
Synthesis of Chromonylthiazolidines and Their Cytotoxicity to Human Cancer Cell Lines

Abstract
:1. Introduction
2. Results and Discussion
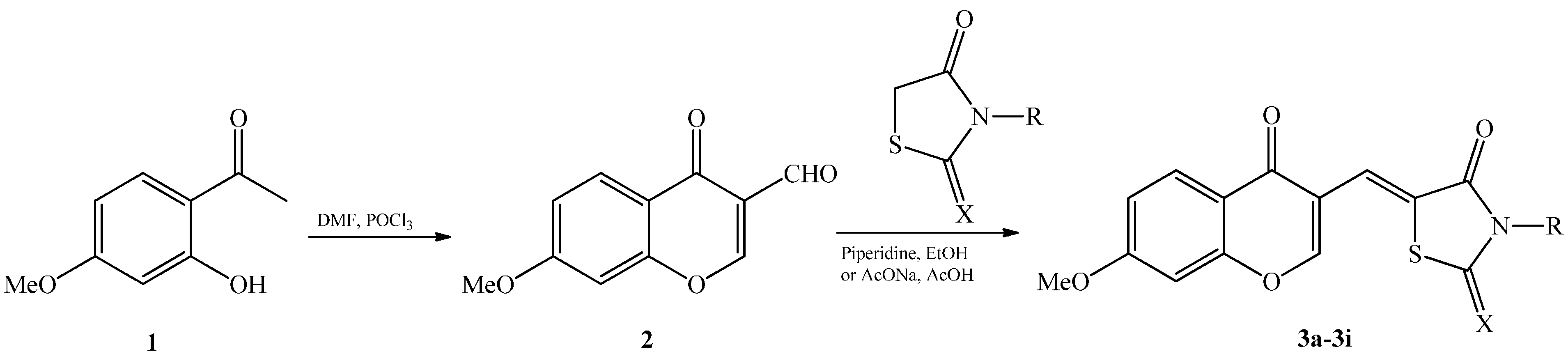

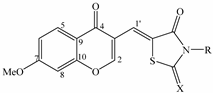
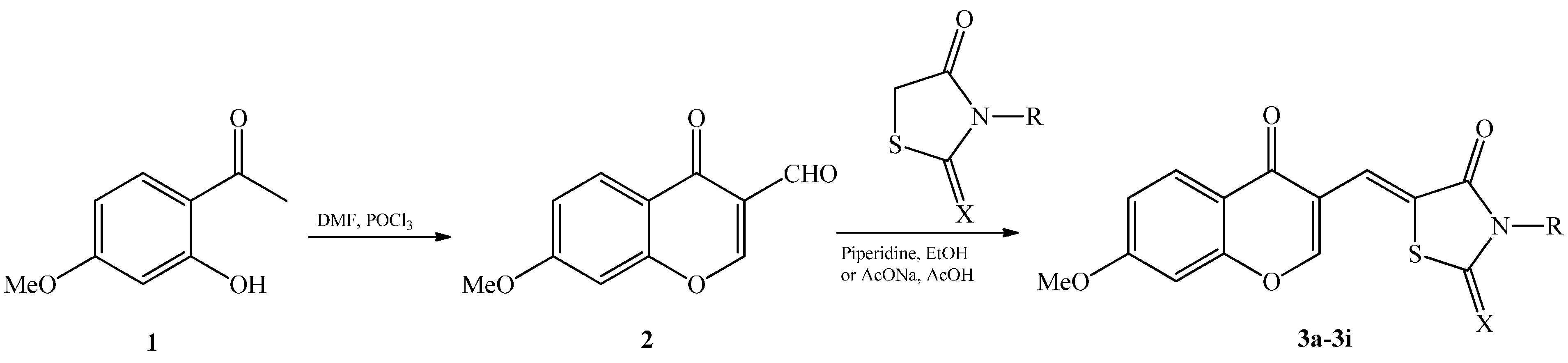{kind=link}
| Compounds | X | R | Yield (%) |
|---|---|---|---|
| 3a | O | H | 72 |
| 3b | O | CH3 | 47 |
| 3c | O | 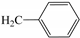 | 38 |
| 3d | S | NH2 | 66 |
| 3e | S | CH3 | 60 |
| 3f | S | H2C-CH=CH2 | 72 |
| 3g | S | H | 55 |
| 3h | S | H2C-COOH | 50 |
| 3i | S | 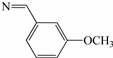 | 66 |
| Comp. | IC50 (μg/mL) | |||||||
|---|---|---|---|---|---|---|---|---|
| HepG2 | HL-60 | KB | LLC | LNCaP | LU-1 | MCF7 | SW480 | |
| 1 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 2 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 3a | >100 | 82.2 ± 4.5 | 44.1 ± 3.6 | 87.4 ± 6.3 | 77.4 ± 5.8 | 52.9 ± 3.4 | 66.0 ± 2.7 | 71.4 ± 3.6 |
| 3b | 86.3 ± 6.4 | 75.3 ± 3.9 | 84.6 ± 4.2 | >100 | 81.6 ± 6.3 | >100 | 32.8 ± 1.4 | 90.1 ± 4.8 |
| 3c | 78.4 ± 5.8 | 92.3 ± 5.2 | 74.1 ± 5.1 | 90.1 ± 7.7 | 84.2 ± 4.1 | 65.5 ± 4.1 | 52.7 ± 3.6 | 85.4 ± 7.4 |
| 3d | 94.5 ± 6.3 | 74.6 ± 4.6 | 90.2 ± 2.4 | >100 | 92.4 ± 5.7 | 59.0 ± 4.3 | 88.4 ± 7.4 | 84.3 ± 6.2 |
| 3e | 81.4 ± 7.4 | 94.8 ± 6.8 | >100 | 90.2 ± 4.9 | >100 | >100 | >100 | 87.6 ± 4.8 |
| 3f | 78.4 ± 3.4 | 87.6 ± 6.7 | >100 | >100 | 91.6 ± 7.6 | >100 | 65.9 ± 4.7 | 81.3 ± 5.6 |
| 3g | 88.4 ± 6.7 | 84.6 ± 5.4 | >100 | 90.8 ± 5.2 | >100 | >100 | >100 | 90.8 ± 6.4 |
| 3h | 91.3 ± 8.2 | 94.2 ± 7.1 | 88.4 ± 3.5 | 87.4 ± 6.4 | 81.9 ± 4.3 | 85.6 ± 5.6 | >100 | 85.7 ± 3.2 |
| 3i | >100 | 84.3 ± 4.2 | 90.2 ± 2.7 | 92.8 ± 6.4 | 85.1 ± 6.1 | >100 | 80.9 ± 6.9 | 92.5 ± 4.1 |
| Ellipticine | 1.45 ± 0.08 | 0.56 ± 0.04 | 0.43 ± 0.05 | 0.98 ± 0.04 | 0.86 ± 0.06 | 1.29 ± 0.11 | 0.49 ± 0.04 | 0.62 ± 0.05 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Isolation of Paeonol (1)
3.3. Synthesis of 3-Formyl-7-methoxychromone (2)
3.4. Synthesis of Chromonylthiazolidines 3a‒i
3.4.1. Procedure 1
3.4.2. Procedure 2
3.5. Cell Culture and Cell Viability Assay
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cho, W.C.S. Contribution of oncoproteomics to cancer biomarker discovery. Mol. Cancer 2007, 6. [Google Scholar] [CrossRef]
- De Martino, J.K.; Boger, D.L. Glycinamide ribonucleotide transformylase (GAR TFase) as a target for cancer therapy. Drug Future 2008, 33, 969–979. [Google Scholar] [CrossRef]
- Curran, W.J. New chemotherapeutic agents: Update of major chemoradiation trials in solid tumors. Oncology 2002, 63, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xu, W. Novel anticancer targets and drug discovery in post genomic age. Curr. Med. Chem. 2005, 5, 53–63. [Google Scholar]
- Li, W.; Lu, Y.; Wang, Z.; Dalton, J.T.; Miller, D.D. Synthesis and antiproliferative activity of thiazolidine analogs for melanoma. Bioorg. Med. Chem. Lett. 2007, 17, 4113–4117. [Google Scholar] [CrossRef] [PubMed]
- Chandrappa, S.; Benaka Prasad, S.B.; Vinaya, K.; Ananda Kumar, C.S.; Thimmegowda, N.R.; Rangappa, K.S. Synthesis and in vitro antiproliferative activity against human cancer cell lines of novel 5-(4-methyl-benzylidene)-thiazolidine-2,4-diones. Investig. New Drugs 2008, 26, 437–444. [Google Scholar] [CrossRef]
- Joshi, H.; Pal, T.; Ramaa, C.S. A new dawn for the use of thiazolidinediones in cancer therapy. Expert Opin. Investig. Drugs 2014, 23, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Blanquicett, C.; Roman, J.; Hart, C.M. Thiazolidinediones as anti-cancer agents. Cancer Ther. 2008, 6, 25–34. [Google Scholar] [PubMed]
- Avupati, V.R.; Yejella, R.P.; Akula, A.; Guntuku, G.S.; Doddi, B.R.; Vutla, V.R.; Anagani, S.R.; Adimulam, L.S.; Vyricharla, A.K. Synthesis, characterization and biological evaluation of some novel 2,4-thiazolidinediones as potential cytotoxic, antimicrobial and antihyperglycemic agents. Bioorg. Med. Chem. Lett. 2012, 22, 6442–6450. [Google Scholar] [CrossRef] [PubMed]
- Hafez, H.N.; El-Gazzar, A.R.B.A. Synthesis and antitumor activity of substituted triazolo[4,3-a]pyrimidin-6-sulfonamide with an incorporated thiazolidinone moiety. Bioorg. Med. Chem. Lett. 2009, 19, 4143–4147. [Google Scholar] [CrossRef] [PubMed]
- Alegaon, S.G.; Alagawadi, K.R. New thiazolidine-2,4-diones as antimicrobial and cytotoxic agent. Med. Chem. Res. 2012, 21, 3214–3223. [Google Scholar] [CrossRef]
- Malikov, V.M.; Yuldashev, M.P. Phenolic compounds of plants of the Scutellaria L. Genus. distribution, structure, and properties. Chem. Nat. Compd. 2003, 38, 358–406. [Google Scholar] [CrossRef]
- Keri, R.S.; Budagumpi, S.; Pai, R.K.; Balakrishna, R.G. Chromones as a privileged scaffold in drug discovery: A review. Eur. J. Med. Chem. 2014, 78, 340–374. [Google Scholar] [CrossRef] [PubMed]
- Beecher, G.R. Overview of dietary flavonoids: Nomenclature, occurrence, and intake. J. Nutr. 2003, 133, 3248S–3254S. [Google Scholar] [PubMed]
- Machado, N.F.L.; Marques, M.P.M. Bioactive chromone derivatives—Structural diversity. Curr. Bioact. Compd. 2010, 6, 76–89. [Google Scholar] [CrossRef]
- Nohara, A.; Umetani, T.; Sanno, Y. Studies on antianaphylactic agents. I. Facile synthesis of 4-oxo-4H-1-benzopyran-3-carboxaldehydes by Vilsmeier reagents. Tetrahedron 1974, 30, 3553–3561. [Google Scholar]
- Stiborová, M.; Černá, V.; Moserová, M.; Mrízová, I.; Arlt, V.; Frei, E. The anticancer drug ellipticine activated with cytochrome P450 mediates DNA damage determining its pharmacological efficiencies: Studies with rats, hepatic cytochrome P450 reductase null (HRN™) mice and pure enzymes. Int. J. Mol. Sci. 2014, 16, 284–306. [Google Scholar] [CrossRef] [PubMed]
- Deane, F.M.; O’Sullivan, E.C.; Maguire, A.R.; Gilbert, J.; Sakoff, J.A.; McCluskey, A.; McCarthy, F.O. Synthesis and evaluation of novel ellipticines as potential anti-cancer agents. Org. Biomol. Chem. 2013, 11, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Azizmohammadi, M.; Khoobi, M.; Ramazani, A.; Emami, S.; Zarrin, A.; Firuzi, O.; Miri, R.; Shafiee, A. 2H-chromene derivatives bearing thiazolidine-2,4-dione, rhodanine or hydantoin moieties as potential anticancer agents. Eur. J. Med. Chem. 2013, 59, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Shivhare, S.; Mishra, R.; Gharia, A.; Gautam, M.D. Synthesis, characterization and antimicrobial studies of Cu(II), Co(II) and Ni(II) complexes with Schiff base derived from N-amino rhodanine and salicylaldehyde. Int. J. Pharm. 2012, 4, 394–397. [Google Scholar]
- Yasuda, T.; Kon, R.; Nakazawa, T.; Ohsawa, K. Metabolism of paeonol in rats. J. Nat. Prod. 1999, 62, 1142–1144. [Google Scholar] [CrossRef] [PubMed]
- Patonay, T.; Kiss-Szikszai, A.; Silva, V.M.L.; Silva, A.M.S.; Pinto, D.C.G.A.; Cavaleiro, J.A.S.; Jeko, J. Microwave-induced synthesis and regio- and stereoselective epoxidation of 3-styrylchromones. Eur. J. Org. Chem. 2008, 1937–1946. [Google Scholar] [CrossRef]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of radio-sensitivity. Cancer Res. 1987, 47, 943–946. [Google Scholar] [PubMed]
- Sample Availability: Samples of the compounds 3a–i are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anh, H.L.T.; Cuc, N.T.; Tai, B.H.; Yen, P.H.; Nhiem, N.X.; Thao, D.T.; Nam, N.H.; Van Minh, C.; Van Kiem, P.; Kim, Y.H. Synthesis of Chromonylthiazolidines and Their Cytotoxicity to Human Cancer Cell Lines. Molecules 2015, 20, 1151-1160. https://doi.org/10.3390/molecules20011151
Anh HLT, Cuc NT, Tai BH, Yen PH, Nhiem NX, Thao DT, Nam NH, Van Minh C, Van Kiem P, Kim YH. Synthesis of Chromonylthiazolidines and Their Cytotoxicity to Human Cancer Cell Lines. Molecules. 2015; 20(1):1151-1160. https://doi.org/10.3390/molecules20011151
Chicago/Turabian StyleAnh, Hoang Le Tuan, Nguyen Thi Cuc, Bui Huu Tai, Pham Hai Yen, Nguyen Xuan Nhiem, Do Thi Thao, Nguyen Hoai Nam, Chau Van Minh, Phan Van Kiem, and Young Ho Kim. 2015. "Synthesis of Chromonylthiazolidines and Their Cytotoxicity to Human Cancer Cell Lines" Molecules 20, no. 1: 1151-1160. https://doi.org/10.3390/molecules20011151
APA StyleAnh, H. L. T., Cuc, N. T., Tai, B. H., Yen, P. H., Nhiem, N. X., Thao, D. T., Nam, N. H., Van Minh, C., Van Kiem, P., & Kim, Y. H. (2015). Synthesis of Chromonylthiazolidines and Their Cytotoxicity to Human Cancer Cell Lines. Molecules, 20(1), 1151-1160. https://doi.org/10.3390/molecules20011151