
Duchenne Muscular Dystrophy: From Diagnosis to Therapy

{kind=link}
{kind=link}
Abstract
:1. Duchenne Muscular Dystrophy
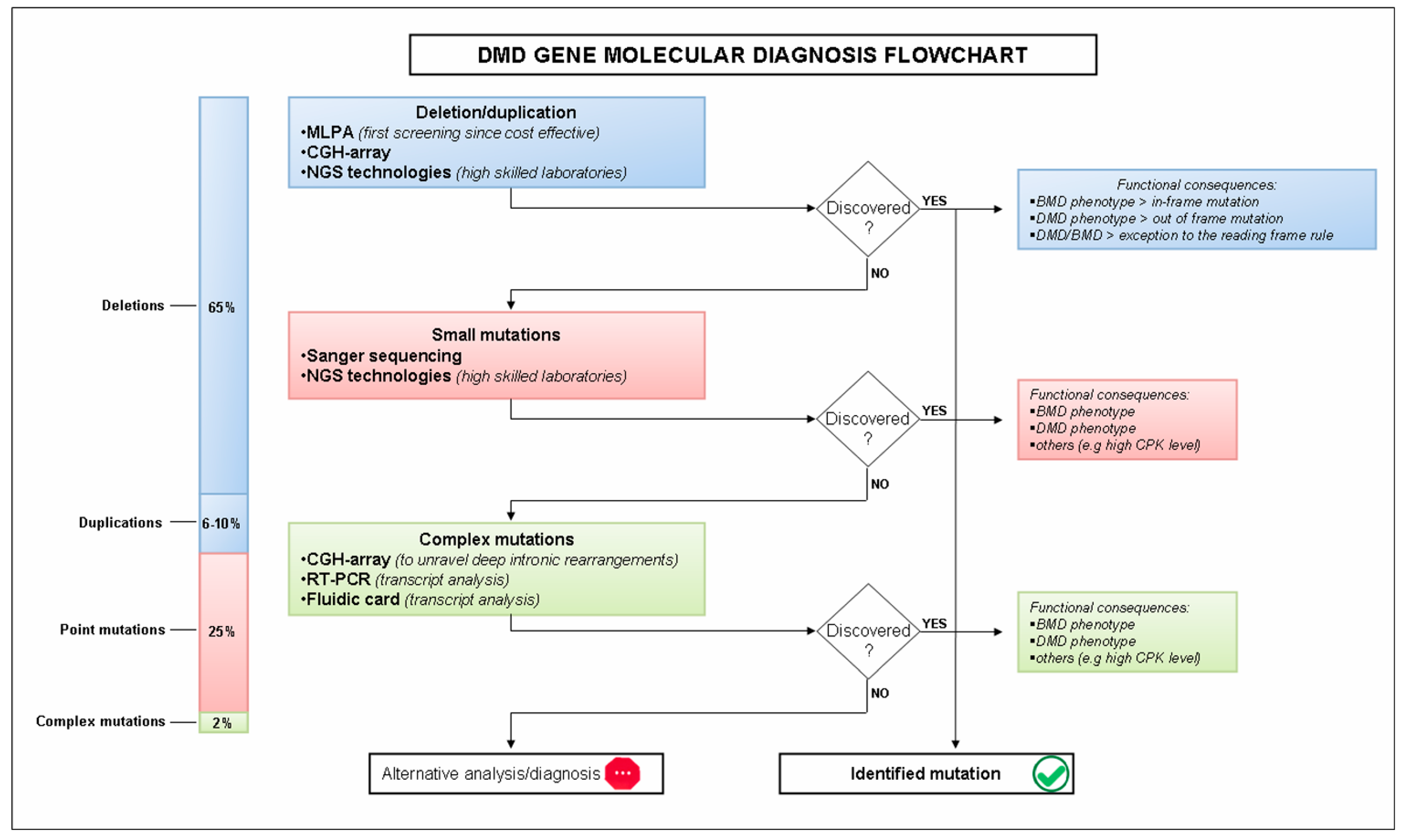
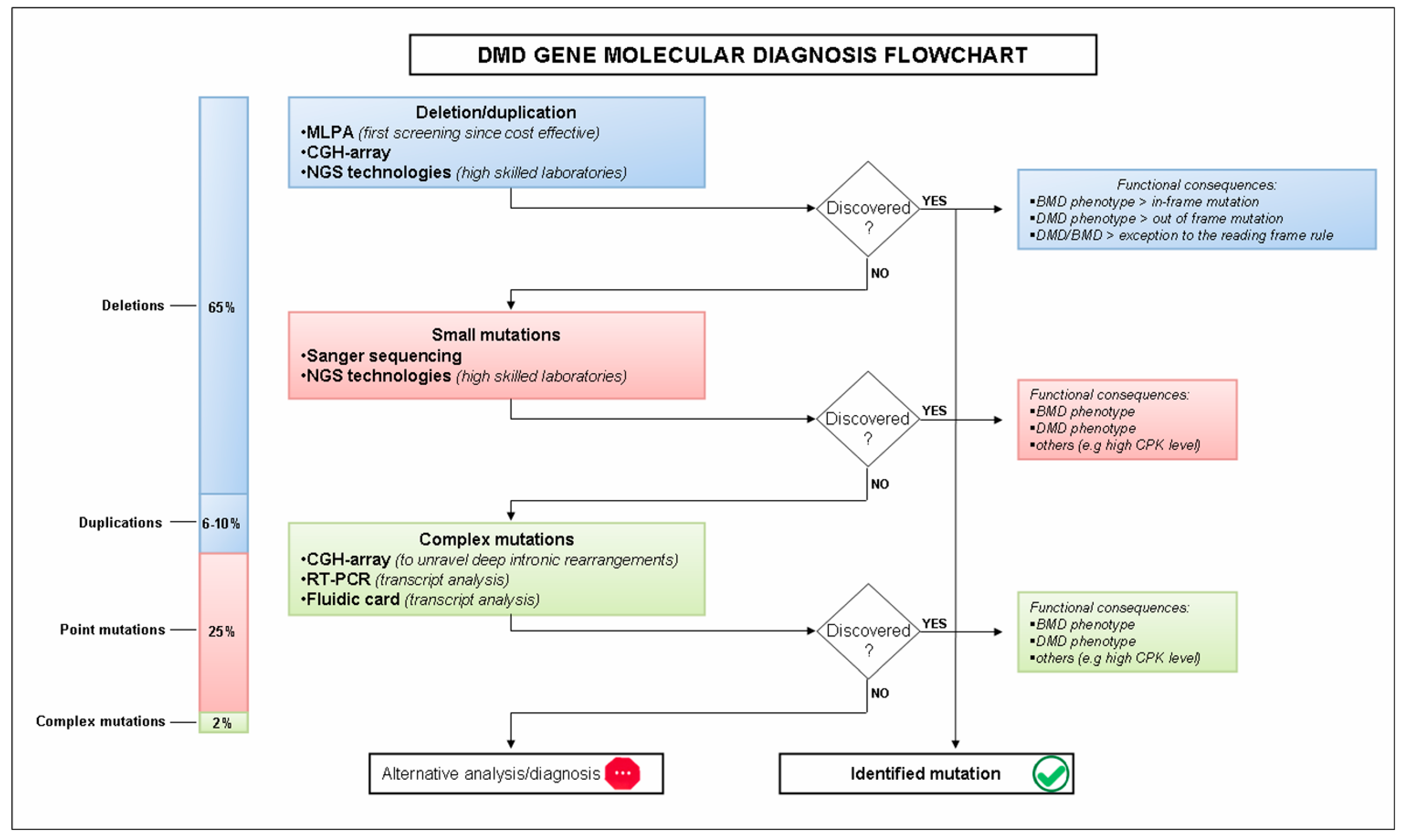
2. Molecular Diagnosis
3. Therapeutic Interventions
3.1. Corticosteroid
3.2. Novel Classical Pharmacological Approaches
3.3. Cell Therapy
3.4. Gene Therapy
3.5. Stop-Codon Read-Through: Mutation Suppression
3.6. Utrophin Modulation
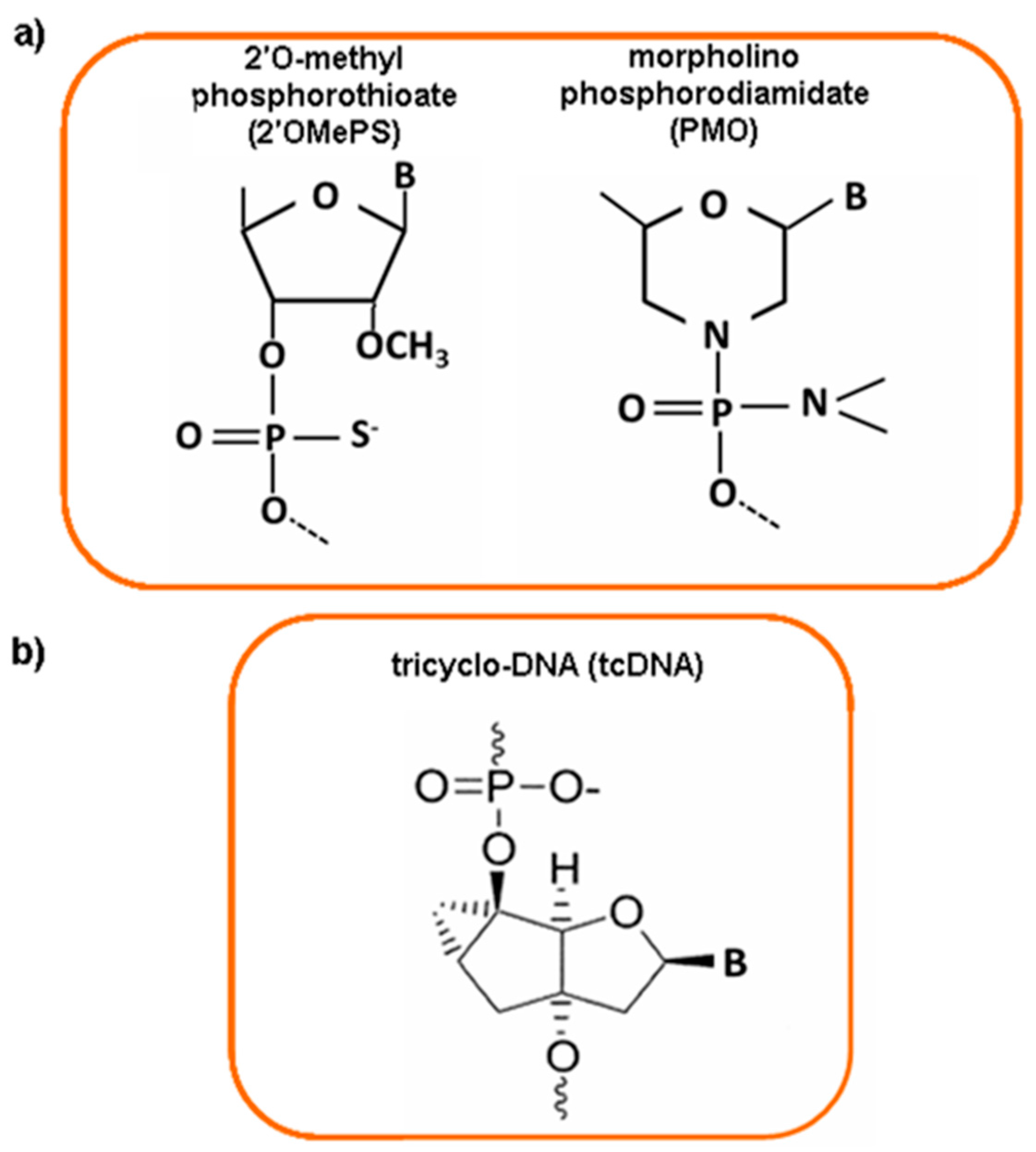
3.7. Antisense Oligonucleotides and Exon Skipping
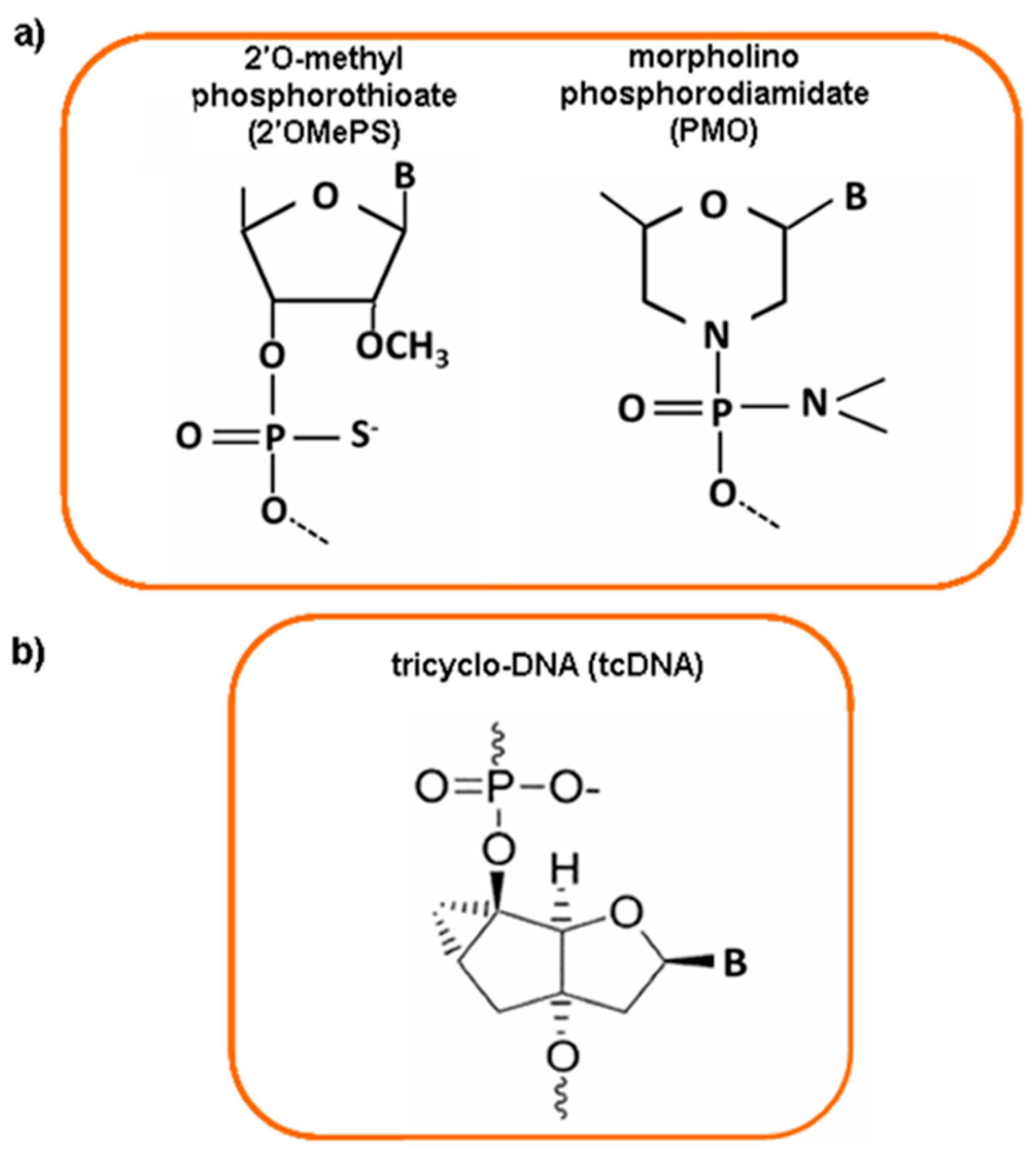
4. Nanoparticles as Delivery System for DMD Therapy
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chung, J.; Smith, A.L.; Hughes, S.C.; Niizawa, G.; Abdel-Hamid, H.Z.; Naylor, E.W.; Hughes, T.; Clemens, P.R. 20-year Follow-up of newborn screening for patients with muscular dystrophy. Muscle Nerve 2015. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Pane, M.; Scalise, R.; Berardinelli, A.; D’Angelo, G.; Ricotti, V.; Alfieri, P.; Moroni, I.; Hartley, L.; Pera, M.C.; Baranello, G.; et al. Early neurodevelopmental assessment in Duchenne muscular dystrophy. Neuromuscul. Disord. 2013, 23, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Cloer, C.; Lu, P.; Milazi, S.; Shaban, M.; Shah, S.N.; Marston-Poe, L.; Moulton, H.M.; Lu, Q.L. Exon skipping restores dystrophin expression, but fails to prevent disease progression in later stage dystrophic dko mice. Gene Ther. 2014, 21, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Van Westering, T.L.E.; Betts, C.A.; Wood, M.J.A. Current understanding of molecular pathology and treatment of cardiomyopathy in Duchenne Muscular Dystrophy. Molecules 2015, 20, 8823–8855. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A. X-Linked Dilated Cardiomyopathy: A Cardiospecific Phenotype of Dystrophinopathy. Pharmaceuticals (Basel) 2015, 8, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Ljubicic, V.; Burt, M.; Jasmin, B.J. The therapeutic potential of skeletal muscle plasticity in Duchenne muscular dystrophy: Phenotypic modifiers as pharmacologic targets. FASEB J. 2014, 28, 548–568. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Tajrishi, M.M.; Sato, S.; Hindi, S.M.; Kumar, A. Therapeutic potential of matrix metalloproteinases in Duchenne muscular dystrophy. Front. Cell Dev. Biol. 2014, 1, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Ferlini, A.; Neri, M.; Gualandi, F. The medical genetics of dystrophinopathies: Molecular genetic diagnosis and its impact on clinical practice. Neuromuscul. Disord. 2013, 23, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Findlay, AR.; Wein, N.; Kaminoh, Y.; Taylor, L.E.; Dunn, D.M.; Mendell, J.R.; King, W.M.; Pestronk, A.; Florence, J.M.; Mathews, K.D.; et al. Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann. Neurol. 2015, 77, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Tajrishi, M.M.; Ogura, Y.; Kumar, A. Wasting mechanisms in muscular dystrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2266–2279. [Google Scholar] [CrossRef] [PubMed]
- Falzarano, M.S.; Passarelli, C.; Ferlini, A. Nanoparticle delivery of antisense oligonucleotides and their application in the exon skipping strategy for Duchenne muscular dystrophy. Nucleic Acid Ther. 2014, 24, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Vlcek, V.; Balabanov, P.; Salmonson, T.; Bakchine, S.; Markey, G.; Weise, M.; Schlosser-Weber, G.; Brohmann, H.; Yerro, C.P.; et al. European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul. Disord. 2015, 25, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Ferlini, A.; Neri, M. Molecular Genetics of Dystrophinopathies; eLS. John Wiley & Sons, Ltd.: Chichester, UK, 2014. [Google Scholar]
- Bovolenta, M.; Neri, M.; Fini, S.; Fabris, M.; Trabanelli, C.; Venturoli, A.; Martoni, E.; Bassi, E.; Spitali, P.; Brioschi, S.; et al. A novel custom high density-comparative genomic hybridization array detects common rearrangements as well as deep intronic mutations in dystrophinopathies. BMC Genom. 2008, 28. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.R.; Chin, E.L.; Mulle, J.G.; Okou, D.T.; Warren, S.T.; Zwick, M.E. Microarray-based mutation detection in the dystrophin gene. Hum. Mutat. 2008, 29, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Ankala, A.; da Silva, C.; Gualandi, F.; Ferlini, A.; Bean, L.J.; Collins, C.; Tanner, A.K.; Hegde, M.R. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann. Neurol. 2015, 77, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Dai, Y.; Yu, P.; Qu, N.; Lan, Z.; Hong, X.; Sun, Y.; Yang, G.; Xie, S.; Shi, Q.; et al. Targeted next-generation sequencing as a comprehensive test for patients with and female carriers of DMD/BMD: A multi-population diagnostic study. Eur. J. Hum. Genet. 2014, 22, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, Y.; Liu, J.; Chen, X.C.; Liu, X.; Wang, C.Z.; He, X.Y. Whole dystrophin gene analysis by next-generation sequencing: A comprehensive genetic diagnosis of Duchenne and Becker muscular dystrophy. Mol. Genet. Genom. 2014, 289, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Bovolenta, M.; Scotton, C.; Falzarano, M.S.; Gualandi, F.; Ferlini, A. Rapid, comprehensive analysis of the dystrophin transcript by a custom micro-fluidic exome array. Hum. Mutat. 2012, 33, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Drachman, D.B.; Toyka, K.V.; Myer, E. Prednisone in Duchenne muscular dystrophy. Lancet 1974, 2, 1409–1412. [Google Scholar] [CrossRef]
- Griggs, R.C.; Moxley, R.T., 3rd; Mendell, J.R.; Fenichel, G.M.; Brooke, M.H.; Pestronk, A.; Miller, J.P.; Cwik, V.A.; Pandya, S.; Robison, J.; et al. Duchenne dystrophy: Randomized, controlled trial of prednisone (18 months) and azathioprine (12 months). Neurology 1993, 43, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Moxley, R.T.; Griggs, R.C.; Brooke, M.H.; Fenichel, G.M.; Miller, J.P.; King, W.; Signore, L.; Pandya, S.; Florence, J.; et al. Randomized, double-blind six- month trial of prednisone in Duchenne’smuscular dystrophy. N. Engl. J. Med. 1989, 320, 1592–1597. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, D.M.; Ruzza, G.; Bonometto, P.; Berardinelli, A.; Gorni, K.; Orcesi, S.; Lanzi, G.; Angelini, C. A multicenter double-blind randomized trial of deflazacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve 2000, 23, 1344–1347. [Google Scholar] [CrossRef]
- Mesa, L.E.; Dubrosvky, A.L.; Corderi, J.; Marco, P.; Flores, D. Steroids in Duchenne muscular dystrophy-deflazacort trial. Neuromuscular. Disord. 1991, 1, 261–266. [Google Scholar] [CrossRef]
- Escolar, D.M.; Hache, L.P.; Clemens, P.R.; Cnaan, A.; McDonald, C.M.; Viswanathan, V.; Kornberg, A.J.; Bertorini, T.E.; Nevo, Y.; Lotze, T.; et al. Randomized, blinded trial of weekend vs. daily prednisone in Duchenne muscular dystrophy. Neurology 2011, 77, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Scully, M.A.; Pandya, S.; Moxley, R.T. Review of Phase II and Phase III clinical trials for Duchenne muscular dystrophy. Expert Opin. Orphan Drugs 2013, 1, 33–46. [Google Scholar] [CrossRef]
- Goemans, N.; Buyse, G. Current Treatment and Management of Dystrophinopathies. Curr. Treat. Options Neurol. 2014, 16, 287. [Google Scholar] [CrossRef] [PubMed]
- Griggs, R.C.; Herr, B.E.; Reha, A.; Elfring, G.; Atkinson, L.; Cwik, V.; McColl, E.; Tawil, R.; Pandya, S.; McDermott, M.P.; et al. Corticosteroids in Duchenne muscular dystrophy: Major variations in practice. Muscle Nerve 2013, 48, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Balaban, B.; Matthews, D.J.; Clayton, G.H.; Carry, T. Corticosteroid treatment and functional improvement in Duchenne muscular dystrophy: Long-term effect. Am. J. Phys. Med. Rehabil. 2005, 84, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Biggar, W.D.; Harris, V.A.; Eliasoph, L.; Alman, B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul. Disord. 2006, 16, 249–255. [Google Scholar] [CrossRef] [PubMed]
- King, W.M.; Ruttencutter, R.; Nagaraja, H.N.; Matkovic, V.; Landoll, J.; Hoyle, C.; Mendell, J.R.; Kissel, J.T. Orthopedic outcomes of long-term daily corticosteroid treatment in Duchenne muscular dystrophy. Neurology 2007, 68, 1607–1611. [Google Scholar] [CrossRef] [PubMed]
- Houde, S.; Filiatrault, M.; Fournier, A.; Dubé, J.; D’Arcy, S.; Bérubé, D.; Brousseau, Y.; Lapierre, G.; Vanasse, M. Deflazacort use in Duchenne muscular dystrophy: an 8-year follow up. Pediatr. Neurol. 2008, 38, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Merlini, L.; Gennari, M.; Malaspina, E.; Cecconi, I.; Armaroli, A.; Gnudi, S.; Talim, B.; Ferlini, A.; Cicognani, A.; Franzoni, E. Early corticosteroid treatment in 4 Duchenne muscular dystrophy patients: 14 year follow-up. Muscle Nerve 2012, 45, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Moxley, R.T.; Pandya, S.; Ciafaloni, E.; Fox, D.J.; Campbell, K. Change in natural history of Duchenne muscular dystrophy with long-term corticosteroid treatment: Implications for management. J. Child Neurol. 2010, 25, 1116–1129. [Google Scholar] [CrossRef] [PubMed]
- Wehling-Henricks, M.; Lee, J.J.; Tidball, J.G. Prednisolone decreases cellular adhesion molecules required for inflammatory cell infiltration in dystrophin-deficient skeletal muscle. Neuromuscul. Disord. 2004, 14, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Weber, M.; Vargas, C. Deflazacort increases laminin expression and myogenic repair, and induces early persistant functional gain in mdx mouse muscular dystrophy. Cell Transplant. 2000, 9, 551–564. [Google Scholar] [PubMed]
- Lim, J.H.; Kim, D.Y.; Bang, M.S. Effects of exercise and steroid on skeletal muscle apoptosis in the mdx mouse. Muscle Nerve 2004, 30, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Guerron, A.D.; Gordish-Dressman, H.; Spurney, C.F.; Iantorno, M.; Hoffman, E.P.; Nagaraju, K. Glucocorticoid-treated mice are an inappropriate positive control for long-term preclinical studies in the mdx mouse. PLoS ONE 2012, 7, e34204. [Google Scholar] [CrossRef] [PubMed]
- Huizenga, N.A.; Koper, J.W.; de Lange, P.; Pols, H.A.; Stolk, R.P.; Burger, H.; Grobbee, D.E.; Brinkmann, A.O.; De Jong, F.H.; Lamberts, S.W. A polymorphism in the glucocorticoid receptor gene may be associated with and increase sensitivity to glucocorticoids in vivo. J. Clin. Endocrinol. Metab. 1998, 83, 144–151. [Google Scholar] [PubMed]
- Bonifati, D.M.; Witchel, S.F.; Ermani, M.; Hoffman, E.P.; Angelini, C.; Pegoraro, E. The glucocorticoid receptor N363S polymorphism and steroid response in Duchenne dystrophy. J. Neurol. Neurosurg. Psychiatry 2006, 77, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K.; Beck, I.M.; Haegeman, G. Classic glucocorticoids versus non-steroidal glucocorticoid receptor modulators: Survival of the fittest regulator of the immune system? Brain Behav. Immun. 2010, 7, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.R.; Damsker, J.M.; Yu, Q.; Dillingham, B.C.; Huynh, T.; van der Meulen, J.H.; Sali, A.; Miller, B.K.; Phadke, A.; Scheffer, L.; et al. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol. Med. 2013, 5, 1569–1585. [Google Scholar] [CrossRef] [PubMed]
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.; Straino, S.; Ragone, G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad Sci. USA 2009, 106, 1679. [Google Scholar] [CrossRef] [PubMed]
- Consalvi, S.; Mozzetta, C.; Bettica, P.; Germani, M.; Fiorentini, F.; del Bene, F.; Rocchetti, M.; Leoni, F.; Monzani, V.; Mascagni, P.; et al. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol. Med. 2013, 19, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Clinical trials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01761292 (accessed on 1 October 2015).
- Cordani, N.; Pisa, V.; Pozzi, L.; Sciorati, C.; Clementi, E. Nitric oxide controls fat deposition in dystrophic skeletal muscle by regulating fibro-adipogenic precursor differentiation. Stem Cells 2014, 32, 874–885. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.G.; Gandossini, S.; Martinelli Boneschi, F.; Sciorati, C.; Bonato, S.; Brighina, E.; Comi, G.P.; Turconi, A.C.; Magri, F.; Stefanoni, G.; et al. Nitric oxide donor and non steroidal anti-inflammatory drugs as a therapy for muscular dystrophies: Evidence from a safety study with pilot efficacy measures in adult dystrophic patients. Pharmacol. Res. 2012, 65, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Bonfanti, C.; Rossi, G.; Tedesco, F.S.; Giannotta, M.; Benedetti, S.; Tonlorenzi, R.; Antonini, S.; Marazzi, G.; Dejana, E.; Sassoon, D.; et al. PW1/Peg3 expression regulates key properties that determine mesoangioblast stem cell competence. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Seto, J.T.; Bengtsson, N.E.; Chamberlain, J.S. Therapy of Genetic Disorders-Novel Therapies for Duchenne Muscular Dystrophy. Curr. Pediatr. Rep. 2014, 2, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Noviello, M.; Tedesco, F.S.; Bondanza, A.; Tonlorenzi, R.; Rosaria Carbone, M.; Gerli, M.F.; Marktel, S.; Napolitano, S.; Cicalese, M.P.; Ciceri, F. Inflammation converts human mesoangioblasts into targets of alloreactive immune responses: Implications for allogeneic cell therapy of DMD. Mol. Ther. 2014, 22, 1342–1352. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, S.; Aartsma-Rus, A.; Vieira, N.M.; Davies, K.E.; van Ommen, G.J.; Kunkel, L.M. The Pathogenesis and Therapy of Muscular Dystrophies. Annu. Rev. Genom. Hum. Genet. 2015, 16, 281–308. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Campbell, K.; Rodino-Klapac, L.; Sahenk, Z.; Shilling, C.; Lewis, S.; Bowles, D.; Gray, S.; Li, C.; Galloway, G.; et al. Dystrophin immunity in Duchenne's muscular dystrophy. N. Engl. J. Med. 2010, 363, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.; Sahenk, Z.; Malik, V.; Kaspar, B.K.; Walker, C.M.; Clark, K.R. Gene therapy for muscular dystrophy: Lessons learned and path forward. Neurosci. Lett. 2012, 527, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Dougherty, J.P. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol. Ther. 2012, 136, 227–266. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Wong, B.; Barohn, R.; Campbell, C.; Comi, G.P.; Connolly, A.M.; Day, J.W.; Flanigan, K.M.; Goemans, N.; et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014, 50, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Clinical trials.gov. Available online: http://www.ptcbio.com/en/pipeline/ataluren-translarna/ (accessed on 1 October 2015).
- Tinsley, J.; Robinson, N.; Davies, K.E. Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to healthy male adult volunteers. J. Clin. Pharmacol. 2015, 55, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Kleopa, K.A.; Drousiotou, A.; Mavrikiou, E.; Ormiston, A.; Kyriakides, T. Naturally occurring utrophin correlates with disease severity in Duchenne muscular dystrophy. Hum. Mol. Genet. 2006, 15, 1623–1628. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, S.; Squire, S.E.; Edwards, B.; Chen, H.; Burns, D.T.; Shah, N.; Babbs, A.; Davies, S.G.; Wynne, G.M.; Russell, A.J.; et al. Second-generation compound for the modulation of utrophin in the therapy of DMD. Hum. Mol. Genet. 2015, 24, 4212–4224. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.M.; Fairclough, R.J.; Storer, R.; Wilkes, F.J.; Potter, A.C.; Squire, S.E.; Powell, D.S.; Cozzoli, A.; Capogrosso, R.F.; Lambert, A.; et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS ONE 2011, 6, e19189. [Google Scholar] [CrossRef] [PubMed]
- Clinical trials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02056808 (accessed on 1 October 2015).
- Wood, M.J.; Gait, M.J.; Yin, H. RNA-targeted splice-correction therapy for neuromuscular disease. Brain 2010, 133, 957–972. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Cirak, S.; Partridge, T. What can we learn from clinical trials of exon skipping for DMD? Mol. Ther. Nucleic Acids 2014, 3, e152. [Google Scholar] [CrossRef] [PubMed]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef]
- Van Deutekom, J.C.; Janson, A.A.; Ginjaar, I.B.; Frankhuizen, W.S.; Aartsma-Rus, A.; Bremmer-Bout, M.; den Dunnen, J.T.; Koop, K.; van der Kooi, A.J.; Goemans, N.M.; et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 2007, 357, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.-A.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 2015, 21, 270–275. [Google Scholar] [CrossRef]
- Jarver, P.; O’Donovan, L.; Gait, M.J. A chemical view of oligonucleotides for exon skipping and related drug applications. Nucleic Acid Ther. 2013, 24, 37–47. [Google Scholar] [PubMed]
- Shen, J.; Wu, X.; Lee, Y.; Wolfram, J.; Yang, Z.; Mao, Z.W.; Ferrari, M.; Shen, H. Porous silicon microparticles for delivery of siRNA therapeutics. J. Vis. Exp. 2015, 15, 52075. [Google Scholar] [CrossRef] [PubMed]
- Bedi, D.; Gillespie, J.W.; Petrenko, V.A., Jr.; Ebner, A.; Leitner, M.; Hinterdorfer, P.; Petrenko, V.A. Targeted delivery of siRNA into breast cancer cells via phage fusion proteins. Mol. Pharmacol. 2013, 10, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Schairer, D.; Martinez, L.R.; Blecher, K.; Chouake, J.; Nacharaju, P.; Gialanella, P.; Friedman, J.M.; Nosanchuk, J.D.; Friedman, A. Nitric oxide nanoparticles: Pre-clinical utility as a therapeutic for intramuscular abscesses. Virulence 2012, 3, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Wu, Y.T.; Lin, J.Y.; Chu, C.H.; Huang, H.Y.; Wang, Y.C.; Chen, J.K.; Yang, C.S. Rapid quantitative analysis of clarithromycin in rat plasma by UPLCMS/MS after intravenous injection of the clarithromycinloadedultrafine PLGA nanoparticles. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 895–896, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Des Rieux, A.; Fievez, V.; Garinot, M.; Schneider, Y.J.; Pre’at, V. Nanoparticles as potential oral delivery systems of proteins and vaccines: A mechanistic approach. J. Control. Release 2006, 116, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, A.; Yamamoto, H.; Takeuchi, H.; Kawashima, Y. Nanoparticles enhance therapeutic efficiency by selectively increased local drug dose in experimental colitis in rats. J. Pharmacol. Exp. Ther. 2005, 315, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Cherif, M.S.; Shuaibu, M.N.; Kurosaki, T.; Helegbe, G.K.; Kikuchi, M.; Yanagi, T.; Tsuboi, T.; Sasaki, H.; Hirayama, K. Immunogenicity of novel nanoparticle-coated MSP-1 C-terminus malaria DNA vaccine using different routes of administration. Vaccine 2011, 29, 9038–9050. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Castranova, V. Toxicology of nanomaterials used in nanomedicine. J. Toxicol. Environ. Health B Crit. Rev. 2011, 14, 593–632. [Google Scholar] [CrossRef] [PubMed]
- Sosnik, A.; Menaker Raskin, M. Polymeric micelles in mucosal drug delivery: Challenges towards clinical translation. Biotechnol. Adv. 2015, 33, 1380–1392. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Gaspar, R. Nanomedicine(s) under the microscope. Mol. Pharm. 2011, 8, 2101–2141. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Zhu, Y.; Oupicky’, D. Recent advances in delivery of drug-nucleic acid combinations for cancer treatment. J. Control. Release 2013, 172, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Woodrow, K.A.; Cu, Y.; Booth, C.J.; Saucier-Sawyer, J.K.; Wood, M.J.; Saltzman, W.M. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat. Mater. 2009, 8, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Breton, A.L.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Wu, T.; Zhang, D.; Zhang, Z. Cell or Cell Membrane-Based Drug Delivery Systems. Theranostics 2015, 5, 863–881. [Google Scholar] [CrossRef] [PubMed]
- Muthu, M.S.; Feng, S.S. Theranostic liposomes for cancer diagnosis and treatment: Current development and pre-clinical success. Expert Opin. Drug Deliv. 2013, 10, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Hu, C.M.J.; Fang, R.H.; Zhang, L. Liposome-like nanostructures for drug delivery. J. Mater. Chem. B 2013, 1, 6569–6585. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falzarano, M.S.; Scotton, C.; Passarelli, C.; Ferlini, A. Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules 2015, 20, 18168-18184. https://doi.org/10.3390/molecules201018168
Falzarano MS, Scotton C, Passarelli C, Ferlini A. Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules. 2015; 20(10):18168-18184. https://doi.org/10.3390/molecules201018168
Chicago/Turabian StyleFalzarano, Maria Sofia, Chiara Scotton, Chiara Passarelli, and Alessandra Ferlini. 2015. "Duchenne Muscular Dystrophy: From Diagnosis to Therapy" Molecules 20, no. 10: 18168-18184. https://doi.org/10.3390/molecules201018168
APA StyleFalzarano, M. S., Scotton, C., Passarelli, C., & Ferlini, A. (2015). Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules, 20(10), 18168-18184. https://doi.org/10.3390/molecules201018168