Conjugates of 1'-Aminoferrocene-1-carboxylic Acid and Proline: Synthesis, Conformational Analysis and Biological Evaluation



 ,
,
Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Hydrogen Bonding Patterns * | |||
|---|---|---|---|---|
| III | 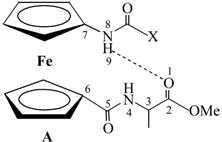 | 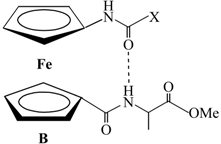 | ||
| IV | 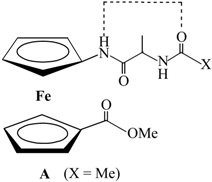 | 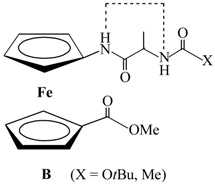 |  | |
2. Results and Discussion
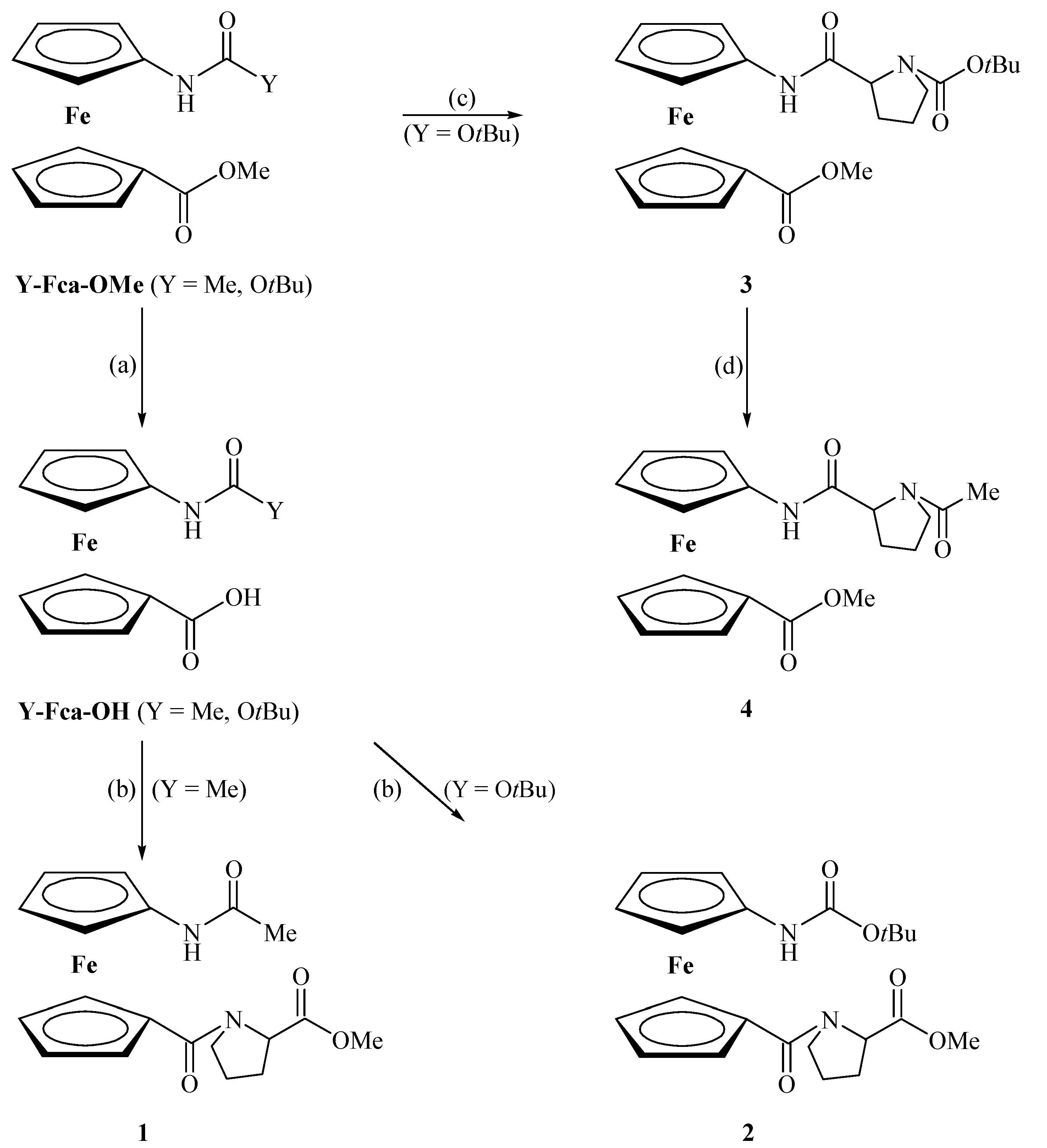
2.1. Chemistry
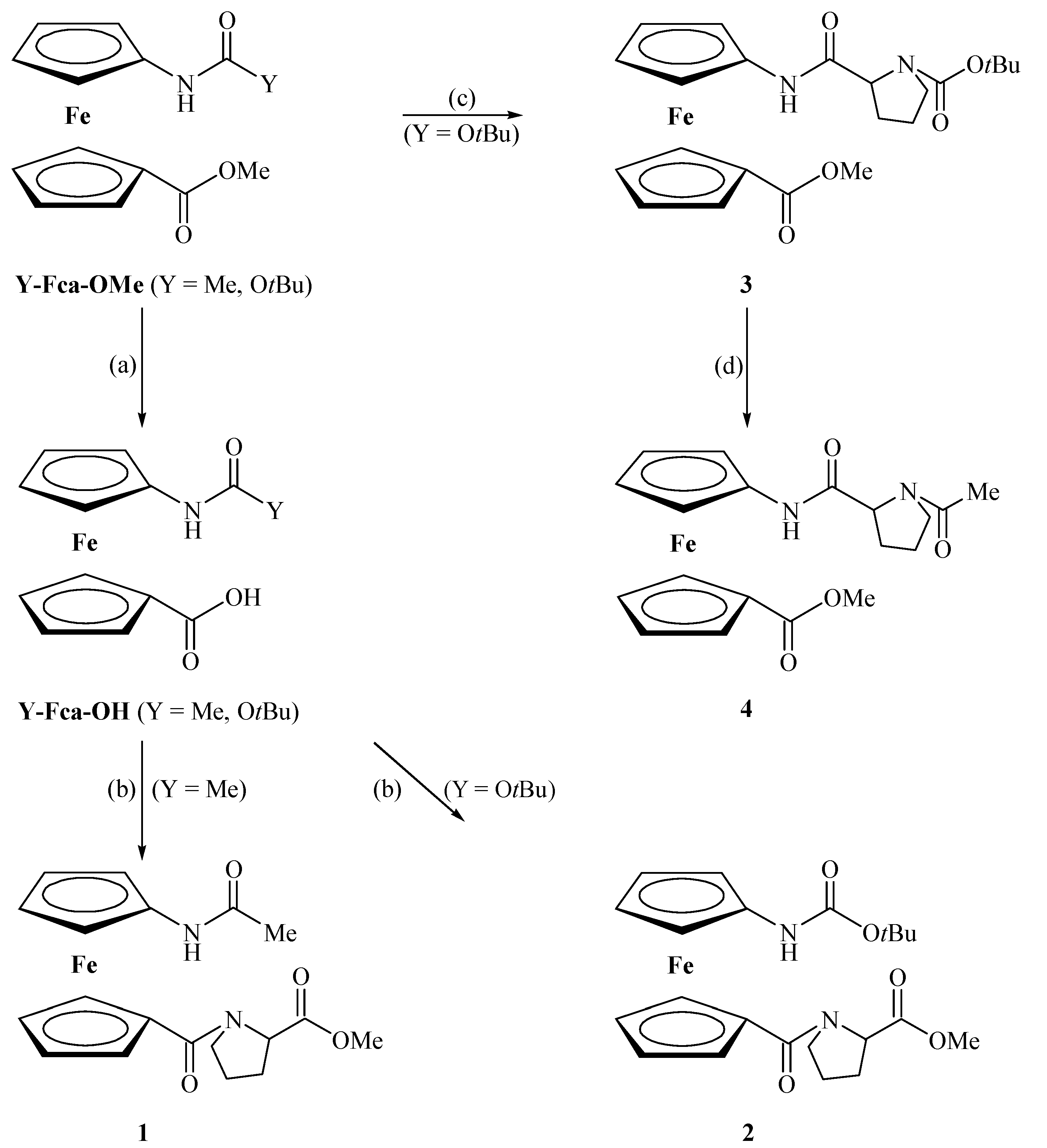
2.2. Conformational Analysis of 1–4 in Solution
| Compound | Formula | νNH (Free) | νNH (Assoc.) | νCO (Ester) | νCO (Amide I) | νCO (Amide II) |
|---|---|---|---|---|---|---|
| IIIa | Ac-Fca-Ala-OMe | 3434 m | 3351 w | 1739 s | 1682 m | 1514 m |
| 1657 m | ||||||
| IIIb | Boc-Fca-Ala-OMe | 3433 m | 3327 m | 1731 s | 1714 s | 1518 m |
| 1655 m | ||||||
| IVa | Boc-Ala-Fca-OMe | 3423 m | 3324 w | 1709 brs | 1557 m | 1504 m |
| 1536 m | ||||||
| IVb | Ac-Ala-Fca-OMe | 3425 m | 3290 w | 1709 s | 1664 m | 1507 m |
| 1562 m | ||||||
| 1538 m | ||||||
| VII | Boc-Phe-Pro-OMe | ~3450 m | [c] | [c] | [c] | |
| VIII | Boc-Pro-Gly-OMe | ~3430 m | ~3310 w | [c] | [c] | [c] |
| IX | Ac-Pro-Gly-OMe | ~3430 m | ~3300 m | [c] | [c] | [c] |
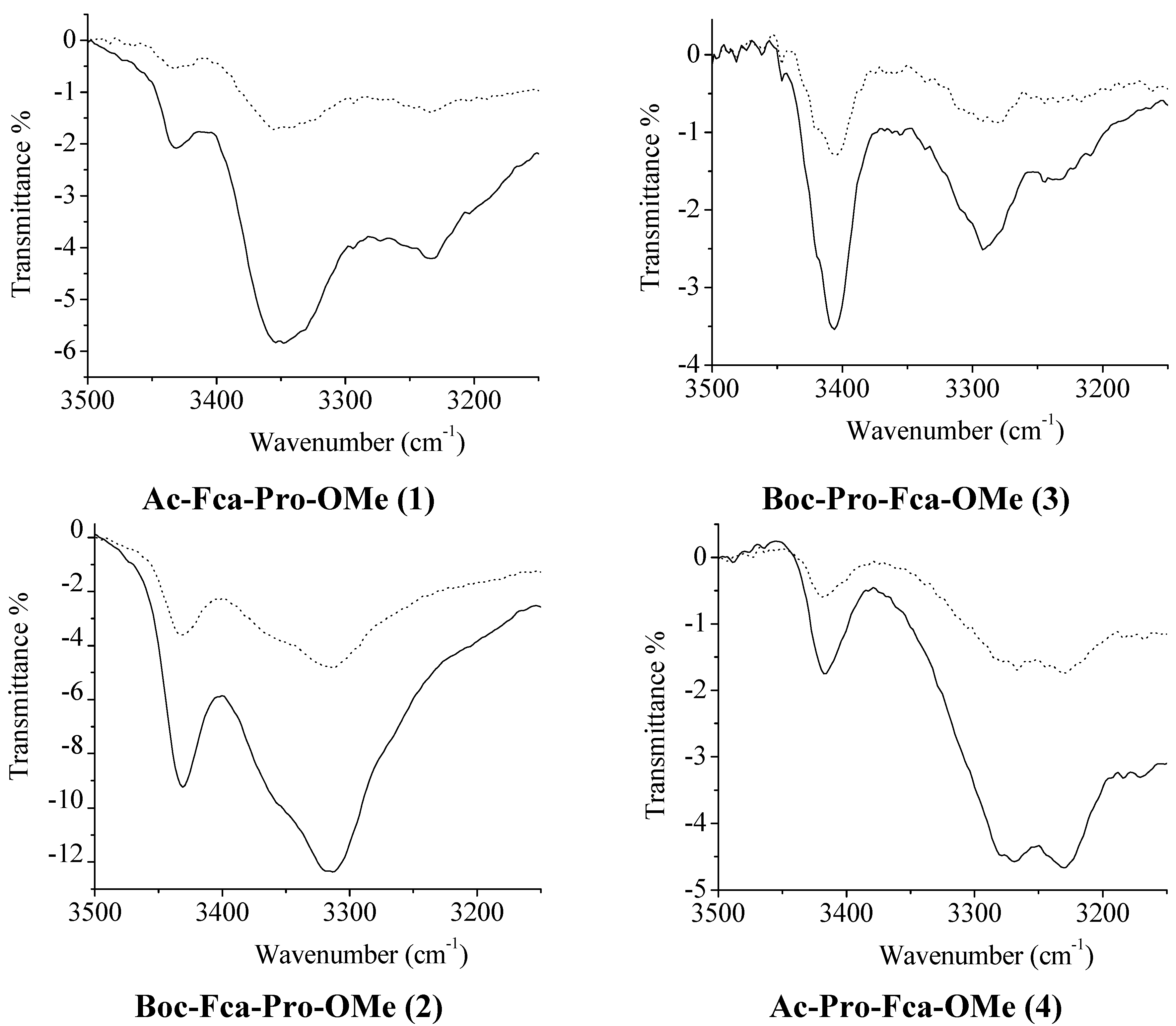
2.2.1. IR Spectroscopy
| Compound | Formula | νNH (Free) | νNH (Assoc.) | νCO (Ester) | νCO (Amide I) | νCO (Amide II) |
|---|---|---|---|---|---|---|
| X | Fc-COOMe | 1711 | ||||
| XI | Fc-NHAc | 3436 m | 1684 | |||
| XII | Fc-NHBoc | 3436 m | 1723 | |||
| 1 | Ac-Fca-Pro-OMe | 3434 w | 3351 m 3231 m | 1741 | 1676 1604 | 1535 |
| 2 | Boc-Fca-Pro-OMe | 3430 m | 3356 sh 3315 m | 1741 sh | 1718 1604 | 1532 |
| 3 | Boc-Pro-Fca-OMe | 3419 sh 3405 m | 3290 m 3234 w | 1706 | 1694 1655 | 1532 |
| 4 | Ac-Pro-Fca-OMe | 3416 w | 3272 m 3229 m | 1709 | 1690 1623 | 1563 |

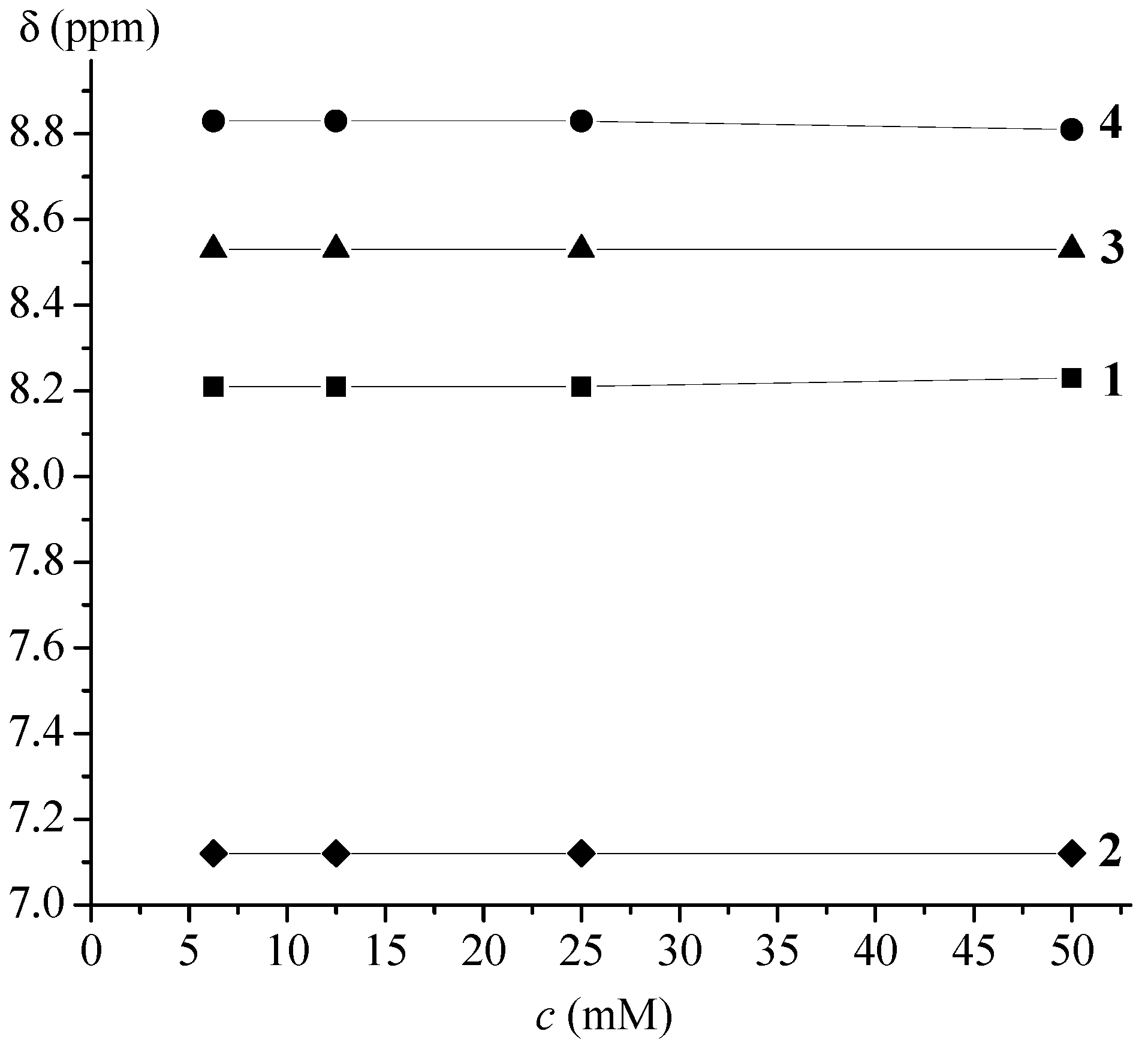
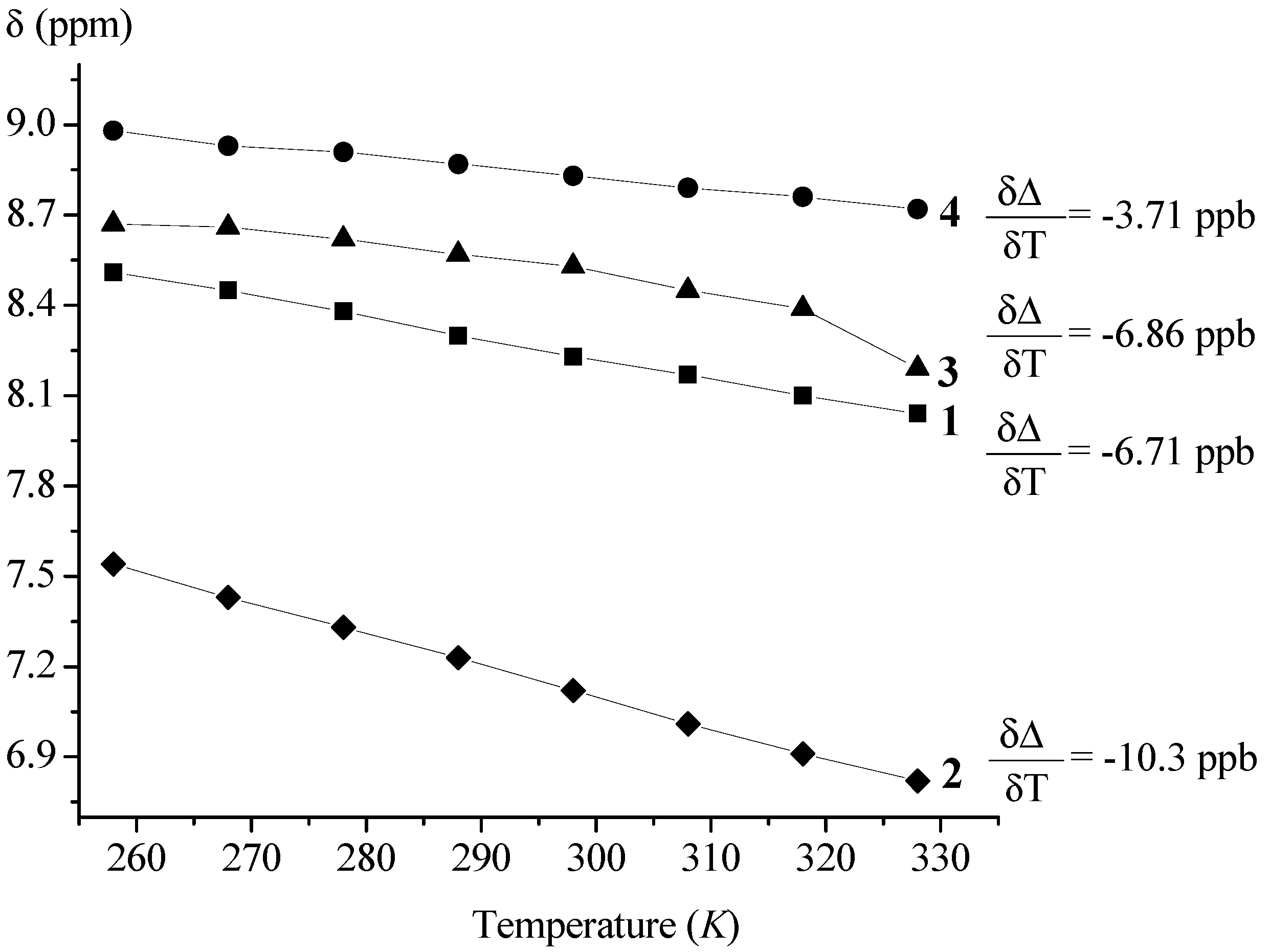
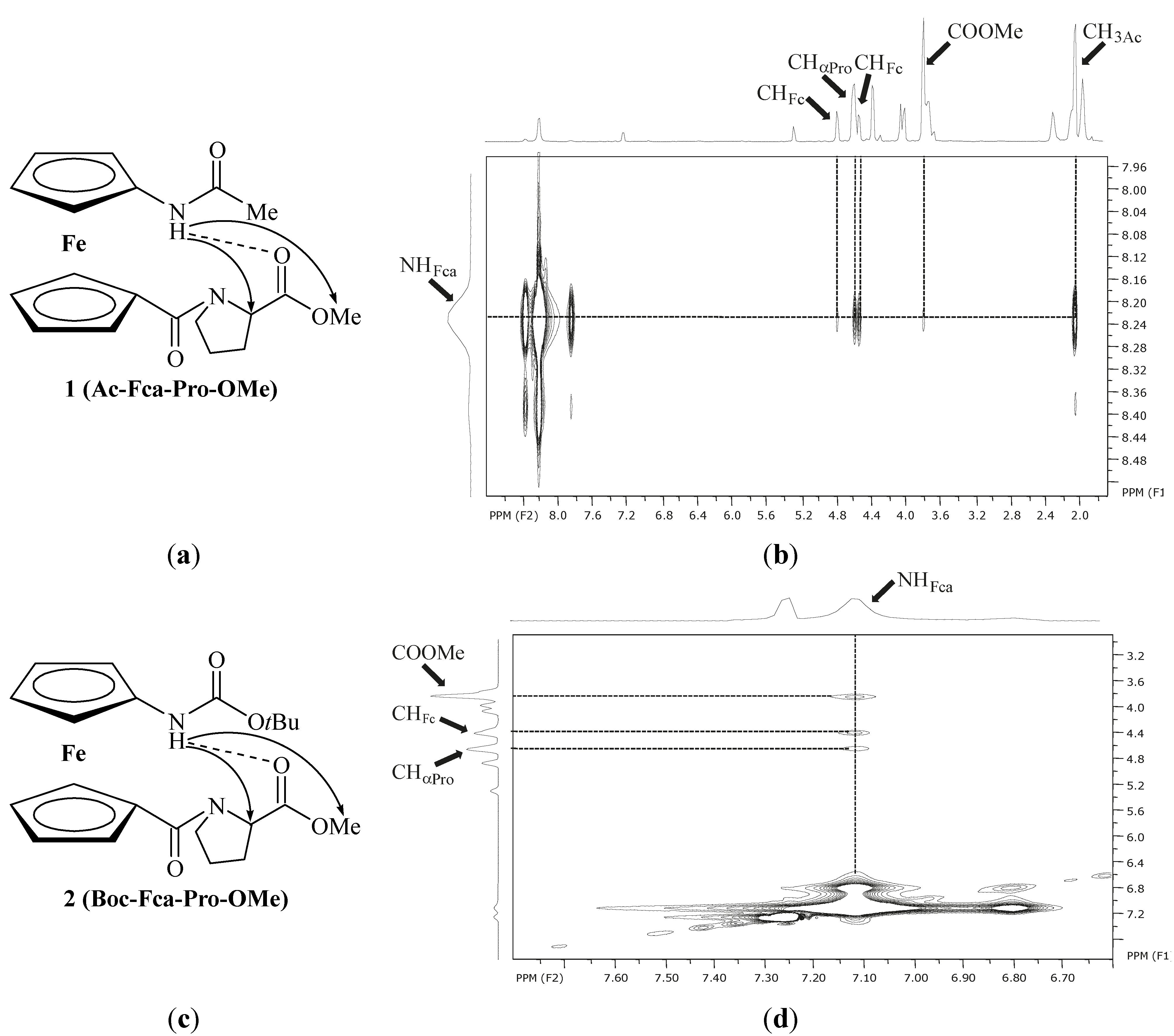
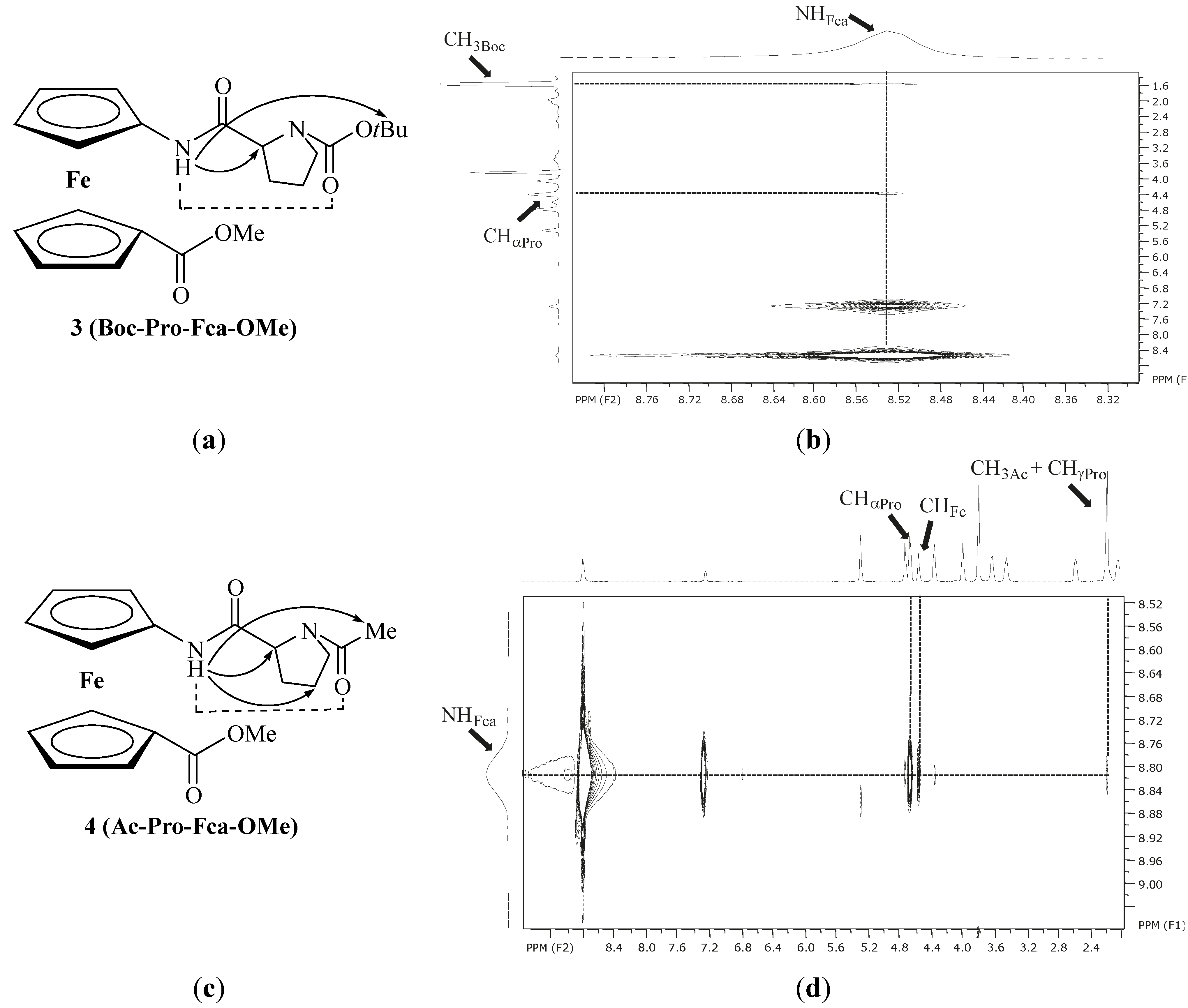
2.2.2. NMR Spectroscopy
| Compound | Formula | δ/ppm (NHAA) [b] | δ/ppm (NHFca/Fc) | IHB Pattern | IHB Ring Size |
|---|---|---|---|---|---|
| IIIa | Ac-Fca-Ala-OMe | 6.71 | 7.93 | NHFca···OCAla | 9-membered |
| NHAla···OCAc | 8-membered | ||||
| IIIb | Boc-Fca-Ala-OMe | 6.77 | 6.40 | NHFca···OCAla | 9-membered |
| NHAla···OCBoc | 8-membered | ||||
| IVa | Boc-Ala-Fca-OMe | 5.14 | 7.64 | NHFca···NAla | 5-membered |
| NHAla···OCFca | 9-membered | ||||
| IVb | Ac-Ala-Fca-OMe | 6.41 | 8.04 | NHFca···OCAc | 7-membered |
| NHFca···NAla | 5-membered | ||||
| NHAla···OCFca | 9-membered | ||||
| VII | Boc-Phe-Pro-OMe | [c] | [d] | ||
| VIII | Ac-Pro-Gly-OMe | 7.25(t) | NHGly···OCAc | 7-membered | |
| 6.23(c) | |||||
| IX | Boc-Pro-Gly-OMe | 7.28(t) | NHGly···OCBoc | 7-membered | |
| 6.49(c) | |||||
| XI | Fc-NHAc | 6.49 | [d] | ||
| XII | Fc-NHBoc | 5.55 | [d] | ||
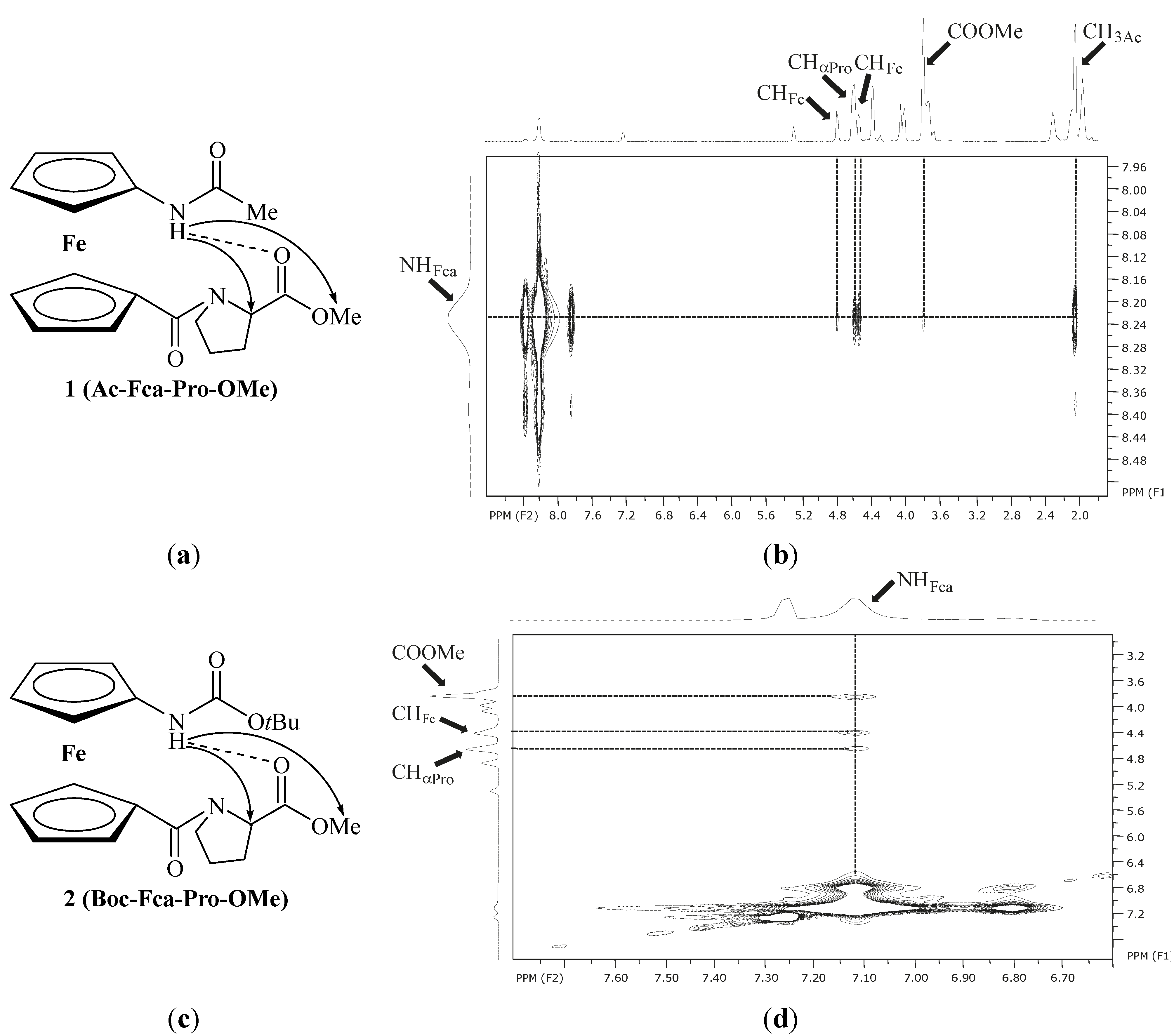
| 1 | Ac-Fca-Pro-OMe [e] | 8.23(t) | NHFca···OCPro | 9-membered | |
| 8.38(c) | |||||
| 2 | Boc-Fca-Pro-OMe [e] | 7.12(t) | NHFca···OCPro | 9-membered | |
| 6.80(c) | |||||
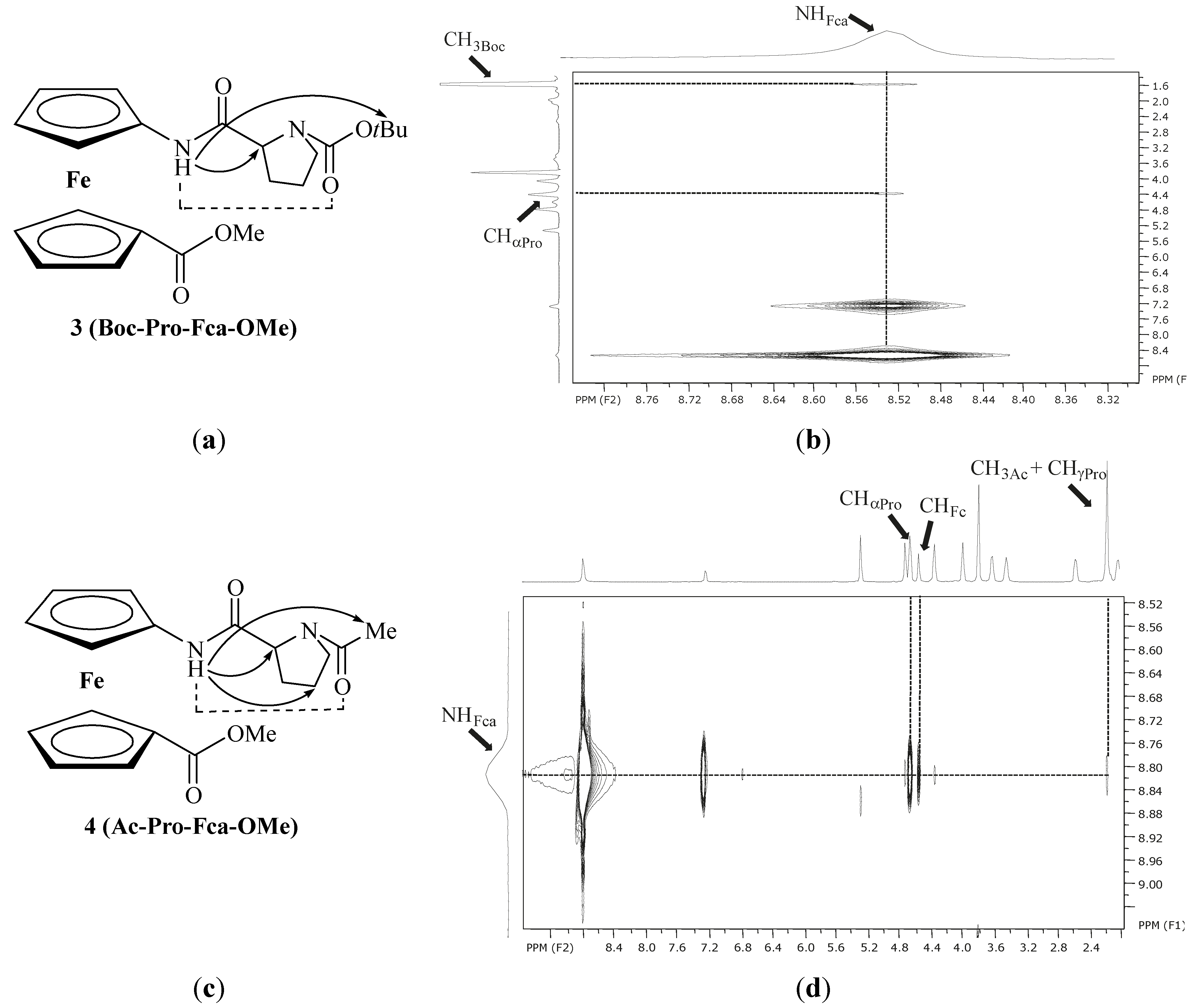
| 3 | Boc-Pro-Fca-OMe [e] | 8.78(t) | NHFca···OCPro | 7-membered | |
| 7.53(c) | |||||
| 4 | Ac-Pro-Fca-OMe [e] | 8.81(t) | NHFca···OCPro | 7-membered | |
| 7.29(c) |
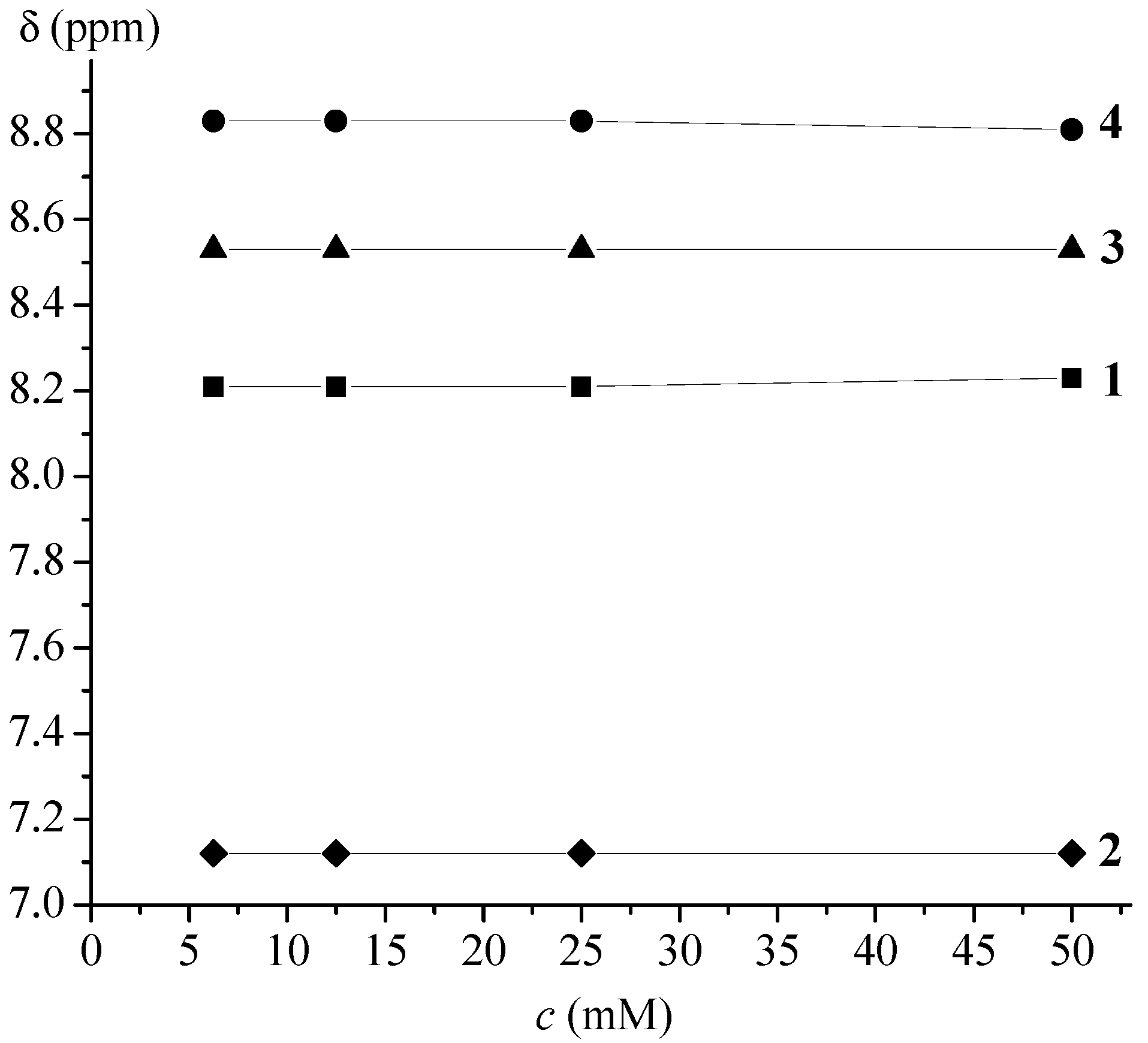
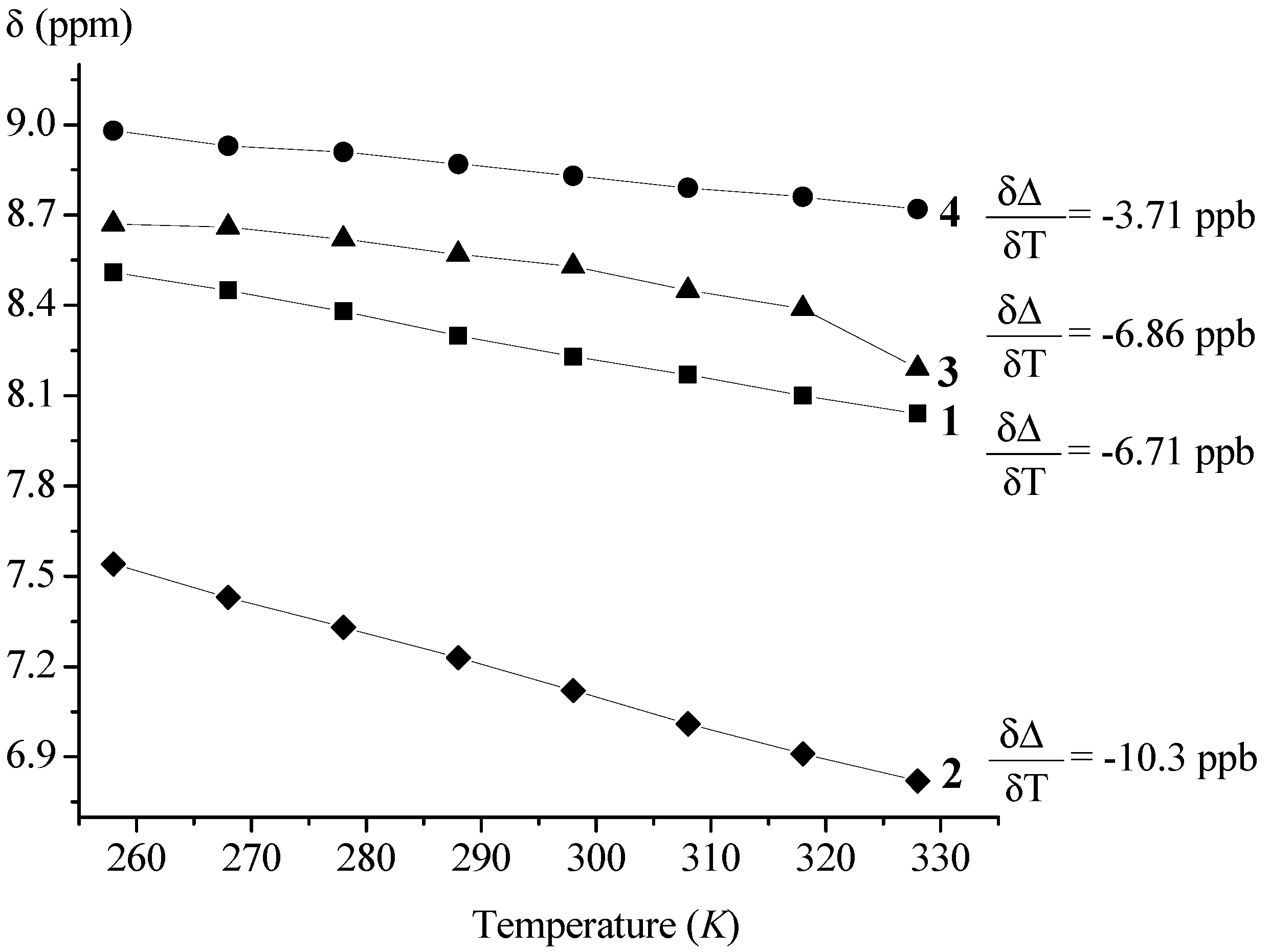
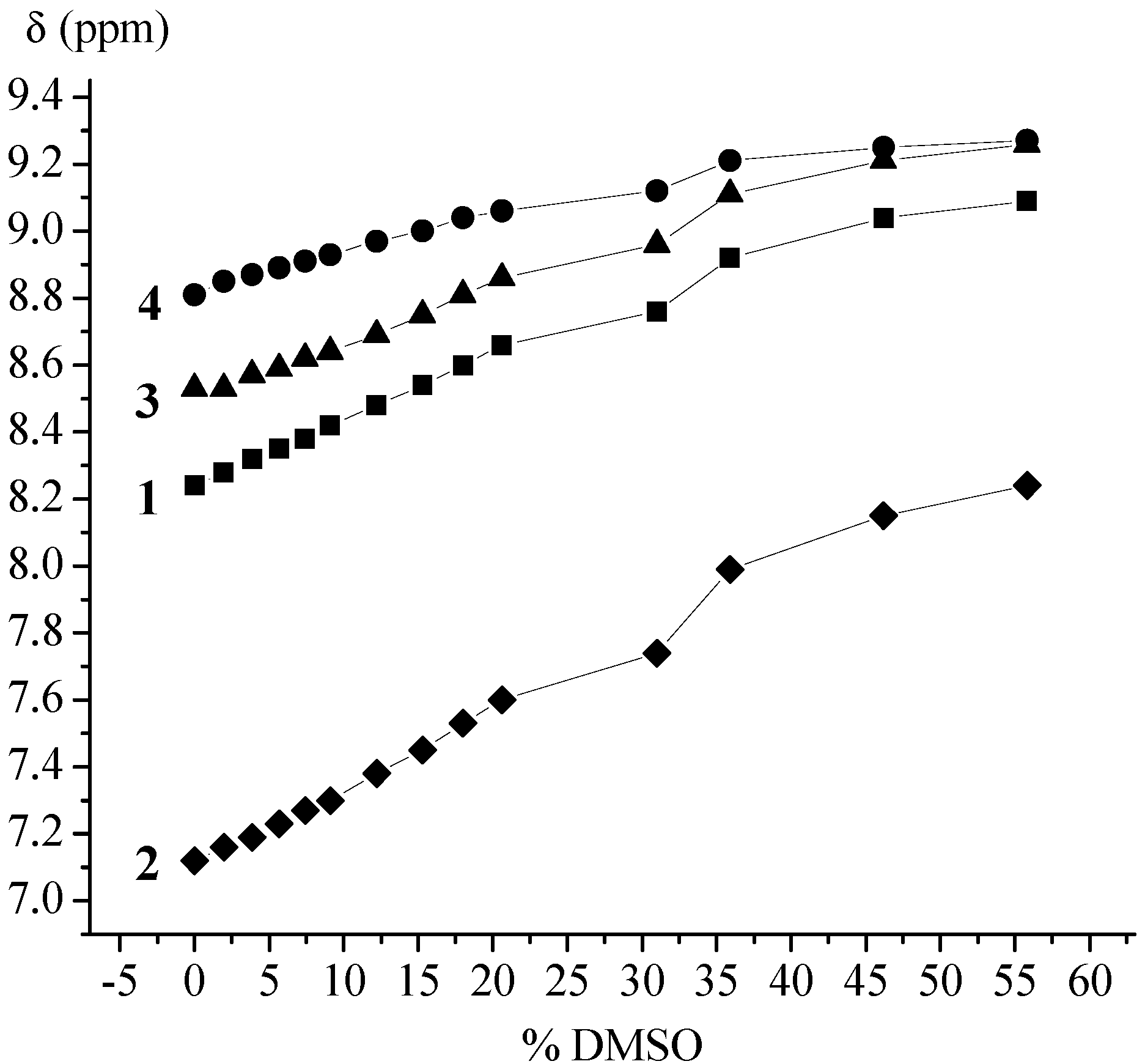
| Ac-Fca-Pro-OMe (1) | T (K) [a] | DMSO (%) [b] | Boc-Fca-Pro-OMe (2) | T (K) [a] | DMSO (%) [b] | ||||||
| 228 | 328 | 0 | 36 | 228 | 328 | 0 | 36 | ||||
| δ (ppm) | cis | 8.67 | 8.04 | 8.39 | 9.00 | δ (ppm) | cis | 7.14 | 6.83 | 6.80 | 7.99 |
| trans | 8.51 | 8.23 | 8.92 | trans | 7.57 | 7.13 | |||||
| trans:cis | 90:10 | 91:9 | 90:10 | trans:cis | 90:10 | 93:7 | |||||
| Boc-Pro-Fca-OMe (3) | T (K) | DMSO (%) [a] | Ac-Pro-Fca-OMe (4) | T (K) | DMSO (%) [a] | ||||||
| 228 | 328 | 0 | 36 | 228 | 328 | 0 | 36 | ||||
| δ (ppm) | cis | 7.38 | 8.21 | 8.53 | 9.00 | δ (ppm) | cis | 7.63 | 8.72 | 7.29 | 9.41 |
| trans | 8.67 | 9.10 | trans | 8.98 | 8.81 | 9.21 | |||||
| trans:cis | 66:33 | 56:44 | trans:cis | 94:6 | 93:7 | 74:26 | |||||
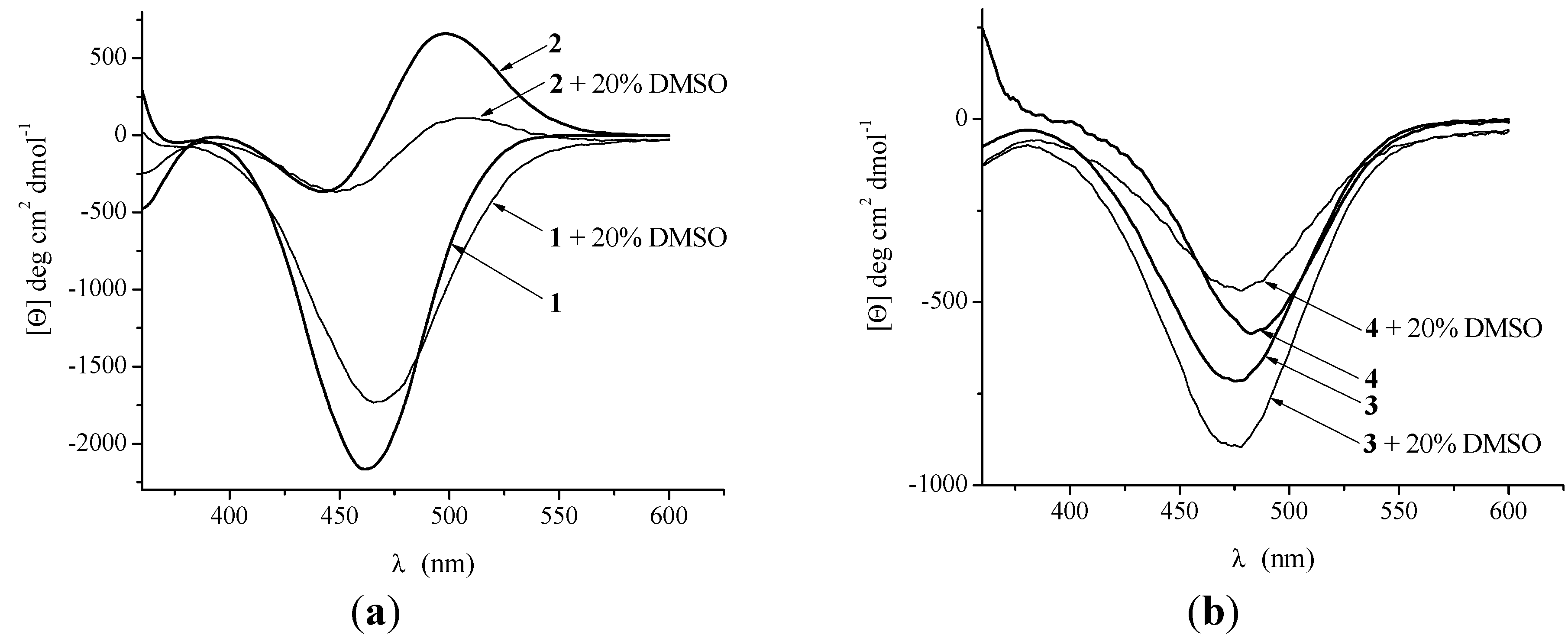
2.2.3. CD Spectroscopy
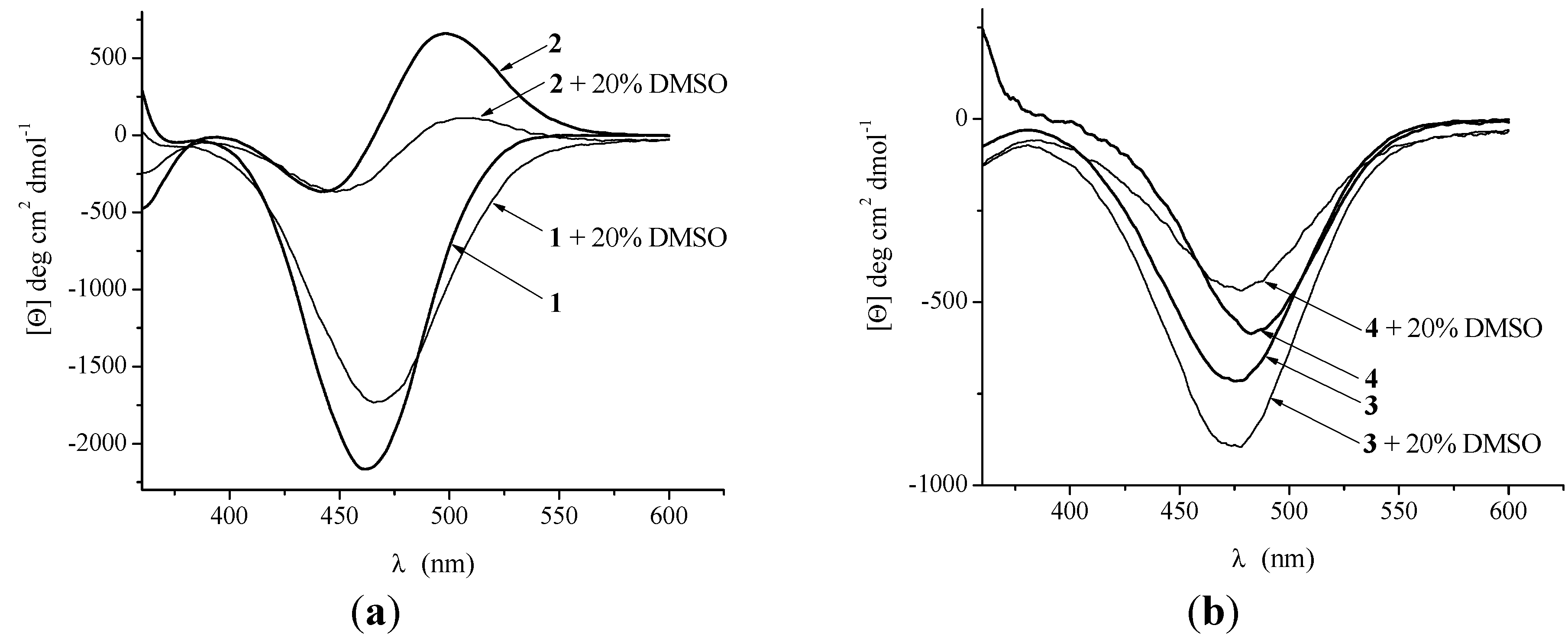
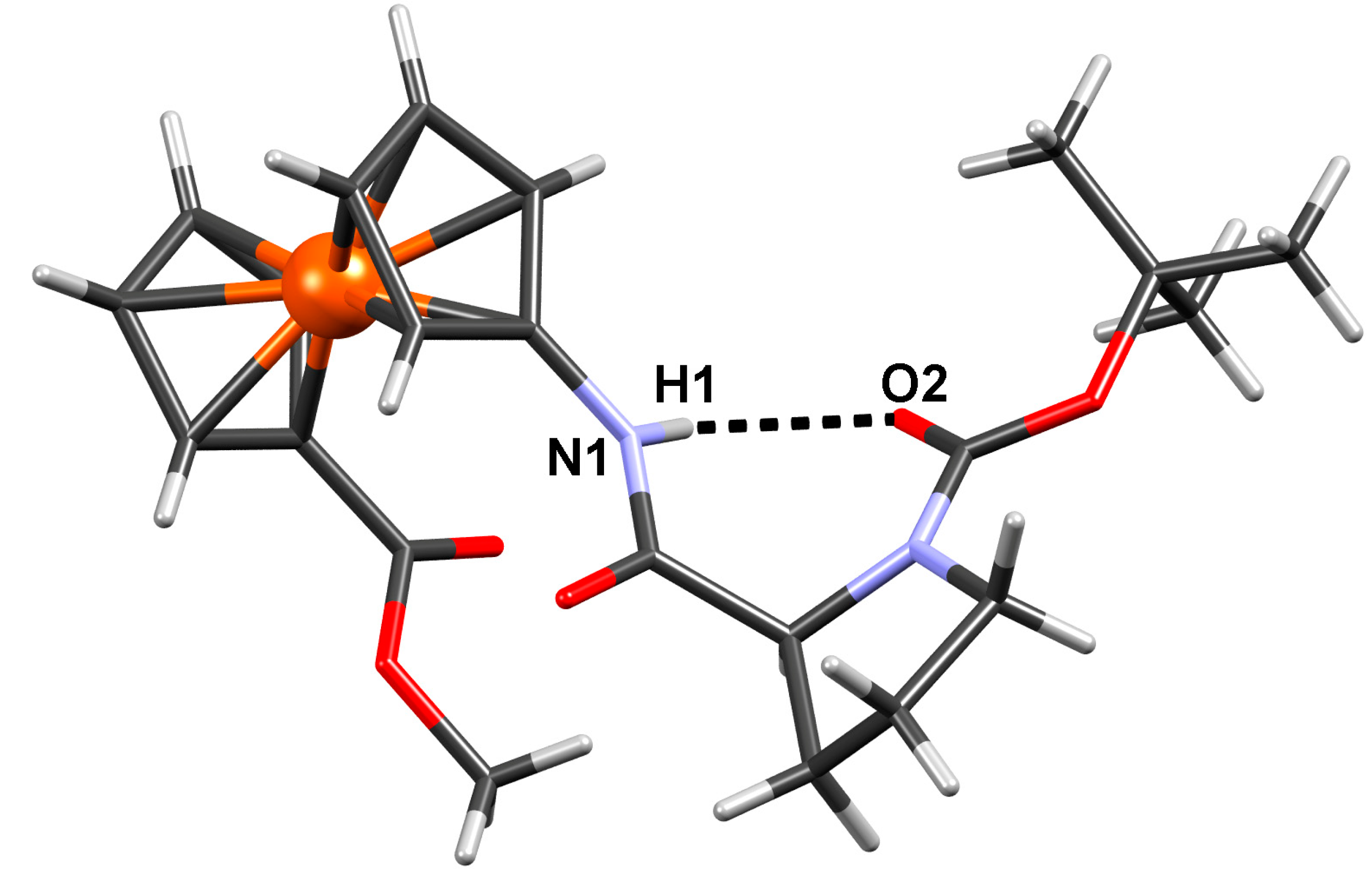
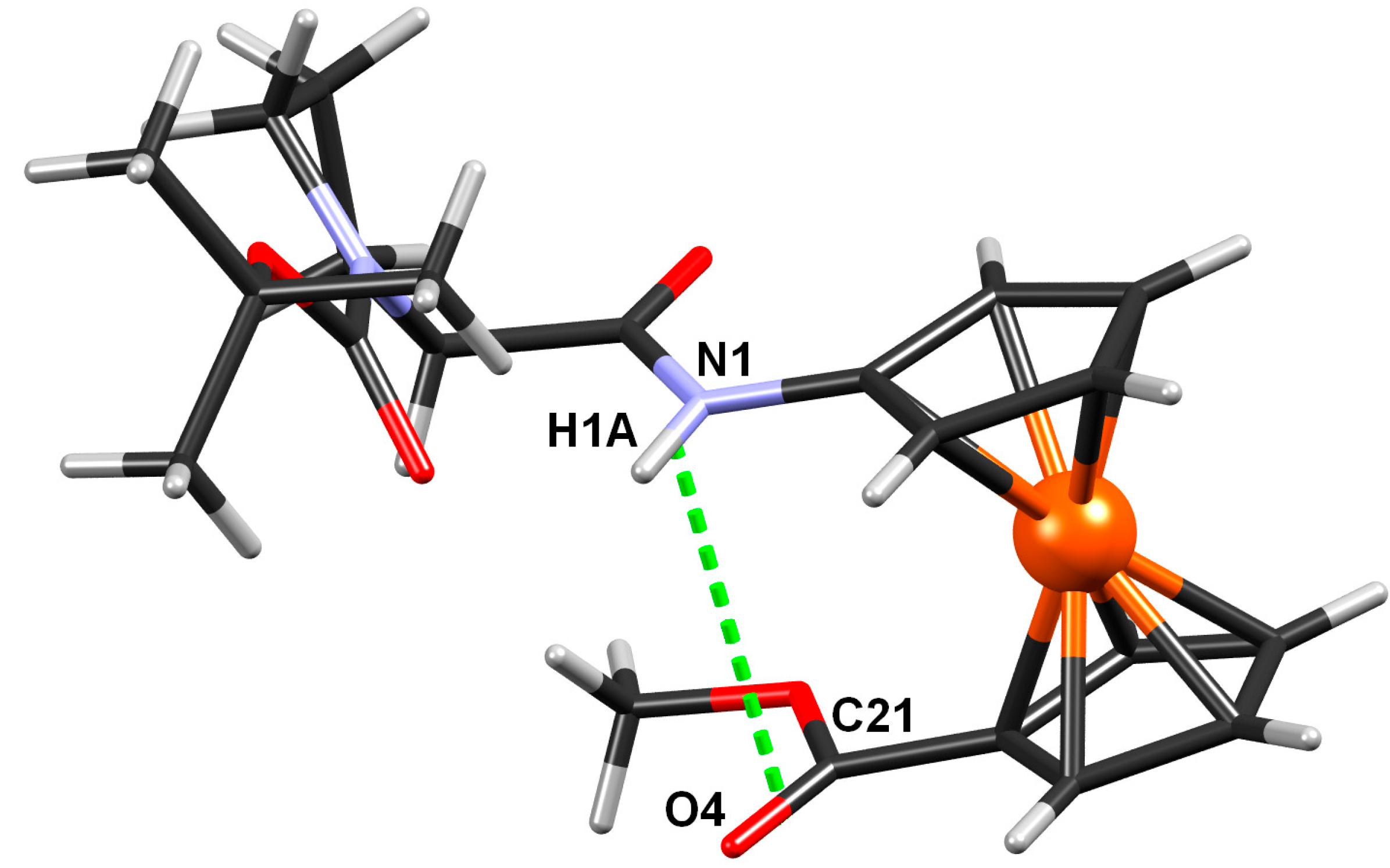
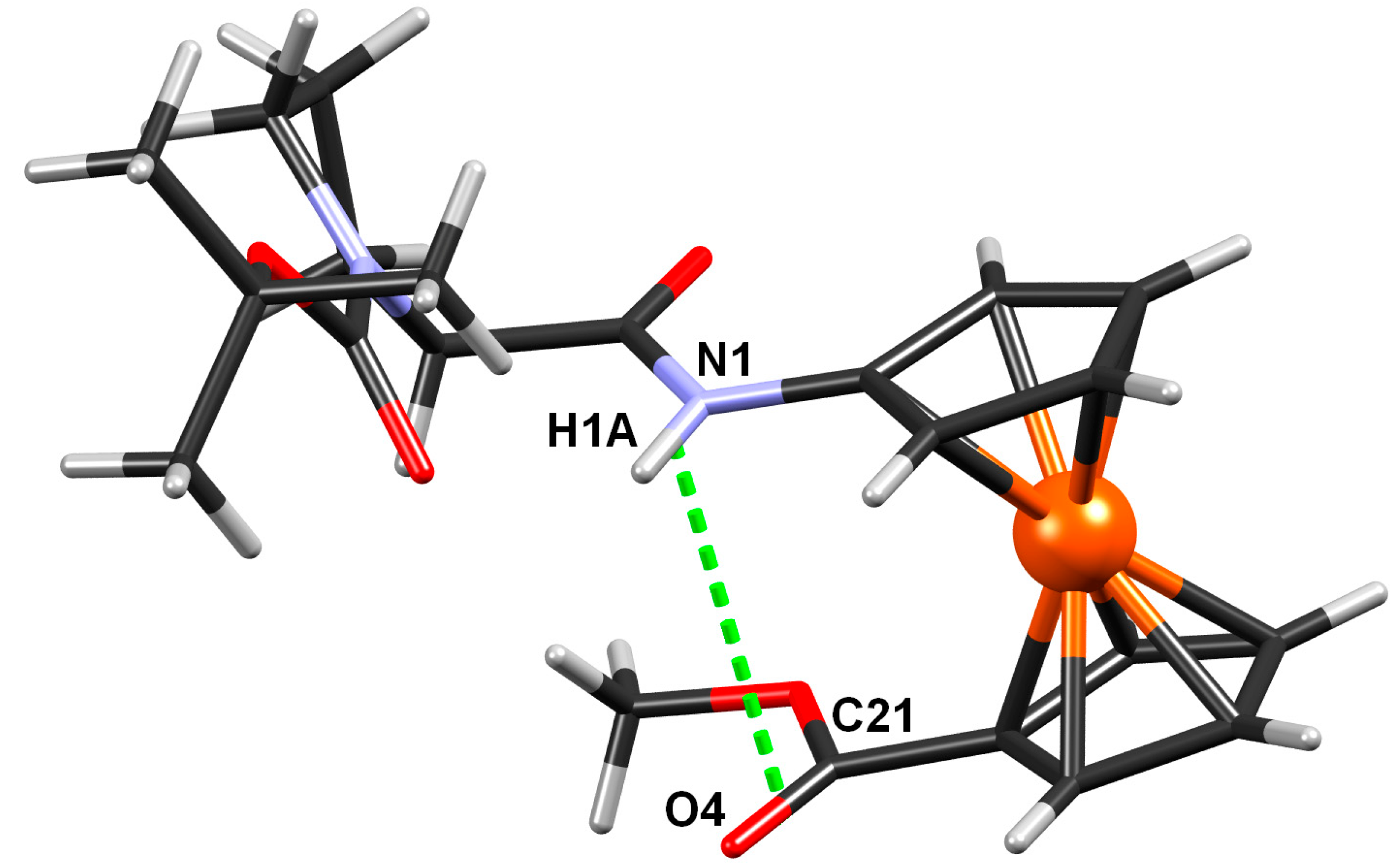
2.2.4. Crystal Structure of Boc-Pro-Fca-OMe (3)
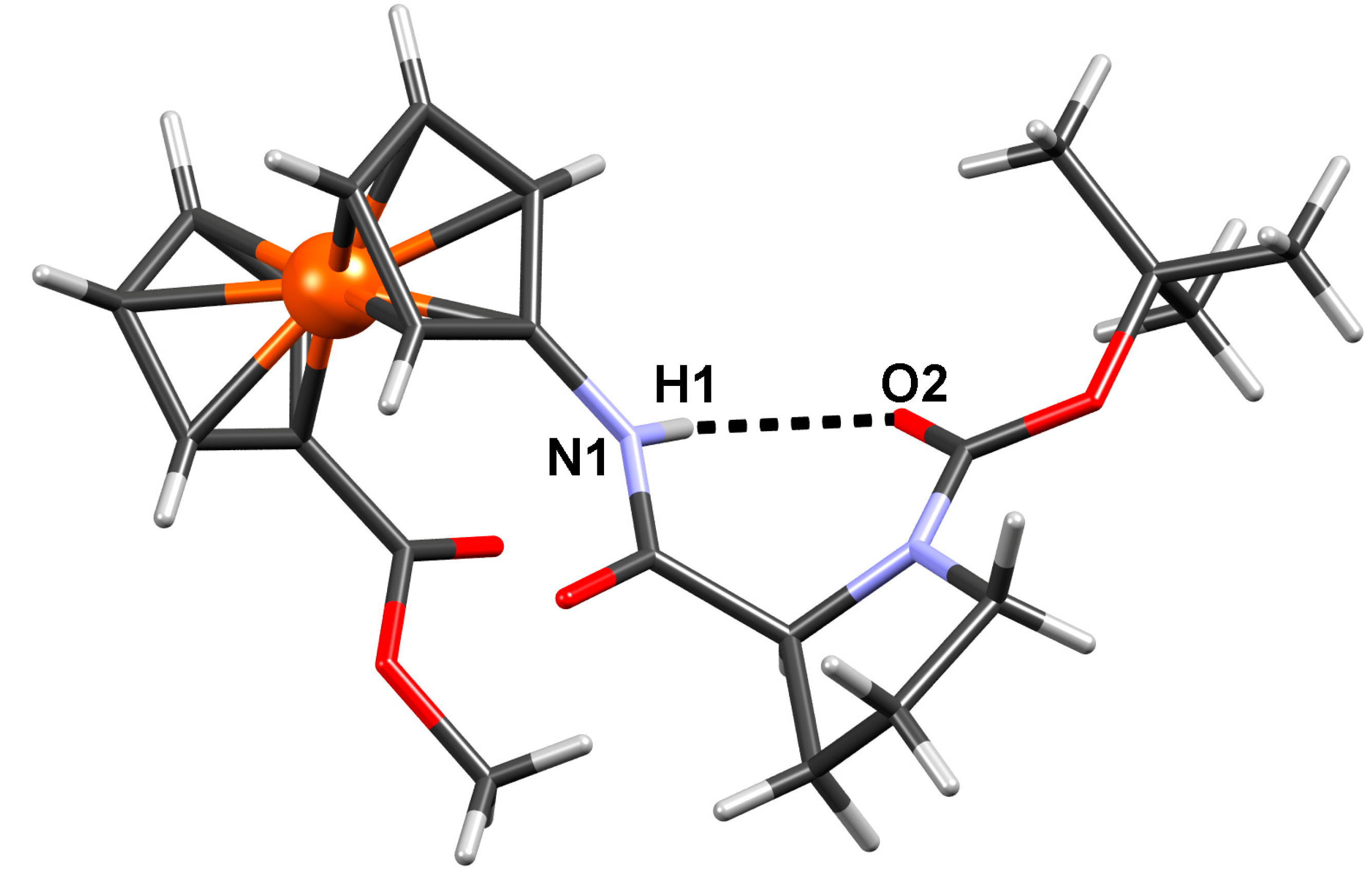
| D–H···A | D–H/Å | H··· A/Å | D···A/Å | D–H···A/° | Symm. op. on A |
|---|---|---|---|---|---|
| N1–H1···O2 | 0.86 | 2.10 | 2.810(10) | 139 | x, y, z |
| C2–H2···O1 | 0.93 | 2.46 | 2.906(12) | 110 | x, y, z |
| C8–H8B···O1 | 0.97 | 2.47 | 2.827(14) | 101 | x, y, z |
| C13–H13C···O2 | 0.96 | 2.37 | 2.969(14) | 120 | x, y, z |
| C15–H15C···O2 | 0.96 | 2.45 | 3.031(16) | 119 | x, y, z |
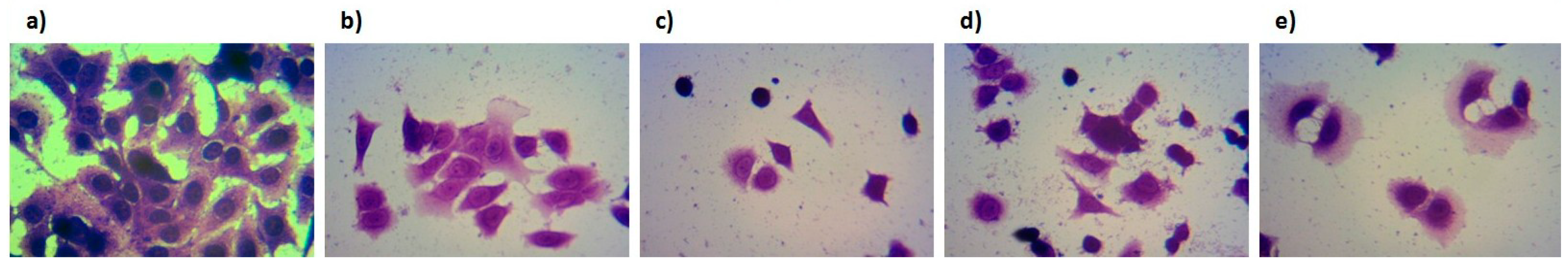
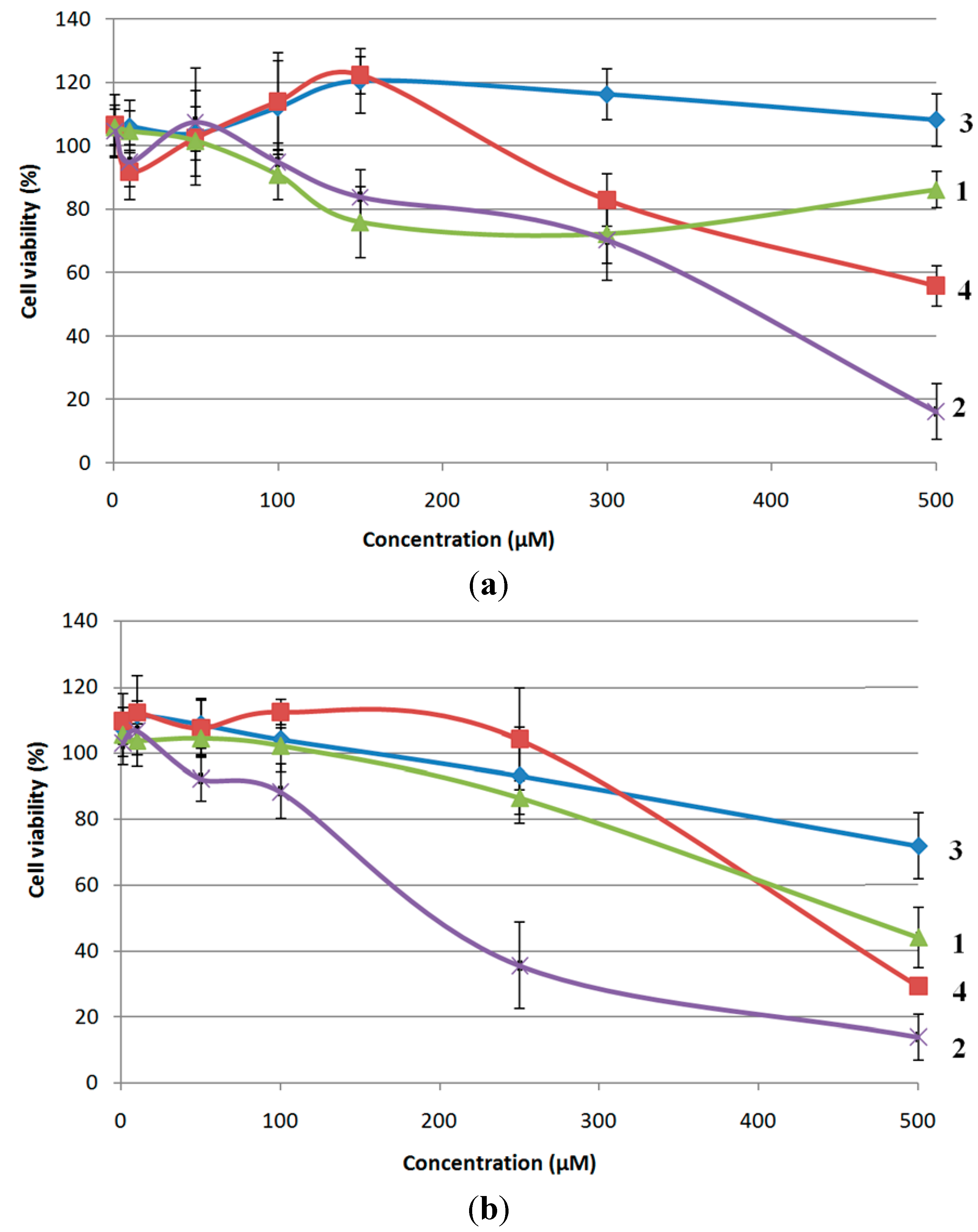
2.3. Biological Evaluation of Bioconjugates 1–4
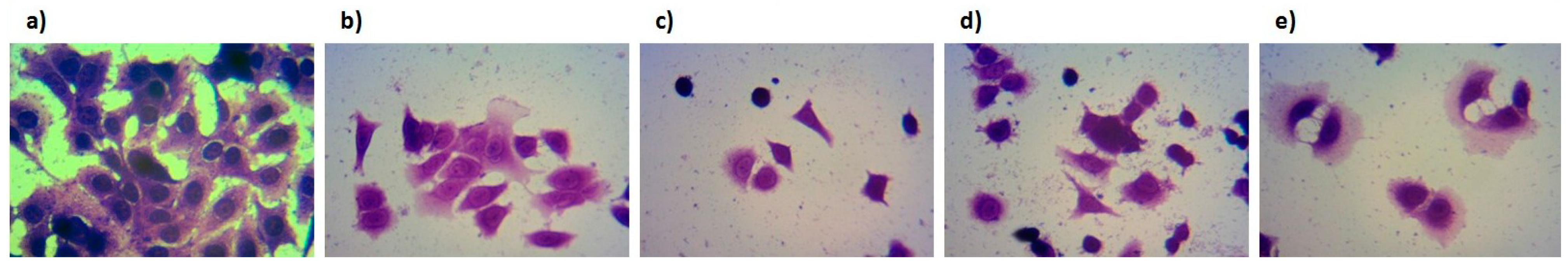
3. Experimental Section
3.1. General Information
3.2. General Procedure for the Preparation of Compounds 1 and 2
3.3. General Procedure for the Preparation of Compounds 3 and 4
3.4. Crystallography
| Compound | Boc-Pro-Fca-OMe(3) |
|---|---|
| Empirical formula | C22H28FeN2O5 |
| Formula wt./g·mol−1 | 456.31 |
| Crystal dimensions/mm | 0.08 × 0.06 × 0.03 |
| Space group | P 21 |
| a/Å | 10.951(5) |
| b/Å | 8.571(5) |
| c/Å | 11.944(5) |
| α/° | 90 |
| β/° | 107.315(5) |
| γ/° | 90 |
| Z | 2 |
| V/Å3 | 1070.3(9) |
| Dcalc/g cm−3 | 1.416 |
| µ/mm−1 | 5.954 |
| Θ range/° | 3.88–75.86 |
| T/K | 293(2) |
| Radiation vawelength | 1.54179 (Cu Kα) |
| Diffractometer type | Xcalibur Nova |
| Range of h, k, l | −13 < h < 13; −10 < k < 10; −11 < l < 14 |
| Reflections collected | 5054 |
| Independent reflections | 3430 |
| Observed reflections ( I ≥ 2σ) | 3108 |
| Absorption correction | Multi-scan |
| Rint | 0.0507 |
| R (F) | 0.1024 |
| Rw(F2) | 0.2806 |
| Goodness of fit | 1.268 |
| H atom treatment | Constrained |
| No. of parameters | 271 |
| No. of restraints | 1 |
| Δρmax, Δρmin (eÅ−3) | 1.3091; −1.272 |
3.5. Biological Evaluation
3.5.1. Cell Culture and Stock Solutions
3.5.2. Cytotoxicity Assay
3.5.3. Staining with Crystal-Violet
4. Conclusions
Supplementary Materials
Supplementary File
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ko, E.; Liu, J.; Burgess, K. Minimalist and universal peptidomimetics. Chem. Soc. Rev. 2011, 40, 4411–4421. [Google Scholar] [CrossRef]
- Giannis, A.; Kolte, T. Peptidomimetics for Receptor Ligands-Discovery, Development, and Medical Perspectives. Angew. Chem. Int. Ed. Engl. 1993, 32, 1244–1267. [Google Scholar] [CrossRef]
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296. [Google Scholar] [CrossRef]
- Kahn, M. Peptide Secondary Structure Mimetics: Recent Advances and Future Challenges. Synlett 1993, 11, 821–826. [Google Scholar] [CrossRef]
- Olson, G.L.; Bolin, D.R.; Bonner, M.P.; Bos, M.; Cook, C.M.; Fry, D.C.; Graves, B.J.; Hatada, M.; Hill, D.E. Concepts and Progress in the Development of Peptide Mimetics. J. Med. Chem. 1993, 136, 3039–3049. [Google Scholar]
- Nomoto, A.; Moriuchi, T.; Yamazaki, S.; Ogawa, A.; Hirao, T. A Highly Ordered Ferrocene System Regulated by Podand Peptide Chains. Chem. Commun. 1998, 1963–1964. [Google Scholar]
- Moriuchi, T.; Nomoto, A.; Yoshida, K.; Ogawa, A.; Hirao, T. Chirality Organization of Ferrocenes Bearing Podand Dipeptide Chains: Synthesis and Structural Characterization. J. Am. Chem. Soc. 2001, 123, 68–75. [Google Scholar] [CrossRef]
- Moriuchi, T.; Nagai, T.; Hirao, T. Chirality Organization of Ferrocenes Bearing Dipeptide Chains of Heterochiral Sequence. Org. Lett. 2005, 7, 5265–5268. [Google Scholar] [CrossRef]
- Moriuchi, T.; Nagai, T.; Hirao, T. Induction of γ-Turn-Like Structure in Ferrocene Bearing Dipeptide Chains via Conformational Control. Org. Lett. 2006, 8, 31–34. [Google Scholar] [CrossRef]
- Moriuchi, T.; Nomoto, A.; Yoshida, K.; Hirao, T. Characterization of Ferrocene Derivatives Bearing Podand Dipeptide Chains (-l-Ala-l-Pro-OR). J. Organomet. Chem. 1999, 589, 50–58. [Google Scholar] [CrossRef]
- Moriuchi, T.; Nomoto, A.; Yoshida, K.; Hirao, T. Intramolecular Conformational Control in Ferrocenes Bearing Podand Dipeptide Chains. Organometallics 2001, 20, 1008–1013. [Google Scholar] [CrossRef]
- Moriuchi, T.; Hirao, T. Design of Ferrocene-Dipeptide Bioorganometallic Conjugates to induce Chirality-Organized Structures. Acc. Chem. Res. 2010, 43, 1040–1051. [Google Scholar] [CrossRef]
- Moriuchi, T.; Hirao, T. Dipeptide-induced chirality organization. J. Inc. Phenom. Macrocycl. Chem. 2012, 74, 23–40. [Google Scholar] [CrossRef]
- Chowdhury, S.; Mahmoud, K.A.; Schatte, G.; Kraatz, H.-B. Amino acid conjugates of 1,1′-diaminoferrocene. Synthesis and chiral organization. Org. Biomol. Chem. 2005, 3, 3018–3023. [Google Scholar] [CrossRef]
- Djaković, S.; Siebler, D.; Čakić Semenčić, M.; Heinze, K.; Rapić, V. Spectroscopic and theoretical study of asymmetric 1,1'-diaminoferrocene conjugates of alpha-amino acids. Organometallics 2008, 27, 1447–1453. [Google Scholar] [CrossRef]
- Barišić, L.; Dropučić, M.; Rapić, V.; Pritzkow, H.; Kirin, S.I.; Metzler-Nolte, N. The first oligopeptide derivative of 1'-aminoferrocene-1-carboxylic acid shows helical chirality with antiparallel strands. Chem. Commun. 2004, 2004–2005. [Google Scholar]
- Barišić, L.; Čakić, M.; Mahmoud, K.A.; Liu, Y.-N.; Kraatz, H.-B.; Pritzkow, H.; Kirin, S.I.; Metzler-Nolte, N.; Rapić, V. Helically chiral ferrocene peptides containing 1'-amino-ferrocene-1-carboxylic acid subunit as turn inducers. Chem. Eur. J. 2006, 12, 4965–4980. [Google Scholar] [CrossRef]
- Barišić, L.; Rapić, V.; Metzler-Nolte, N. Incorporation of the unnatural organometallic amino acid 1'-aminoferrocene-1-carboxylic acid (Fca) into oligopeptides by a combination of Fmoc and Boc solid phase synthetic methods. Eur. J. Inorg. Chem. 2006, 2006, 4019–4021. [Google Scholar]
- Čakić Semenčić, M.; Siebler, D.; Heinze, K.; Rapić, V. Bis- and Trisamides Derived from 1'-Aminoferrocene-1-carboxylic Acid and alpha-Amino Acids: Synthesis and Conformational Analysis. Organometallics 2009, 28, 2028–2037. [Google Scholar] [CrossRef]
- akić Semenčić, M.; Heinze, K.; Förster, C.; Rapić, V. Bioconjugates of 1'-Aminoferrocene-1-carboxylic Acid with (S)-3-Amino-2-methylpropanoic Acid and l-Alanine. Eur. J. Inorg. Chem. 2010, 2010, 1089–1097. [Google Scholar]
- Lapić, J.; Siebler, D.; Heinze, K.; Rapić, V. Conformational Analysis of Heteroannularly Substituted Ferrocene Oligoamides. Eur. J. Inorg. Chem. 2007, 14, 2014–2024. [Google Scholar]
- Barišić, L.; Kovačević, M.; Mamić, M.; Kodrin, I.; Mihalić, Z.; Rapić, V. Synthesis and Conformational Analysis of Methyl N-Alanyl-1'-aminoferrocene-1-carboxylate. Eur. J. Inorg. Chem. 2012, 11, 1810–1822. [Google Scholar]
- Donoli, A.; Marcuzzo, V.; Moretto, A.; Cardena, R.; Santi, S.; Toniolo, C. Charge mapping in peptide chains by oxidation of the terminal ferrocenyl group. Org. Lett. 2011, 13, 1282–1285. [Google Scholar] [CrossRef]
- Donoli, A.; Marcuzzo, V.; Moretto, A.; Crisma, M.; Toniolo, C.; Cardena, R.; Bisello, A.; Santi, S. New bis-Ferrocenyl End-Capped Peptides: Synthesis and Charge Transfer Properties. J. Pept. Sci. 2013, 100, 14–24. [Google Scholar] [CrossRef]
- Barišić, L.; Rapić, V.; Kovač, V. Ferrocene Compounds. XXIX Efficient Syntheses of 1'-Aminoferrocene-1-carboxylic Acid Derivatives. Croat. Chem. Acta 2002, 75, 199–210. [Google Scholar]
- Kirin, S.I.; Kraatz, H.-B.; Metzler-Nolte, N. Systematizing structural motifs and nomenclature in 1,n'-disubstituted ferrocene peptides. Chem. Soc. Rev. 2006, 35, 348–354. [Google Scholar] [CrossRef]
- Vanhoof, G.; Goossens, F.; De Meester, I.; Hendriks, D.; Scharpé, S. Proline motifs in peptides and their biological processing. FASEB J. 1995, 9, 736–744. [Google Scholar]
- Kay, B.K.; Williamson, M.P.; Sudol, M. The importance of being proline: The interaction of proline-rich motifs in signalling proteins with their cognate domains. FASEB J. 2000, 14, 231–241. [Google Scholar]
- Troganis, A.; Gerothanassis, I.P.; Athanassiou, Z.; Mavromoustakos, T.; Hawkes, G.E.; Sakarellos, C. Thermodynamic origin of cis/trans isomers of a proline-containing beta-turn model dipeptide in aqueous solution: A combined variable temperature 1H-NMR, two-dimensional 1H,1H gradient enhanced nuclear Overhauser effect spectroscopy (NOESY), one-dimensional steady-state intermolecular 13C,1H NOE, and molecular dynamics study. Biopolymers 2000, 53, 72–83. [Google Scholar] [CrossRef]
- Ganesh, S.; Jayakumar, R. Role of N-t-Boc group in helix initiation in a novel tetrapeptide. J. Peptide Res. 2003, 59, 249–256. [Google Scholar] [CrossRef]
- Ishimoto, B.; Tonan, K.; Ikawa, S. Coupling of intramolecular hydrogen bonding to the cis-to-trans isomerization of a proline imide bond of small model peptides. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1999, 56, 201–209. [Google Scholar] [CrossRef]
- Sugawara, M.; Tonan, K.; Ikawa, S. Effect of solvent on the cis–trans conformational. equilibrium of a proline imide bond of short model peptides. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2001, 57, 1305–1316. [Google Scholar] [CrossRef]
- Yusuke, J.; Tonan, K.; Ikawa, S. Competitive formation of 10- and 7-membered hydrogen-bonded rings of proline-containing model peptides. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2002, 58, 2795–2802. [Google Scholar] [CrossRef]
- Tonan, K.; Ikawa, S. Intramolecular Hydrogen Bonding and Conformation of Small Peptides: Variable-Temperature FTIR Study on N-Acetyl-L-Pro-L-Leu-Gly-NH2 and Related Compounds. J. Am. Chem. Soc. 1996, 118, 6960–6965. [Google Scholar] [CrossRef]
- Ananthanarayanan, V.S.; Cameron, T.S. Proline-containing beta-turns. IV. Crystal and solution conformations of tert-butyloxycarbonyl-l-prolyl-d-alanine and tert-butyloxycarbonyl-l-prolyl-d-alanyl-l-alanine. Int. J. Peptide Protein Res. 1988, 31, 399–411. [Google Scholar] [CrossRef]
- Ning, L.; de-Ning, W.; Sheng-Kang, Y. Hydrogen bonding between urethane and urea: band assignment for the carbonyl region of FTi.r. spectrum. Polymer 1996, 37, 3045–3047. [Google Scholar] [CrossRef]
- Dorman, D.E.; Bovey, F.A. Carbon-13 magnetic resonance spectroscopy. Spectrum of proline in oligopeptides. J. Org. Chem. 1973, 38, 2379–2383. [Google Scholar] [CrossRef]
- Dumy, P.; Keller, M.; Ryan, D.E.; Rohwedder, B.; Wohr, T.; Mutter, M. Pseudo-Prolines as a Molecular Hinge: Reversible Induction of cis Amide Bonds into Peptide Backbones. J. Am. Chem. Soc. 1997, 119, 918–925. [Google Scholar] [CrossRef]
- Llinás, M.; Klein, M.P. Charge relay at the peptide bond. A proton magnetic resonance study of solvation effects on the amide electron density distribution. J. Am.Chem.Soc. 1975, 97, 4731–4737. [Google Scholar] [CrossRef]
- Kessler, H. Conformation and Biological Activity of Cyclic Peptides. Angew. Chem. Int. Ed. Engl. 1982, 21, 512–523. [Google Scholar] [CrossRef]
- Iqbal, M.; Balaram, P. Aggregation of apolar peptides in organic solvents. Concentration dependence of 1H-nmr parameters for peptide NH groups in 310 helical decapeptide fragment of suzukacillin. Biopolymers 1982, 21, 1427–1433. [Google Scholar] [CrossRef]
- Vijayakumar, E.K.S.; Balaram, P. Stereochemistry of α-Aminoisobutyric Acid Peptides in Solution: Helical Conformations of Protected Decapeptides with Repeating Aib-l-Ala and Aib-l-Val Sequences. Biopolymers 1983, 22, 2133–2140. [Google Scholar] [CrossRef]
- Andersen, N.H.; Neidigh, J.W.; Harris, S.M.; Lee, G.M.; Liu, Z.; Tong, H. Extracting Information from the Temperature Gradients of Polypeptide NH Chemical Shifts. 1. The Importance of Conformational Averaging. J. Am. Chem. Soc. 1997, 119, 8547–8561. [Google Scholar] [CrossRef]
- Baxter, N.J.; Williamson, M.P. Temperature dependence of 1H chemical shifts in proteins. J. Biomol. NMR 1997, 9, 359–369. [Google Scholar] [CrossRef]
- Lee, H.-J.; Park, H.-M.; Lee, K.-B. The beta-turn scaffold of tripeptide containing an azaphenylalanine residue. Biophys. Chem. 2007, 125, 117–126. [Google Scholar] [CrossRef]
- Appoh, F.E.; Sutherland, T.C.; Kraatz, H.-B. Changes in the hydrogen bonding pattern in ferrocene peptides. J. Organomet. Chem. 2004, 689, 4669–4677. [Google Scholar] [CrossRef]
- Lapić, J.; Pavlović, G.; Siebler, D.; Heinze, K.; Rapić, V. Structural, spectroscopic and theoretical study of ferrocene ureidopeptides. Organometallics 2008, 27, 726–735. [Google Scholar] [CrossRef]
- Stevens, E.S.; Sugawara, N.; Bonora, G.M.; Toniolo, C. Conformational analysis of linear peptides. 3. Temperature dependence of NH chemical shifts in chloroform. J. Am. Chem. Soc. 1980, 102, 7048–7050. [Google Scholar] [CrossRef]
- Srivastava, K.R.; Kumar, A.; Goyal, B.; Durani, S. Stereochemistry and solvent role in protein folding: Nuclear magnetic resonance and molecular dynamics studies of poly-l and alternating-l,d homopolypeptides in dimethyl sulfoxide. J. Phys. Chem. B. 2011, 26, 6700–6708. [Google Scholar] [CrossRef]
- Sladojevich, F.; Guarna, A.; Trabocchi, A. Evaluation of stereochemically dense morpholine-based scaffolds as proline surrogates in β-turn peptides. Org. Biomol. Chem. 2010, 8, 916–924. [Google Scholar] [CrossRef]
- Madison, V.; Schellman, J. Location of proline derivatives in conformational space. I. Conformational calculations; optical activity and NMR experiments. Biopolymers 1970, 9, 511–567. [Google Scholar] [CrossRef]
- Montagut, M.; Lemanceau, B.; Bellocq, A.-M. Conformational analysis of thyrotropin releasing factor by proton magnetic resonance spectroscopy. Biopolymers 1974, 13, 2615–2629. [Google Scholar] [CrossRef]
- Higasijima, T.; Tasumi, M.; Miyazawa, T. 1H nuclear magnetic resonance studies of N-acetyl-l-proline N-methylamide. Molecular conformations, hydrogen bondings, and thermodynamic quantities in various solvents. Biopolymers 1977, 16, 1259–1270. [Google Scholar] [CrossRef]
- Madison, V.; Kopple, K.D. Solvent-dependent conformational distributions of some dipeptides. J. Am. Chem. Soc. 1980, 102, 4855–4863. [Google Scholar] [CrossRef]
- Eberhardt, E.S.; Loh, S.N.; Hinck, A.P.; Raines, R.T. Solvent Effects on the Energetics of Prolyl Peptide Bond Isomerization. J. Am. Chem. Soc. 1992, 114, 5437–5439. [Google Scholar] [CrossRef]
- Eberhardt, E.S.; Loh, S.N.; Raines, R.T. Thermodynamic Origin of Prolyl Peptide Bond Isomers. Tetrahedron Lett. 1993, 34, 3055–3056. [Google Scholar] [CrossRef]
- Eberhardt, E.S.; Raines, R.T. Amide-Amide and Amide-Water Hydrogen Bonds: Implications for Protein Folding and Stability. J. Am. Chem. Soc. 1994, 116, 2149–2150. [Google Scholar] [CrossRef]
- McDonald, D.Q.; Still, W.C. Molecular Mechanics Parameters and Conformational Free Energies of Proline-Containing Peptides. J. Org. Chem. 1996, 61, 1385–1391. [Google Scholar] [CrossRef]
- Eberhardt, E.S.; Panisik, N., Jr.; Raines, R.T. Inductive Effects on the Energetics of Prolyl Peptide Bond Isomerization: Implications for Collagen Folding and Stability. J. Am. Chem. Soc. 1996, 118, 12261–5439. [Google Scholar] [CrossRef]
- Deetz, M.J.; Fahey, J.E.; Smith, B.D. NMR studies of hydrogen bonding interactions with secondary amide and urea groups. J. Phys. Org. Chem. 2001, 14, 463–467. [Google Scholar] [CrossRef]
- Sarkar, S.K.; Young, P.E.; Sullivan, C.E.; Torchia, D.A. Detection of cis and trans X-Pro peptide bonds in proteins by 13C NMR: Application to collagen. Proc. Natl. Acad. Sci. USA 1984, 81, 4800–4803. [Google Scholar] [CrossRef]
- O’Neal, K.D.; Chari, M.V.; Mcdonald, C.H.; Cook, R.G.; Yu-Lee, L.Y.; Morrisett, J.D.; Shearer, W.T. Multiple cis-trans conformers of the prolactin receptor proline-rich motif (PRM) peptide detected by reverse-phase HPLC, CD and NMR spectroscopy. Biochem J. 1996, 315, 833–844. [Google Scholar]
- Husain, R.D.; McCandless, J.; Stevenson, P.J.; Large, T.; Guthrie, D.J.S.; Walker, B. Detection of cis-trans isomers of a synthetic peptide fragment of Erythropoietin. J. Chromatogr. Sci. 2002, 40, 1–6. [Google Scholar] [CrossRef]
- Bragg, R.A.; Clayden, J.; Morris, G.A.; Pink, J.H. Stereodynamics of bond rotation in tertiary aromatic amides. Chem. Eur. J. 2002, 8, 1279–1289. [Google Scholar] [CrossRef]
- Berggren, K.; Vindebro, R.; Bergström, C.; Spoerry, C.; Persson, H.; Fex, T.; Kihlberg, J.; von Pawel-Rammingen, U.; Luthman, K. 3-Aminopiperidine-Based Peptide Analogues as the First Selective Noncovalent Inhibitors of the Bacterial Cysteine Protease IdeS. J. Med. Chem. 2012, 55, 2549–2560. [Google Scholar] [CrossRef]
- Kelly, S.M.; Price, N.C. The Use of Circular Dichroism in the Investigation of Protein Structure and Function. Curr. Protein Pept. Sci. 2000, 1, 349–384. [Google Scholar] [CrossRef]
- Kovač, V.; Čakić Semenčić, M.; Kodrin, I.; Roca, S.; Rapić, V. Ferrocene-dipeptide conjugates derived from aminoferrocene and 1-acetyl-1'-aminoferrocene: Synthesis and conformational studies. Tetrahedron 2013, 69, 10497–10506. [Google Scholar] [CrossRef]
- Jios, J.L.; Kirin, S.I.; Buceta, N.N.; Weyhermüller, T.; della Védova, C.O.; Metzler-Nolte, N. Synthesis and structural characterization of metallated bioconjugates: C-terminal labeling of amino acids with aminoferrocene. J. Organomet. Chem. 2007, 692, 4209–4214. [Google Scholar] [CrossRef]
- Byun, Y.S.; Lightner, D.A. Exciton coupling from dipyrrinone chromophores. J. Org. Chem. 1991, 56, 6027–6033. [Google Scholar] [CrossRef]
- Kharb, R.; Rana, M.; Sharma, P.C.; Yar, M.S. Therapeutic importance of peptidomimetics in medicinal chemistry. J. Chem. Pharm. Res. 2011, 3, 173–186. [Google Scholar]
- Li, L.; Thomas, R.M.M.; Suzuki, H.; de Brabander, J.K.; Wang, X.; Harran, P.G. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science 2004, 305, 1471–1474. [Google Scholar] [CrossRef]
- Liao, Y.-F.; Wang, B.-J.; Hsu, W.-M.; Lee, H.; Liao, C.-Y.; Wu, S.-Y.; Cheng, H.-T.; Hu, M.K. Unnatural Amino Acid-Substituted (Hydroxyethyl)urea Peptidomimetics Inhibit γ-Secretase and Promote the Neuronal Differentiation of Neuroblastoma Cells. Mol. Pharmacol. 2007, 71, 588–601. [Google Scholar]
- Boyd, M.R.; Paull, K.D. Some Practical Considerations and Applications of the National Cancer Institute in Vitro Anticancer Drug Discovery Screen. Drug. Develop. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Cortés, R.; Crespo, M.; Davin, L.; Martín, R.; Quirante, J.; Ruiz, D.; Messeguer, R.; Calvis, C.; Baldomà, L.; Badia, J.; et al. Seven-membered cycloplatinated complexes as a new family of anticancer agents. X-ray characterization and preliminary biological studies. Eur. J. Med. Chem. 2012, 54, 557–566. [Google Scholar] [CrossRef]
- Bayoumi, A.H. Antiproliferative Properties of Vinyl Dipeptides: Synthesis and MCF-7 Cell Line Testing. Open J. Med. Chem. 2012, 2, 105–111. [Google Scholar] [CrossRef]
- CrysAlis PRO; Oxford Diffraction Ltd.: Oxford, UK, 2007.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Farrugia, L.J. ORTEP-3 for Windows-A version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Cryst. 1997, 30, 565. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: visualization and analysis of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival, application to proliferation and cytotoxic assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–4 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kovačević, M.; Molčanov, K.; Radošević, K.; Srček, V.G.; Roca, S.; Čače, A.; Barišić, L. Conjugates of 1'-Aminoferrocene-1-carboxylic Acid and Proline: Synthesis, Conformational Analysis and Biological Evaluation. Molecules 2014, 19, 12852-12880. https://doi.org/10.3390/molecules190812852
Kovačević M, Molčanov K, Radošević K, Srček VG, Roca S, Čače A, Barišić L. Conjugates of 1'-Aminoferrocene-1-carboxylic Acid and Proline: Synthesis, Conformational Analysis and Biological Evaluation. Molecules. 2014; 19(8):12852-12880. https://doi.org/10.3390/molecules190812852
Chicago/Turabian StyleKovačević, Monika, Krešimir Molčanov, Kristina Radošević, Višnja Gaurina Srček, Sunčica Roca, Alan Čače, and Lidija Barišić. 2014. "Conjugates of 1'-Aminoferrocene-1-carboxylic Acid and Proline: Synthesis, Conformational Analysis and Biological Evaluation" Molecules 19, no. 8: 12852-12880. https://doi.org/10.3390/molecules190812852
APA StyleKovačević, M., Molčanov, K., Radošević, K., Srček, V. G., Roca, S., Čače, A., & Barišić, L. (2014). Conjugates of 1'-Aminoferrocene-1-carboxylic Acid and Proline: Synthesis, Conformational Analysis and Biological Evaluation. Molecules, 19(8), 12852-12880. https://doi.org/10.3390/molecules190812852