Abstract
The conformation of dimethyl (R,R)-tartrate has been analyzed on the basis of the single crystal X-ray diffraction method as well as by ab-initio quantum chemical studies. The results showed that the extended T conformation containing two planar hydroxyester moieties predominates in both ab-initio and X-ray studies. The lowest energy conformer in ab-initio calculations has C2 symmetry and hydrogen bonds between a hydroxyl group and the nearest carbonyl oxygen. The second in energetical sequence, with an energy difference of only 1.2 kcal/mol, is the asymmetrical conformer, which differs from the lowest energy form by the rotation of one of the ester groups by 180°. Intramolecular OH…O hydrogen bonds observed in this rotamer again involve only proximal functional groups. This conformer is present in the crystal structure of the studied compound, although its conformation in the solid state is no longer stabilized by intramolecular hydrogen bonds of the type mentioned above. In the crystal, hydroxyl groups are mostly involved in intermolecular hydrogen bonds and form only a weak intramolecular hydrogen bond with each other. The planar arrangement of the α− hydroxyester moieties combined with the extended conformation of the carbon chain seems to be stabilized by the intramolecular hydrogen bonds between neighboring functional groups and by the long range dipole-dipole interactions between two pairs of CO and (β)C-H bonds.
Introduction
This work is a part of our studies on the packing and conformational behavior of (R,R)-tartaric acid derivatives, both symmetrically and unsymmetrically substituted. Available X-ray data for (R,R)-tartaric acid esters are limited to the publications of Eggli et al. [1] and Sharples and co-workers [2].
There have been several attempts to predict conformation of dimethyl (R,R)-tartrate on the basis of vibrational circular dichroism (VCD) results. Marcott et al. [3] suggested that dimethyl (R,R)-tartrate exists in two different hydrogen bonded conformers – one with internal hydrogen bonding between (β)OH and O=C groups, and the other with internal hydrogen bonding between (α)OH and O=C groups. Su and Keiderling [4] on the basis of their VCD and NMR studies concluded that the G+ conformer predominates (see Figure 1). Nafie and co-workers [5] interpretation of VCD results required hydrogen bonds between (α)OH and O=C groups, and they pointed to the G+ conformer in preference to the T one. Polvarapu et al. [6] performed further VCD measurements and stated, that their results could be interpreted in favor of the T conformation of dimethyl (R,R)-tartrate only when allowing the charge flow along the central C*-C* bond. They concluded however, that the G+ form could not be ruled out only on the basis of VCD results and may exist in equilibrium with the T conformer. They performed ab-initio calculations up to RHF/6-31//RHF/STO-3G level [7] for seven chosen structures of (R,R)-tartaric acid and found, that within these conformers the T form is energetically favored. By analogy they postulated the same for dimethyl (R,R)-tartrate. The T conformation was indicated by 13C NMR data for tartaric acid [8] and its dimethyl diester [9]. Raman optical activity studies (ROA) also indicated the T conformer for these molecules [10]. Semiempirical studies on (R,R)-tartaric acid and its dimethyl diester [11] also pointed to the T conformer as the favored structure.
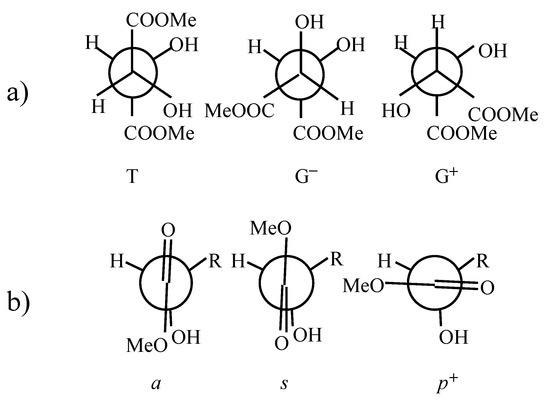
Figure 1.
Explanation of notation used to describe conformations of (R,R)-tartaric acid dimethyl diester. a) T (trans), G+ (gauche+) and G− (gauche−) conformers describing the rotation about C*-C* bond. b) s (synplanar), a (antiplanar), p+ (perpendicular) conformers describing the rotation around C*-C(sp2) bond.
These results combined with their diverse interpretations stimulated our interest in dimethyl (R,R)-tartrate and prompted us to study the conformations of this compound using X-ray analysis and ab-initio quantum chemical methods.
Methods
X-ray diffraction
The title compound was synthesized in the Laboratory of Natural Products, A. Mickiewicz University [12]. The crystals were needle shaped. Unit-cell parameters were determined on a Syntex P21 diffractometer by a least- squares fitting of the setting angles of 15 reflections. Crystal data and some details concerning data collection and structure refinement are given in Table 1.

Table 1.
Crystal data for dimethyl (R,R)-(+)-tartrate.
Intensities of reflections were measured using θ-2θ scan technique on a Syntex P21 diffractometer, with the scan rate depending directly on the net count obtained on rapid pre-scan for each reflection. Two standard reflections were monitored after collection of every 100 reflections as a check of electronic reliability and crystal stability. Integrated intensities were obtained by peak profile analysis according to Lehmann and Larsen [13]. Data were corrected for Lorentz and polarization effects but not for absorption.
The structure was solved by direct methods with the program SHELXS86 [14]. Full-matrix least squares refinement was carried out on F 2 with SHELXL93 [15]. All non-hydrogen atoms were refined anisotropically. The hydroxyl hydrogens were located from a difference Fourier map and their positions and isotropic displacement parameters were allowed to vary. Methyl hydrogens were treated as follows: one methyl hydrogen was located from a difference Fourier map and positions of the remaining two were calculated assuming sp3 hybridization. Hydrogens bonded to the chiral carbon atoms C(2) and C(3) were also placed in calculated positions. During the refinement all the H atoms followed the shifts of (“ridig” on) the atoms to which they were attached. Methyl H atoms were assigned a common isotropic temperature factor of the value U = 0.09 Å2, while for H2 and H3 the U value was 0.03 Å2. At the end of the refinement an empirical isotropic extinction parameter x was introduced to correct the calculated structure factors by multiplying them by a factor k [[1+xFc2λ3/sin(2θ)]−1/4. Atomic scattering factors used were those included in SHELXL93 [15]. The final atomic coordinates with equivalent isotropic temperature factors are given in Table 2. Pertinent torsion angles are compared in Table 3.
Table 2.
Atomic coordinates (x 104) and equivalent iso- tropic displacement parameters (Å 2 x 103) for dimethyl (R,R)-(+)-tartrate. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
Table 3.
Torsion angles (°).
The geometry of the hydrogen bonds is given in Table 4. Tables of anisotropic displacement parameters, bond lengths and angles and H-atom coordinates have been deposited at the Cambridge Crystallographic Data Centre.
Table 4.
Geometry of hydrogen bonds.
Ab-initio calculations
Results of semi-empirical studies described elsewhere [11] were utilized as starting points for ab-initio studies on dimethyl (R,R)-tartrate. All stable geometries from AM1 (36 unique structures) and from PM3 (58 unique conformers) were optimized at Hartree-Fock level at 3-21G basis set (RHF/3-21G). The 30 stable conformers obtained were further examined at 6-31G* basis set (RHF/6-31G*). The five lowest energy structures, at RHF/6-31G* level, were studied using the Möller-Plesset perturbation theory at MP2/6-31G* level. Optimization of all geometrical parameters was performed. The Gaussian 94 program package [16] on the CRAY J916 was used to obtain the results.
Results and Discussion
X-ray crystallography
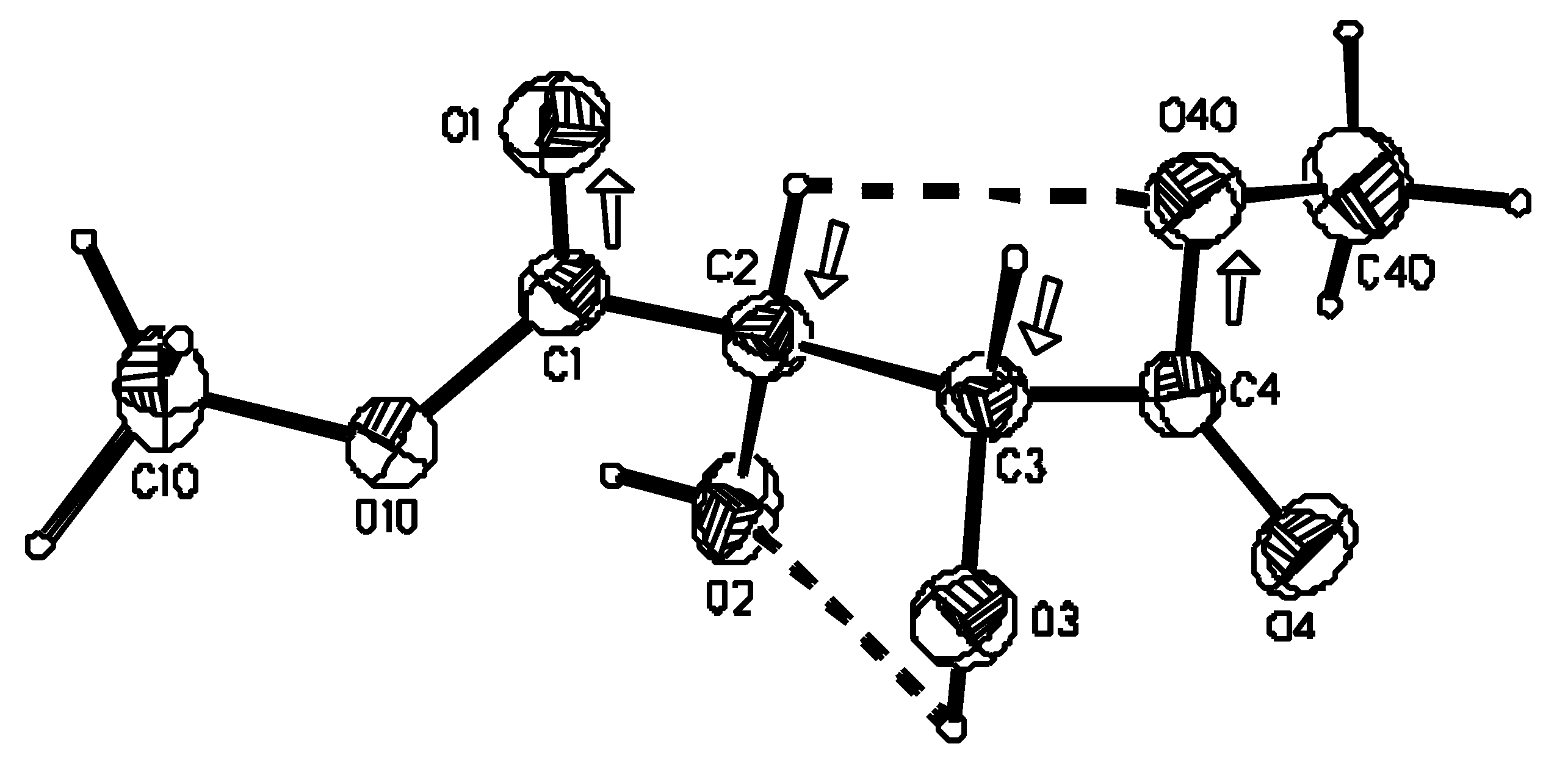
The atom numbering scheme is displayed in Figure 2 [17].
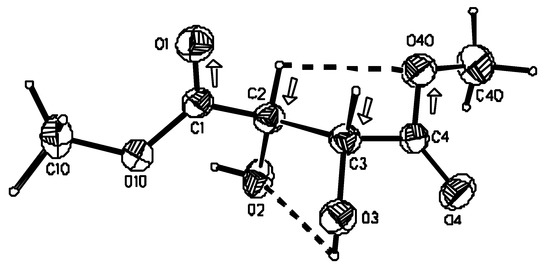
Figure 2.
Perspective view of the molecule of (R,R)-tartaric acid dimethyl diester and the atom numbering scheme. Thermal ellipsoids were drawn at 40% probability level. Molecular conformation is stabilized by intramolecular hydrogen bonds (dashed lines) and attractive dipole-dipole interactions (dipoles marked with arrows).
In the investigated dimethyl (R,R)-tartrate molecule, as in the vast majority of (R,R)-tartaric acid crystalline esters [1,18] the conformation around the C*-C* bond, linking the two chiral centers, is staggered and such that two carboxyl groups are trans (T) and two adjacent C-OH bonds are minus gauche. The only exception reported in the literature constitutes the titanium coordinated diisopropyl (R,R)-tartrate in which two ester groups are in gauche minus (G-) orientation, and consequently, the four atom carbon chain is bent [2]. The value of the C-C*-C*-C torsion angle in the investigated compound differs from the ideal value of 180° and amounts to −169.2(1)°, while the (H)O-C*-C*-O(H) torsion angle is −58.4(2)°. The corresponding values for 11 other crystal structures (15 independent measurements) listed in the Cambridge Structural Database [19] give average torsion angle moduli of 170.1(4.2)° and 70.1(5.8)°.
The conformation around the C*-C bond is such that the hydroxyl group lies nearly in the plane of the neighboring ester group. However, at one end of the molecule the α-hydroxyl oxygen eclipses the carbonyl oxygen (s form, Figure 1b), while at the other end the carbonyl oxygen is on the opposite side of the α-hydroxyl oxygen (a conformer, Figure 1b), which now eclipses the methylated oxygen. Consequently the O=C-C*-O(H) torsion angle has the value of 0.2(2)° at the C(4) end of the molecule and a value of −176.8(2)° at the C(1) end of the molecule. Although the molecule still consists of two planar fragments, its C2 molecular symmetry is lost (Figure 2). The observed asymmetry of the molecule is reflected in the crystal packing as the two carbonyl groups have totally different surroundings (vide infra). Our studies on (R,R)-tartaric acid derivatives seem to indicate that conformational freedom about the C*--C bond is characteristic for an ester group and is not observed within a α-hydroxycarboxyl or amide moiety [12,18,20]. For example in the crystal structure of the methyl ester of (R,R)-tartaric acid monoamide, methyl ester groups from two independent molecules adopt alternate orientations, i.e. they are rotated by 180° around the C-C* bond [20]. The α-hydroxyester moiety exhibits a planar conformation in all tartaric acid esters studied so far by X-ray diffraction methods [1,12,18,20]. Such tendency also prevails in isolated molecules as indicated by ab initio methods (vide infra). Interestingly, while in the isolated molecule such conformation is stabilized by intramolecular hydrogen bonds in which hydroxyl groups act as proton donors to one of the oxygen atoms from the neighboring ester groups, in the crystalline state there is no indication of the presence of this type of bond. An analogous situation exists in (R,R)-tartaric acid if one compares X-ray [21] and quantum chemical results [6]. In the investigated compound, however, we observe weak intramolecular hydrogen bond interaction between two vicinal hydroxyl groups [O(3)-H…O(2)] and between the C(β)-H and OMe group [C(2)-H…O(40)]. Geometrical parameters describing these and other types of hydrogen bonds observed in the crystal are listed in Table 4. Although the interaction between C(2)-H and O(40) satisfies the criteria for intramolecular carbon oxygen hydrogen bond, the antiparallel arrangement of the C-H and C-O(Me) bonds might suggest the presence of dipole-dipole interactions. Similar electrostatic interactions might be expected to occur between the other pair of antiparallel dipoles, namely C(3)-H and C(1)=O which, arranged in only a slightly different way, do not satisfy the criteria for the presence of the intramolecular hydrogen bond. The angles between the two pairs of dipoles C(2)-H and C(4)-O(40), and C(3)-H and C(1)=O(1) amount to 4.1 and 3.1°, respectively. Undoubtedly, the conformation characteristic of the investigated compound and of the vast majority of other (R,R) tartaric acid esters is stabilized not only by the formation of intramolecular hydrogen bonds but, to a significant extent, by the electrostatic dipole/dipole interactions.
Crystal structure
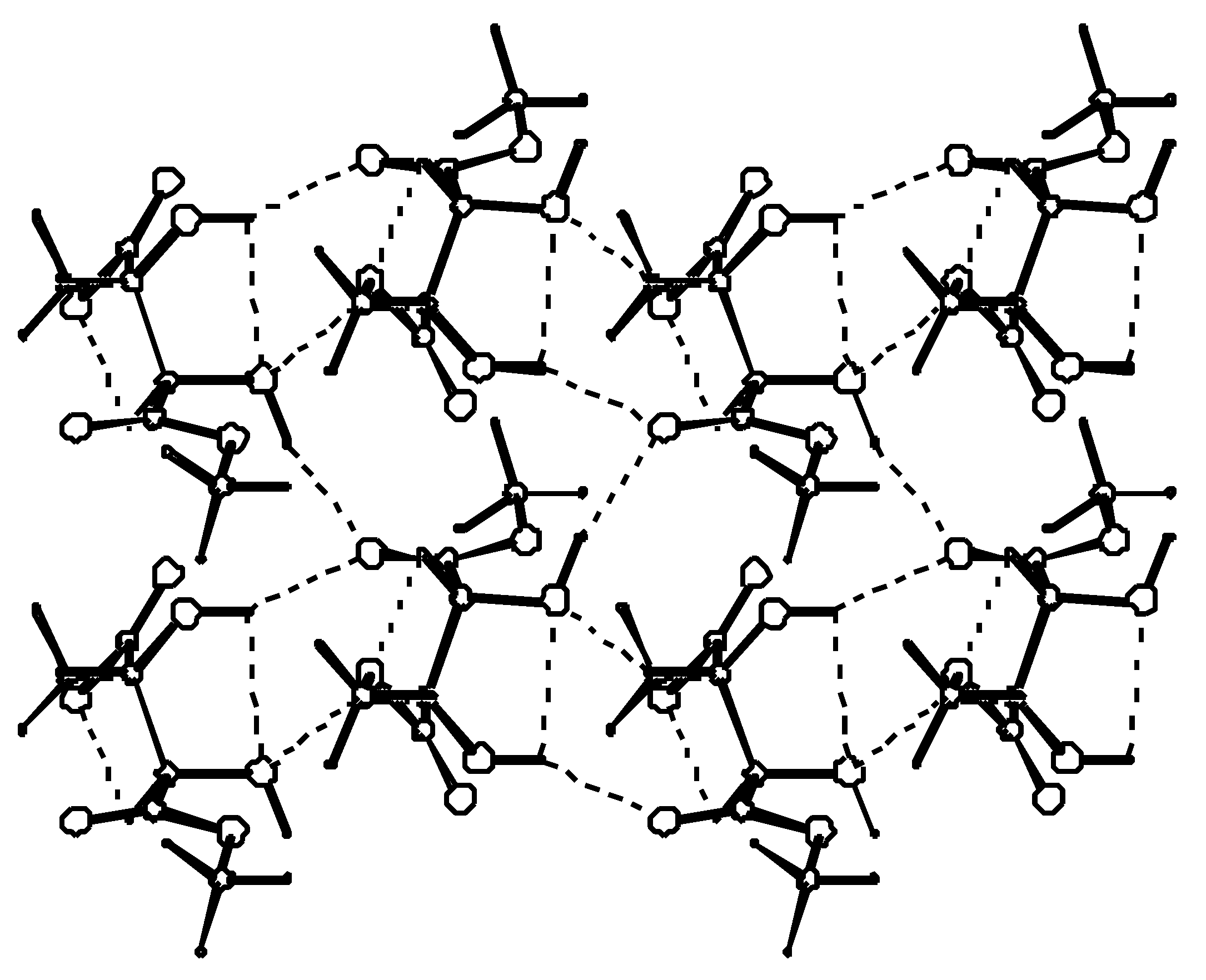
In the crystal, molecules connected by hydrogen bonds, lie roughly in the plane parallel to the (001) lattice plane. Methyl groups are stacked above and below the layer. This is illustrated in Figure 3.
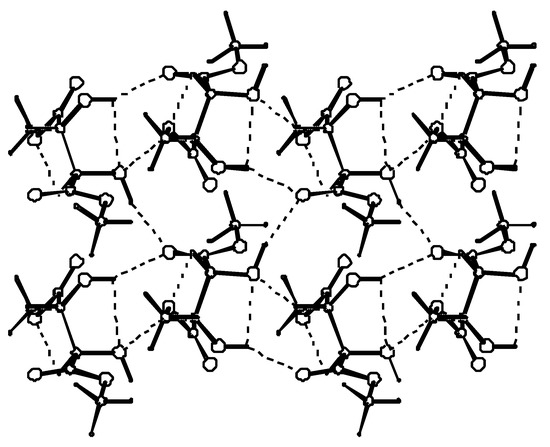
Figure 3.
A layer formed by hydrogen-bonded molecules. Each molecule uses two of its hydroxyl groups to form hydrogen bonds with only one of its two carbonyl oxygens. The second carbonyl oxygen acts as an acceptor in a weak C-H…O bond.
Hydrogen bond parameters are listed in Table 4. The two ends of a symmetrically substituted molecule are involved in hydrogen bonding in a different manner. While the carbonyl oxygen at C(1) is involved as a double acceptor in a relatively strong hydrogen bond from two neighboring hydroxyl groups, the carbonyl group at C(4) acts as an acceptor of a weak hydrogen bond from the proximal methyl ester group. Also the involvement of the hydroxyl group as an acceptor of a proton in hydrogen bonding is seen only for O(2) and not for O(3). These differences in hydrogen-bond functionality do not affect the C=O or C*--OH bond lengths which are the same at both ends of the molecule. To describe hydrogen bond topology we may use the graph set notation proposed by Etter and developed by Bernstein [22]. The first level graph set is N1: C(6)C(5)S(5)S(5)C(4)C(5). The molecules connected by C(6) [O(3)-H…O(1)] and C(4) [C(3)-H…O(2)] chains extend along the y direction. Combination of the two chains gives rise to the formation of the (10) motifs which are fused together to form a ribbon. The ribbons are further connected via C(5) chains to form a layer parallel to the (001) lattice plane. Combination of C(4), C(5) and C(6) chains gives rise to the formation of (12) rings as the third order networks. Among the (R,R)-tartaric acid derivatives packing patterns described by C(6), C(5) and S(5) packing designators always involve at least one hydroxyl group. They seem to be characteristic of (R,R)-tartaric acid and of the majority of its esters and amides.
Ab initio quantum chemical studies
The relative energies of all stable conformers, with respect to the lowest energy T(ss) form, are in the range up to 15.8 kcal/mol and 11.2 kcal/mol for RHF/3-21G and RHF/6-31G* levels, respectively. The 30 and 26 stable structures were localized at 3-21G and 6-31G* basis sets, respectively. The energetic differences between conformers tend to decrease when a set of polarization functions is added to the basis set. The same effect can be seen when an electron correlation is taken into account i.e. when a higher level of theory (MP2 scheme) is utilized.
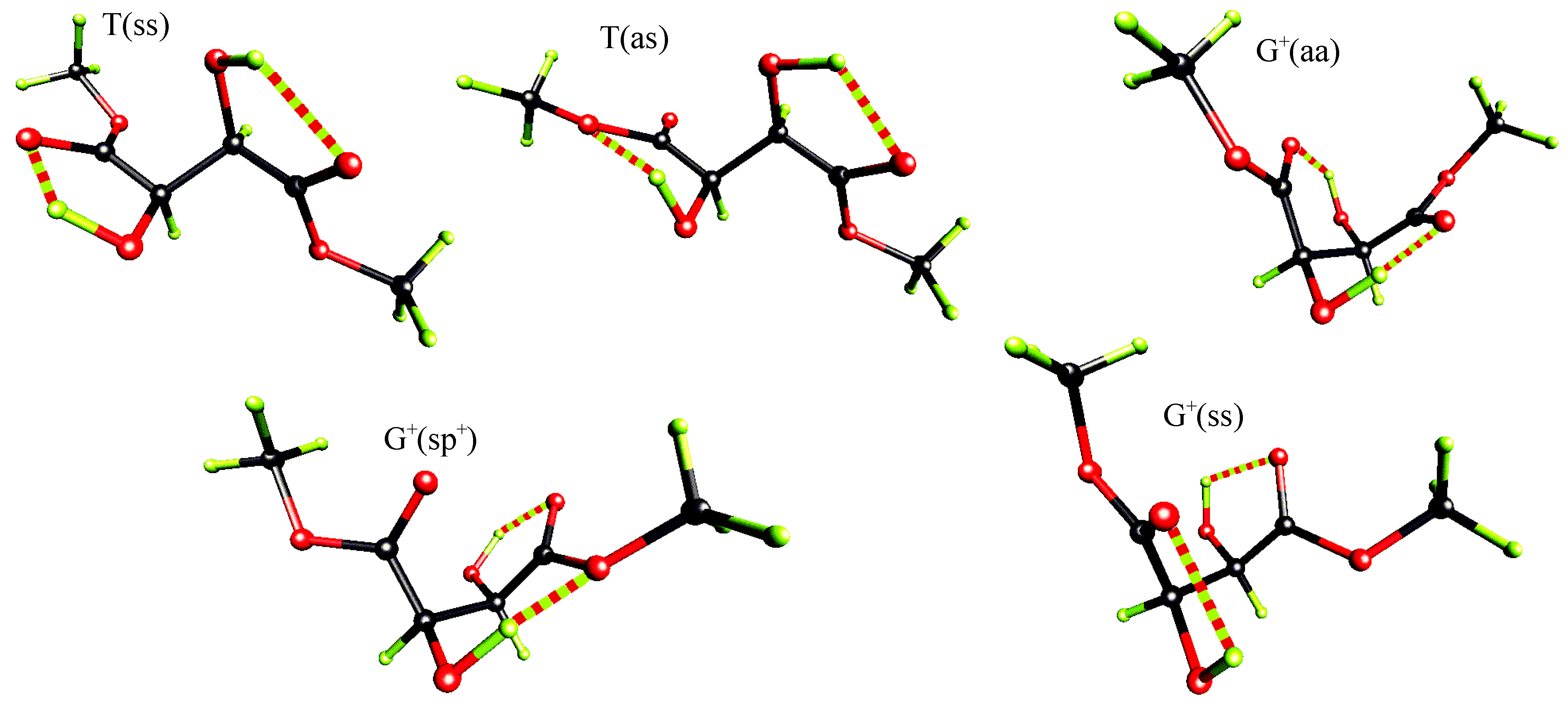
Table 5 presents the results of our ab-initio calculations. For simplicity only five conformers (see Figure 4) of the lowest energy calculated at RHF/6-31G* level were included in the table for all three levels of accuracy considered.
Table 5.
Ab-initio results for dimethyl (R,R)-tartrate.
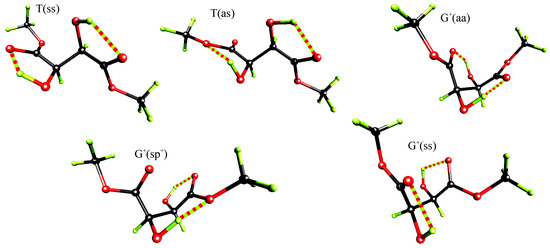
Figure 4.
Perspective view of the five lowest energy conformers of (R,R)-tartaric acid dimethyl diester at RHF/6-31G* level.
Independently of the level of theory, the extended T(ss) conformer with hydrogen bonds between C=O and (α)O-H groups is of the lowest energy. The second in the energetical sequence is the T(as) conformer, which differs from the T(ss) form by the rotation of one of the ester groups by 180°. Intramolecular OH…O hydrogen bonds observed in this T(as) rotamer again involve only proximal functional groups but at one end of the molecule the acceptor is the carbonyl oxygen, while at the other end it is the methylated oxygen.
These T conformers are followed by the G+ ones: the G+(aa), the G+(sp+) and the G+(ss). At MP2/6-31G* level the energy differences between the examined G+ conformers are smaller then 0.22 kcal/mol and their relative energy varies from 1.38 to 1.60 kcal/mol. The corresponding relative energies of these conformers at RHF/6-31G* and RHF/3-21G levels are in the range 2.19 - 3.38 and 2.45 - 5.98 kcal/mol, respectively.
The G+(ss) conformer, the third in the energetical sequence at MP2/6-31G*, is stabilized by hydrogen bonds each closing five membered rings between (α)O-H and the carbonyl oxygen. The G+(aa) rotamer is stabilized by hydrogen bonds between (β)O-H and O=C groups of S(6) motif. The energy difference between the G+(aa) and the G+(ss) conformers, at MP2/6-31G* level, is relatively small and equals 0.11 kcal/mol.
Ab-initio calculations clearly indicate that the extended T conformation with both carbonyl oxygens eclipsed by α-hydroxyls is preferred for the isolated molecules. This T(ss) conformation being stabilized by intramolecular hydrogen bonds between α-hydroxyls and the nearest carbonyl oxygens and by the attraction of two pairs of antiparallel dipoles formed by (β)C-H and C-O(Me) bonds. The T(as) conformer which differs from the lowest energy form by 180° rotation of one of the ester groups and which is present in the crystal, may also exist in equilibrium with the T(ss) conformer. At MP2/6-31G* level this conformer has a relative energy equal to 1.2 kcal/mol. However this conformer is asymmetrical and the lowest energy form possesses C2 symmetry. Because of entropy reasons, the asymmetrical conformers are preferred to symmetrical ones by RTlnω, where R is the gas constant, T is the temperature in Kelvin and ω is a degeneracy of the state. For the analyzed systems we obtain the value of 0.41 kcal/mol and by this amount any symmetrical conformer of dimethyl (R,R)-tartrate is destabilized in comparison with an asymmetrical one [23].
Conclusions
Both X-ray and ab-initio studies indicate that dimethyl (R,R)-tartrate tends to adopt the extended T conformation with ester groups mutually trans, hydroxyls minus gauche, and hydrogens plus gauche. The ester groups do not form an extension of the carbon chain but their oxygens tend to eclipse α-hydroxyl groups. In isolated molecules such a conformation is always stabilized by intramolecular hydrogen bonds between the hydroxyl and the neighboring ester groups. Quantum chemical calculations point out the T(ss) and T(as) conformers as the two lowest energy forms. The latter conformer has also been found in the crystal structure. However, due to the engagement of the hydroxyl groups in intermolecular rather than intramolecular hydrogen bonds, the T(as) conformer in the solid state differs from its analog in the gas phase in the orientation of O-H bonds. Common to all these conformers is an antiparallel arrangement of two pairs of (β)CH/CO bonds leading to attractive dipole-dipole interactions. This type of interaction seems therefore to be a meaningful factor in stabilizing a particular conformation of dimethyl (R,R)-tartrate.
Acknowledgements
The authors thank Prof. J. Gawronski for providing the sample for X-ray analysis and Poznan Super Computer and Networking Center for a computing grant. This work was partially supported by KBN grant No T11F010 08p01.
References
- Eggli, M.; Dobler, M. Helv. Chim. Acta 1989, 72, 1136.
- Pedersen, S. F.; Dewan, J. C.; Eckman, R. R.; Sharpless, K. B. J. Am. Chem. Soc. 1987, 109, 1279. [CrossRef]
- Marcott, C.; Blackburn, C. C.; Faulkner, T. R.; Moscowitz, A.; Overend, J. J. Am. Chem. Soc. 1978, 100, 5262. [CrossRef]
- Su, C. N.; Keiderling, T. A. J. Am. Chem. Soc. 1980, 102, 511. [CrossRef]
- Freedman, T. B.; Balukjian, G. A.; Nafie, L. A. J. Am. Chem. Soc. 1985, 107, 6213. [CrossRef]
- Polvarapu, P. L.; Ewig, C. S.; Chandramouly, T. J. Am. Chem. Soc. 1985, 109, 6213.
- For references for the various split-valence basis sets, explanations of basis set notation, and both the RHF and MP2 methods see: Hehre, W. J.; Radom, L.; Schleyer, P. v. R.; Pople, J. A. Ab Initio Molecular Orbital Theory; Wiley: New York, 1986. [Google Scholar]
- Ascenso, J.; Gil, V. M. S. Can. J. Chem. 1980, 58, 1376.
- Hasan, M. Org. Magn. Res. 1980, 14, 309.
- (a) Barron, L. D. Tetrahedron 1978, 34, 607. (b) Barron, L. D.; Gargano, A. R.; Hecht, L.; Polvarapu, P. L.; Sugeta, H. Spectrochim. Acta 1992, 48A, 1051.
- Hasan, M.; Rychlewski, J.; Rychlewska, U. Computational Methods in Science and Technology 1996, 2, 51.
- Gawronski, J.; Gawronska, K.; Skowronek, P.; Rychlewska, U.; Warzajtis, B.; Rychlewski, J.; Hoffmann, M.; Szarecka, A. Tetrahedron. submitted for publication.
- Lehmann, M. S.; Larsen, F. K. Acta Crystallogr. 1974, A30, 580.
- Sheldrick, G. M. SHELXS86. In Program for the solution of crystal structures; Univ. of Göttingen: Germany, 1993. [Google Scholar]
- Sheldrick, G. M. SHELXL93. In Program for the Refinement of Crystal Structure; Univ. of Göttingen: Germany, 1986. [Google Scholar]
- Gaussian 94, Revision C.3, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Gill, P. M. W.; Johnson, B. G.; Robb, M. A.; Cheeseman, J. R.; Keith, T.; Petersson, G. A.; Montgomery, J. A.; Raghavachari, K.; Al-Laham, M. A.; Zakrzewski, V. G.; Ortiz, J. V.; Foresman, J. B.; Cioslowski, J.; Stefanov, B. B.; Nanayakkara, A.; Challacombe, M.; Peng, C. Y.; Ayala, P. Y.; Chen, W.; Wong, M. W.; Andres, J. L.; Replogle, E. S.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Binkley, J. S.; Defrees, D. J.; Baker, J.; Stewart, J. P.; Head-Gordon, M.; Gonzalez, C.; Pople, J. A. Gaussian Inc.: Pittsburgh, PA, 1995.
- Stereochemical Workstation Operation Manual, Release 3.4, Siemens Analytical X-Ray Instruments, Inc., Madison, Wisconsin, USA, 1989.
- Szczepanska, B.; Rychlewska, U. Correlations, Transformations and Interactions in Organic Chemistry; pp. 233–244. Jones, D.W., Katrusiak, A., Eds.; Oxford University Press, 1994. [Google Scholar]
- Allen, F. H.; Kennard, O. 3D Search and Research using the Cambridge Structural Database. Chemical Design Automation News, 1993; 8, 131. [Google Scholar]
- Szarecka, A.; Hoffmann, M.; Rychlewski, J.; Rychlewska, U. J. Mol. Struct. 1996, 374, 363.
- (+)-Tartaric acid: (a) Stern, F.; Beevers, C. A. Acta Crystallogr. 1950, 3, 341. (b) Okaya, Y.; Stemple, N. R.; Kay, M. I. ibid. 1996, 21, 237. (c) Albertsson, J.; Oskarsson, A.; Stahl, K. J. Appl. Crystallogr. 1979, 12, 537.
- For explanation of graph set notation see: (a) Etter, M. C. J. Am. Chem. Soc. 1982, 104, 1095. (b) Etter, M. C.; MacDonald, J. C.; Bernstein, J. Acta Crystallogr. 1990, B46, 256. (c) Etter, M. C. Acc. Chem. Res. 1990, 23, 120. (d) Bernstein, J.; Etter, M. C.; MacDonald, J. C. J. Chem. Soc. Perkin Trans. 2. 1990, 695. (e) Bernstein, J.; Davis, R. E.; Shimoni, L.; Chang, N. L. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555.
- Cramer, C. J.; Truhlar, D. G. J. Am. Chem. Soc. 1994, 99, 3892.
- Sample Availability: not available
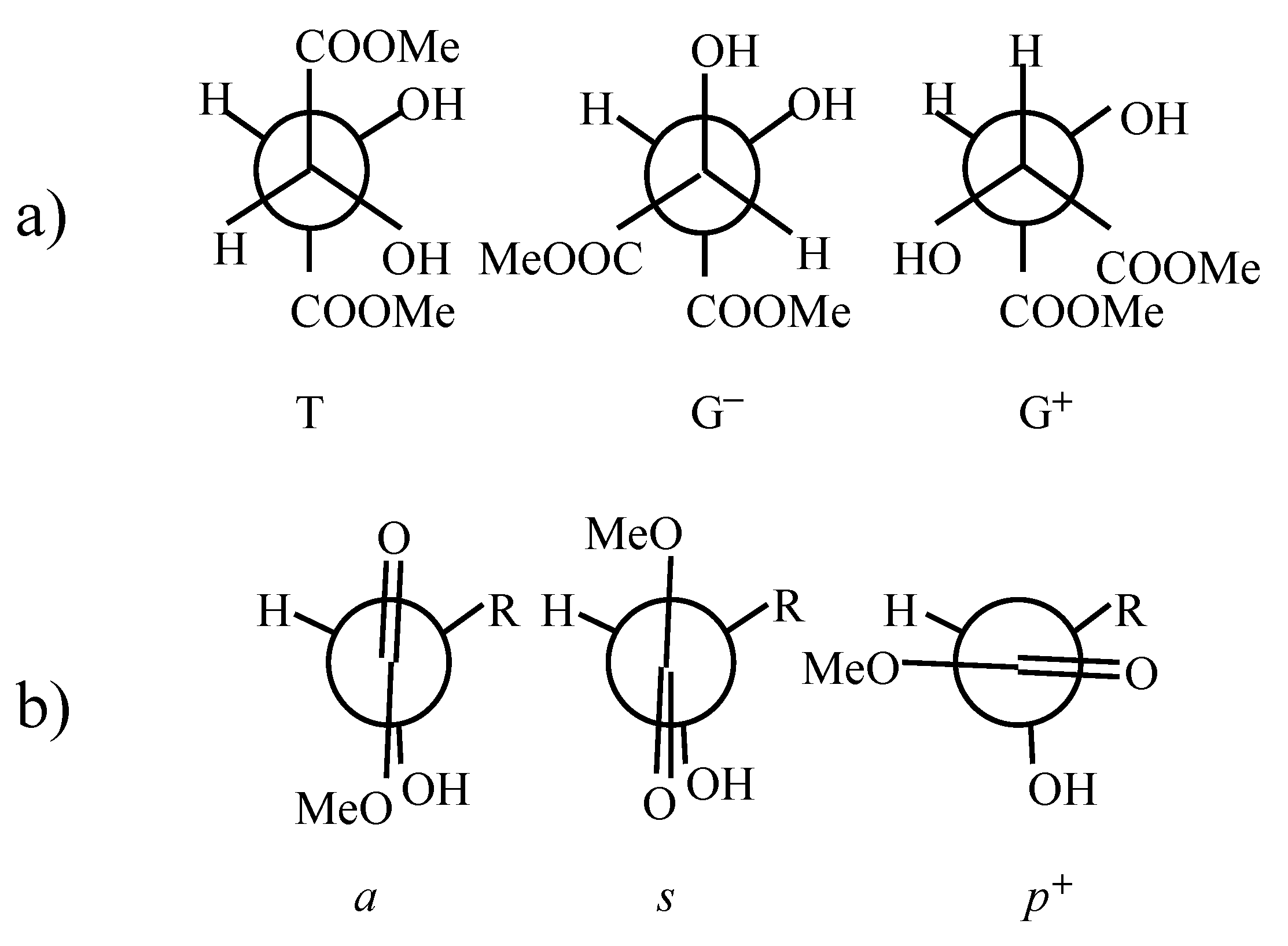
© 1997 MDPI. All rights reserved