Access to Optically Pure β-Hydroxy Esters via Non-Enzymatic Kinetic Resolution by a Planar-Chiral DMAP Catalyst

Abstract
:
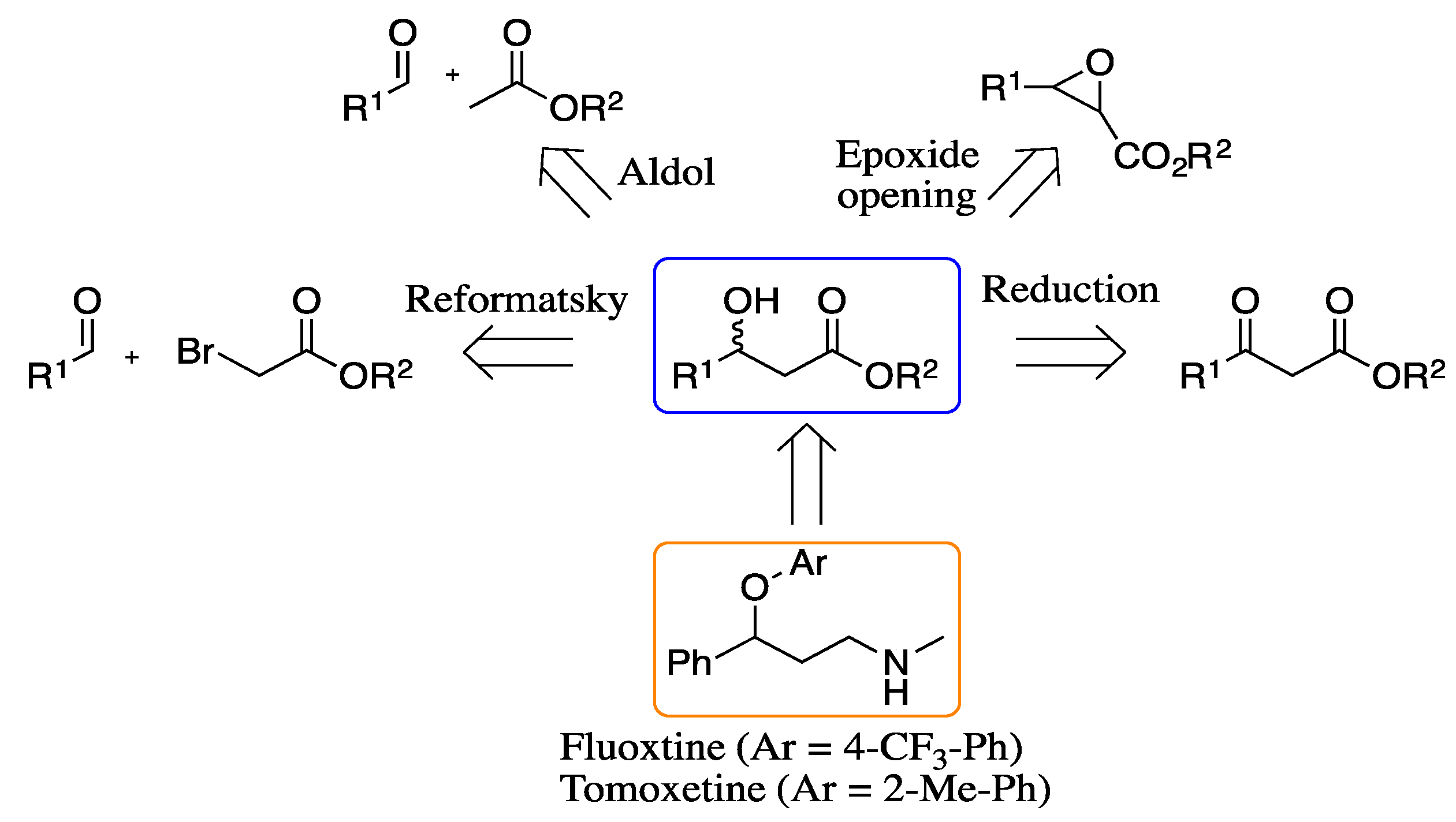
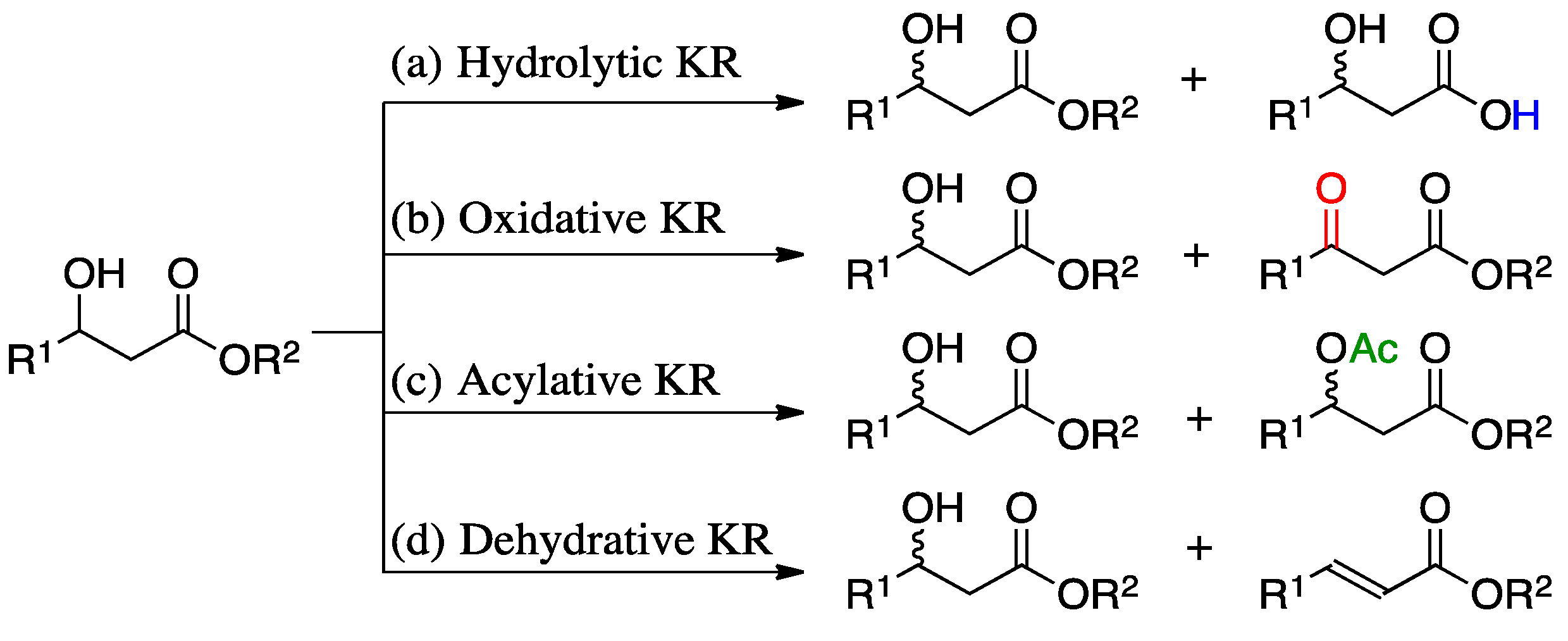
1. Introduction
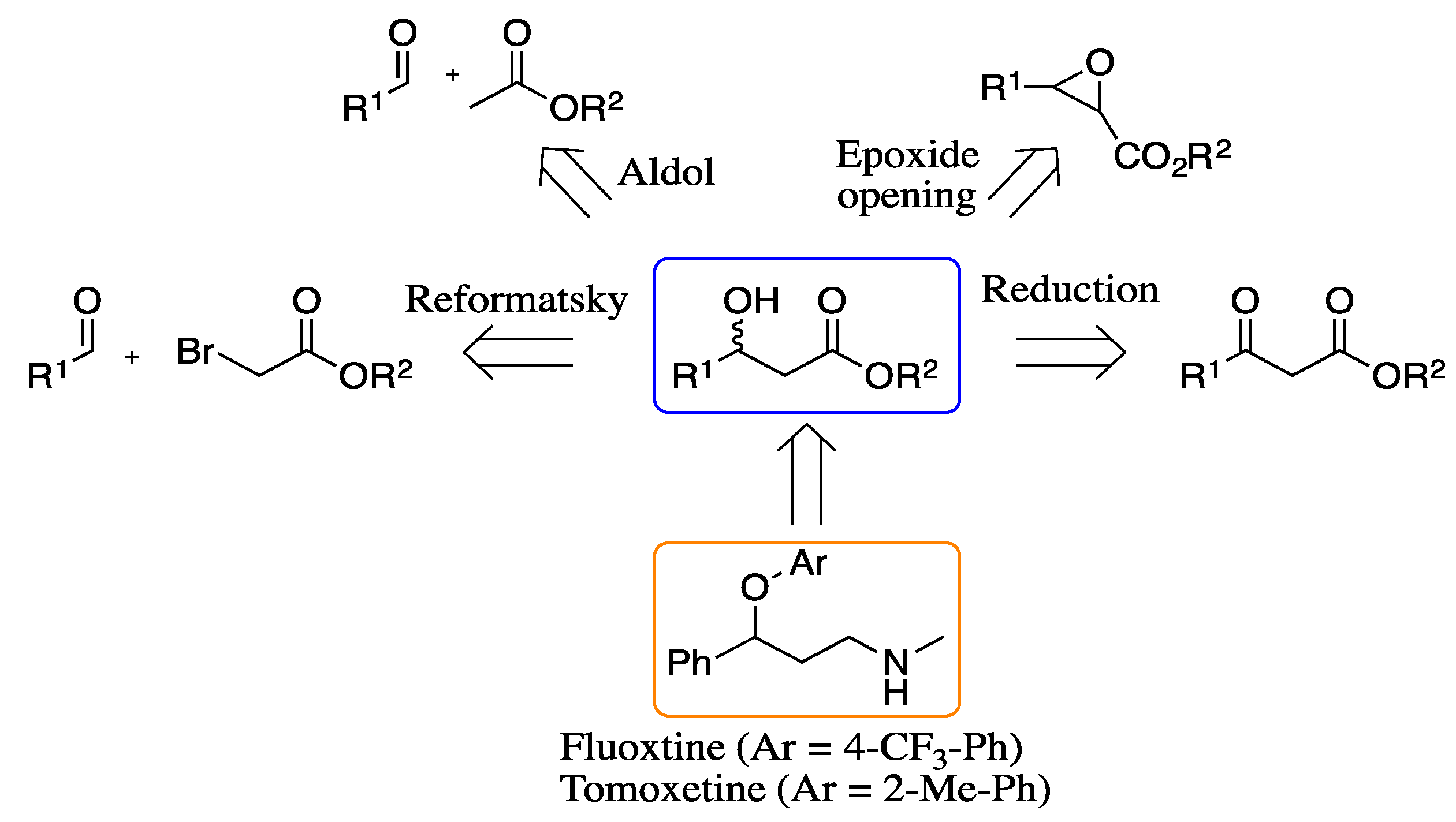
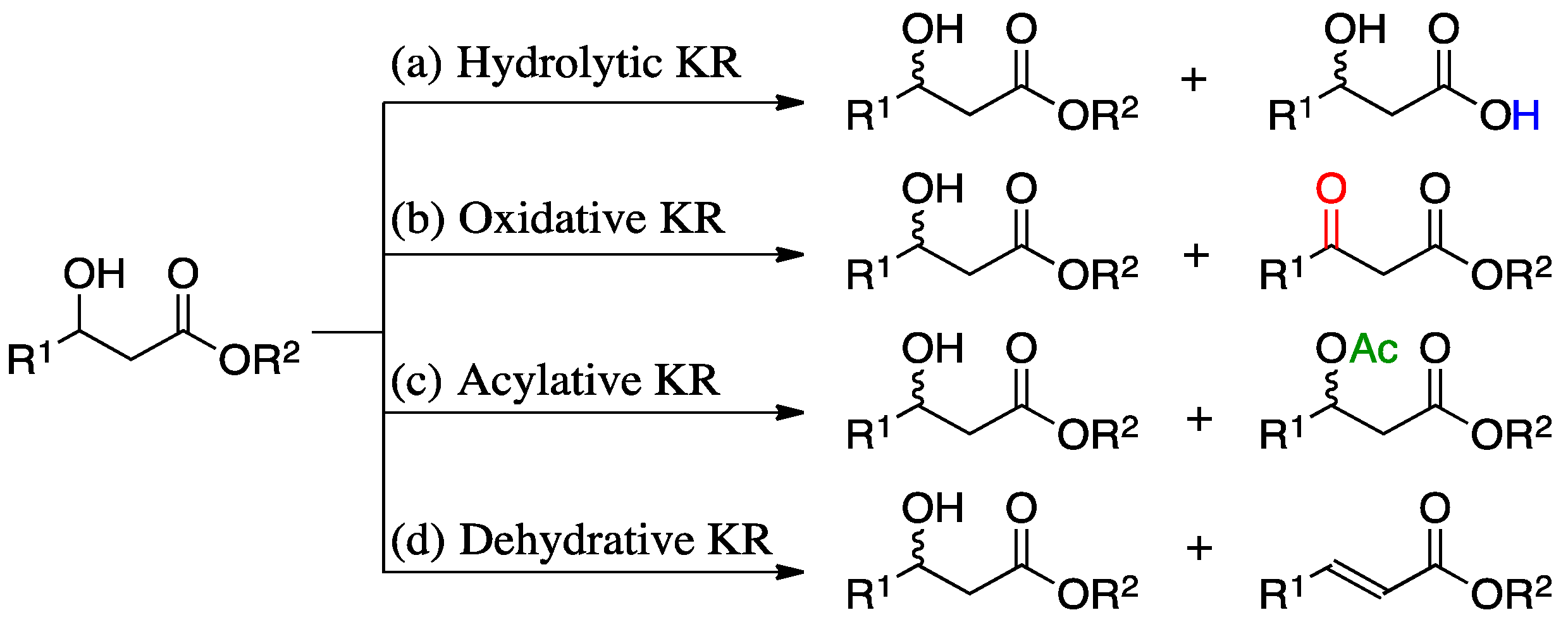
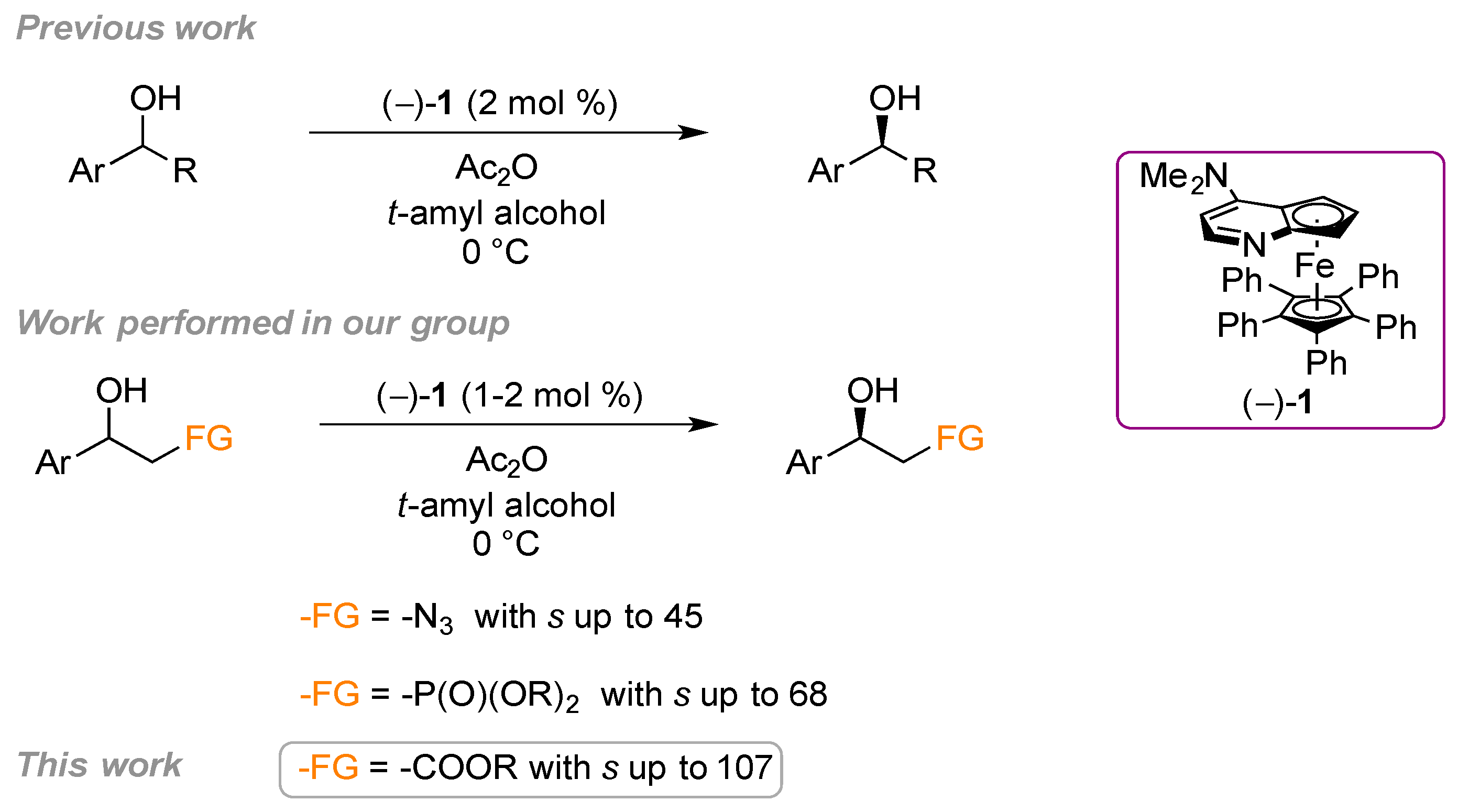
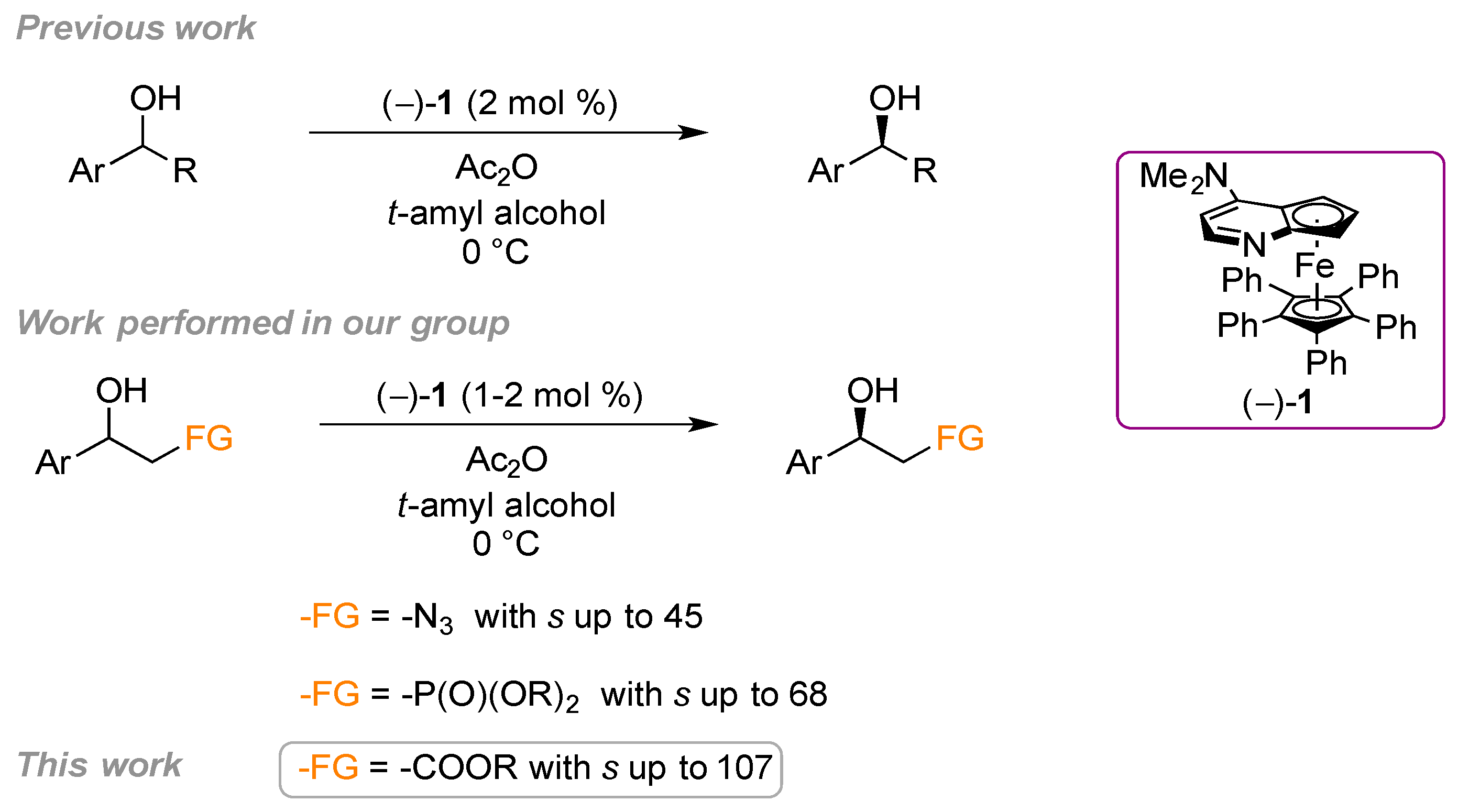
2. Results and Discussion
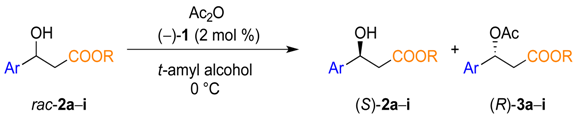
2.1. Substrate Screening

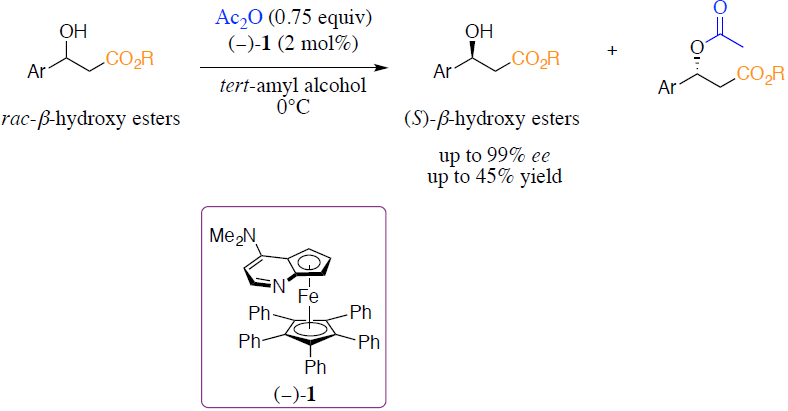{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R | Ar | Time/h | eeROH (%) b | eeROAc (%) b | Conv. (%) c | sc |
|---|---|---|---|---|---|---|---|
| 1 | Et | Ph (2a) | 3 | 32.4 | 95.4 | 25.4 | 58 |
| 2 | Et | 4-NO2-Ph (2b) | 3 | 84.2 | 92.8 | 44.6 | 71 |
| 3 | Et | 4-MeO-Ph (2c) | 3 | 39.8 | 94.6 | 29.6 | 53 |
| 4 | tert-Bu | Ph (2d) | 3 | 28.8 | 95.2 | 23.2 | 54 |
| 5 | tert-Bu | 4-NO2-Ph (2e) | 3 | 88.4 | 93.6 | 48.6 | 89 |
| 6 | tert-Bu | 4-MeO-Ph (2f) | 3 | 39.4 | 96.0 | 29.1 | 72 |
| 7 | tert-Bu | 2-Naphthyl (2g) | 3 | 83.4 | 95.2 | 46.7 | 107 |
| 8 | tert-Bu | 4-Cl-Ph (2h) | 3 | 75.0 | 95.2 | 44.1 | 92 |
| 9 | tert-Bu | 2,6-Cl2-Ph (2i) | 3 | 40.4 | 96.6 | 29.5 | 86 |
| Entry | R | Ar | Conv. (%) c | Yield (%) d | eeROH (%) e |
|---|---|---|---|---|---|
| 1 | Et | Ph (2a) | 57 | 41 | 99 |
| 2 | Et | 4-NO2-Ph (2b) | 79 | 23 | 99 f |
| 3 | Et | 4-MeO-Ph (2c) | 55 | 32 | 99 f |
| 4 b | tert-Bu | Ph (2d) | 56 | 39 | 95 |
| 5 | tert-Bu | 4-NO2-Ph (2e) | 66 | 33 | 99 f |
| 6 | tert-Bu | 4-MeO-Ph (2f) | 55 | 41 | 99 |
| 7 | tert-Bu | 2-Naphthyl (2g) | 54 | 45 | 98 |
| 8 | tert-Bu | 4-Cl-Ph (2h) | 65 | 34 | 99 f |
| 9 b | tert-Bu | 2,6-Cl2-Ph (2i) | 63 | 31 | 99 f |
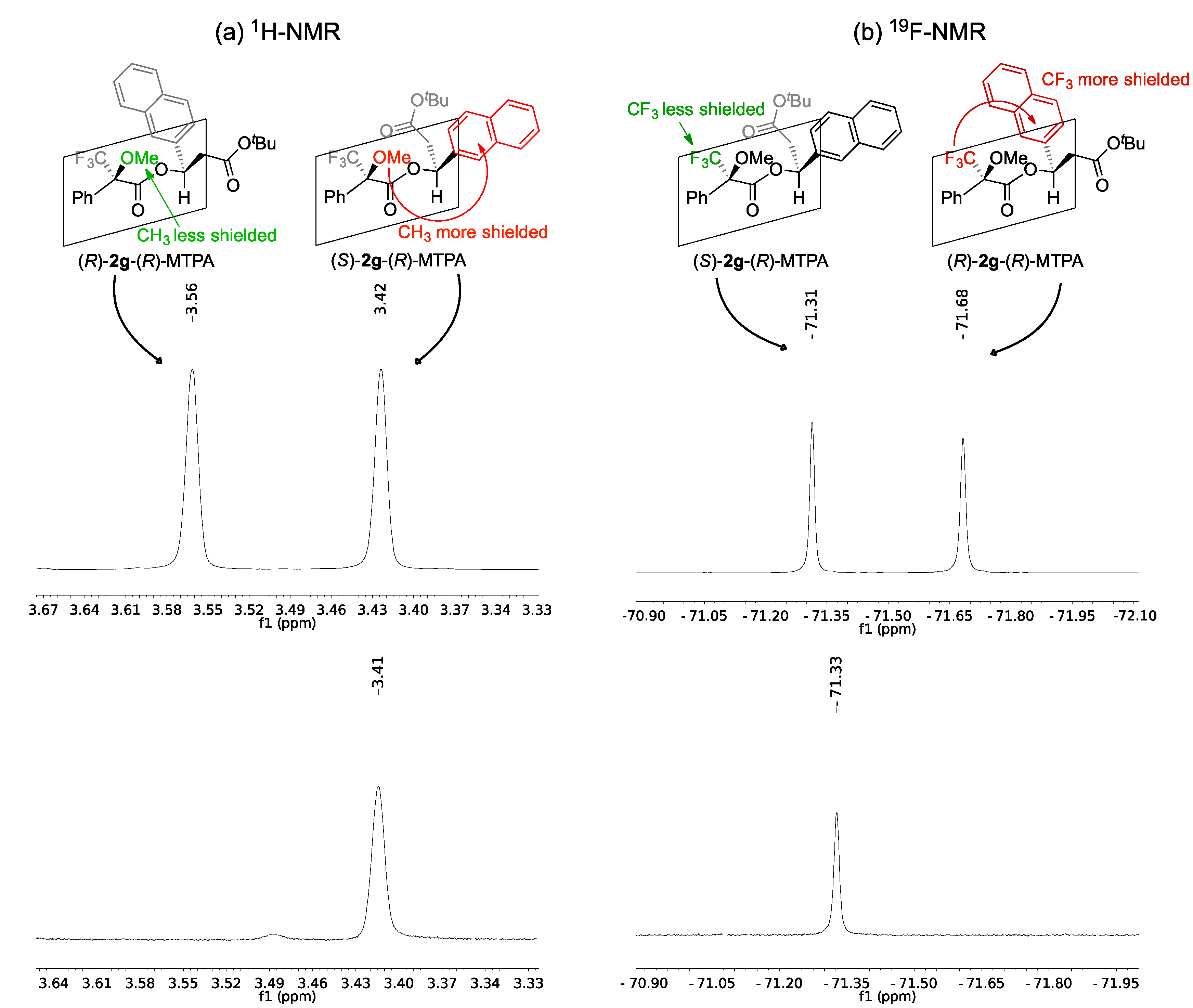
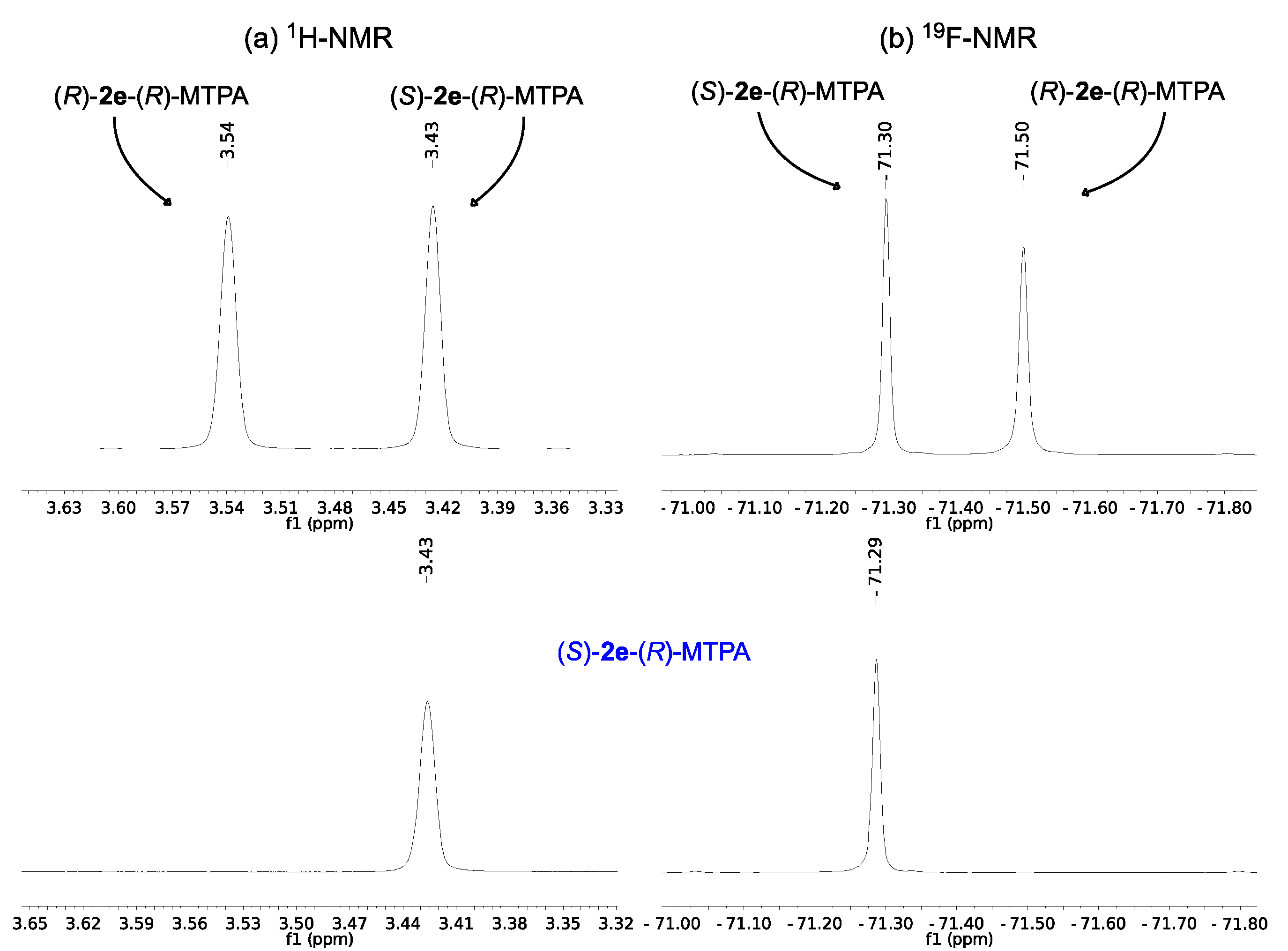
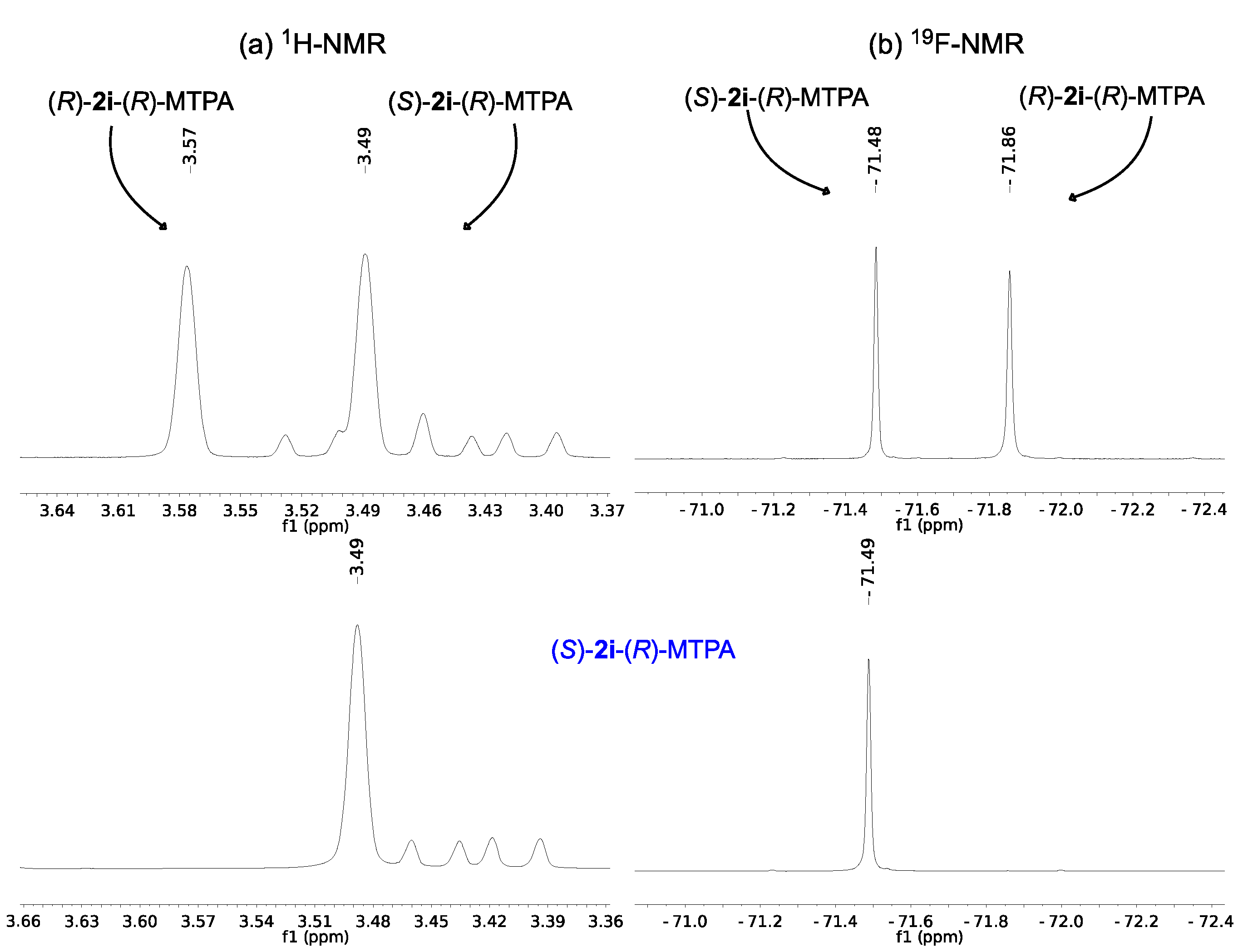
2.2. Assignment of the Absolute Configuration of 2e and 2i after KR
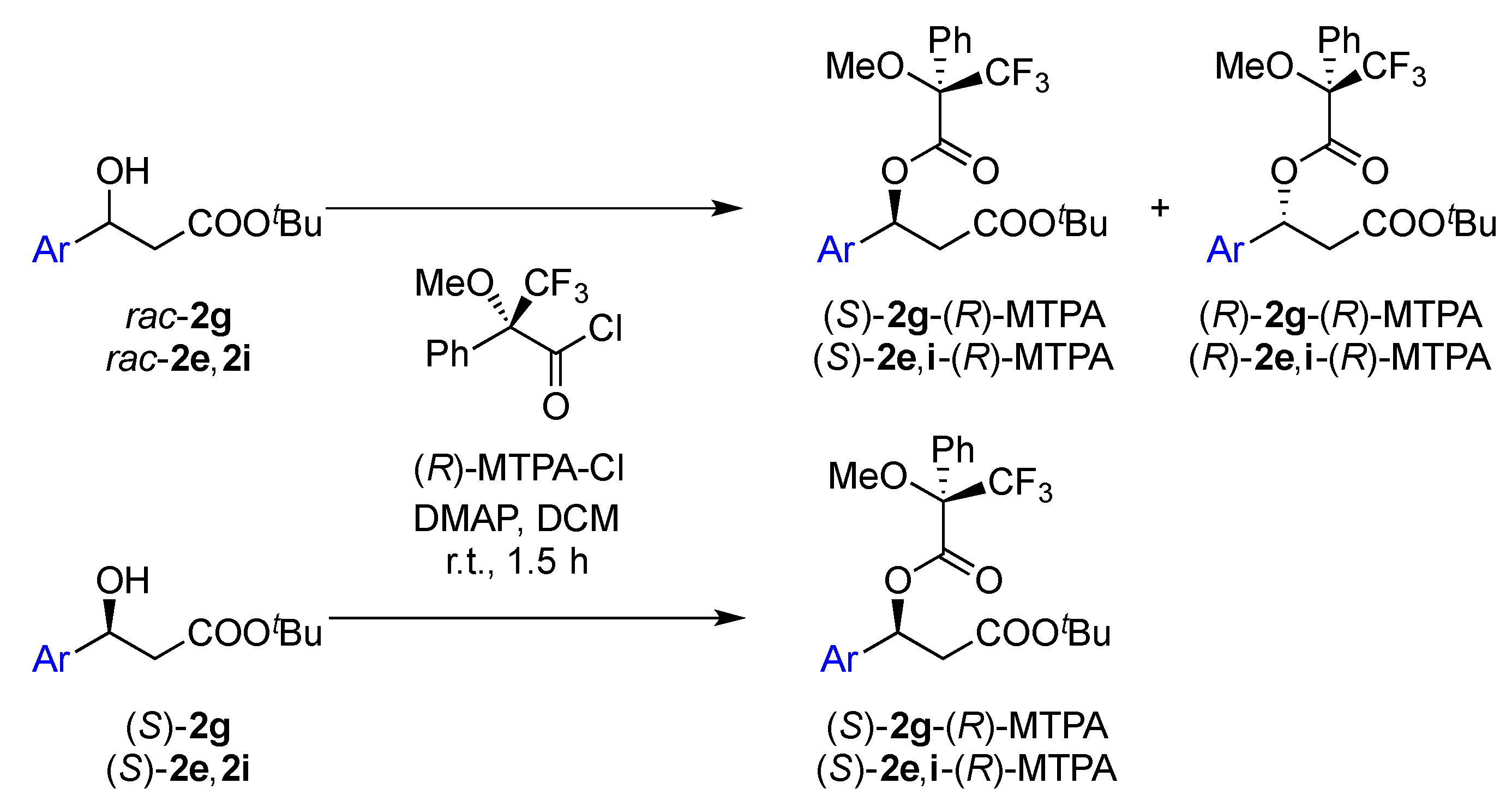
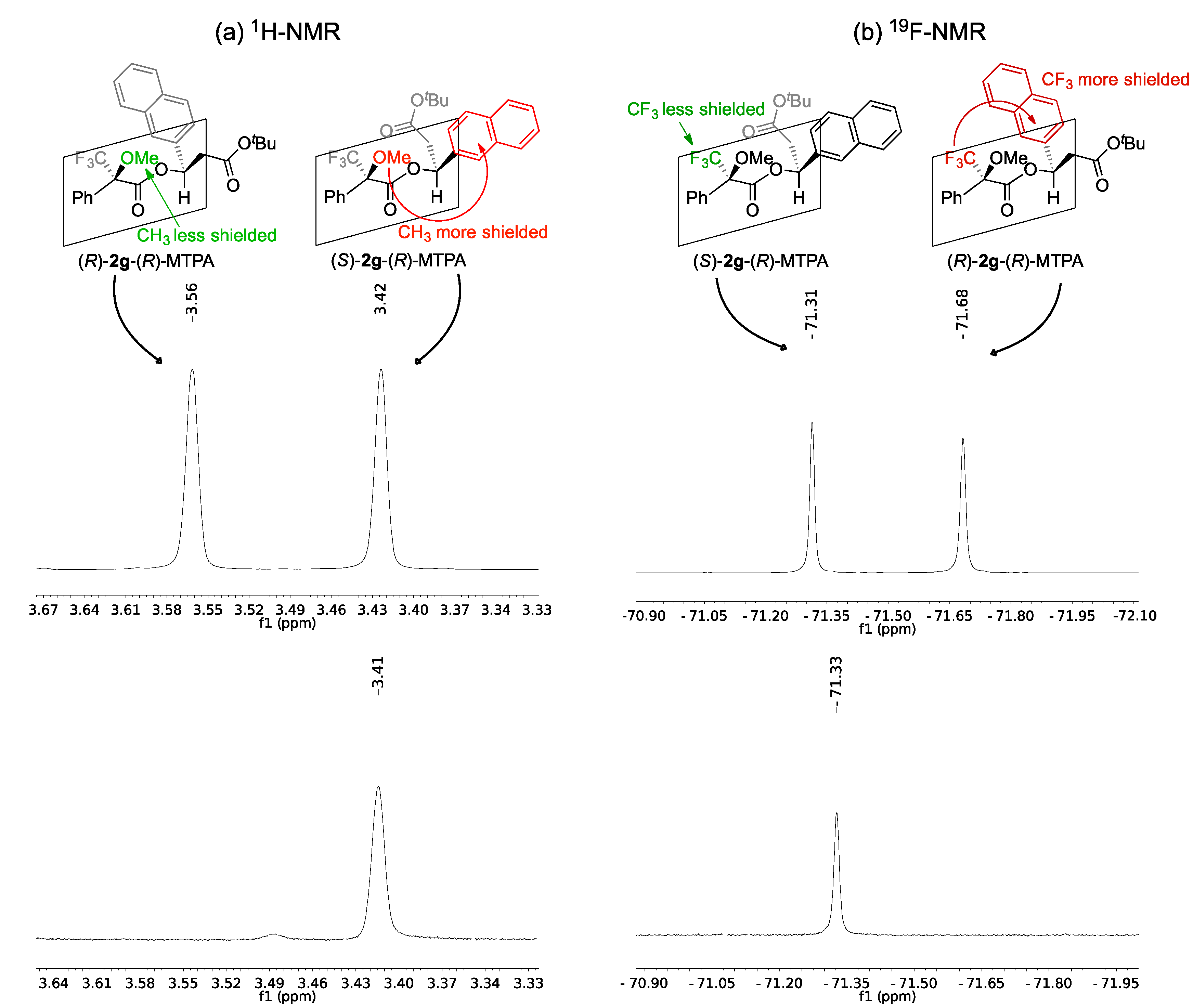
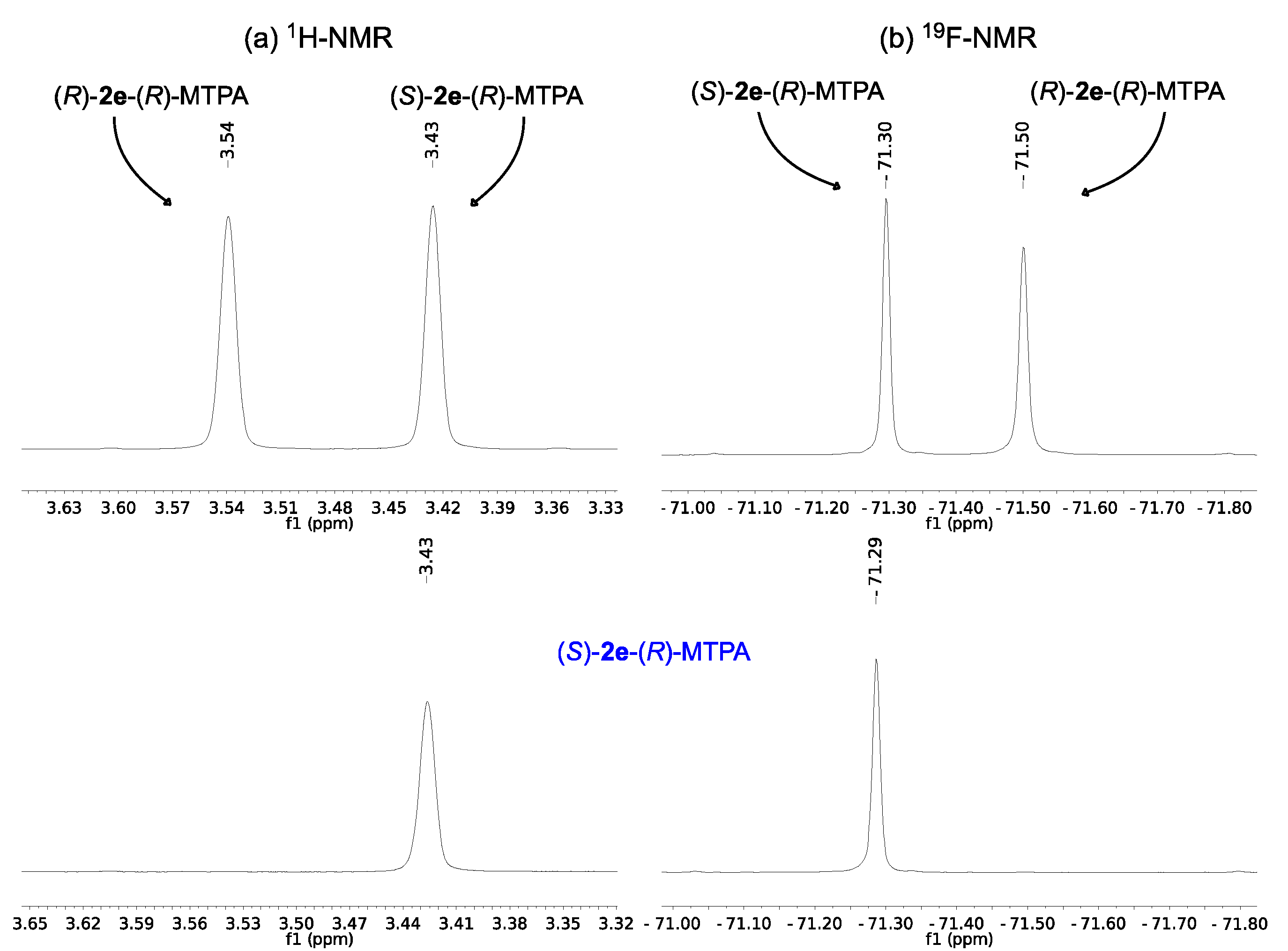
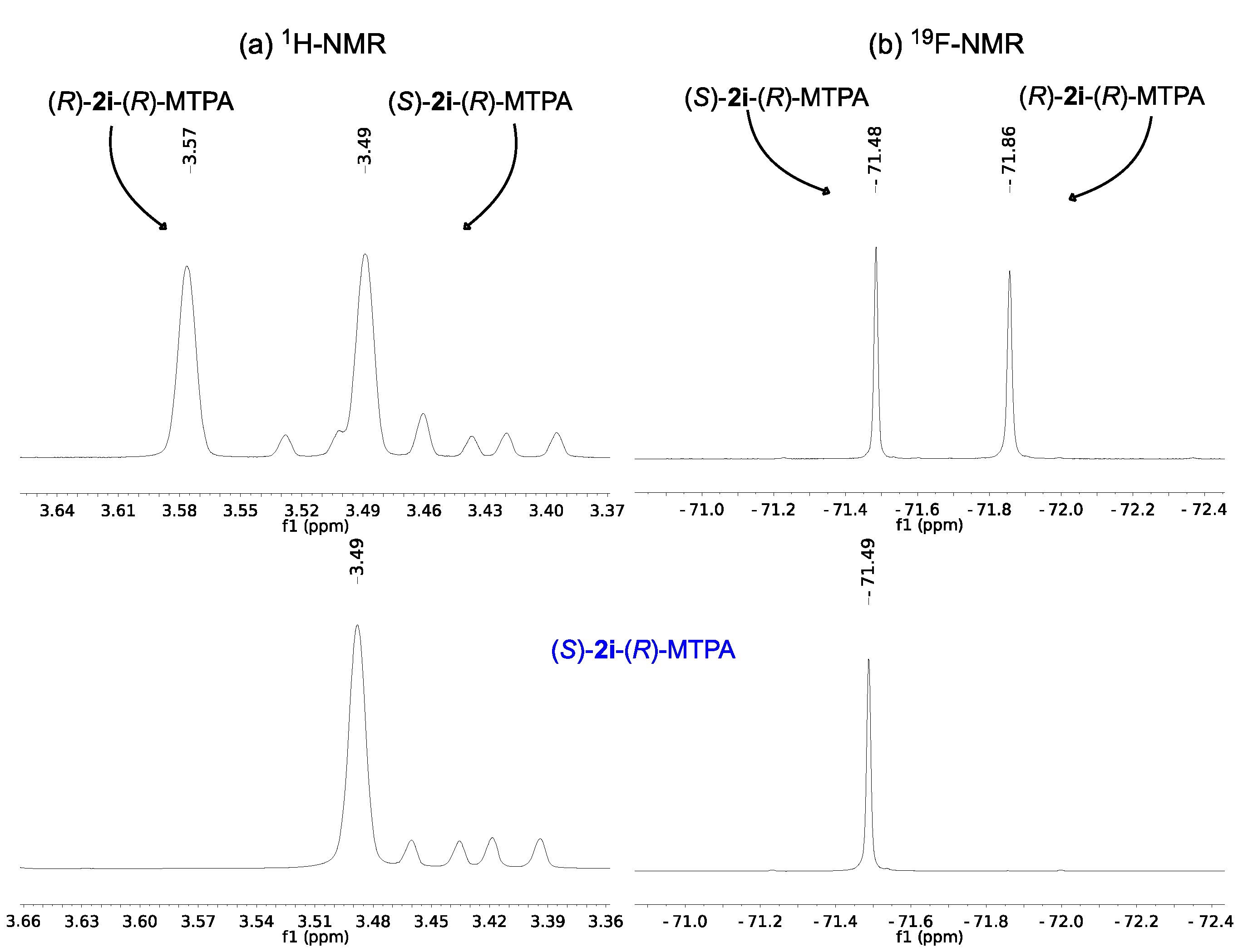
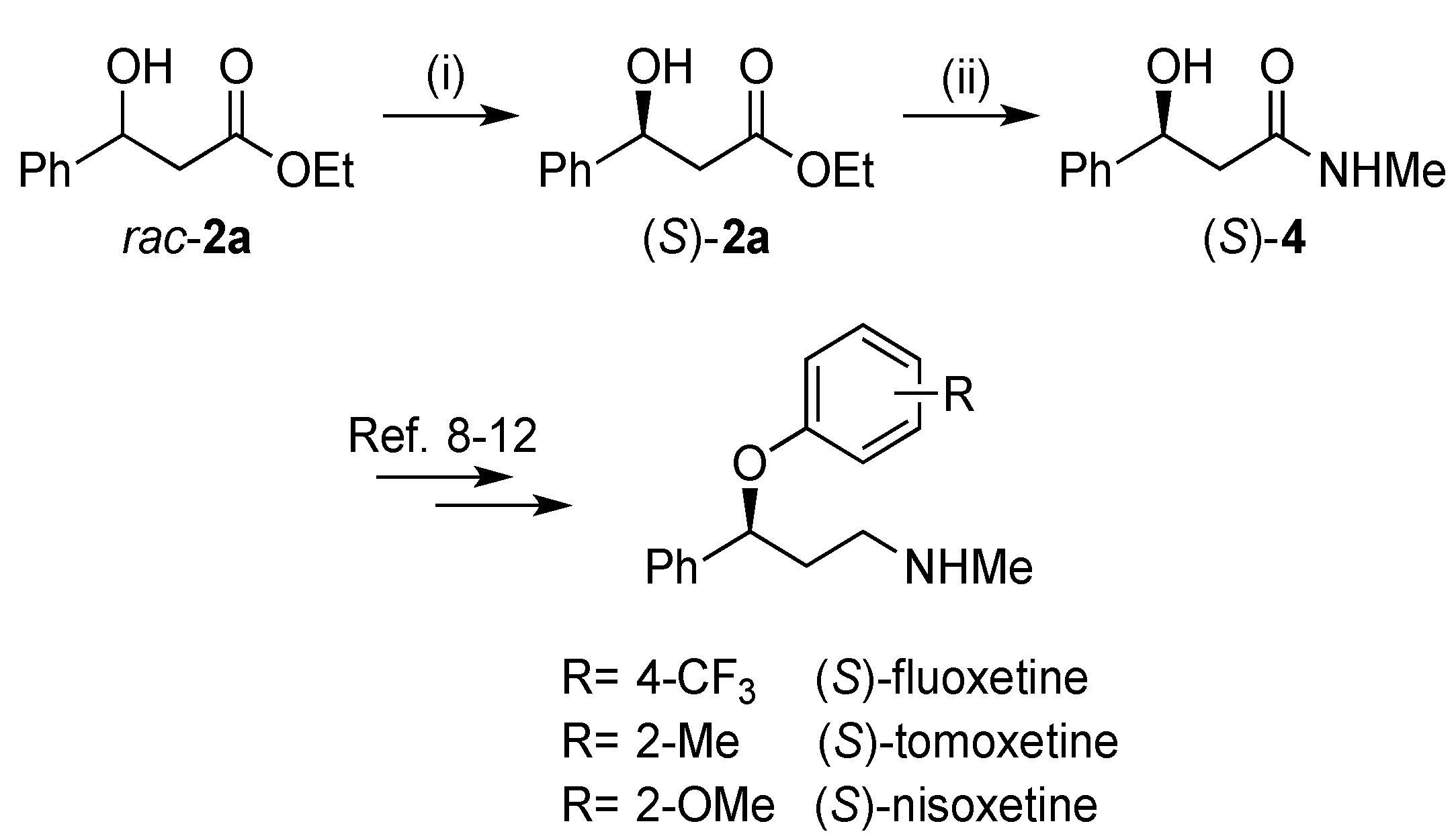
2.3. Synthesis of (S)-3-Hydroxy-N-methyl-3-phenylpropanamide
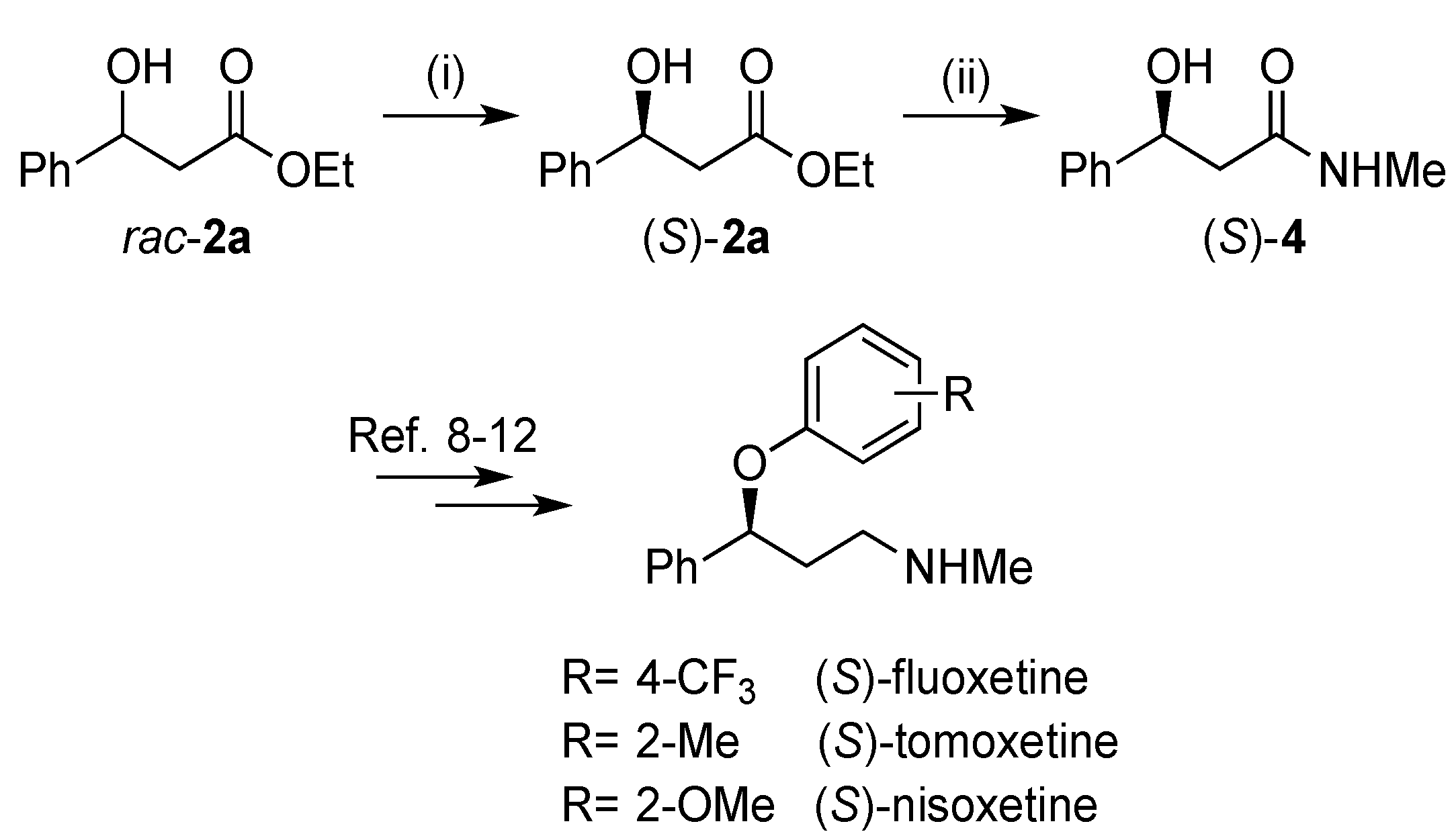
3. Experimental Section
3.1. General Information
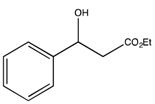
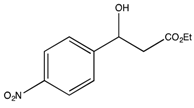
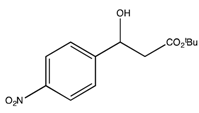
3.2. Preparation of the Racemic Substrates
3.2.1. General Procedure
3.2.2. Characterization Data


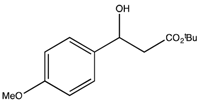
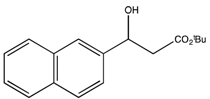
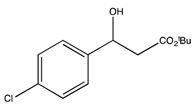

3.3. General Procedure for the Kinetic Resolution
3.3.1. Selectivity Factor for the KR of 2a–i after 3 h
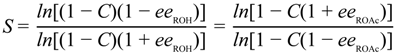
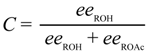
3.3.2. Synthesis of Optically Pure Alcohols
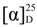 : −45.3 (c 3.7, CDCl3) (Lit. [71] [ : = −46.5 (c 1.04, CHCl3)). 1H-NMR (CDCl3) δ 7.28–7.17 (m, 5H, ArH), 5.04 (dd, J = 7.8, 4.2 Hz, 1H, CH), 4.10 (m, J = 6.9 Hz, 2H, CH2), 3.22 (bs, 1H, OH), 2.64–2.50 (m, 2H, CH2), and 1.17 (t, J = 6.9 Hz, 3H, CH3). 13C{1H}-NMR (CDCl3) δ 172.6, 142.8, 128.8 (2C), 128.0, 125.9 (2C), 70.6, 61.1, 43.6 and 14.4.
: −45.3 (c 3.7, CDCl3) (Lit. [71] [ : = −46.5 (c 1.04, CHCl3)). 1H-NMR (CDCl3) δ 7.28–7.17 (m, 5H, ArH), 5.04 (dd, J = 7.8, 4.2 Hz, 1H, CH), 4.10 (m, J = 6.9 Hz, 2H, CH2), 3.22 (bs, 1H, OH), 2.64–2.50 (m, 2H, CH2), and 1.17 (t, J = 6.9 Hz, 3H, CH3). 13C{1H}-NMR (CDCl3) δ 172.6, 142.8, 128.8 (2C), 128.0, 125.9 (2C), 70.6, 61.1, 43.6 and 14.4.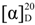 : −58.0 (c 0.23, CHCl3) (Lit. [64] : −59.5 (c 1.5, CHCl3)). 1H-NMR (CDCl3) δ 7.87 (d, 2H, ArH), 7.46 (d, 2H, ArH), 5.25 (dd, J = 7.8, 4.2 Hz, 1H, CH), 4.29–4.11 (m, 2H, CH2), 3.62 (bs, 1H, OH), 2.71 (q, J = 6.9 Hz, 2H, CH2) and 1.24 (t, J = 6.9 Hz, 3H, CH3). 13C{1H}-NMR (CDCl3) δ 172.0, 149.9, 147.4, 126.2 (2C), 123.9 (2C), 69.2, 61.3, 43.1 and 14.1.: −45.9 (c 0.75, CHCl3) (Lit. [71]
: −58.0 (c 0.23, CHCl3) (Lit. [64] : −59.5 (c 1.5, CHCl3)). 1H-NMR (CDCl3) δ 7.87 (d, 2H, ArH), 7.46 (d, 2H, ArH), 5.25 (dd, J = 7.8, 4.2 Hz, 1H, CH), 4.29–4.11 (m, 2H, CH2), 3.62 (bs, 1H, OH), 2.71 (q, J = 6.9 Hz, 2H, CH2) and 1.24 (t, J = 6.9 Hz, 3H, CH3). 13C{1H}-NMR (CDCl3) δ 172.0, 149.9, 147.4, 126.2 (2C), 123.9 (2C), 69.2, 61.3, 43.1 and 14.1.: −45.9 (c 0.75, CHCl3) (Lit. [71] 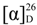 : −28.6 (c 1, CHCl3)). 1H-NMR (CDCl3) δ 7.29 (d, 2H, J = 7.5 Hz, ArH), 6.86 (d, 2H, J = 7.5 Hz, ArH), 5.08 (dd, J = 8.8, 4.0 Hz, 1H, CH), 4.19–4.09 (m, 2H, CH2), 3.80 (s, 3H, OCH3), 2.76–2.69 (m, 2H, CH2) and 1.27 (t, J = 6.9 Hz, 3H, CH3). 13C{1H}-NMR (CDCl3) δ 172.3, 159.1, 134.8, 126.9 (2C), 113.8 (2C), 69.9, 60.7, 55.2, 43.3 and 14.1.: −85.0 (c 0.1, CHCl3) (Lit. [71] : −37.7 (c 1.2, CHCl3)). 1H-NMR (CDCl3) δ 7.39–7.22 (m, 5H, ArH), 5.07 (dd, 1H, J = 7.8, 4.2 Hz, CH), 3.42 (bs, 1H, OH), 2.67 (dd, 1H, J = 16.0, 4.8 Hz, CH2), 2.61 (dd, 1H, J = 16.0, 4.8 Hz, CH2) and 1.43 (s, 9H, 3 CH3). 13C{1H}-NMR (CDCl3) δ 171.9, 142.6, 128.4 (2C), 127.6, 125.7 (2C), 81.5, 70.4, 44.3 and 28.0.: −31.4 (c 0.19, CHCl3). 1H-NMR (CDCl3) δ 8.20 (d, 2H, J = 8.1 Hz, ArH), 7.55 (d, 2H, J = 8.1 Hz, ArH), 5.18 (dd, J = 8.3, 4.1 Hz, 1H, CH), 2.73–2.57 (m, 2H, CH2) and 1.49 (s, 9H, 3 CH3). 13C{1H}-NMR (CDCl3) δ 171.3, 150.0, 147.3, 126.5 (2C), 123.6 (2C), 82.1, 69.4, 43.8 and 28.0. IR (neat): v(cm−1) 3438, 2941, 2977, 2903, 1700, 1510, 1343, 1148, 843. HRMS (ESI, m/z) Calcd for C13H18NO5+ [M+H+]: 268.1185, found: 268.1189.: −34.5 (c 1.6, CHCl3) (Lit. value for the (R)-enantiomer [46] : +28.0 (c 0.15, CHCl3)). 1H-NMR (CDCl3) δ 7.29 (d, 2H, J = 7.5 Hz, ArH), 6.86 (d, 2H, J = 7.5 Hz, ArH), 5.01 (dd, J = 8.8, 4.0 Hz, 1H, CH), 3.78 (s, 3H, OCH3), 3.15 (bs, 1H, OH), 2.71–2.54 (m, 2H, CH2) and 1.44 (s, 9H, 3 CH3). 13C{1H}-NMR (CDCl3) δ 171.9, 159.1, 134.9, 127.0 (2C), 113.8 (2C), 81.4, 70.0, 55.2, 44.3, and 28.0.: −36.9 (c 0.42, CHCl3) (Lit. [21] : −24.8 (c 1.1, CHCl3)). 1H-NMR (CDCl3) δ 7.85–7.81 (m, 4H, ArH), 7.50–7.26 (m, 3H, ArH), 5.26 (dd, J = 7.3, 5.5 Hz, 1H, CH), 2.76–2.74 (m, 2H, CH2) and 1.46 (s, 9H, 3 CH3). 13C{1H}-NMR (CDCl3) δ 171.9, 140.1, 133.3, 133.0, 128.3, 128.0, 127.7, 126.1, 125.9, 124.5, 123.9, 81.6, 70.5, 44.3 and 28.1.: −34.1 (c 1.9, CHCl3) (Lit. [21]
: −28.6 (c 1, CHCl3)). 1H-NMR (CDCl3) δ 7.29 (d, 2H, J = 7.5 Hz, ArH), 6.86 (d, 2H, J = 7.5 Hz, ArH), 5.08 (dd, J = 8.8, 4.0 Hz, 1H, CH), 4.19–4.09 (m, 2H, CH2), 3.80 (s, 3H, OCH3), 2.76–2.69 (m, 2H, CH2) and 1.27 (t, J = 6.9 Hz, 3H, CH3). 13C{1H}-NMR (CDCl3) δ 172.3, 159.1, 134.8, 126.9 (2C), 113.8 (2C), 69.9, 60.7, 55.2, 43.3 and 14.1.: −85.0 (c 0.1, CHCl3) (Lit. [71] : −37.7 (c 1.2, CHCl3)). 1H-NMR (CDCl3) δ 7.39–7.22 (m, 5H, ArH), 5.07 (dd, 1H, J = 7.8, 4.2 Hz, CH), 3.42 (bs, 1H, OH), 2.67 (dd, 1H, J = 16.0, 4.8 Hz, CH2), 2.61 (dd, 1H, J = 16.0, 4.8 Hz, CH2) and 1.43 (s, 9H, 3 CH3). 13C{1H}-NMR (CDCl3) δ 171.9, 142.6, 128.4 (2C), 127.6, 125.7 (2C), 81.5, 70.4, 44.3 and 28.0.: −31.4 (c 0.19, CHCl3). 1H-NMR (CDCl3) δ 8.20 (d, 2H, J = 8.1 Hz, ArH), 7.55 (d, 2H, J = 8.1 Hz, ArH), 5.18 (dd, J = 8.3, 4.1 Hz, 1H, CH), 2.73–2.57 (m, 2H, CH2) and 1.49 (s, 9H, 3 CH3). 13C{1H}-NMR (CDCl3) δ 171.3, 150.0, 147.3, 126.5 (2C), 123.6 (2C), 82.1, 69.4, 43.8 and 28.0. IR (neat): v(cm−1) 3438, 2941, 2977, 2903, 1700, 1510, 1343, 1148, 843. HRMS (ESI, m/z) Calcd for C13H18NO5+ [M+H+]: 268.1185, found: 268.1189.: −34.5 (c 1.6, CHCl3) (Lit. value for the (R)-enantiomer [46] : +28.0 (c 0.15, CHCl3)). 1H-NMR (CDCl3) δ 7.29 (d, 2H, J = 7.5 Hz, ArH), 6.86 (d, 2H, J = 7.5 Hz, ArH), 5.01 (dd, J = 8.8, 4.0 Hz, 1H, CH), 3.78 (s, 3H, OCH3), 3.15 (bs, 1H, OH), 2.71–2.54 (m, 2H, CH2) and 1.44 (s, 9H, 3 CH3). 13C{1H}-NMR (CDCl3) δ 171.9, 159.1, 134.9, 127.0 (2C), 113.8 (2C), 81.4, 70.0, 55.2, 44.3, and 28.0.: −36.9 (c 0.42, CHCl3) (Lit. [21] : −24.8 (c 1.1, CHCl3)). 1H-NMR (CDCl3) δ 7.85–7.81 (m, 4H, ArH), 7.50–7.26 (m, 3H, ArH), 5.26 (dd, J = 7.3, 5.5 Hz, 1H, CH), 2.76–2.74 (m, 2H, CH2) and 1.46 (s, 9H, 3 CH3). 13C{1H}-NMR (CDCl3) δ 171.9, 140.1, 133.3, 133.0, 128.3, 128.0, 127.7, 126.1, 125.9, 124.5, 123.9, 81.6, 70.5, 44.3 and 28.1.: −34.1 (c 1.9, CHCl3) (Lit. [21]  : −25.4 (c 2.0, CHCl3)). 1H-NMR (CDCl3) δ 7.34–7.31 (m, 4H, ArH), 5.09–5.05 (m, 1H, CH), 2.63–2.60 (m, 2H, CH2) and 1.45 (s, 9H, 3 CH3). 13C{1H}-NMR (CDCl3) δ 171.7, 141.1, 133.3, 128.6 (2C), 127.1 (2C), 81.7, 69.7, 44.1 and 28.0.: +21.8 (c 2.0, CHCl3). 1H-NMR (CDCl3) δ 7.32–7.12 (m, 3H, ArH), 5.90 (dd, 1H, J = 10.5, J = 4.2 Hz, 1H, CH), 3.23–2.62 (m, 2H, CH2) and 1.46 (s, 9H, 3 CH3). 13C{1H}-NMR (CDCl3) δ 170.5, 136.2, 134.6, 129.4 (2C), 129.2 (2C), 81.3, 68.2, 40.7 and 28.0. IR (neat): v(cm−1) 3514, 2987, 2938, 1700, 1561, 1144, 766. HRMS (ESI, m/z) Calcd for C13H17Cl2O3+ [M+H+]: 291.0555, found: 291.0551.
: −25.4 (c 2.0, CHCl3)). 1H-NMR (CDCl3) δ 7.34–7.31 (m, 4H, ArH), 5.09–5.05 (m, 1H, CH), 2.63–2.60 (m, 2H, CH2) and 1.45 (s, 9H, 3 CH3). 13C{1H}-NMR (CDCl3) δ 171.7, 141.1, 133.3, 128.6 (2C), 127.1 (2C), 81.7, 69.7, 44.1 and 28.0.: +21.8 (c 2.0, CHCl3). 1H-NMR (CDCl3) δ 7.32–7.12 (m, 3H, ArH), 5.90 (dd, 1H, J = 10.5, J = 4.2 Hz, 1H, CH), 3.23–2.62 (m, 2H, CH2) and 1.46 (s, 9H, 3 CH3). 13C{1H}-NMR (CDCl3) δ 170.5, 136.2, 134.6, 129.4 (2C), 129.2 (2C), 81.3, 68.2, 40.7 and 28.0. IR (neat): v(cm−1) 3514, 2987, 2938, 1700, 1561, 1144, 766. HRMS (ESI, m/z) Calcd for C13H17Cl2O3+ [M+H+]: 291.0555, found: 291.0551.3.3.3. Methods Used to Determine Enantiomeric Excess
3.4. Synthesis of (S)-3-Hydroxy-N-methyl-3-phenylpropanamide ((S)-4)
: −26.7 (c 1.0, CH3OH) (Lit. [36] : −26.2 (c 1.25, CH3OH)). 1H-NMR (CDCl3) δ 7.49–7.14 (m, 5H, ArH), 5.82 (br s, 1H, NH), 5.16-5.05 (m, 1H, CHOH), 2.82 (d, J = 4.5 Hz, 3H, NHCH3), 2.59–2.52 (m, 2H, CH2). 13C{1H}-NMR (CDCl3) δ 172.56, 143.16, 128.67, 127.84, 125.71, 71.07, 44.72, 26.37.4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Noyori, R. Asymmetric catalysis: Science and opportunities (Nobel Lecture). Angew. Chem. Int. Ed. 2002, 41, 2008–2022. [Google Scholar]
- Thomassigny, C.; Sineriz, F.; Greck, C.; Lou, J.-D. β-Hydroxy esters as synthetic precursors of natural compounds and analogues. Recent Res. Dev. Org. Chem. 2004, 8, 377–400. [Google Scholar]
- Mori, K. Synthesis of optically active pheromones. Tetrahedron 1989, 45, 3233–3298. [Google Scholar] [CrossRef]
- Cui, J.; Zhang, M.J.; Wang, X.Q.; Cui, N.J. The stereoselective preparation of β-keto esters using baker’s yeast in the presence of rice wine koji. Chin. Chem. Lett. 2008, 19, 311–313. [Google Scholar] [CrossRef]
- Wong, D.T.; Bymaster, F.P.; Reid, L.R.; Fuller, R.W.; Perry, K.W. Inhibition of serotonin uptake by optical isomers of fluoxetine. Drug Dev. Res. 1985, 6, 397–403. [Google Scholar] [CrossRef]
- Robertson, D.W.; Krushinski, J.H.; Fuller, R.W.; Leander, J.D. The absolute configurations and pharmacological activities of the optical isomers of fluoxetine, a selective serotonin-uptake inhibitor. J. Med. Chem. 1988, 31, 1412–1417. [Google Scholar] [CrossRef]
- Fuller, R.W.; Snoddy, H.D. Fluoxetine enantiomers as antagonists of p-chloroamphetamine effects in rats. Pharmacol. Biochem. Behav. 1986, 24, 281–284. [Google Scholar] [CrossRef]
- Chai, L.; Chen, H.; Li, Z.; Wang, Q.; Tao, F. Enantioselective hydrogenation of β-keto esters using a MeO-PEG-supported Biphep ligand under atmospheric pressure: A practical synthesis of (S)-fluoxetine. Synlett 2006, 2395–2398. [Google Scholar]
- Chea, H.; Sim, H.-S.; Yun, J. Copper-catalyzed conjugate addition of diboron reagents to α,β-unsaturated amides: Highly reactive copper-1,2-bis(diphenylphosphino)benzene catalyst system. Adv. Synth. Catal. 2009, 351, 855–858. [Google Scholar] [CrossRef]
- Kakei, H.; Nemoto, T.; Ohshima, T.; Shibasaki, M. Efficient synthesis of chiral α- and β-hydroxy amides: Application to the synthesis of (R)-fluoxetine. Angew. Chem. Int. Ed. 2004, 43, 317–320. [Google Scholar]
- Khatik, G.L.; Kumar, V.; Nair, V.A. Reversal of selectivity in acetate aldol reactions of N-acetyl-(S)-4-isopropyl-1-[(R)-1-phenylethyl]imidazolidin-2-one. Org. Lett. 2012, 14, 2442–2445. [Google Scholar] [CrossRef]
- Kumar, A.; Ner, D.H.Y.; Dike, S. A new chemoenzymatic enantioselective synthesis of R-(−)-tomoxetine, (R)- and (S)-fluoxetine. Tetrahedron Lett. 1991, 32, 1901–1904. [Google Scholar] [CrossRef]
- Mukaiyama, T. Organic Reactions; John Wiley & Sons, Inc.: New York, NY, USA, 1982; Volume 28. [Google Scholar]
- Braun, M. Stereoselective aldol reactions with α-unsubstituted chiral enolates. Angew. Chem. Int. Ed. Engl. 1987, 26, 24–37. [Google Scholar] [CrossRef]
- Solladié, G.; Bauder, C.; Arce-Dubois, E.; Pasturel-Jacopé, Y. Asymmetric synthesis of β-hydroxyesters by aldol type condensation of enantiomerically pure t-butyl p-tolyl sulfinyl acetate: Unexpected substituent effect on the absolute configuration of the product. Tetrahedron Lett. 2001, 42, 2923–2925. [Google Scholar]
- Howell, G.P.; Fletcher, S.P. Catalytic asymmetric synthesis of acyclic arrays by tandem 1,4-addition-aldol reactions. J. Am. Chem. Soc. 2006, 128, 14977–14985. [Google Scholar] [CrossRef]
- Guetté, M.; Capillon, J.; Guetté, J.P. Synthese asymetrique de β-hydroxyesters par reaction de reformatsky en presence de (−) sparteine. Tetrahedron 1973, 29, 3659–3667. [Google Scholar]
- Soai, K.; Kawase, Y. Asymmetric synthesis of β-hydroxyesters by the enantioselective Reformatsky reaction in the presence of chiral aminoalcohols. Tetrahedron Asymmetry 1991, 2, 781–784. [Google Scholar] [CrossRef]
- Mi, A.; Wang, Z.; Zhang, J.; Jiang, Y. Asymmetric Syntheses XXVI: Catalytic enantioselective syntheses of β-hydroxy esters via double chiral induction in asymmetric Reformatsky reactions. Synth. Commun. 1997, 27, 1469–1473. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, W. Enantioselective synthesis of β-hydroxy esters by Reformatsky reactions in chiral micelles. Tetrahedron Asymmetry 1997, 8, 3575–3578. [Google Scholar] [CrossRef]
- Andrés, J.M.; Martín, Y.; Pedrosa, R.; Pérez-Encabo, A. Enantioselective Reformatsky reaction induced by chiral β-amino alcohols. Tetrahedron 1997, 53, 3787–3794. [Google Scholar]
- Fernández-Ibáñez, M.A.; Maciá, B.; Minnaard, A.J.; Feringa, B.L. Catalytic enantioselective Reformatsky reaction with aldehydes. Angew. Chem. Int. Ed. 2008, 47, 1317–1319. [Google Scholar]
- Carlsen, P.H.J.; Katsuki, T.; Martín, V.S.; Sharpless, K.B. A greatly improved procedure for ruthenium tetroxide catalyzed oxidations of organic compounds. J. Org. Chem. 1981, 46, 3936–3938. [Google Scholar] [CrossRef]
- Chong, J.M.; Sharpless, K.B. Regioselective openings of 2,3-epoxy acids with organocuprates. Tetrahedron Lett. 1985, 26, 4683–4686. [Google Scholar] [CrossRef]
- Otsubo, K.; Inanaga, J.; Yamaguchi, M. SmI2-induced highly regioselective reduction of α,β-epoxy esters and γ,δ-epoxy-α,β-insaturated esters. An efficient route to optically active β-hydroxy and γ-hydroxy esters. Tetrahedron Lett. 1987, 28, 4437–4440. [Google Scholar]
- Van der Baan, J.L.; Barnick, J.W.F.K.; Bickelhaupt, F. Synthesis of β-hydroxy esters by lithium/ammonia reduction of α,β-epoxy esters. Synthesis 1990, 897–899. [Google Scholar]
- Servi, S. Baker’s yeast as a reagent in organic synthesis. Synthesis 1990, 1990, 1–25. [Google Scholar] [CrossRef]
- Rodríguez, S.; Schroeder, K.T.; Kayser, M.M.; Stewart, J.D. Asymmetric synthesis of β-hydroxy esters and α-alkyl-β-hydroxy esters by recombinant Escherichia coli expressing enzymes from baker’s yeast. J. Org. Chem. 2000, 65, 2586–2587. [Google Scholar]
- Kalaitzakis, D.; Rozzell, J.D.; Kambourakis, S.; Smonou, I. Highly stereoselective reductions of α-alkyl-1,3-diketones and α-alkyl-β-keto esters catalyzed by isolated NADPH-dependent ketoreductases. Org. Lett. 2005, 7, 4799–4801. [Google Scholar] [CrossRef]
- Kaluzna, I.A.; Feske, B.D.; Wittayanan, W.; Ghiviriga, I.; Stewart, J.D. Stereoselective, biocatalytic reductions of α-chloro-β-keto esters. J. Org. Chem. 2005, 70, 342–345. [Google Scholar] [CrossRef]
- Noyori, R.; Ohkuma, T.; Kitamura, M.; Takaya, H.; Sayo, N.; Kumobayashi, H.; Akutagawa, S. Asymmetric hydrogenation of β-keto carboxylic esters. A practical, purely chemical access to β-hydroxy esters in high enantiomeric purity. J. Am. Chem. Soc. 1987, 109, 5856–5858. [Google Scholar]
- Ager, D.J.; Laneman, S.A. Reductions of 1,3-dicarbonyl systems with ruthenium-biarylbisphosphine catalysts. Tetrahedron Asymmetry 1997, 8, 3327–3355. [Google Scholar] [CrossRef]
- Ohkuma, T.; Noyori, R. Enantioselective ketone and β-ketone ester hydrogenations (including mechanisms). In Handbook of Homogeneous Hydrogenation; De Vries, J.G., Elsevier, C.J., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2006; Volume 3, pp. 1105–1163. [Google Scholar]
- Fan, W.; Li, W.; Ma, X.; Tao, X.; Li, X.; Yao, Y.; Xie, X.; Zhang, Z. Ru-catalyzed asymmetric hydrogenation of γ-heteroatom substituted β-keto esters. J. Org. Chem. 2011, 76, 9444–9451. [Google Scholar] [CrossRef]
- Ariger, M.A.; Carreira, E.M. pH-Independent transfer hydrogenation in water: Catalytic, enantioselective reduction of β-keto esters. Org. Lett. 2012, 14, 4522–4524. [Google Scholar] [CrossRef]
- Xie, J.-H.; Liu, X.-Y.; Yang, X.-H.; Xie, J.-B.; Wang, L.-X.; Zhou, Q.-L. Chiral iridium catalysts bearing spiro pyridine-aminophosphine ligands enable highly efficient asymmetric hydrogenation of β-aryl β-ketoesters. Angew. Chem. Int. Ed. 2012, 51, 201–203. [Google Scholar] [CrossRef]
- b>Ribeiro, C.M.R.; Passaroto, E.N.; Brenelli, E.C.S. Enzymatic resolution of ethyl 3-hydroxy-3-phenylpropanoate and analogs using hydrolases. J. Braz. Chem. Soc. 2001, 12, 742–746. [Google Scholar]
- Ribeiro, C.M.R.; Passaroto, E.N.; Brenelli, E.C.S. Ultrasound in enzymatic resolution of ethyl 3-hydroxy-3-phenylpropanoate. Tetrahedron Lett. 2001, 42, 6477–6479. [Google Scholar] [CrossRef]
- Chaubey, A.; Parshad, R.; Koul, S.; Taneja, S.C.; Qazi, G.N. Enantioselectivity modulation through immobilization of Arthrobacter sp. lipase: Kinetic resolution of fluoxetine intermediate. J. Mol. Catal. B: Enzym. 2006, 42, 39–44. [Google Scholar]
- Wang, P.-Y.; Tsai, S.-W. Hydrolytic resolution of (R,S)-3-hydroxy-3-phenylpropionates by esterase from Klebsiella oxytoca: Effects of leaving alcohol, covalent immobilization and aqueous pH. J. Mol. Catal. B: Enzym. 2009, 59, 70–75. [Google Scholar]
- Ali, I.S.; Sudalai, A. Pd-catalyzed kinetic resolution of benzylic alcohols: A practical synthesis of (R)-tomoxetine and (S)-fluoxetine hydrochlorides. Tetrahedron Lett. 2002, 43, 5435–5436. [Google Scholar] [CrossRef]
- Mandal, S.K.; Jensen, D.R. Scope of enantioselective palladium (II)-catalyzed aerobic alcohol oxidations with (−)-sparteine. J. Org. Chem. 2003, 68, 4600–4603. [Google Scholar] [CrossRef]
- Xu, C.; Yuan, C. Candida Rugosa lipase-catalyzed kinetic resolution of β-hydroxy-β-arylpropionates and δ-hydroxy-δ-aryl-β-oxo-pentanoates. Tetrahedron 2005, 61, 2169–2186. [Google Scholar] [CrossRef]
- Brem, J.; Naghi, M.; Toşa, M.-I.; Boros, Z.; Poppe, L.; Irimie, F.-D.; Paizs, C. Lipase mediated sequential resolution of aromatic β-hydroxy esters using fatty acid derivatives. Tetrahedron Asymmetry 2011, 22, 1672–1679. [Google Scholar] [CrossRef]
- Brem, J.; Turcu, M.C.; Paizs, C.; Lundell, K.; Toşa, M.-I.; Irimie, F.-D.; Kanerva, L.T. Immobilization to improve the properties of Pseudomonas fluorescens lipase for the kinetic resolution of 3-aryl-3-hydroxy esters. Process Biochem. 2012, 47, 119–126. [Google Scholar] [CrossRef]
- Choi, E.T.; Lee, M.H.; Kim, Y.; Park, Y.S. Asymmetric dehydration of β-hydroxy esters and application to the syntheses of flavane derivatives. Tetrahedron 2008, 64, 1515–1522. [Google Scholar] [CrossRef]
- Müller, C.E.; Schreiner, P.R. Organocatalytic enantioselective acyl transfer onto racemic as well as meso alcohols, amines, and thiols. Angew. Chem. Int. Ed. 2011, 50, 6012–6042. [Google Scholar]
- Vedejs, E.; Chen, X. Kinetic resolution of secondary alcohols. Enantioselective acylation mediated by a chiral (dimethylamino)pyridine derivative. J. Am. Chem. Soc. 1996, 118, 1809–1810. [Google Scholar]
- Vedejs, E.; Chen, X. Parallel kinetic resolution. J. Am. Chem. Soc. 1997, 119, 2584–2585. [Google Scholar] [CrossRef]
- Ruble, J.C.; Latham, H.A.; Fu, G.C. Effective kinetic resolution of secondary alcohols with a planar-chiral analogue of 4-(dimethylamino)pyridine. Use of the Fe(C5Ph5) group in asymmetric catalysis. J. Am. Chem. Soc. 1997, 119, 1492–1493. [Google Scholar]
- Ruble, J.C.; Tweddell, J.; Fu, G.C. Kinetic resolution of arylalkylcarbinols catalyzed by a planar-chiral derivative of DMAP: A new benchmark for nonenzymatic acylation. J. Org. Chem. 1998, 63, 2794–2795. [Google Scholar] [CrossRef]
- Bellemin-Laponnaz, S.; Tweddell, J.; Ruble, J.C.; Breitling, F.M.; Fu, G.C. The kinetic resolution of allylic alcohols by a non-enzymatic acylation catalyst; application to natural product synthesis. Chem. Commun. 2000, 1009–1010. [Google Scholar]
- Tao, B.; Ruble, J.C.; Hoic, D.A.; Fu, G.C. Nonenzymatic kinetic resolution of propargylic alcohols by a planar-chiral DMAP derivative: Crystallographic characterization of the acylated catalyst. J. Am. Chem. Soc. 1999, 121, 5091–5092. [Google Scholar] [CrossRef]
- Lee, S.Y.; Murphy, J.M.; Ukai, A.; Fu, G.C. Nonenzymatic dynamic kinetic resolution of secondary alcohols via enantioselective acylation: synthetic and mechanistic studies. J. Am. Chem. Soc. 2012, 134, 15149–15153. [Google Scholar] [CrossRef]
- Díaz-Álvarez, A.E.; Mesas-Sánchez, L.; Dinér, P. Non-enzymatic dynamic kinetic resolution of secondary aryl alcohols: Planar chiral ferrocene and ruthenium catalysts in cooperation. Angew. Chem. Int. Ed. 2013, 52, 502–504. [Google Scholar]
- Mesas-Sánchez, L.; Díaz-Álvarez, A.E.; Dinér, P. Non-enzymatic kinetic resolution of 1,2-azidoalcohols using a planar-chiral DMAP derivative catalyst. Tetrahedron 2013, 69, 753–757. [Google Scholar]
- Mesas-Sánchez, L.; Díaz-Álvarez, A.E.; Koukal, P.; Dinér, P. Kinetic resolution of 2-hydroxy-2-aryl-ethylphosphonates by a non-enzymatic acylation catalyst. Tetrahedron 2014, 70, 3807–3811. [Google Scholar]
- Kawabata, T.; Nagato, M.; Takasu, K.; Fuji, K. Nonenzymatic kinetic resolution of racemic alcohols through an “induced fit” process. J. Am. Chem. Soc. 1997, 119, 3169–3170. [Google Scholar] [CrossRef]
- Wei, Y.; Held, I.; Zipse, H. Stacking interactions as the principal design element in acyl-transfer catalysts. Org. Biomol. Chem. 2006, 4, 4223–4230. [Google Scholar] [CrossRef]
- Li, X.; Liu, P.; Houk, K.N.; Birman, V.B. Origin of enantioselectivity in CF3−PIP-catalyzed kinetic resolution of secondary benzylic alcohols. J. Am. Chem. Soc. 2008, 130, 13836–13837. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Chisholm, C.D. Assignment of absolute configuration using chiral reagents and NMR spectroscopy. Chirality 2011, 23, 190–214. [Google Scholar] [CrossRef]
- Dale, J.A.; Mosher, H.S. Nuclear magnetic resonance enantiomer regents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and α-methoxy-α-trifluoromethylphenylacetate (MTPA) esters. J. Am. Chem. Soc. 1973, 95, 512–519. [Google Scholar]
- Baskar, B.; Pandian, N.G.; Priya, K.; Chadha, A. Asymmetric reduction of alkyl 2-oxo-4-arylbutanoates and -but-3-enoates by Candida parapsilosis ATCC 7330: Assignment of the absolute configuration of ethyl 2-hydroxy-4-(p-methylphenyl)but-3-enoate by 1H-NMR. Tetrahedron Asymmetry 2004, 15, 3961–3966. [Google Scholar] [CrossRef]
- Padhi, S.K.; Chadha, A. Deracemization of aromatic β-hydroxy esters using immobilized whole cells of Candida parapsilosis ATCC 7330 and determination of absolute configuration by 1H-NMR. Tetrahedron Asymmetry 2005, 16, 2790–2798. [Google Scholar] [CrossRef]
- Padhi, S.K.; Titu, D.; Pandian, N.G.; Chadha, A. Deracemisation of β-hydroxy esters using immobilised whole cells of Candida parapsilosis ATCC 7330: Substrate specificity and mechanistic investigation. Tetrahedron 2006, 62, 5133–5140. [Google Scholar] [CrossRef]
- Silva, F.A.; Gouverneur, V. Elongation of β-hydroxyenones by cross-metathesis. Tetrahedron Lett. 2005, 46, 8705–8709. [Google Scholar] [CrossRef]
- Uccello-Barretta, G.; Pini, D.; Mastantuono, A.; Salvadori, P. Direct NMR assay of enantiomeric purity of chiral β-hydroxy esters by using quinine as chiral solvating agent. Tetrahedron Asymmetry 1995, 6, 1965–1972. [Google Scholar]
- Iwasaki, G.; Saeki, S.; Hamana, M. A novel nucleophilic substitution of the formyl group in p-nitrobenzaldehyde with some carbanions. Chem. Lett. 1986, 173–176. [Google Scholar] [CrossRef]
- Sgreccia, L.; Bandini, M.; Morganti, S.; Quintavalla, A.; Umani-Ronchi, A.; Cozzi, P.G. Titanium-catalyzed Reformatsky-type reaction. J. Organomet. Chem. 2007, 692, 3191–3197. [Google Scholar] [CrossRef]
- Pini, D.; Mastantuosno, A.; Salvadori, P. New chiral ligand for optically active β-hydroxy esters synthesis by enantioselective Reformatsky reactions. Tetrahedron Asymmetry 1994, 5, 1875–1876. [Google Scholar] [CrossRef]
- Shiomi, T.; Adachi, T.; Toribatake, K.; Zhou, L.; Nishiyama, H. Asymmetric β-boration of α,β-unsaturated carbonyl compounds promoted by chiral rhodium-bisoxazolinylphenyl catalysts. Chem. Commun. 2009, 5987–5989. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Díazlvarez, A.E.; Mesas-Sánchez, L.; Dinér, P. Access to Optically Pure β-Hydroxy Esters via Non-Enzymatic Kinetic Resolution by a Planar-Chiral DMAP Catalyst. Molecules 2014, 19, 14273-14291. https://doi.org/10.3390/molecules190914273
Díazlvarez AE, Mesas-Sánchez L, Dinér P. Access to Optically Pure β-Hydroxy Esters via Non-Enzymatic Kinetic Resolution by a Planar-Chiral DMAP Catalyst. Molecules. 2014; 19(9):14273-14291. https://doi.org/10.3390/molecules190914273
Chicago/Turabian StyleDíazlvarez, Alba E., Laura Mesas-Sánchez, and Peter Dinér. 2014. "Access to Optically Pure β-Hydroxy Esters via Non-Enzymatic Kinetic Resolution by a Planar-Chiral DMAP Catalyst" Molecules 19, no. 9: 14273-14291. https://doi.org/10.3390/molecules190914273
APA StyleDíazlvarez, A. E., Mesas-Sánchez, L., & Dinér, P. (2014). Access to Optically Pure β-Hydroxy Esters via Non-Enzymatic Kinetic Resolution by a Planar-Chiral DMAP Catalyst. Molecules, 19(9), 14273-14291. https://doi.org/10.3390/molecules190914273