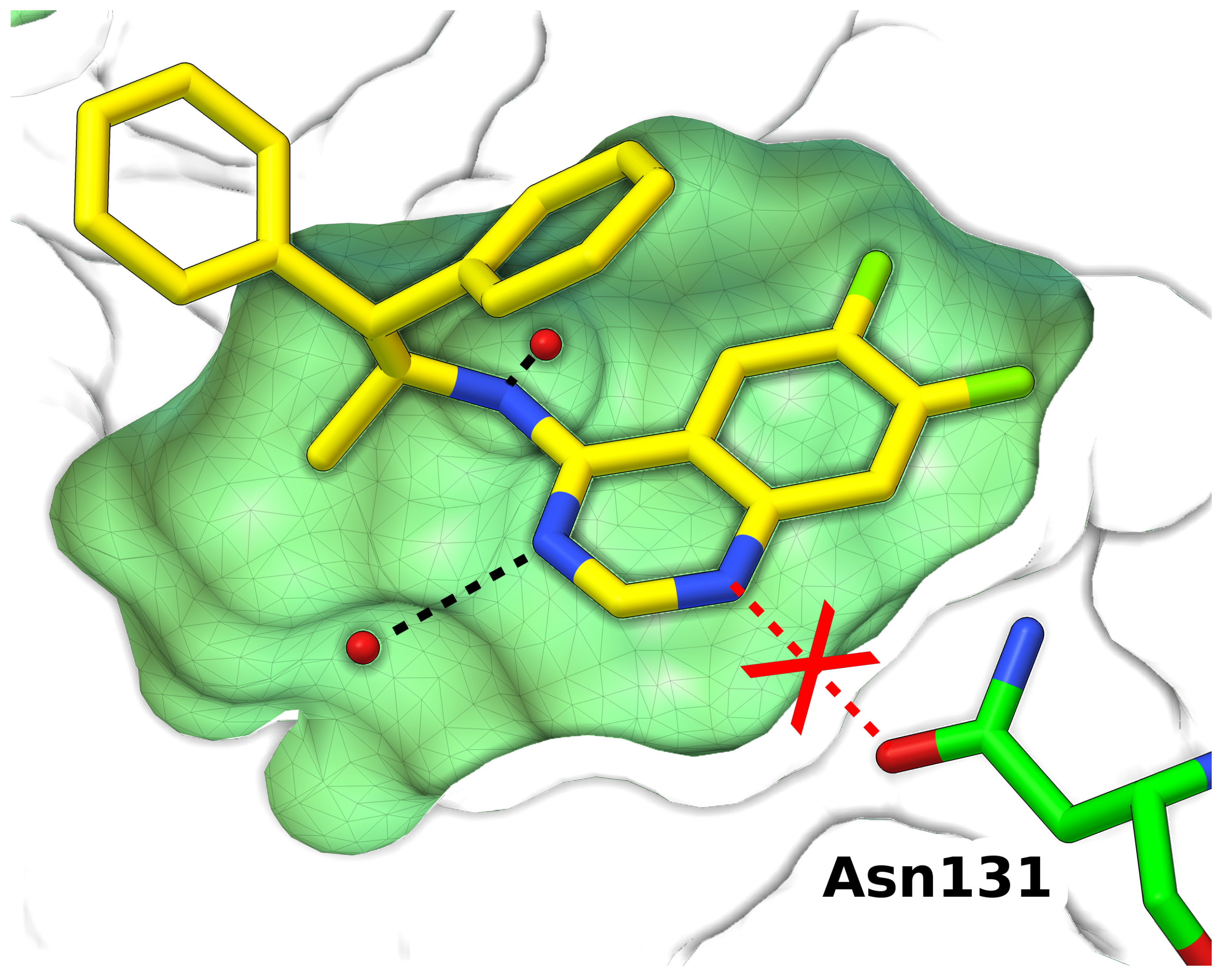{kind=link}
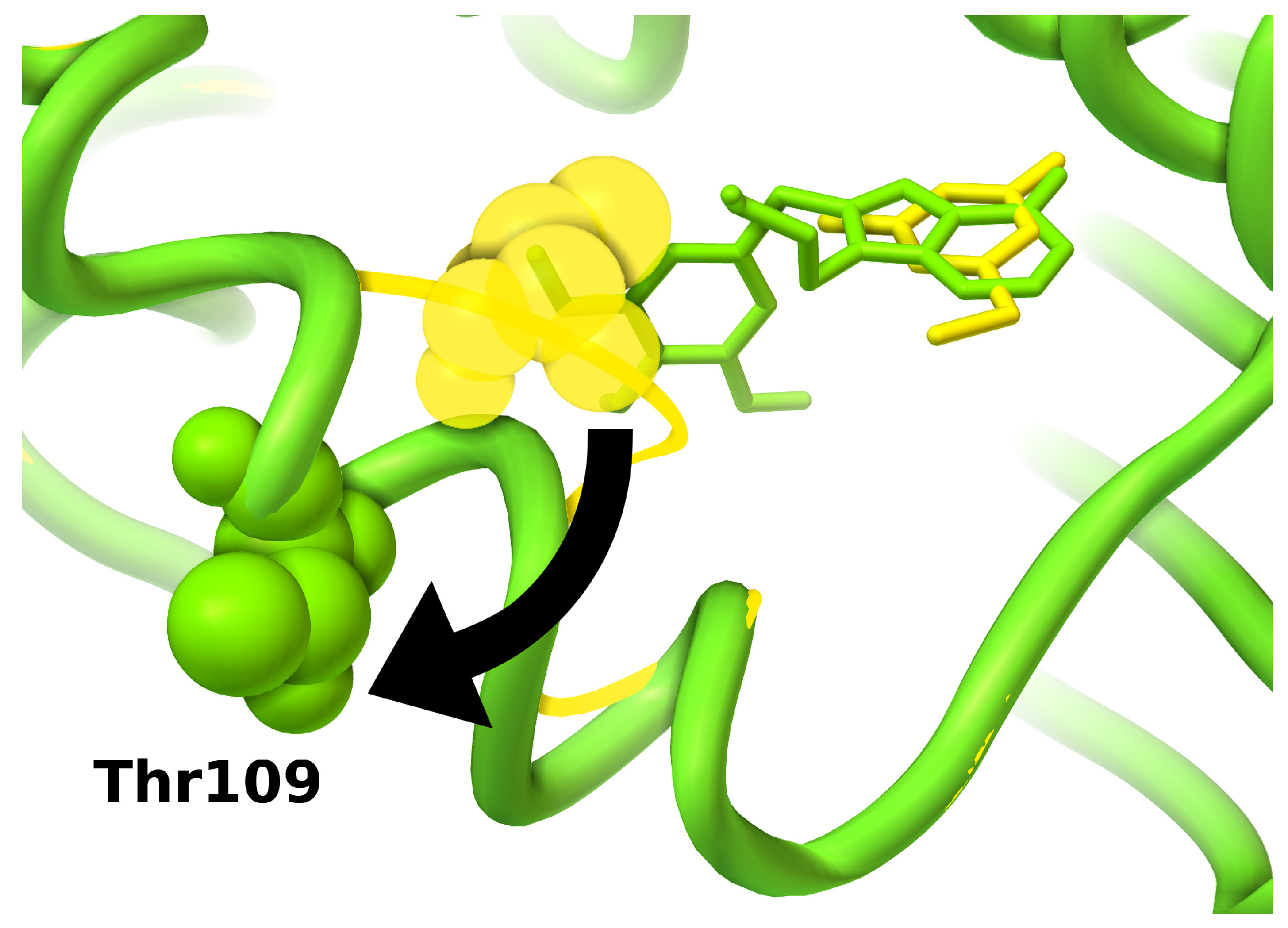{kind=link}
1. Introduction

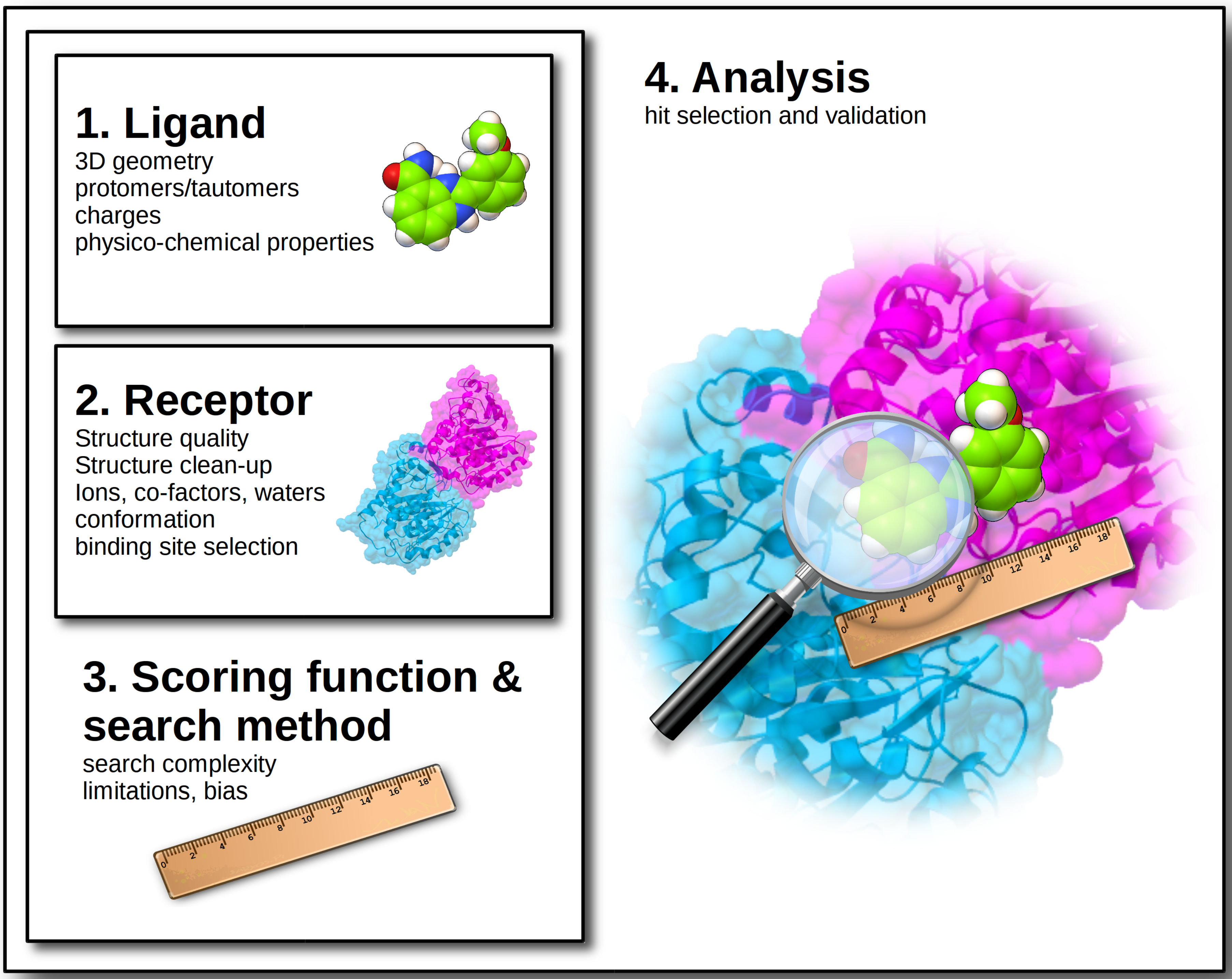
2. Ligand Structures
2.1. Accurate 3D Geometries
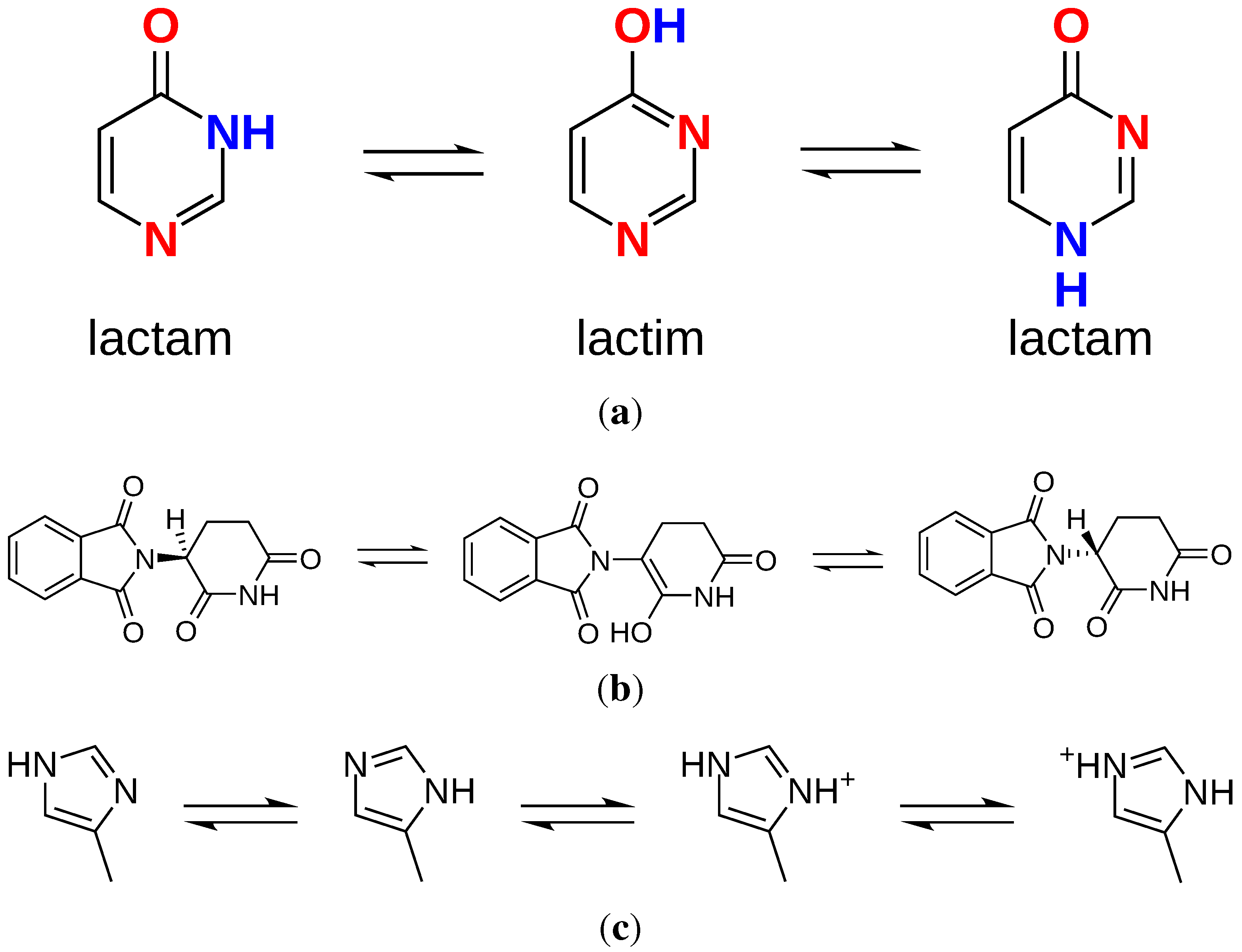
2.2. Tautomers and Protonation States
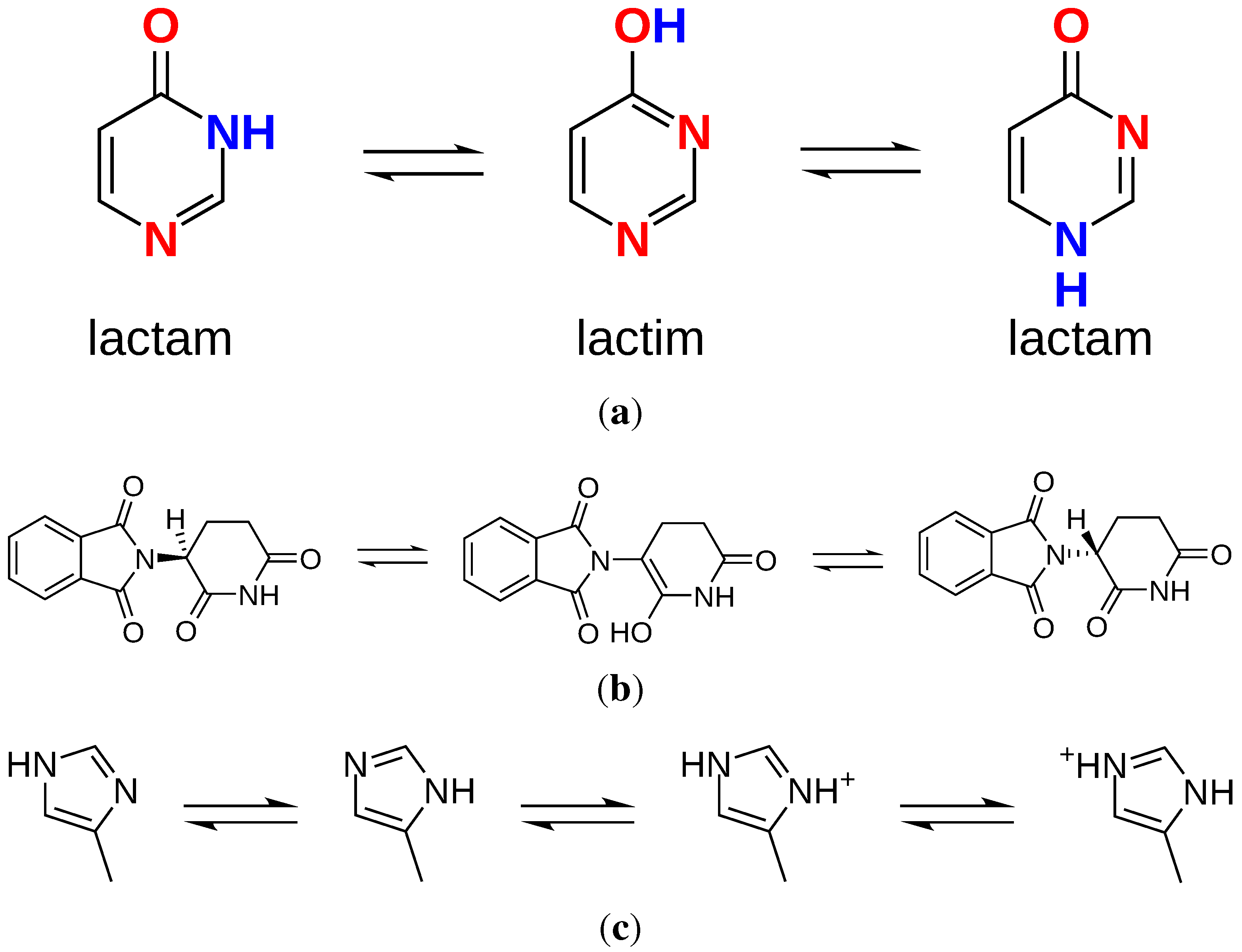
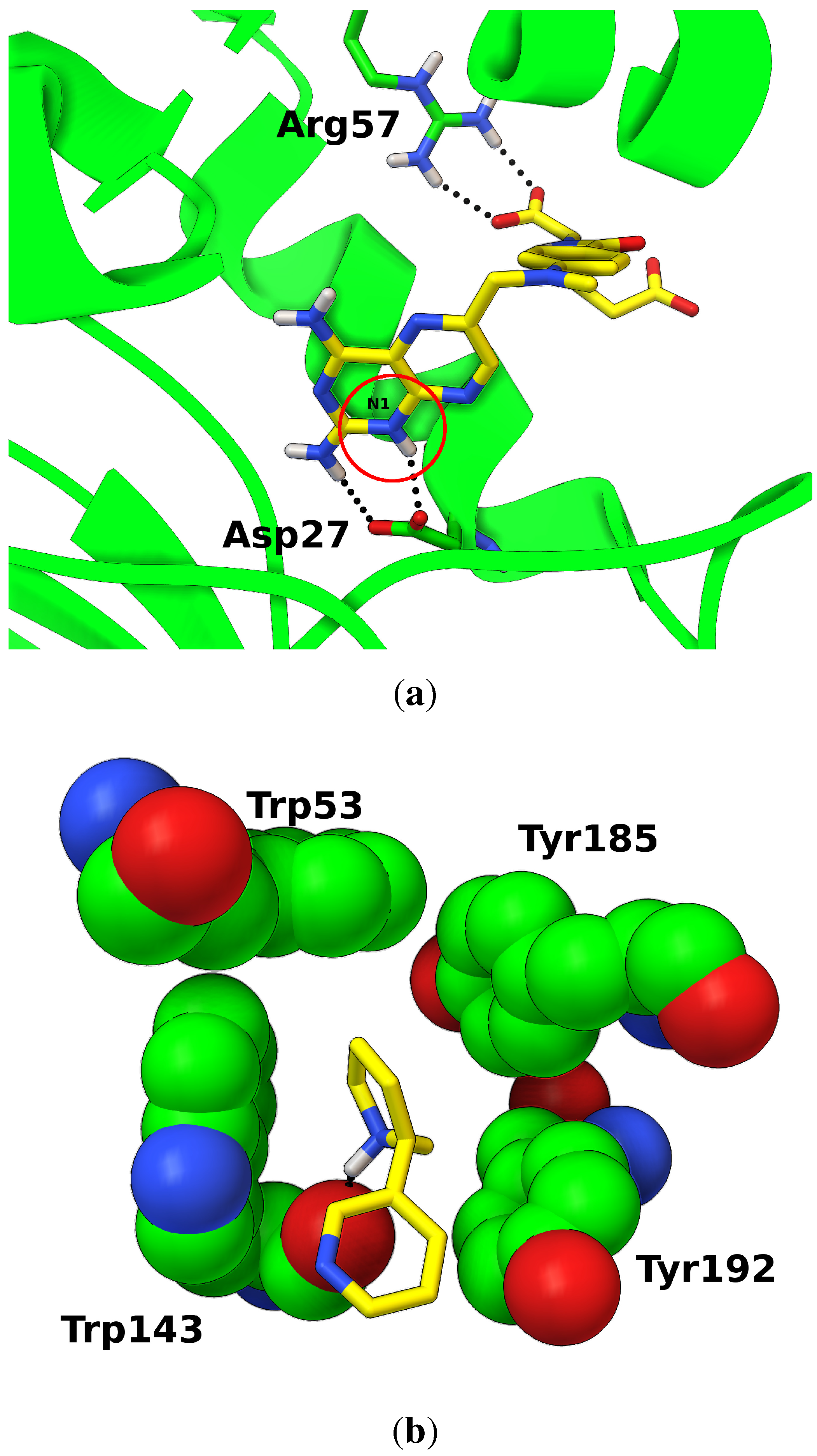
2.3. Charges
2.4. Physicochemical Properties
3. Target Structure
3.1. Structure Quality
3.2. Structure Clean-Up
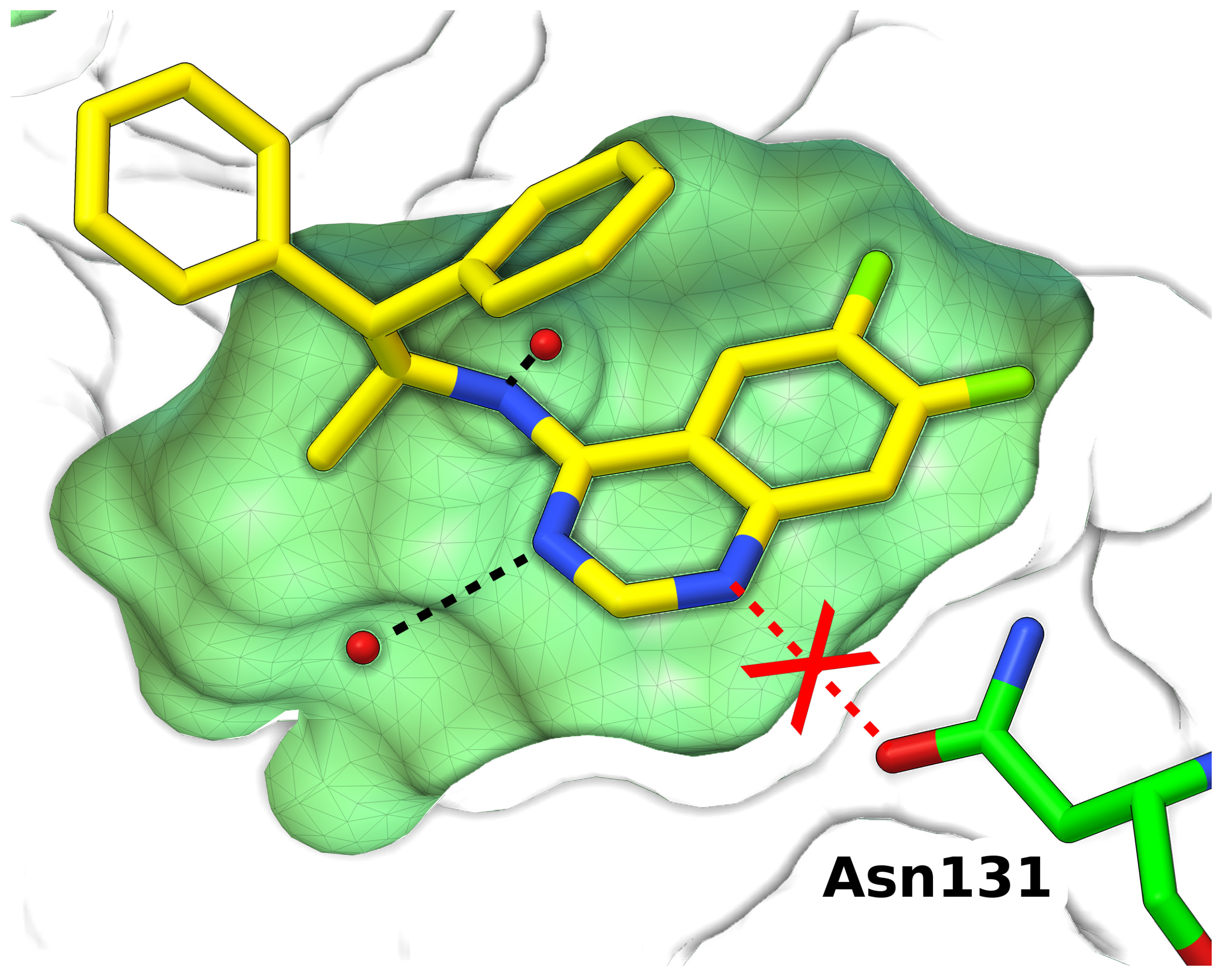
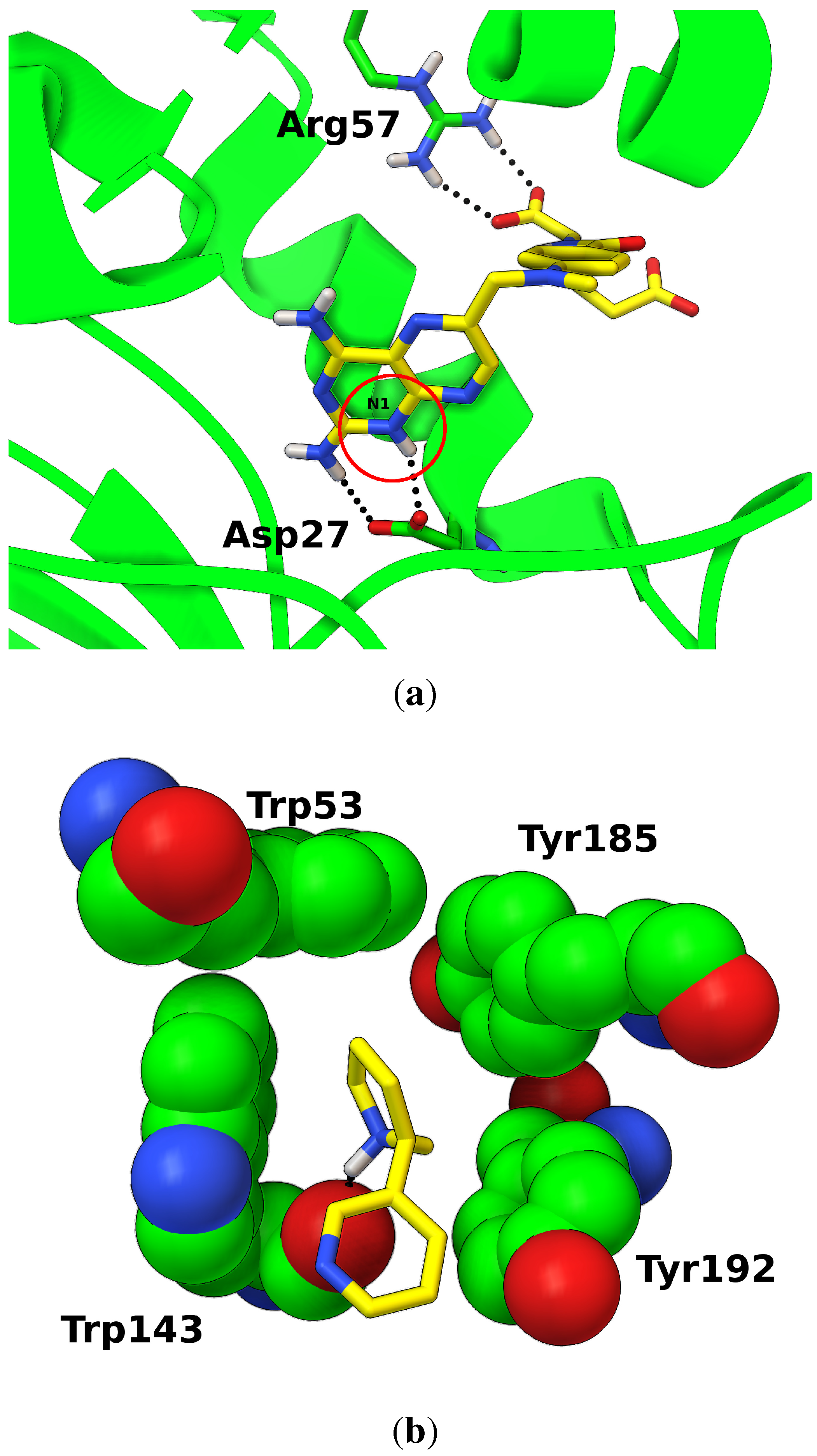
3.3. Protonation States
3.4. Coordinating Metal Ions, Co-Factors and Waters
3.5. Structure Conformation
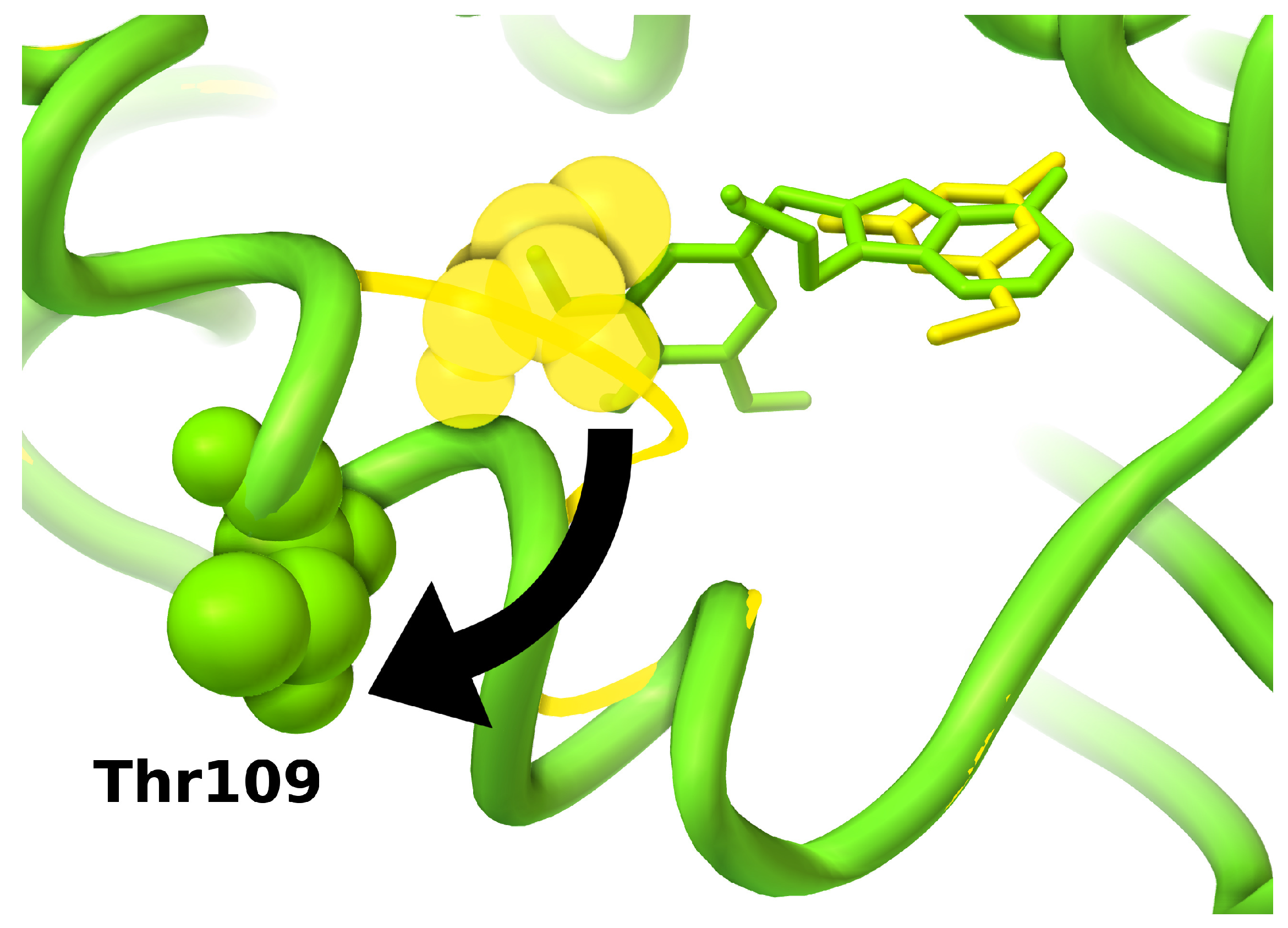
3.6. Binding Site Selection
4. Scoring Function and Search Method
5. Results and Assay
5.1. Hit Selection
5.2. Hit Validation
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Walters, W.P.; Namchuk, M. Designing screens: How to make your hits a hit. Nat. Rev. Drug Discov. 2003, 2, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Perola, E.; Xu, K.; Kollmeyer, T.M.; Kaufmann, S.H.; Prendergast, F.G.; Pang, Y.P. Successful virtual screening of a chemical database for farnesyltransferase inhibitor leads. J. Med. Chem. 2000, 43, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Grüneberg, S.; Stubbs, M.T.; Klebe, G. Successful virtual screening for novel inhibitors of human carbonic anhydrase: Strategy and experimental confirmation. J. Med. Chem. 2002, 45, 3588–3602. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Evers, A.; Klabunde, T. Structure-based drug discovery using GPCR homology modeling: Successful virtual screening for antagonists of the alpha1A adrenergic receptor. J. Med. Chem. 2005, 48, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.; Ferreira, R.S.; Irwin, J.J.; Shoichet, B.K. Docking and chemoinformatic screens for new ligands and targets. Curr. Opin. Biotechnol. 2009, 20, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Barelier, S.; Eidam, O.; Fish, I.; Hollander, J.; Figaroa, F.; Nachane, R.; Irwin, J.J.; Shoichet, B.K.; Siegal, G. Increasing Chemical Space Coverage by Combining Empirical and Computational Fragment Screens. ACS Chem. Biol. 2014, 9, 1528–1535. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Coleman, R.G.; Fraser, J.S.; Shoichet, B.K. Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat. Chem. 2014, 6, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Al Olaby, R.R.; Cocquerel, L.; Zemla, A.; Saas, L.; Dubuisson, J.; Vielmetter, J.; Marcotrigiano, J.; Khan, A.G.; Catalan, F.V.; Perryman, A.L.; et al. Identification of a novel drug lead that inhibits HCV infection and cell-to-cell transmission by targeting the HCV E2 glycoprotein. PLoS ONE 2014, 9, e111333. [Google Scholar] [CrossRef] [PubMed]
- Perryman, A.L.; Yu, W.; Wang, X.; Ekins, S.; Forli, S.; Li, S.G.; Freundlich, J.S.; Tonge, P.J.; Olson, A.J. A Virtual Screen Discovers Novel, Fragment-Sized Inhibitors of Mycobacterium tuberculosis InhA. J. Chem. Inf. Model. 2015, 55, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Babbage, C. Passages from the Life of a Philosopher; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar]
- Irwin, J.J.; Shoichet, B.K. ZINC-a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Uitdehaag, J.C.; Mosi, R.; Kalk, K.H.; van der Veen, B.A.; Dijkhuizen, L.; Withers, S.G.; Dijkstra, B.W. X-ray structures along the reaction pathway of cyclodextrin glycosyltransferase elucidate catalysis in the α-amylase family. Nat. Struct. Mol. Biol. 1999, 6, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Malloci, G.; Vargiu, A.V.; Serra, G.; Bosin, A.; Ruggerone, P.; Ceccarelli, M. A Database of Force-Field Parameters, Dynamics, and Properties of Antimicrobial Compounds. Molecules 2015, 20, 13997–14021. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, A.; William, A.; Blanchard, S.; Nagaraj, H.; Williams, M.; Wang, H.; Lee, A.; Sun, E.; Teo, E.L.; Tan, E.; et al. Structure-based design of nitrogen-linked macrocyclic kinase inhibitors leading to the clinical candidate SB1317/TG02, a potent inhibitor of cyclin dependant kinases (CDKs), Janus kinase 2 (JAK2), and Fms-like tyrosine kinase-3 (FLT3). J. Mol. Model. 2013, 19, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Watts, K.S.; Dalal, P.; Tebben, A.J.; Cheney, D.L.; Shelley, J.C. Macrocycle conformational sampling with MacroModel. J. Chem. Inf. Model. 2014, 54, 2680–2696. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Botta, M. Lennard-Jones potential and dummy atom settings to overcome the AutoDock limitation in treating flexible ring systems. J. Chem. Inf. Model. 2007, 47, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. RDKit: Open-Source Cheminformatics, 2013. Available online: http://www.rdkit.org (accessed on 14 July 2015).
- ChemAxon. Standardizer, JChem 15.7.13.0, 2015. Available online: http://www.chemaxon.com (accessed on 14 July 2015).
- Miteva, M.A.; Guyon, F.; Tufféry, P. Frog2: Efficient 3D conformation ensemble generator for small compounds. Nucleic Acids Res. 2010, 38, W622–W627. [Google Scholar] [CrossRef] [PubMed]
- CACTVS-Server. Online SMILES Translator and Structure File Generator, 2010. Available online: http://cactus.nci.nih.gov/translate/ (accessed on 20 July 2015).
- Ebejer, J.P.; Morris, G.M.; Deane, C.M. Freely available conformer generation methods: How good are they? J. Chem. Inf. Model. 2012, 52, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, P.M.; Vignesh, N.; Kumar, G.R.; Doble, M. Computational approaches to enhance activity of taxanes as antimitotic agent. Med. Chem. Res. 2012, 21, 2557–2570. [Google Scholar] [CrossRef]
- Sayle, R.A. So you think you understand tautomerism? J. Comput.-Aided Mol. Des. 2010, 24, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Sitzmann, M.; Ihlenfeldt, W.D.; Nicklaus, M.C. Tautomerism in large databases. J. Comput.-Aided Mol. Des. 2010, 24, 521–551. [Google Scholar] [CrossRef]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure; John Wiley & Sons: New York, NY, USA, 2007. [Google Scholar]
- Martin, Y.C. Let’s not forget tautomers. J. Comput.-Aided Mol. Des. 2009, 23, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Xiu, P.; Meng, Y.; Zhao, W.; Wang, Z.; Zhou, R. Enantiomerization mechanism of thalidomide and the role of water and hydroxide ions. Chem.-A Eur. J. 2012, 18, 14305–14313. [Google Scholar] [CrossRef] [PubMed]
- Brik, A.; Wong, C.H. HIV-1 protease: Mechanism and drug discovery. Org. Biomol. Chem. 2003, 1, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Barman, A.; Prabhakar, R. Protonation states of the catalytic dyad of β-secretase (BACE1) in the presence of chemically diverse inhibitors: A molecular docking study. J. Chem. Inf. Model. 2012, 52, 1275–1287. [Google Scholar] [CrossRef] [PubMed]
- Stams, T.; Chen, Y.; Christianson, D.W.; Boriack-Sjodin, P.; Hurt, J.D.; Laipis, P.; Silverman, D.N.; Liao, J.; May, J.A.; Dean, T. Structures of murine carbonic anhydrase IV and human carbonic anhydrase II complexed with brinzolamide: Molecular basis of isozyme-drug discrimination. Protein Sci. 1998, 7, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E.C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; de Francesco, R.; Gallinari, P.; et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 15064–15069. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.; Langan, P.; Coates, L.; Mustyakimov, M.; Schoenborn, B.; Howell, E.E.; Dealwis, C. Neutron diffraction studies of Escherichia coli dihydrofolate reductase complexed with methotrexate. Proc. Natl. Acad. Sci. USA 2006, 103, 18493–18498. [Google Scholar] [CrossRef] [PubMed]
- Cocco, L.; Roth, B.; Temple, C.; Montgomery, J.A.; London, R.E.; Blakley, R.L. Protonated state of methotrexate, trimethoprim, and pyrimethamine bound to dihydrofolate reductase. Arch. Biochem. Biophys. 1983, 226, 567–577. [Google Scholar] [CrossRef]
- Hurst, R.; Rollema, H.; Bertrand, D. Nicotinic acetylcholine receptors: From basic science to therapeutics. Pharmacol. Ther. 2013, 137, 22–54. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.P.; Lester, H.A.; Dougherty, D.A. Nicotinic pharmacophore: The pyridine N of nicotine and carbonyl of acetylcholine hydrogen bond across a subunit interface to a backbone NH. Proc. Natl. Acad. Sci. USA 2010, 107, 13206–13211. [Google Scholar] [CrossRef] [PubMed]
- Park, M.S.; Gao, C.; Stern, H.A. Estimating binding affinities by docking/scoring methods using variable protonation states. Proteins Struct. Funct. Bioinform. 2011, 79, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Heinz, H.; Suter, U.W. Atomic charges for classical simulations of polar systems. J. Phys. Chem. B 2004, 108, 18341–18352. [Google Scholar] [CrossRef]
- Ponder, J.W.; Wu, C.; Ren, P.; Pande, V.S.; Chodera, J.D.; Schnieders, M.J.; Haque, I.; Mobley, D.L.; Lambrecht, D.S.; DiStasio, R.A., Jr.; et al. Current status of the AMOEBA polarizable force field. J. Phys. Chem. B 2010, 114, 2549–2564. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.M. Polarizable force fields for molecular dynamics simulations of biomolecules. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2015, 5, 241–254. [Google Scholar] [CrossRef]
- Slater, J.C. A simplification of the Hartree-Fock method. Phys. Rev. 1951, 81, 385. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Jones, T.A.; Zou, J.Y.; Cowan, S.T.; Kjeldgaard, M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. Sect. A Found. Crystallogr. 1991, 47, 110–119. [Google Scholar] [CrossRef]
- Dewar, M.J.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Universal quantum mechanical model for solvation free energies based on gas-phase geometries. J. Phys. Chem. B 1998, 102, 3257–3271. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187217. [Google Scholar]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Basma, M.; Sundara, S.; Çalgan, D.; Vernali, T.; Woods, R.J. Solvated ensemble averaging in the calculation of partial atomic charges. J. Comput. Chem. 2001, 22, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Olson, R.M.; Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Self-consistent reaction field model for aqueous and nonaqueous solutions based on accurate polarized partial charges. J. Chem. Theory Comput. 2007, 3, 2011–2033. [Google Scholar] [CrossRef]
- Woods, R.; Chappelle, R. Restrained electrostatic potential atomic partial charges for condensed-phase simulations of carbohydrates. J. Mol. Struct. THEOCHEM 2000, 527, 149–156. [Google Scholar] [CrossRef]
- Pierpont, C.G. Studies on charge distribution and valence tautomerism in transition metal complexes of catecholate and semiquinonate ligands. Coord. Chem. Rev. 2001, 216, 99–125. [Google Scholar] [CrossRef]
- Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Mittal, R.R.; Harris, L.; McKinnon, R.A.; Sorich, M.J. Partial charge calculation method affects CoMFA QSAR prediction accuracy. J. Chem. Inf. Model. 2009, 49, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A “rule of three” for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Abad-Zapatero, C. A Sorcerer’s apprentice and the rule of five: From rule-of-thumb to commandment and beyond. Drug Discov. Today 2007, 12, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Köster, H.; Craan, T.; Brass, S.; Herhaus, C.; Zentgraf, M.; Neumann, L.; Heine, A.; Klebe, G. A small nonrule of 3 compatible fragment library provides high hit rate of endothiapepsin crystal structures with various fragment chemotypes. J. Med. Chem. 2011, 54, 7784–7796. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Lorber, D.M.; Weston, G.S.; Baase, W.A.; Matthews, B.W.; Shoichet, B.K. Docking molecules by families to increase the diversity of hits in database screens: Computational strategy and experimental evaluation. Proteins Struct. Funct. Bioinform. 2001, 42, 279–293. [Google Scholar] [CrossRef]
- Huang, D.; Caflisch, A. Library screening by fragment-based docking. J. Mol. Recognit. 2010, 23, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shoichet, B.K. Molecular docking and ligand specificity in fragment-based inhibitor discovery. Nat. Chem. Biol. 2009, 5, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Feyfant, E.; Cross, J.B.; Paris, K.; Tsao, D.H. Fragment-based drug design. In Chemical Library Design; Humana Press: Totowa, NJ, USA, 2011; pp. 241–252. [Google Scholar]
- Siegal, G.; Eiso, A.; Schultz, J. Integration of fragment screening and library design. Drug Discov. Today 2007, 12, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Graves, A.P.; Shivakumar, D.M.; Boyce, S.E.; Jacobson, M.P.; Case, D.A.; Shoichet, B.K. Rescoring docking hit lists for model cavity sites: Predictions and experimental testing. J. Mol. Biol. 2008, 377, 914–934. [Google Scholar] [CrossRef] [PubMed]
- Bisignano, P.; Doerr, S.; Harvey, M.J.; Favia, A.D.; Cavalli, A.; de Fabritiis, G. Kinetic characterization of fragment binding in AmpC β-lactamase by high-throughput molecular simulations. J. Chem. Inf. Model. 2014, 54, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A.; Miller, M.C.; Brennan, R.G. Structural mechanism of the simultaneous binding of two drugs to a multidrug-binding protein. Embo J. 2004, 23, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.M.; St-Gallay, S.A.; Kleywegt, G.J. Limitations and lessons in the use of X-ray structural information in drug design. Drug Discov. Today 2008, 13, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.R.; Porebski, P.J.; Chruszcz, M.; Minor, W. X-ray crystallography: Assessment and validation of protein-small molecule complexes for drug discovery. Expert Opin. Drug Discov. 2011, 6, 771–782. [Google Scholar] [PubMed]
- Debye, P. Interferenz von Röntgenstrahlen und Wärmebewegung. Ann. Phys. 1913, 348, 49–92. [Google Scholar] [CrossRef]
- Miller, G. Scientific publishing. A scientist’s nightmare: Software problem leads to five retractions. Science 2006, 314, 1856–1857. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.W. Five retracted structure reports: Inverted or incorrect? Protein Sci. 2007, 16, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Hillisch, A.; Pineda, L.F.; Hilgenfeld, R. Utility of homology models in the drug discovery process. Drug Discov. Today 2004, 9, 659–669. [Google Scholar] [CrossRef]
- Evers, A.; Klebe, G. Successful virtual screening for a submicromolar antagonist of the neurokinin-1 receptor based on a ligand-supported homology model. J. Med. Chem. 2004, 47, 5381–5392. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Bernard, P.; Hibert, M.; Rognan, D. Protein-based virtual screening of chemical databases. II. Are homology models of g-protein coupled receptors suitable targets? Proteins Struct. Funct. Bioinform. 2003, 50, 5–25. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Rueda, M.; Katritch, V.; GPCR Dock 2010 participants; Stevens, R.C.; Abagyan, R. Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure 2011, 19, 1108–1126. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.W.; Sweet, R.M. Macromolecular Crystallography; Gulf Professional Publishing: Oxford, UK, 2003; Volume 374. [Google Scholar]
- Wawrzak, Z.; Sandalova, T.; Steffens, J.J.; Basarab, G.S.; Lundqvist, T.; Lindqvist, Y.; Jordan, D.B. High-resolution structures of scytalone dehydratase-inhibitor complexes crystallized at physiological pH. Proteins Struct. Funct. Bioinform. 1999, 35, 425–439. [Google Scholar] [CrossRef]
- Piccoli, P.M.; Koetzle, T.F.; Schultz, A.J. Single crystal neutron diffraction for the inorganic chemist—A practical guide. Comments Inorg. Chem 2007, 28, 3–38. [Google Scholar] [CrossRef]
- Blakeley, M.P.; Langan, P.; Niimura, N.; Podjarny, A. Neutron crystallography: Opportunities, challenges, and limitations. Curr. Opin. Struct. Biol. 2008, 18, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Meilleur, F.; Weiss, K.L.; Myles, D.A. Deuterium labeling for neutron structure-function-dynamics analysis. In Micro and Nano Technologies in Bioanalysis; Humana Press: Totowa, NJ, USA, 2009; pp. 281–292. [Google Scholar]
- Kear, J.L.; Blackburn, M.E.; Veloro, A.M.; Dunn, B.M.; Fanucci, G.E. Subtype polymorphisms among HIV-1 protease variants confer altered flap conformations and flexibility. J. Am. Chem. Soc. 2009, 131, 14650–14651. [Google Scholar] [PubMed]
- Heaslet, H.; Rosenfeld, R.; Giffin, M.; Lin, Y.C.; Tam, K.; Torbett, B.E.; Elder, J.H.; McRee, D.E.; Stout, C.D. Conformational flexibility in the flap domains of ligand-free HIV protease. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Tiefenbrunn, T.; Forli, S.; Baksh, M.M.; Chang, M.W.; Happer, M.; Lin, Y.C.; Perryman, A.L.; Rhee, J.K.; Torbett, B.E.; Olson, A.J.; et al. Small molecule regulation of protein conformation by binding in the flap of HIV protease. ACS Chem. Biol. 2013, 8, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Strub, M.P.; Hoh, F.; Sanchez, J.F.; Strub, J.M.; Böck, A.; Aumelas, A.; Dumas, C. Selenomethionine and selenocysteine double labeling strategy for crystallographic phasing. Structure 2003, 11, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, W.A. Maturation of MAD phasing for the determination of macromolecular structures. J. Synchrotron Radiat. 1999, 6, 845–851. [Google Scholar] [CrossRef]
- Johansson, L.; Gafvelin, G.; Arnér, E.S. Selenocysteine in proteins—Properties and biotechnological use. Biochim. Biophys. Acta (Bba)-Gen. Subj. 2005, 1726, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jaakola, V.P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.; Lane, J.R.; Ijzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Do, R.K.G.; Šali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [PubMed]
- Read, R.J.; Adams, P.D.; Arendall, W.B.; Brunger, A.T.; Emsley, P.; Joosten, R.P.; Kleywegt, G.J.; Krissinel, E.B.; Lütteke, T.; Otwinowski, Z.; et al. A new generation of crystallographic validation tools for the protein data bank. Structure 2011, 19, 1395–1412. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Word, J.M.; Lovell, S.C.; Richardson, J.S.; Richardson, D.C. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 1999, 285, 1735–1747. [Google Scholar] [CrossRef]
- Vriend, G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Harte, W.E., Jr.; Beveridge, D.L. Prediction of the protonation state of the active site aspartyl residues in HIV-1 protease-inhibitor complexes via molecular dynamics simulation. J. Am. Chem. Soc. 1993, 115, 3883–3886. [Google Scholar] [CrossRef]
- Wang, Y.X.; Freedberg, D.I.; Yamazaki, T.; Wingfield, P.T.; Stahl, S.J.; Kaufman, J.D.; Kiso, Y.; Torchia, D.A. Solution NMR evidence that the HIV-1 protease catalytic aspartyl groups have different ionization states in the complex formed with the asymmetric drug KNI-272. Biochemistry 1996, 35, 9945–9950. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Bolon, B.; Kahn, S.; Bennett, B.D.; Babu-Khan, S.; Denis, P.; Fan, W.; Kha, H.; Zhang, J.; Gong, Y.; et al. Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation. Nat. Neurosci. 2001, 4, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Polgár, T.; Keserü, G.M. Virtual screening for β-secretase (BACE1) inhibitors reveals the importance of protonation states at Asp32 and Asp228. J. Med. Chem. 2005, 48, 3749–3755. [Google Scholar] [CrossRef] [PubMed]
- Vallee, B.L.; Williams, R. Metalloenzymes: The entatic nature of their active sites. Proc. Natl. Acad. Sci. USA 1968, 59, 498–505. [Google Scholar] [PubMed]
- Englebienne, P.; Fiaux, H.; Kuntz, D.A.; Corbeil, C.R.; Gerber-Lemaire, S.; Rose, D.R.; Moitessier, N. Evaluation of docking programs for predicting binding of Golgi α-mannosidase II inhibitors: A comparison with crystallography. Proteins Struct. Funct. Bioinform. 2007, 69, 160–176. [Google Scholar] [CrossRef] [PubMed]
- Moitessier, N.; Englebienne, P.; Lee, D.; Lawandi, J.; Corbeil, C. Towards the development of universal, fast and highly accurate docking/scoring methods: A long way to go. Br. J. Pharmacol. 2008, 153, S7–S26. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Balaz, S.; Shelver, W.H. A practical approach to docking of zinc metalloproteinase inhibitors. J. Mol. Graph. Model. 2004, 22, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Raushel, F.M.; Shoichet, B.K. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry 2005, 44, 12316–12328. [Google Scholar] [CrossRef] [PubMed]
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4Zn: An Improved Autodock Force Field for Small-Molecule Docking to Zinc Metalloproteins. J. Chem. Inf. Model. 2014, 54, 2371–2379. [Google Scholar] [CrossRef] [PubMed]
- Seebeck, B.; Reulecke, I.; Kämper, A.; Rarey, M. Modeling of metal interaction geometries for protein–ligand docking. Proteins Struct. Funct. Bioinform. 2008, 71, 1237–1254. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Read, J.A.; Wilkinson, K.W.; Tranter, R.; Sessions, R.B.; Brady, R.L. Chloroquine Binds in the Cofactor Binding Site ofPlasmodium falciparum Lactate Dehydrogenase. J. Biol. Chem. 1999, 274, 10213–10218. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, C. Multiple ligand simultaneous docking: Orchestrated dancing of ligands in binding sites of protein. J. Comput. Chem. 2010, 31, 2014–2022. [Google Scholar] [CrossRef] [PubMed]
- Villacanas, O.; Madurga, S.; Giralt, E.; Belda, I. Explicit treatment of water molecules in protein-ligand docking. Curr. Comput.-Aided Drug Des. 2009, 5, 145–154. [Google Scholar] [CrossRef]
- Baldwin, E.T.; Bhat, T.N.; Gulnik, S.; Liu, B.; Topol, I.A.; Kiso, Y.; Mimoto, T.; Mitsuya, H.; Erickson, J.W. Structure of HIV-1 protease with KNI-272, a tight-binding transition-state analog containing allophenylnorstatine. Structure 1995, 3, 581–590. [Google Scholar] [CrossRef]
- Forli, S.; Olson, A.J. A force field with discrete displaceable waters and desolvation entropy for hydrated ligand docking. J. Med. Chem. 2012, 55, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Shoichet, B.K. Exploiting ordered waters in molecular docking. J. Med. Chem. 2008, 51, 4862–4865. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Chessari, G.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Nissink, J.W.M.; Taylor, R.D.; Taylor, R. Modeling water molecules in protein-ligand docking using GOLD. J. Med. Chem. 2005, 48, 6504–6515. [Google Scholar] [CrossRef] [PubMed]
- De Beer, S.; Vermeulen, N.P.; Oostenbrink, C. The role of water molecules in computational drug design. Curr. Top. Med. Chem. 2010, 10, 55–66. [Google Scholar] [CrossRef]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, C.R.; Mathiowetz, A.M. Addressing limitations with the MM-GB/SA scoring procedure using the WaterMap method and free energy perturbation calculations. J. Chem. Inf. Model. 2010, 50, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Fenimore, P.W.; Frauenfelder, H.; McMahon, B.; Young, R. Bulk-solvent and hydration-shell fluctuations, similar to α-and β-fluctuations in glasses, control protein motions and functions. Proc. Natl. Acad. Sci. USA 2004, 101, 14408–14413. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Levinson, N.M.; Kuchment, O.; Shen, K.; Young, M.A.; Koldobskiy, M.; Karplus, M.; Cole, P.A.; Kuriyan, J. A Src-like inactive conformation in the abl tyrosine kinase domain. PLoS Biol. 2006, 4, 753. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Arlow, D.H.; Borhani, D.W.; Jensen, M.Ø.; Piana, S.; Shaw, D.E. Identification of two distinct inactive conformations of the β2-adrenergic receptor reconciles structural and biochemical observations. Proc. Natl. Acad. Sci. USA 2009, 106, 4689–4694. [Google Scholar] [CrossRef] [PubMed]
- Standfuss, J.; Edwards, P.C.; D’Antona, A.; Fransen, M.; Xie, G.; Oprian, D.D.; Schertler, G.F. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature 2011, 471, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Gouldson, P.R.; Kidley, N.J.; Bywater, R.P.; Psaroudakis, G.; Brooks, H.D.; Diaz, C.; Shire, D.; Reynolds, C.A. Toward the active conformations of rhodopsin and the β2-adrenergic receptor. Proteins Struct. Funct. Bioinform. 2004, 56, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Frauenfelder, H.; Chen, G.; Berendzen, J.; Fenimore, P.W.; Jansson, H.; McMahon, B.H.; Stroe, I.R.; Swenson, J.; Young, R.D. A unified model of protein dynamics. Proc. Natl. Acad. Sci. USA 2009, 106, 5129–5134. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.; Barril, X.; Dymock, B.; Sheridan, L.; Surgenor, A.; Beswick, M.; Drysdale, M.; Collier, A.; Massey, A.; Davies, N.; et al. Structure-activity relationships in purine-based inhibitor binding to HSP90 isoforms. Chem. Biol. 2004, 11, 775–785. [Google Scholar] [CrossRef]
- Koshland, D.E. The key-lock theory and the induced fit theory. Angew. Chem. Int. Ed. Engl. 1995, 33, 2375–2378. [Google Scholar] [CrossRef]
- McGovern, S.L.; Shoichet, B.K. Information decay in molecular docking screens against holo, apo, and modeled conformations of enzymes. J. Med. Chem. 2003, 46, 2895–2907. [Google Scholar] [CrossRef] [PubMed]
- Brough, P.A.; Barril, X.; Borgognoni, J.; Chene, P.; Davies, N.G.; Davis, B.; Drysdale, M.J.; Dymock, B.; Eccles, S.A.; Garcia-Echeverria, C.; et al. Combining hit identification strategies: Fragment-based and in silico approaches to orally active 2-aminothieno [2,3-d] pyrimidine inhibitors of the Hsp90 molecular chaperone. J. Med. Chem. 2009, 52, 4794–4809. [Google Scholar] [CrossRef] [PubMed]
- Weik, M.; Colletier, J.P. Temperature-dependent macromolecular X-ray crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 437–446. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A. Introduction to protein crystallization. Methods 2004, 34, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Eyal, E.; Gerzon, S.; Potapov, V.; Edelman, M.; Sobolev, V. The limit of accuracy of protein modeling: Influence of crystal packing on protein structure. J. Mol. Biol. 2005, 351, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Kornev, A.P.; Haste, N.M.; Taylor, S.S.; Ten Eyck, L.F. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 17783–17788. [Google Scholar] [CrossRef] [PubMed]
- Lexa, K.W.; Carlson, H.A. Protein flexibility in docking and surface mapping. Q. Rev. Biophys. 2012, 45, 301–343. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Holien, J.; Ramsland, P.A. Improvements, trends, and new ideas in molecular docking: 2012–2013 in review. J. Mol. Recognit. 2015, 28, 581–604. [Google Scholar] [CrossRef] [PubMed]
- Craig, I.R.; Essex, J.W.; Spiegel, K. Ensemble docking into multiple crystallographically derived protein structures: An evaluation based on the statistical analysis of enrichments. J. Chem. Inf. Model. 2010, 50, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Perryman, A.L.; Schames, J.R.; McCammon, J.A. Computational drug design accommodating receptor flexibility: The relaxed complex scheme. J. Am. Chem. Soc. 2002, 124, 5632–5633. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Kovacs, J.A.; Abagyan, R.A. Representing receptor flexibility in ligand docking through relevant normal modes. J. Am. Chem. Soc. 2005, 127, 9632–9640. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Sanschagrin, P.C.; Greenwood, J.R.; Repasky, M.P.; Sherman, W.; Farid, R. Improving database enrichment through ensemble docking. J. Comput.-Aided Mol. Des. 2008, 22, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, S.T.; Liu, J.; Estiu, G.; Oltvai, Z.; Wiest, O. Identification of novel bacterial histidine biosynthesis inhibitors using docking, ensemble rescoring, and whole-cell assays. Bioorg. Med. Chem. 2010, 18, 5148–5156. [Google Scholar] [CrossRef] [PubMed]
- Totrov, M.; Abagyan, R. Flexible ligand docking to multiple receptor conformations: A practical alternative. Curr. Opin. Struct. Biol. 2008, 18, 178–184. [Google Scholar] [CrossRef] [PubMed]
- IBM. World Community Grid, 2007. Available online: http://www.worldcommunitygrid.org/about_us/viewAboutUs.do (accessed on 20 July 2015).
- Hetényi, C.; van der Spoel, D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 2002, 11, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Hetényi, C.; van der Spoel, D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef]
- Davis, I.W.; Raha, K.; Head, M.S.; Baker, D. Blind docking of pharmaceutically relevant compounds using RosettaLigand. Protein Sci. 2009, 18, 1998–2002. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, D.; Sanchez, R. Improving accuracy and efficiency of blind protein-ligand docking by focusing on predicted binding sites. Proteins: Struct. Funct. Bioinform. 2009, 74, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, D.; Sanchez, R. Beyond structural genomics: Computational approaches for the identification of ligand binding sites in protein structures. J. Struct. Funct. Genomics 2011, 12, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lu, Y.; Wang, S. Comparative evaluation of 11 scoring functions for molecular docking. J. Med. Chem. 2003, 46, 2287–2303. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Andrews, C.W.; Capelli, A.M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein–ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899–12908. [Google Scholar] [CrossRef] [PubMed]
- Moitessier, N.; Westhof, E.; Hanessian, S. Docking of aminoglycosides to hydrated and flexible RNA. J. Med. Chem. 2006, 49, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Perola, E.; Walters, W.P.; Charifson, P.S. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins Struct. Funct. Bioinform. 2004, 56, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.A.; Morris, G.M.; Biggin, P.C. One size does not fit all: The limits of structure-based models in drug discovery. J. Chem. Theory Comput. 2013, 9, 4266–4274. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Gasch, T.; Stahl, M. Scoring functions for protein–ligand interactions: A critical perspective. Drug Discov. Today Technol. 2004, 1, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Chia-en, A.C.; Chen, W.; Gilson, M.K. Ligand configurational entropy and protein binding. Proc. Natl. Acad. Sci. USA 2007, 104, 1534–1539. [Google Scholar]
- Merlitz, H.; Wenzel, W. Comparison of stochastic optimization methods for receptor–ligand docking. Chem. Phys. Lett. 2002, 362, 271–277. [Google Scholar] [CrossRef]
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martínez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing pitfalls in virtual screening: A critical review. J. Chem. Inf. Model. 2012, 52, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Bleicher, K.H.; Böhm, H.J.; Müller, K.; Alanine, A.I. Hit and lead generation: Beyond high-throughput screening. Nat. Rev. Drug Discov. 2003, 2, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Li, X.; Li, Y.; Liu, Z.; Wang, R. Comparative assessment of scoring functions on a diverse test set. J. Chem. Inf. Model. 2009, 49, 1079–1093. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual screening with AutoDock: Theory and practice. Expert Opin. Drug Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.A.; Congreve, M.; Murray, C.W.; Rees, D.C. Fragment-based lead discovery: Leads by design. Drug Discov. Today 2005, 10, 987–992. [Google Scholar] [CrossRef]
- Balius, T.E.; Mukherjee, S.; Rizzo, R.C. Implementation and evaluation of a docking-rescoring method using molecular footprint comparisons. J. Comput. Chem. 2011, 32, 2273–2289. [Google Scholar] [CrossRef] [PubMed]
- Perryman, A.L.; Santiago, D.N.; Forli, S.; Santos-Martins, D.; Olson, A.J. Virtual screening with AutoDock Vina and the common pharmacophore engine of a low diversity library of fragments and hits against the three allosteric sites of HIV integrase: Participation in the SAMPL4 protein–ligand binding challenge. J. Comput.-Aided Mol. Des. 2014, 28, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Chapsal, B.D.; Weber, I.T.; Mitsuya, H. Design of HIV protease inhibitors targeting protein backbone: An effective strategy for combating drug resistance. Acc. Chem. Res. 2007, 41, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Houston, D.R.; Walkinshaw, M.D. Consensus docking: Improving the reliability of docking in a virtual screening context. J. Chem. Inf. Model. 2013, 53, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Plewczynski, D.; Łażniewski, M.; Grotthuss, M.V.; Rychlewski, L.; Ginalski, K. VoteDock: Consensus docking method for prediction of protein–ligand interactions. J. Comput. Chem. 2011, 32, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P.; Gleeson, D. QM/MM as a tool in fragment based drug discovery. A cross-docking, rescoring study of kinase inhibitors. J. Chem. Inf. Model. 2009, 49, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Rio, A.D.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Gallicchio, E.; Deng, N.; He, P.; Wickstrom, L.; Perryman, A.L.; Santiago, D.N.; Forli, S.; Olson, A.J.; Levy, R.M. Virtual screening of integrase inhibitors by large scale binding free energy calculations: The SAMPL4 challenge. J. Comput.-Aided Mol. Des. 2014, 28, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Slynko, I.; Scharfe, M.; Rumpf, T.; Eib, J.; Metzger, E.; Schüle, R.; Jung, M.; Sippl, W. Virtual screening of PRK1 inhibitors: Ensemble docking, rescoring using binding free energy calculation and QSAR model development. J. Chem. Inf. Model. 2014, 54, 138–150. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, C.; Pospisil, P.; Pos, W.; Folkers, G.; Vermeulen, N.P. Binding mode prediction of cytochrome p450 and thymidine kinase protein-ligand complexes by consideration of water and rescoring in automated docking. J. Med. Chem. 2005, 48, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Forli, S.; He, P.; Perryman, A.; Wickstrom, L.; Vijayan, R.; Tiefenbrunn, T.; Stout, D.; Gallicchio, E.; Olson, A.J.; et al. Distinguishing Binders from False Positives by Free Energy Calculations: Fragment Screening Against the Flap Site of HIV Protease. J. Phys. Chem. B 2014, 119, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Zartler, E.R. Quantum Tessera Consulting, 2013. Available online: http://www.quantumtessera.com/your-computation-is-only-as-good-as-your-experimental-follow-up/ (accessed on 14 July 2015).
- Lo, M.C.; Aulabaugh, A.; Jin, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Lea, W.A.; Simeonov, A. Fluorescence polarization assays in small molecule screening. Expert Opin. Drug Discov. 2011, 6, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.M.; Van Molle, I.; Baud, M.G.; Galdeano, C.; Geraldes, C.F.; Ciulli, A. Is NMR fragment screening fine-tuned to assess druggability of protein–protein interactions? ACS Med. Chem. Lett. 2013, 5, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Wielens, J.; Headey, S.J.; Rhodes, D.I.; Mulder, R.J.; Dolezal, O.; Deadman, J.J.; Newman, J.; Chalmers, D.K.; Parker, M.W.; Peat, T.S.; et al. Parallel Screening of Low Molecular Weight Fragment Libraries Do Differences in Methodology Affect Hit Identification? J. Biomol. Screen. 2013, 18, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forli, S. Charting a Path to Success in Virtual Screening. Molecules 2015, 20, 18732-18758. https://doi.org/10.3390/molecules201018732
Forli S. Charting a Path to Success in Virtual Screening. Molecules. 2015; 20(10):18732-18758. https://doi.org/10.3390/molecules201018732
Chicago/Turabian StyleForli, Stefano. 2015. "Charting a Path to Success in Virtual Screening" Molecules 20, no. 10: 18732-18758. https://doi.org/10.3390/molecules201018732
APA StyleForli, S. (2015). Charting a Path to Success in Virtual Screening. Molecules, 20(10), 18732-18758. https://doi.org/10.3390/molecules201018732