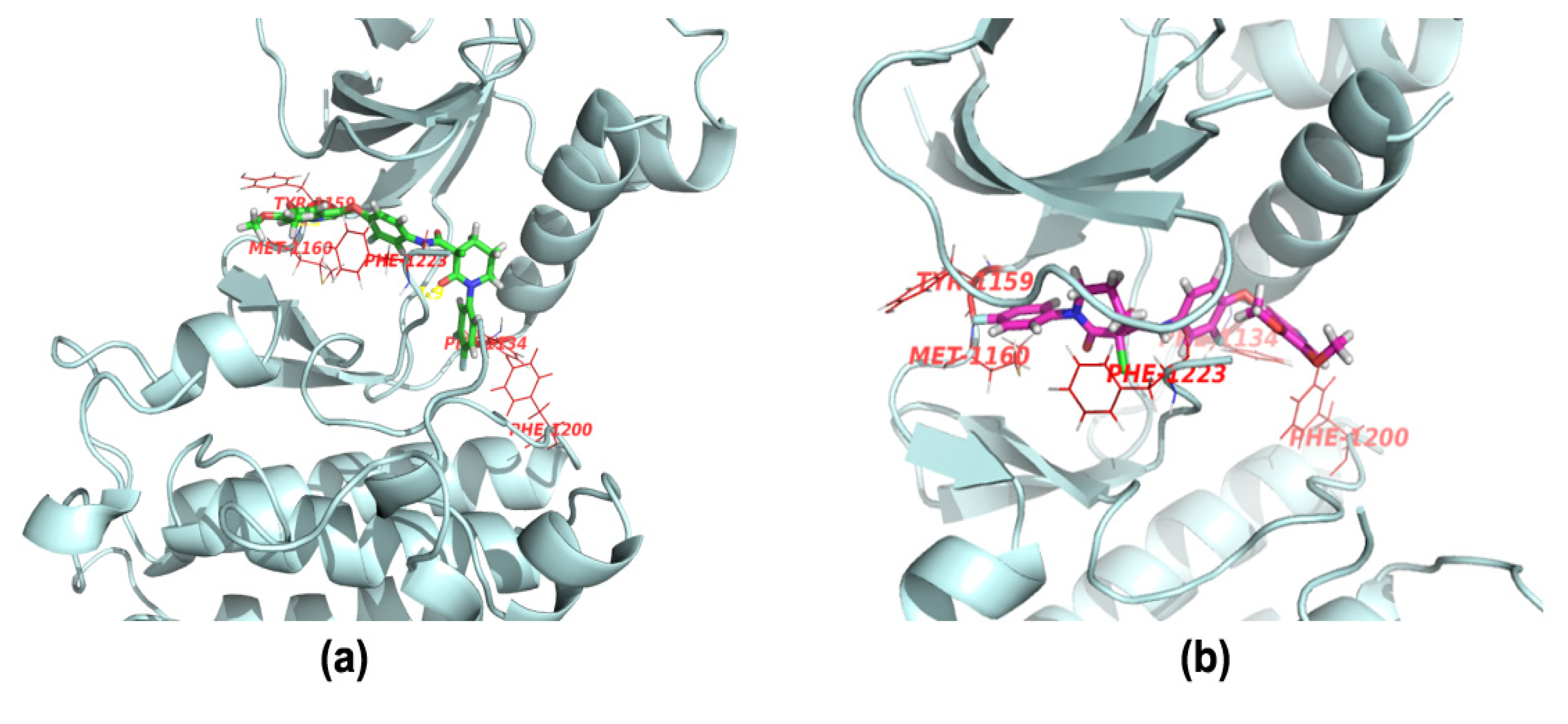
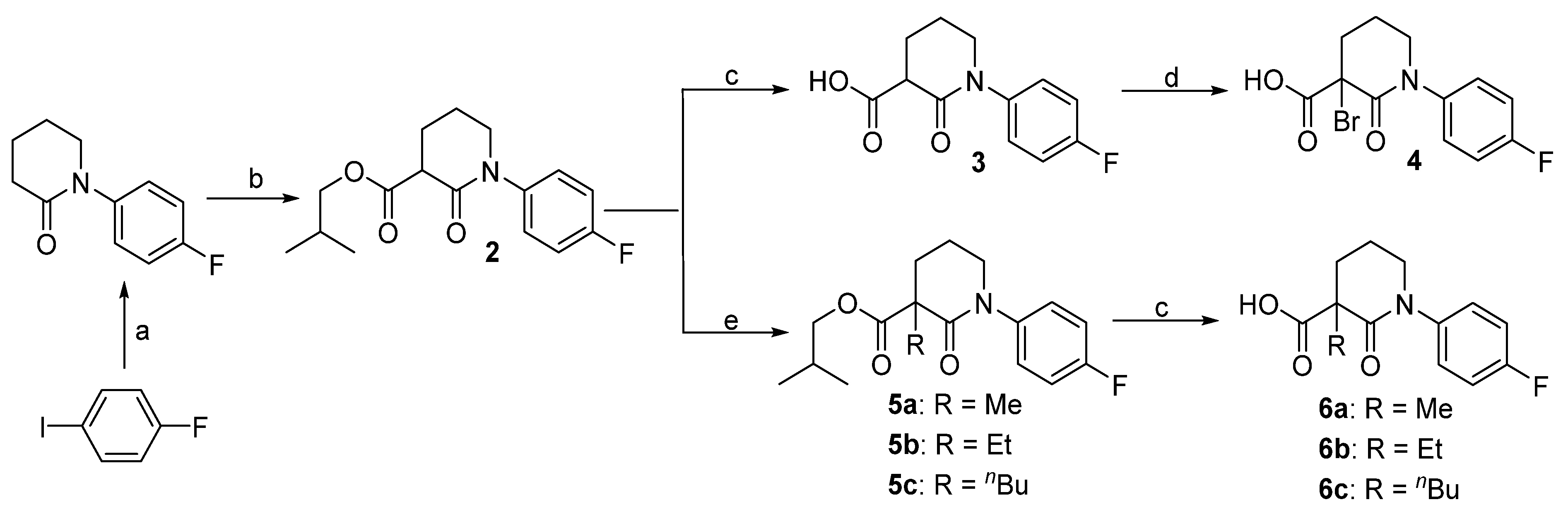
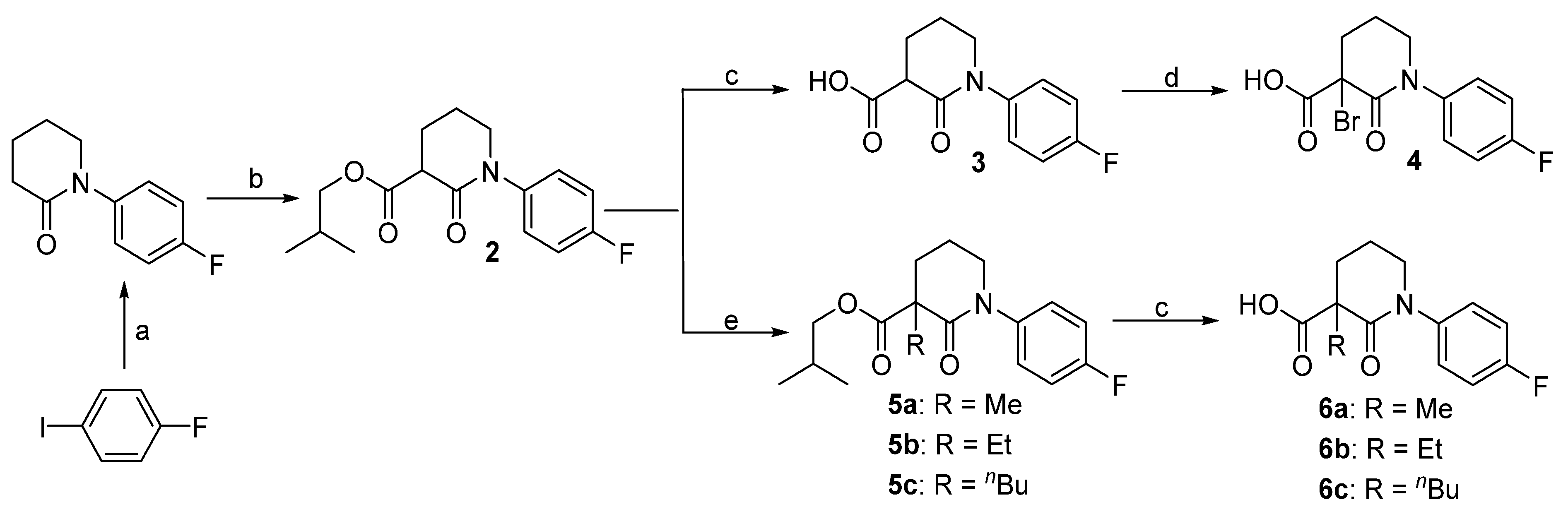
3.2. Synthesis
Isobutyl 1-(4-fluorophenyl)-2-oxopiperidine-3-carboxylate (2). 1-Fluoro-4-iodobenzene (2.22 g, 10 mmol) and piperidin-2-one (1.2 g, 12 mmol) were added to dry DMF (30 mL), followed by the addition of K3PO4 (6.36 g, 30 mmol) and CuI (190 mg, 0.1 mmol). The mixture was heated to 100 °C for 12 h before filtering through Celite. After washing with ethyl acetate (3 × 10 mL), the combined organic phase was concentrated and the residue was purified by column chromatography to give 1-(4-fluorophenyl)piperidin-2-one (1.73 g, 90%) as a yellow solid. This N-arylpiperidin-2-one (386 mg, 2 mmol) was dissolved in dry THF (20 mL) and cooled to −78 °C. After the addition of tert-BuLi (1.4 mL, 1.6 M in THF, 2.2 mmol) and stirring at this temperature for 4 h, isobutyl chlorofomate (400 μL, 2 mmol) was added. Ten min later, the reaction was quenched by addition of saturated aq. NH4Cl (2 mL). The mixture was diluted with water (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography to give compound 2 (480 mg, 82%) as a yellow wax. 1H-NMR (600 MHz, CDCl3) δ 7.25–7.20 (m, 2H, ArH), 7.09–7.03 (m, 2H,ArH), 3.99 (dd, 1H, J = 10.6, 6.7 Hz, CH), 3.83 (d, 1 H, J = 6.7 Hz, CHH), 3.70–3.61 (m, 1H, CHH), 3.58 (t, 1 H, J = 6.9 Hz, CH), 2.32–2.24 (m, 1H, CHH), 2.23–2.16 (m, 1H, CHH), 2.12–2.04 (m, 1H, CHH), 2.02–1.87 (m, 2H, CH, CHH), 0.94 (d, 6 H, J = 6.6 Hz, CH3); 13C-NMR (150 MHz, CDCl3) δ 171.0, 166.3, 162.1, 160.4, 138.8, 127.9, 116.2, 100.0, 71.5, 51.6, 49.6, 27.8, 25.3, 21.4, 19.1; HR-MS (ESI) Calcd for C16H21FNO3 [M + H]+ 294.1506, Found 294.1518.
1-(4-Fluorophenyl)-2-oxopiperidine-3-carboxylic acid (3). To a solution of 2 (217 mg, 0.74 mmol) in THF/MeOH/H2O (1/1/1, 3 mL in total) at 0 °C was added LiOH monohydrate (94 mg, 2.2 mmol). The reaction mixture was warmed to room temperature and stirred for 5 h. The solution was acidified to pH 1 with 1 mol/L HCl and extracted with EtOAc (3 × 20 mL). The organic extracts were combined and washed with brine (2 × 5 mL). Evaporation of the solvent gave the corresponding acid 3 (152 mg, 87%) as a white solid. 1H-NMR (600 MHz, CDCl3) δ 7.33–7.28 (m, 1H, ArH), 7.25–7.19 (m, 1H, ArH), 3.69–3.55 (m, 2H, NCH2), 3.43 (dd, 1H, J = 8.2, 6.5 Hz, CH), 2.16–2.10 (m, 1 H, CHH), 2.08–2.02 (m, 1H, CHH), 1.98–1.91 (m, 1H, CHH), 1.91–1.83 (m, 1H, CHH); 13C-NMR (150 MHz, CDCl3) δ 174.3, 170.2, 161.3, 159.6, 138.7, 127.9, 115.6, 51.8, 50.3, 27.5, 21.6; HR-MS (ESI) Calcd for C12H13FNO3 238.0880 [M + H]+, found 238.0910.
3-Bromo-1-(4-fluorophenyl)-2-oxopiperidine-3-carboxylic acid (4). To a solution of acid 3 (220 mg, 0.93 mmol) in Et2O (5 mL) was added liquid Br2 (48 μL, 0.93 mmol) at 0 °C. The reaction mixture was stirred for 2 h before concentrated in vacuo. The residue was purified by column chromatography, giving compound 4 (265 mg, 91%) as white solid. 1H-NMR (600 MHz, acetone-d6) δ 13.03 (s, 1H, OH), 7.44–7.40 (m, 2H, ArH), 7.25–7.20 (m, 2H, ArH), 4.04 (td, 1H, J = 12.1, 4.6 Hz, NCHH), 3.82 (ddt, 1H, J = 13.0, 6.3, 2.4 Hz, NCHH), 2.77–2.69 (m, 1H, CHH), 2.62–2.56 (m, 1H, CHH), 2.53–2.43 (m, 1H, CHH), 2.19–2.12 (m, 1H, CHH); 13C-NMR (150 MHz, acetone-d6) δ 166.4, 162.5, 160.9, 140.6, 140.6, 129.0, 129.0, 116.2, 52.1, 32.3, 20.4.
3.2.1. General Procedure for the Synthesis of Isobutyl 1-(4-Fluorophenyl)-3-alkyl-2-oxopiperidine-3-carboxylates 5a–c
To a solution of compound 2 (586 mg, 2 mmol) in dry THF (10 mL) at 0 °C was added NaH (72 mg, 80% suspension in mineral oil, 2.4 mmol) in portions. Thirty min later, alkyl halide (MeI, EtBr, or n-BuBr, 2.6 mmol) was added slowly and the reaction mixture was stirred at this temperature for another 5 h. When TLC showed all the starting material consumed, the reaction mixture was quenched with 0.5 mol/L HCl, diluted with water (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layer was washed with brine, dried over Na2SO4 and concentrated. The residue was purified by column chromatography to give the desired compound as pale yellow oil.
Isobutyl 1-(4-fluorophenyl)-3-methyl-2-oxopiperidine-3-carboxylate (5a). 87% yield; 1H-NMR (600 MHz, CDCl3) δ 7.25–7.19 (m, 1 H, ArH), 7.12–7.01 (m, 1H, ArH), 4.06–3.88 (m, 2H, OCH2), 3.78–3.58 (m, 1H, CHH), 2.47–2.31 (m, 1H, CHH), 2.10–1.94 (m, 3H, CH2, CH), 1.94–1.84 (m, 1H, CHH), 1.57 (d, 3H, J = 2.4 Hz, CH3), 0.97 (d, 3H, J = 2.0 HzCH3), 0.96 (d, 3H, J = 2.1 Hz, CH3); 13C-NMR (150 MHz, CDCl3) δ 173.8, 170.1, 161.9, 160.2, 139.3, 127.8, 116.0, 115.9, 71.5, 51.9, 51.3, 33.6, 27.8, 22.9, 20.4, 19.2; HR-MS (ESI) Calcd for C17H23FNO3 308.1662 [M + H]+, found 308.1599.
Isobutyl 3-ethyl-1-(4-fluorophenyl)-2-oxopiperidine-3-carboxylate (5b). 76% yield; 1H-NMR (600 MHz, CDCl3) δ 7.24–7.18 (m, 2H, ArH), 7.08–7.03 (m, 2H, ArH), 4.01–3.90 (m, 2H, OCH2), 3.72–3.65 (m, 1 H, CHH), 3.63–3.56 (m, 1H, CHH), 2.31–2.24 (m, 1H, CHH), 2.16–2.09 (m, 1H, CHH), 2.10–2.04 (m, 1H, CHH), 2.02–1.91 (m, 4H, CH2), 0.98 (t, 3H, J = 7.4 Hz, CH3), 0.96 (d, 6H, J = 2.1 Hz, 2 × CH3); 13C-NMR (150 MHz, CDCl3) δ 173.6, 170.0, 161.8, 160.2, 139.3, 127.8, 116.0, 71.5, 51.9, 51.3, 33.6, 30.1, 27.8, 22.9, 20.4, 19.8; HR-MS (ESI) Calcd for C18H25FNO3 322.1819 [M + H]+, found 322.1830.
Isobutyl 3-butyl-1-(4-fluorophenyl)-2-oxopiperidine-3-carboxylate (5c). 83% yield; 1H-NMR (600 MHz, CDCl3) δ 7.23–7.18 (m, 2H, ArH), 7.08–7.02 (m, 2H, ArH), 4.00–3.89 (m, 2H, OCH2), 3.72–3.66 (m, 1H, CHH), 3.61–3.56 (m, 1H, CHH), 2.32–2.26 (m, 1H, CHH), 2.10–1.86 (m, 6H), 1.46–1.38 (m, 1H, CHH), 1.37–1.29 (m, 2H, CH2), 1.30–1.21 (m, 1H, CHH), 0.96 (d, 6H, J = 2.1 Hz, 2 × CH3), 0.90 (t, 3H, J = 7.2 Hz, CH3); 13C-NMR (150 MHz, CDCl3) δ 173.5, 169.4, 161.8, 160.2, 139.4, 127.8, 115.9, 71.5, 54.9, 51.6, 35.7, 30.1, 27.0, 23.2, 20.7, 19.2, 14.1; HR-MS (ESI) Calcd for C20H29FNO3 350.2132, [M + H]+, found 350.2122.
3.2.2. 1-(4-Fluorophenyl)-3-alkyl-2-oxopiperidine-3-carboxylic Acids 6a–c were Prepared by a Procedure Similar to that of Compound 3.
3-Methyl-1-(4-fluorophenyl)-2-oxopiperidine-3-carboxylic acid (6a). White solid; 88% yield; 1H-NMR (600 MHz, DMSO-d6) δ 12.59 (s, 1 H, OH), 7.31–7.25 (m, 2H, ArH), 7.24–7.17 (m, 2H, ArH), 3.65 (dt, 1H, J = 12.1, 6.1 Hz, NCHH), 3.59 (dt, 1H, J = 11.9, 5.8 Hz, NCHH), 2.25–2.18 (m, 1H, CHH), 1.95–1.87 (m, 2H, CH2), 1.87–1.80 (m, 1H, CHH), 1.37 (s, 3H, CH3); 13C-NMR (150 MHz, DMSO-d6) δ 174.9, 169.7, 160.9, 159.3, 139.8, 128.2, 115.6, 51.2, 50.4, 32.8, 22.6, 19.8; HR-MS (ESI) Calcd for C13H15FNO3 252.1036, [M + H]+, found 252.1040.
3-Ethyl-1-(4-fluorophenyl)-2-oxopiperidine-3-carboxylic acid (6b). white solid; 79% yield; 1H-NMR (600 MHz, DMSO-d6) δ 12.79 (s, 1 H, OH), 7.26–7.23 (m, 2H, ArH), 7.16–7.12 (m, 2H, ArH), 3.92 (dt, J = 12.7, 6.5 Hz, 1H, NCHH), 3.74 (dt, J = 12.6, 6.4 Hz, 1H, NCHH), 2.56–2.50 (m, 1H, CHH), 2.20 (q, J = 6.7 Hz, 1H, CHH), 2.05–2.01 (m, 1H, CHH), 2.01–1.91 (m, 2H, CHH), 1.84 (dt, J = 12.3, 7.0 Hz, 1 H, CHH), 0.71 (t, J = 6.6 Hz, 3H, CH3); 13C-NMR (150 MHz, DMSO-d6) δ 174.8, 169.8, 161.2, 159.6, 139.5, 128.4, 115.8, 51.7, 50.9, 33.6, 29.7, 22.6, 18.8; HR-MS (ESI) Calcd for C14H17FNO3 266.1193, [M + H]+, found 266.1201.
3-Butyl-1-(4-fluorophenyl)-2-oxopiperidine-3-carboxylic acid (6c). White solid; 84% yield; 1H-NMR (600 MHz, DMSO-d6) δ 12.66 (s, 1H, OH), 7.25–7.20 (m, 2H, ArH), 7.14 (dd, 2H, J = 8.6, 7.0 Hz, ArH), 3.96–3.87 (m, 1H, CHH), 3.78–3.72 (m, 1H, CHH), 2.49–2.42 (m, 1H, CHH), 2.22 (dt, J = 19.0, 7.5 Hz, 2H, CH2), 2.03–1.91 (m, 3H, CHH, CH2), 1.38–1.21 (m, 4H, CH2CH2), 0.88 (t, 3 H, J = 6.4 Hz, CH3); 13C-NMR (150 MHz, DMSO-d6) δ 174.7, 169.5, 161.6, 160.0, 139.6, 127.9, 115.6, 54.6, 51.3, 35.4, 30.3, 23.2, 20.7, 14.2; HR-MS (ESI) Calcd for C16H21FNO3 294.1506, [M + H]+, found 294.1496.
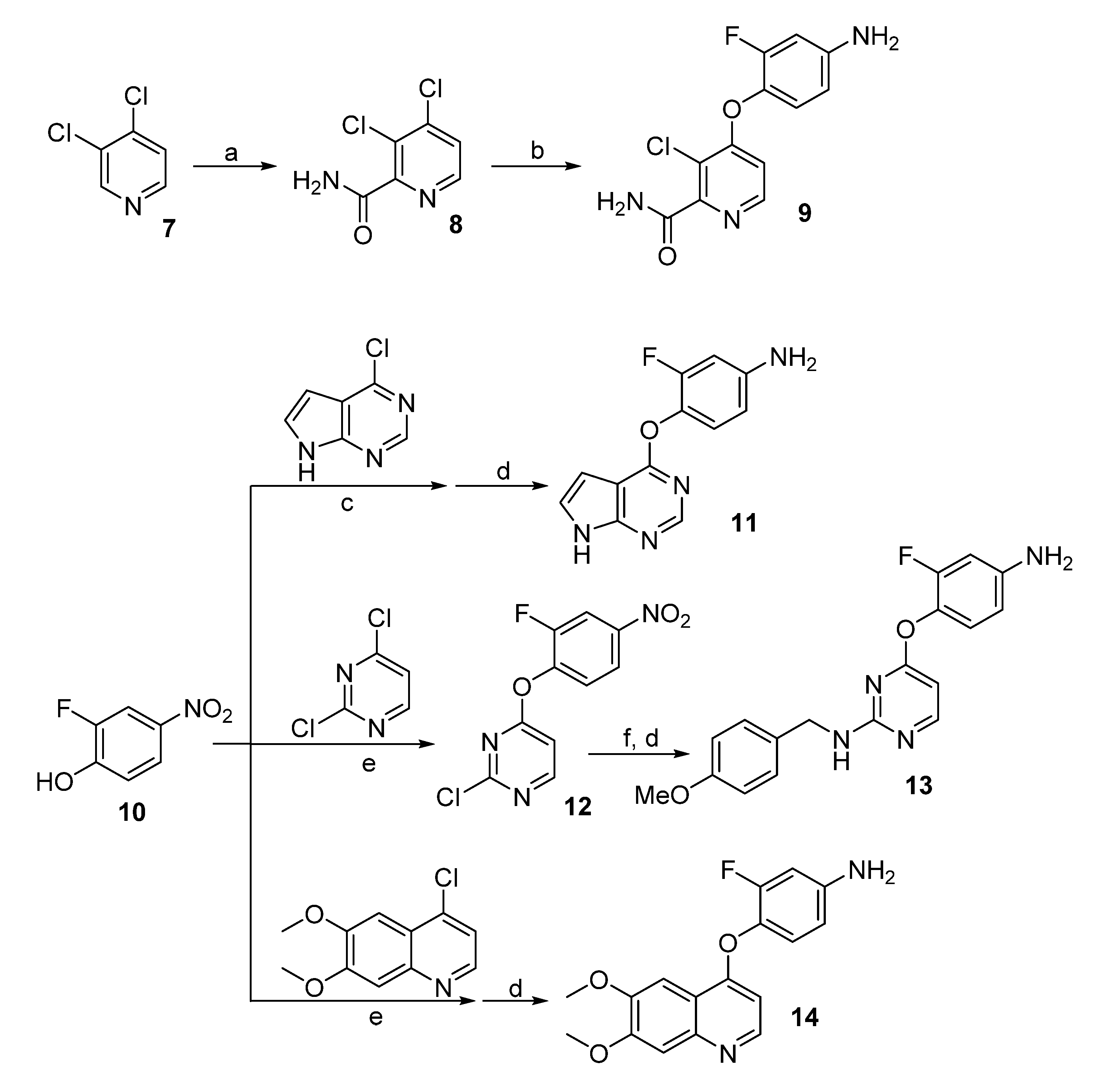
3,4-Dichloropicolinamide (8). To a solution of 2,2,6,6-tetramethylpiperidine (1.56 g, 11 mmol) in dry ether (20 mL) at 0 °C was added n-BuLi (4.4 mL, 2.5 M in THF, 11 mmol) slowly. The reaction mixture was stirred at this temperature for 30 min before cooled to −78 °C. A solution of 3,4-dichloropyridine (1.48 g, 10 mmol) in dry ether (5 mL) was injected via syringe to the above reaction mixture and stirred for 2 h before trimethylsilyl isothiocyanate (15 mmol) was added. After warmed to room temperature, the reaction was quenched by the addition of HOAc (2 mL) and water (10 mL), and then let to stir overnight. The suspension was filtered and washed with cold water, giving the title compound as a gray solid (686 mg, 40%). 1H-NMR (DMSO-d6, 600 MHz) δ 8.50 (d, 1H, J = 5.2 Hz, ArH), 8.12 (br s, 1H, CONH2), 7.83 (d, 1H, J = 5.2 Hz, ArH), 7.82 (br s, 1H, CONH2).
4-(4-Amino-2-fluorophenoxy)-3-chloropicolinamide (9). To a solution of 4-amino-2-fluorophenol (465 mg, 3.65 mmol) in DMF (10 mL) was added potassium tert-butoxide (440 mg, 3.95 mmol). Thirty min later, 3,4-dichloropicolinamide (8) was added and the solution was heated to 50 °C. When TLC showed all the starting materials consumed, the reaction mixture was diluted with EtOAc (50 mL), washed with saturated NaHCO3, brine, and dried over Na2SO4. After concentration, the residue was purified by column chromatography giving the title compound as a pale yellow solid (580 mg, 79%). 1H-NMR (DMSO-d6, 600 MHz) δ 8.29 (d, 1 H, J = 5.6 Hz, ArH), 7.00 (t, 1H, J = 8.8 Hz, ArH), 6.79 (d, 1H, J = 5.6 Hz, ArH), 6.63–6.55 (m, 2H, ArH); 13C-NMR (DMSO-d6, 150 MHz) δ 166.6, 160.8, 154.1, 153.9, 149.0, 148.7, 128.5, 123.7, 115.9, 110.1, 110.0, 101.3.
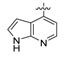
4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)oxy)-3-fluoroaniline (11). The solution of 4-chloro-7H-pyrrolo-[2,3-d]pyrimidine (1.0 g, 6.5 mmol) and 2-fluoro-4-nitrophenol (1.5 g, 9.5 mmol) in bromobenzene (5 mL) was heated at 130 °C for 4 h in a sealed tube. After that, the reaction mixture was cooled to room temperature, diluted with Et2O (5 mL) and filtered. Recrystallization in MeOH gave 4-(2-fluoro-4-nitrophenoxy)-7H-pyrrolo[2,3-d]pyrimidine as a yellow solid (1.6 g, 85%), which was used for the next step directly. To a solution of this nitro compound in THF (5 mL) and MeOH (5 mL) was added zinc powder (130 mg, 2 mmol) and NH4Cl (270 mg, 5 mmol). The reaction mixture was stirred at room temperature for 5 h before filtered through a Celite pad. The filtrate was diluted with EtOAc, washed with water, dried over Na2SO4 and concentrated. The residue was purified by column chromatography giving compound 11 (115 mg, 69%) as a brown solid. 1H-NMR (600 MHz, DMSO-d6) δ 12.19 (s, 1H, NH), 8.28 (s, 1H, CH), 7.44 (t, 1H, J = 3.1 Hz, ArH), 7.00 (t, 1H, J = 8.9 Hz, ArH), 6.48 (dd, 1H, J = 13.0, 2.6 Hz, ArH), 6.45 (dd, 1H, J = 3.4, 1.5 Hz, CH), 6.42–6.39 (m, 1H, CH), 5.35 (s, 2H, NH2).
2-Chloro-4-(2-fluoro-4-nitrophenoxy)pyrimidine (12). 2-Fluoro-4-nitrophenol (314 mg, 2 mmol), K2CO3 (304 mg, 2.2 mmoo) and 2,4-dichloropyrimidine (300 mg, 2 mmol) were dissolved in DMF (20 mL) and heated at 100 °C for 2 h. the reaction mixture was concentrated, diluted with EtOAc (100 mL), washed with water, brine, and concentrated in vacuo. The residue was purified by column chromatography giving compound 12 (324 mg, 65%) as a white solid. 1H-NMR (600 MHz, DMSO-d6) δ 8.78 (d, J = 5.7 Hz, 1H, ArH), 8.44 (dd, 1H J = 10.2, 2.7 Hz, ArH), 8.25 (ddd, 1H, J = 9.0, 2.7, 1.3 Hz, ArH), 7.82 (dd, 1H, J = 9.0, 7.7 Hz, ArH), 7.49 (d, 1H, J = 5.7 Hz, ArH).
4-(4-Amino-2-fluorophenoxy)-N-(4-methoxybenzyl)pyrimidin-2-amine (13). To the solution of compound 12 (239 mg 1mmol) and 4-methoxybenzylamine (192 mg, 1.4 mmol) in DMF (8 mL) was added K2CO3 (152 mg, 1.1 mmol). The reaction mixture was heated at 100 °C for 1 h before concentrated in vacuo. After diluted with EtOAc, the solution was washed with water and brine, and then concentrated. The residue was purified by column chromatography giving a yellow solid (231 mg, 68%), which was treated by zinc powder and NH4Cl as described for the preparation of compound 11. After workup and purification, compound 13 was obtained as a brown solid (115 mg, 69%). 1H-NMR (600 MHz, CDCl3) δ 8.06 (br s, 1H, ArH), 7.12 (br s, 2H, ArH), 6.92 (t, 1H, J = 8.6 Hz, ArH), 6.80 (d, 2H, J = 8.2 Hz, ArH), 6.47 (dd, 1H, J = 11.8, 2.7 Hz, ArH), 6.45–6.39 (m, 1H, ArH), 6.15 (d, 1H, J = 5.8 Hz, ArH), 4.36 (br s, 2H, NH2), 3.92 (br s, 2H, CH2), 3.78 (s, 3H, CH3).
4-((6,7-Dimethoxyquinolin-4-yl)oxy)-3-fluoroaniline (14). The procedure used was similar to that used for the synthesis of compound 11. Compound 14 was obtained as a brown solid in 76% yield. 1H-NMR (600 MHz, CDCl3) δ 8.47 (d, 1H, J = 5.1 Hz, ArH), 7.58 (s, 1H, ArH), 7.40 (s, 1H, ArH), 7.02 (t, 1H, J = 8.6 Hz, ArH), 6.55 (dd, 1H, J = 12.0, 2.7 Hz, ArH), 6.49 (dd, 1H, J = 8.9, 2.6 Hz, ArH), 6.40 (d, 1H, J = 5.0 Hz, ArH), 4.05 (s, 3H, OCH3), 4.03 (s, 3H, OCH3), 3.84 (br s, 2H, NH2).
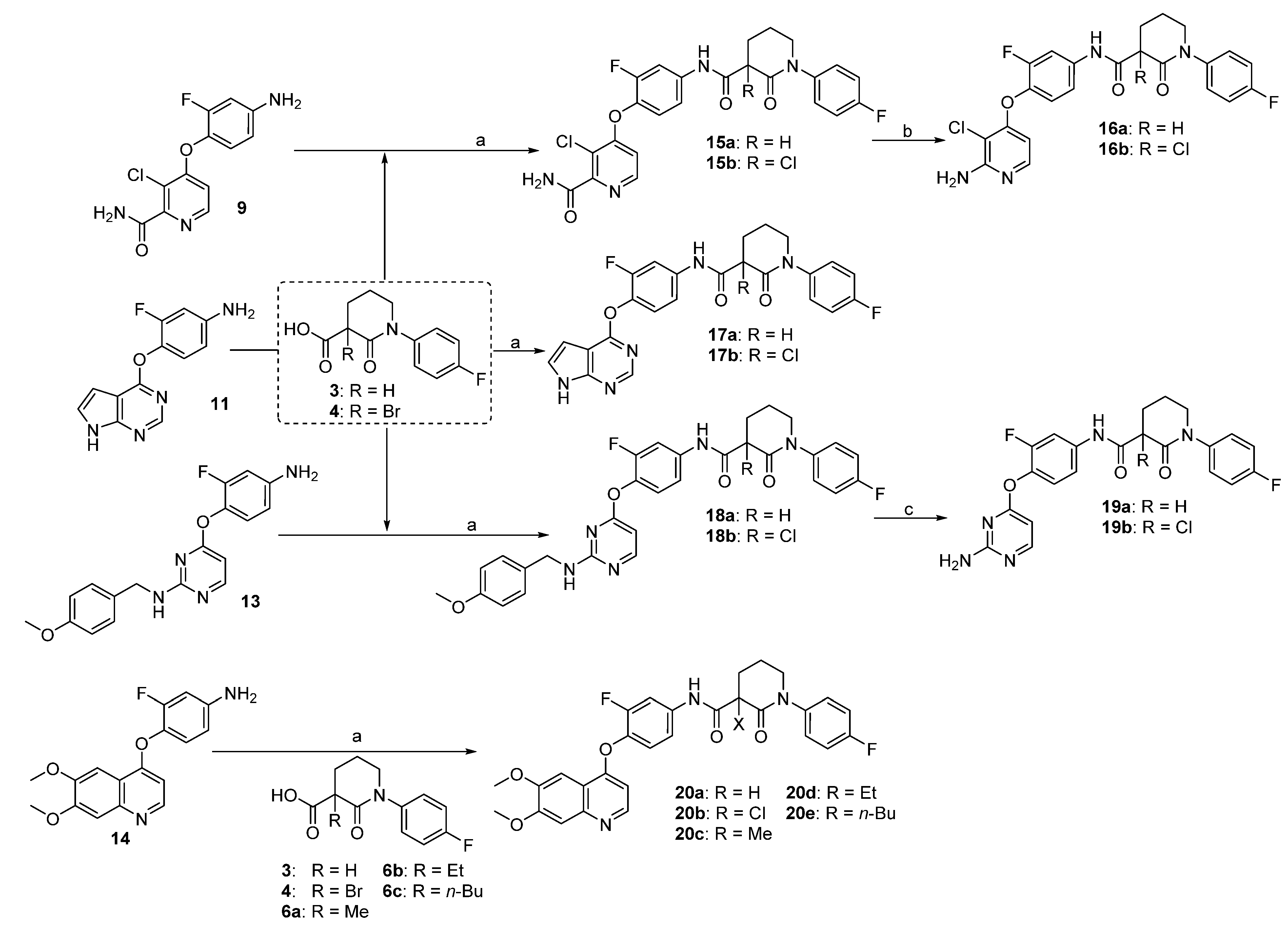
3.2.3. General Procedure for the Preparation of 15a–b, 17a–b, 18a–b and 20a–b
EDC-HCl (1.2 g, 6.25 mmol) was added to a suspension of the carboxylic acid (2.5 mol of 3, 4, or 6a–c) and the amine (2.5 mmol of 9, 11, 13 or 14) in THF (25 mL) at 0 °C followed by DMAP (30 mg, 0.25 mmol). The reaction mixture was warmed to room temperature and stirred overnight. After diluted with EtOAc (150 mL), the whole mixture was washed with 1 M HCl (3 × 10 mL), 5% NaHCO3 (3 × 10 mL), and brine (3 × 10 mL), dried over Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography to give corresponding amide.
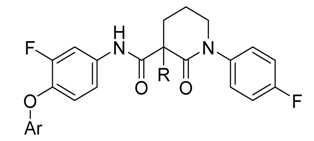
3-Chloro-4-(2-fluoro-4-(1-(4-fluorophenyl)-2-oxopiperidine-3-carboxamido)phenoxy)picolinamide (15a, from 9 and 3): 76% yield; 1H-NMR (600 MHz, DMSO-d6) δ 10.56 (s, 1H, NH), 8.33 (d, 1H, J = 5.5 Hz, ArH), 8.07 (s, 1H, NH), 7.91 (dd, 1H, J = 12.9, 2.4 Hz, ArH), 7.77 (s, 1H, NH), 7.44 (dd, 1H, J = 8.9, 2.4 Hz, ArH), 7.41 (t, 1H, J = 8.8 Hz, ArH), 7.36–7.32 (m, 2H, ArH), 7.26–7.20 (m, 2H, ArH), 6.84 (dd, 1H, J = 5.5, 1.1 Hz, ArH), 3.76–3.68 (m, 1H, CH), 3.65–3.57 (m, 2H, CH2), 2.21–2.13 (m, 2H, CH2), 2.12–2.04 (m, 1H, CHH), 1.96–1.87 (m, 1H, CHH); 13C-NMR (150 MHz, DMSO-d6) δ 169.3, 166.5, 160.9, 160.0, 159.3, 154.2, 153.8, 152.2, 148.7, 139.4, 138.5, 138.4, 134.8, 134.7, 128.3, 128.2, 123.7, 116.3, 115.9, 115.6, 115.4, 110.6, 107.9, 107.8, 54.9, 51.2, 50.5, 48.6, 24.8, 21.2; MS (ESI pos ion) m/z: calcd for C24H19ClF2N4O4 500.1, found 501.1 [M + H]+; HR-MS (ESI) Calcd for C24H20ClF2N4O4 501.1141 [M + H]+, found 501.1160.
3-Chloro-4-(4-(3-chloro-1-(4-fluorophenyl)-2-oxopiperidine-3-carboxamido)-2-fluorophenoxy)-picolinamide (15b, from 9 and 4) 68% yield; 1H-NMR (600 MHz, CDCl3) δ 10.11 (s, 1H, NH), 8.24 (d, 1H, J = 5.5 Hz, ArH), 7.80 (dd, 1H, J = 11.9, 2.5 Hz, ArH), 7.54 (d, 1H, J = 3.9 Hz, ArH), 7.33–7.22 (m, 3H, ArH), 7.18–7.11 (m, 3H, NH), 6.68 (dd, 1H, J = 5.5, 1.1 Hz, ArH), 6.15 (d, 1H, J = 3.4 Hz, ArH), 3.85–3.78 (m, 1H, CHH), 3.73–3.68 (m, 1H, CHH), 2.96–2.87 (m, 1H, CHH), 2.65–2.56 (m, 1H, CHH), 2.45–2.34 (m, 1H, CHH), 2.17–2.07 (m, 1H, CHH); 13C-NMR (150 MHz, CDCl3) δ 166.8, 166.0, 164.9, 162.6, 161.8, 160.9, 154.7, 153.0, 148.3, 147.0, 137.9, 136.8, 136.8, 136.8, 127.9, 127.8, 123.4, 121.2, 116.7, 116.6, 116.5, 111.7, 109.8, 109.6, 64.4, 52.6, 33.8, 19.4; MS (ESI pos ion) m/z: calcd for C24H18Cl2F2N4O4 534.1 found 535.1 [M + H]+; HR-MS (ESI) Calcd for C24H19Cl2F2N4O4 535.0752 [M + H]+, found 535.0764.
N-(4-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)-3-fluorophenyl)-1-(4-fluorophenyl)-2-oxopiperidine-3-carboxamide (17a, from 11 and 3): 31% yield, 1H-NMR (600 MHz, CDCl3) δ 11.02 (s, 1H, NH), 10.19 (s, 1H, NH), 8.87 (s, 1H, ArH), 8.54 (s, 1H, ArH), 8.13–7.93 (m, 1H, ArH), 7.81 (d, 1H, J = 11.6 Hz, ArH), 7.42 (s, 1H, ArH), 7.31–7.20 (m, 2H, ArH), 7.16–7.03 (m, 2H, ArH), 6.92 (s, 1H, ArH), 3.74–3.59 (m, 2H, NCH2), 3.59–3.47 (m, 1H, CH), 2.63–2.51 (m, 1H, CHH), 2.26–2.16 (m, 1 H, CHH), 2.15–2.07 (m, 1 H, CHH), 2.08–1.98 (m, 1 H, CHH); 13C-NMR (150 MHz, CDCl3) δ 169.5, 168.3, 162.5, 160.9, 156.2, 155.9, 154.6, 152.4, 151.1, 139.8, 138.5, 134.5, 134.2, 128.2, 128.1, 126.2, 123.0, 116.9, 116.8, 116.7, 116.6, 115.8, 115.6, 109.4, 109.2, 100.3, 53.1, 47.8, 47.7, 29.7, 22.8, 21.6; MS (ESI pos ion) m/z: calcd for C24H19F2N5O3 463.1, found 464.2 [M + H]+; HR-MS (ESI) Calcd for C24H20F2N5O3 464.1534 [M + H]+, found 464.1547.
N-(4-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)-3-fluorophenyl)-3-chloro-1-(4-fluorophenyl)-2-oxo-piperidine-3-carboxamide (17b, from 11 and 4): 42% yield; 1H-NMR (600 MHz, CDCl3) δ 8.25 (s, 1H, ArH), 7.89 (dd, J = 12.5, 2.5 Hz, 1H, ArH), 7.50 (d, J = 3.6 Hz, 1H, ArH), 7.48 (dd, J = 2.5, 1.3 Hz, 1H, ArH), 7.46–7.42 (m, 3H, ArH), 7.38 (t, J = 8.7 Hz, 1H, ArH), 7.25–7.19 (m, 2H, ArH), 6.64 (d, J = 3.6 Hz, 1H, ArH), 3.98–3.88 (m, 1H. CHH), 3.84–3.78 (m, 1H. CHH), 2.96 (ddd, J = 14.8, 11.6, 3.2 Hz, 1H. CHH), 2.59–2.52 (m, 1H. CHH), 2.40–2.31 (m, 1H. CHH), 2.21–2.11 (m, 1H. CHH); 13C-NMR (150 MHz, CDCl3) δ 169.7, 168.5, 162.7, 160.9, 156.3, 155.9, 154.7, 152.4, 151.3, 139.8, 138.6, 134.7, 134.5, 128.4, 128.3, 126.4, 123.6, 117.0, 116.8, 116.7, 116.6, 115.9, 115.7, 109.6, 109.4, 100.6, 64.5, 53.5, 47.9, 47.7, 29.9, 22.8, 21.8; MS (ESI pos ion) m/z: calcd for C24H18ClF2N5O3 497.1, found 498.0 [M + H]+; HR-MS (ESI) Calcd for C24H19ClF2N5O3 498.1145 [M + H]+, found 498.1155.
N-(3-fluoro-4-((2-((4-methoxybenzyl)amino)pyrimidin-4-yl)oxy)phenyl)-1-(4-fluorophenyl)-2-oxo-piperidine-3-carboxamide (18a, from 13 and 3): 69% yield, 1H-NMR (600 MHz, CDCl3) δ 10.19 (s, 1H, NH), 8.06 (s, 1H, ArH), 7.75 (d, 1H, J = 12.2 Hz, ArH), 7.24–7.18 (m, 3H, ArH), 7.11 (t, 2H, J = 8.3 Hz, ArH), 7.06 (t, 1H, J = 8.4 Hz, ArH), 7.00 (d, 1H, J = 8.6 Hz, ArH), 6.78 (s, 2H, ArH), 6.23 (s, 1H, ArH), 4.24 (s, 2H, CH2), 3.76 (s, 3H, OCH3), 3.71–3.62 (m, 2H, NCH2), 3.62–3.57 (m, 1H, CH), 2.58–2.48 (m, 1H, CHH), 2.25–2.14 (m, 1H, CHH), 2.13–2.05 (m, 1H, CHH), 2.05–1.97 (m, 1H, CHH); 13C-NMR (150 MHz, CDCl3) δ 169.3, 166.0, 162.4, 160.8, 159.0, 156.7, 154.8, 138.4, 129.3, 128.2, 128.1, 123.7, 116.6, 116.5, 115.3, 114.0, 55.4, 52.7, 47.7, 22.9, 21.7; MS (ESI pos ion) m/z: calcd for C30H27F2N5O4 559.2, found 560.2 [M + H]+; HR-MS (ESI) Calcd for C30H28F2N5O4 560.2109 [M + H]+, found 560.2125.
3-Chloro-N-(3-fluoro-4-((2-((4-methoxybenzyl)amino)pyrimidin-4-yl)oxy)phenyl)-1-(4-fluorophenyl)-2-oxopiperidine-3-carboxamide (18b, from 13 and 4):57% yield; 1H-NMR (600 MHz, CDCl3) δ 9.96 (s, 1H, NH), 8.12 (s, 1H, ArH), 7.69 (dd, J = 11.8, 2.5 Hz, 1H, ArH), 7.25–7.22 (m, 2H, ArH), 7.19 (dd, 1H, J = 8.9, 2.4 Hz, ArH), 7.15–7.08 (m, 4H, ArH), 6.80 (d, 2H, J = 8.0 Hz, ArH), 6.18 (d, 1H, J = 5.7 Hz, ArH), 4.32 (s, 2H, CH2), 3.81–3.75 (m, 1H, CHH), 3.77 (s, 3H, OCH3), 3.69 (dt, 1H, J = 12.4, 4.9 Hz, CHH), 2.90 (ddd, 1H, J = 14.6, 11.3, 3.0 Hz, CHH), 2.63–2.54 (m, 1H, CHH), 2.41–2.31 (m, 1H, CHH), 2.15–2.06 (m, 1H, CHH); 13C-NMR (150 MHz, CDCl3) δ 166.9, 164.6, 162.6, 162.1, 160.9, 159.5, 158.9, 157.7, 155.3, 153.70, 138.0, 136.4, 136.3, 135.8, 135.8, 130.9, 129.1, 127.9, 127.8, 124.2, 116.7, 116.5, 115.7, 113.9, 64.4, 55.4, 55.3, 52.7, 45.0, 33.9, 19.5; MS (ESI pos ion) m/z: calcd for C30H26ClF2N5O4 593.2, found 594.2 [M + H]+; HR-MS (ESI) Calcd for C30H27ClF2N5O4 594.1720 [M + H]+, found 594.1733.
N-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-3-fluorophenyl)-1-(4-fluorophenyl)-2-oxopiperidine-3-carbox-amide (20a, from 14 and 3): 61% yield; 1H-NMR (600 MHz, CDCl3) δ 10.30 (s, 1H, NH), 8.51 (s, 1 H, ArH), 7.84 (dd, 1H, J = 12.1, 2.5 Hz, ArH), 7.69 (s, 1H, ArH), 7.59 (s, 1H, ArH), 7.29–7.26 (m, 1H, ArH), 7.25–7.21 (m, 2H, ArH), 7.18 (t, 1H, J = 8.6 Hz, ArH), 7.12 (t, 2H, J = 8.5 Hz, ArH), 6.49 (d, 1H, J = 5.5 Hz, ArH), 4.08 (s, 3H, OCH3), 4.06 (s, 3H, OCH3), 3.75–3.60 (m, 2H, CHH), 3.58–3.49 (m, 1H, CH), 2.60–2.47 (m, 1H, CHH), 2.29–2.19 (m, 1H, CHH), 2.14–1.97 (m, 2H, CH2); 13C-NMR (150 MHz, CDCl3) δ 169.3, 166.4, 154.6, 150.6, 145.7, 138.4, 128.1, 123.5, 116.6, 116.5, 116.2, 99.7, 56.7, 56.4, 52.7, 47.8, 29.8, 23.0, 22.8, 21.7; MS (ESI pos ion) m/z: calcd for C29H25F2N3O5 533.2, found 534.2 [M + H]+; HR-MS (ESI) Calcd for C29H26F2N3O5 534.1841 [M + H]+, found 534.1850.
3-Chloro-N-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-3-fluorophenyl)-1-(4-fluorophenyl)-2-oxopiperidine-3-carboxamide (20b, from 14 and 4): 54% yield; 1H-NMR (600 MHz, CDCl3) δ 10.05 (s, 1H, NH), 8.48 (d, 1H, J = 5.4 Hz, ArH), 7.79 (dd, 1H, J = 12.0, 2.5 Hz, ArH), 7.57 (s, 1H, ArH), 7.44 (s, 1H, ArH), 7.29–7.23 (m, 4H, ArH), 7.20 (t, 1H, J = 8.6 Hz, ArH), 7.13 (t, 2H, J = 8.4 Hz, ArH), 6.39 (d, 1H, J = 5.3 Hz, ArH), 4.05 (s, 3H, OCH3), 4.04 (s, 3H, OCH3), 3.80 (ddd, 1H, J = 14.5, 10.3, 4.7 Hz, CHH), 3.74–3.66 (m, 1H, CHH), 2.97–2.83 (m, 1H, CHH), 2.67–2.56 (m, 1H, CHH), 2.45–2.34 (m, 1H, CHH), 2.15–2.07 (m, 1H, CHH); 13C-NMR (150 MHz, CDCl3) δ 166.9, 164.8, 162.6, 161.0, 160.2, 155.3, 153.6, 153.1, 149.8, 148.6, 146.7, 137.9, 137.7, 137.6, 136.3, 136.2, 127.9, 127.8, 123.8, 116.6, 116.5, 116.4, 115.6, 113.0, 109.8, 109.7, 109.6, 109.6, 107.8, 107.74, 99.5, 99.5, 64.3, 56.3, 56.2, 52.7, 33.8, 29.8, 19.5; MS (ESI pos ion) m/z: calcd for C29H24ClF2N3O5 567.1, found 568.1 [M + H]+; HR-MS (ESI) Calcd for C29H25ClF2N3O5 568.1451 [M + H]+, found 568.1461.
N-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-3-fluorophenyl)-1-(4-fluorophenyl)-3-methyl-2-oxopiperidine-3-carboxamide (20c, from 14 and 6a): 68% yield; 1H-NMR (600 MHz, CDCl3) δ 10.01 (s, 1H, NH), 8.47 (d, 1H, J = 5.3 Hz, ArH), 7.82 (dd, 1H, J = 12.2, 2.5 Hz, ArH), 7.58 (s, 1H, ArH), 7.42 (s, 1H, ArH), 7.25–7.20 (m, 3H, ArH), 7.19 (t, 1H, J = 8.5 Hz, ArH), 7.15–7.11 (m, 2H, ArH), 6.38 (d, 1H, J = 5.1 Hz, ArH), 4.06 (s, 3H, OCH3), 4.04 (s, 3H, OCH3), 3.71–3.62 (m, 2H, NCH2), 2.83–2.76 (m, 1H, CHH), 2.07–1.97 (m, 2H, CH2), 1.87–1.81 (m, 1H, CHH), 1.71 (s, 3H, CH3); 13C-NMR (150 MHz, CDCl3) δ 173.7, 170.1, 162.4, 160.8, 160.2, 153.7, 153.0, 149.7, 148.9, 146.9, 138.7, 137.1, 128.2, 123.8, 116.6, 116.5, 116.0, 116.0, 115.6, 109.2, 107.9, 102.3, 99.5, 56.3, 56.2, 53.1, 50.5, 30.9, 27.7, 20.7, 14.3; MS (ESI pos ion) m/z: calcd for C30H27F2N3O5 547.2, found 548.2 [M + H]+; HR-MS (ESI) Calcd for C30H28F2N3O5 548.1997 [M + H]+, found 548.2012.
N-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-3-fluorophenyl)-3-ethyl-1-(4-fluorophenyl)-2-oxopiperidine-3-carboxamide (20d, from 14 and 6b): 58% yield; 1H-NMR (600 MHz, CDCl3) δ 9.99 (s, 1H, NH), 8.47 (d, 1H, J = 5.4 Hz, ArH), 7.82 (dd, 1H, J = 12.1, 2.4 Hz, ArH), 7.58 (s, 1H, ArH), 7.44 (s, 1H, ArH), 7.24–7.16 (m, 4H, ArH), 7.15–7.11 (m, 2H, ArH), 6.39 (d, 1H, J = 5.2 Hz, ArH), 4.06 (s, 3H, OCH3), 4.05 (s, 3H, OCH3), 3.71–3.59 (m, 2H, NCH2), 2.81 (1H, ddd, J = 13.9, 6.4, 2.7 Hz, CHH), 2.22–2.14 (m, 1H, CHH), 2.12–1.93 (m, 4H, CH2CH3), 1.82–1.76 (m, 1H, CHH), 1.02 (t, 3H, J = 7.4 Hz, CH2CH3); 13C-NMR(150 MHz, CDCl3) δ 173.3, 169.0, 165.8, 162.5, 160.4, 155.3, 153.7, 153.1, 149.7, 148.7, 146.8, 138.8, 137.1, 137.1, 137.0, 136.9, 128.3, 115.7, 107.8, 102.3, 99.6, 56.3, 55.2, 53.1, 33.8, 27.0, 20.8; MS (ESI pos ion) m/z: calcd for C31H29F2N3O5 561.2, found 562.2 [M + H]+; HR-MS (ESI) Calcd for C31H30F2N3O5 562.2154 [M + H]+, found 562.2160.
N-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-3-fluorophenyl)-3-butyl-1-(4-fluorophenyl)-2-oxopiperidine-3-carboxamide (20e, from 14 and 6c): 64% yield; 1H-NMR (600 MHz, CDCl3) δ 9.98 (s, 1H, NH), 8.47 (d, 1H, J = 5.4 Hz, ArH), 7.82 (dd, 1H, J = 12.5, 2.6 Hz, ArH), 7.58 (s, 1H, ArH), 7.42 (s, 1H, ArH), 7.25–7.16 (m, 4H, ArH), 7.16–7.11 (m, 2H, ArH), 6.39 (d, 1H, J = 5.1 Hz, ArH), 4.06 (s, 3H, OCH3), 4.05 (s, 3H, OCH3), 3.71–3.59 (m, 2H, NCH2), 2.82 (ddd, 1H, J = 14.0, 6.1, 2.7 Hz, CHH), 2.13–1.92 (m, 4H, 2 × CH2), 1.84–1.77 (m, 1H, CHH), 1.42–1.28 (m, 4H, 2 × CH2), 0.95–0.89 (m, 3H, CH2CH3); 13C-NMR (150 MHz, CDCl3) δ 173.3, 169.1, 160.3, 153.0, 149.7, 148.9, 147.0, 138.8, 137.1, 128.3, 128.2, 123.8, 116.7, 116.5, 116.0, 115.7, 109.4, 108.0, 102.3, 99.6, 56.3, 56.3, 54.9, 53.1, 40.5, 27.5, 27.0, 23.0, 20.9, 14.1; MS (ESI pos ion) m/z: calcd for C33H33F2N3O5 589.2, found 590.2 [M + H]+; HR-MS (ESI) Calcd for C33H34F2N3O5 590.2467 [M + H]+, found 590.2478.
3.2.4. Preparation of 16a and 16b
To amide 15a or 15b (0.2 mmol) in ethyl acetate (2 mL), acetonitrile (2 mL), and water (1 mL) at 0 °C was added iodobenzene diacetate (82 mg, 0.26 mmol). After stirring at room temperature for 2 h, saturated NaHCO3 (3 mL) was added, followed by 30 mL of ethyl acetate. The mixture was filtered, and the filtrate was washed with brine (3 × 5 mL), dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography on silica gel to give compounds 16a–b.
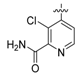
N-(4-((2-amino-3-chloropyridin-4-yl)oxy)-3-fluorophenyl)-1-(4-fluorophenyl)-2-oxopiperidine-3-carboxamide (16a). white solid; yield 72%; 1H-NMR (600 MHz, CDCl3) δ 10.00 (s, 1H, NH), 7.71 (dd, 1H, J = 12.6, 2.5 Hz, ArH), 7.24–7.18 (m, 3H, ArH), 7.17–7.10 (m, 3H, ArH), 7.02 (dt, 1H, J = 9.0, 1.8 Hz, ArH), 6.62 (t, 1H, J = 8.9 Hz, ArH), 5.01 (s, 2H, NH2), 3.65 (dq, 2H, J = 7.2, 4.3, 3.4 Hz, NCH2), 3.54 (t, J = 6.3 Hz, 1H, CH), 2.59–2.49 (m, 1H, CHH), 2.21–2.15 (m, 1H, CHH), 2.10–2.05 (m, 1H, CHH), 2.04–1.98 (m, 1H, CHH); 13C-NMR (150 MHz, CDCl3) 169.4, 165.7, 162.5, 160.9, 155.6, 154.7, 151.1, 146.5, 139.9, 138.5, 134.5, 134.4, 128.2, 128.2, 123.3, 116.9, 116.8, 116.7, 116.6, 116.6, 115.3, 115.2, 109.4, 109.2, 108.9, 52.8, 47.6, 47.5, 29.8, 22.9, 21.8; MS (ESI pos ion) m/z: calcd for C23H19ClF2N4O3 472.1, found 473.1 [M + H]+; HR-MS (ESI) Calcd for C23H20ClF2N4O3 473.1192 [M + H]+, found 473.1214.
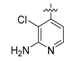
N-(4-((2-amino-3-chloropyridin-4-yl)oxy)-3-fluorophenyl)-3-chloro-1-(4-fluorophenyl)-2-oxo-piperidine-3-carboxamide (16b). white solid; yield 76%; 1H-NMR (600 MHz, CDCl3) δ 10.02 (s, 1H, NH), 7.77 (d, 1H, J = 5.8 Hz, ArH), 7.74 (dd, 1H, J = 12.0, 2.5 Hz, ArH), 7.26–7.23 (m, 2H, ArH), 7.23–7.20 (m, 1H, ArH), 7.16–7.11 (m, 3H, ArH), 5.99 (dd, 1H, J = 5.8, 1.0 Hz, ArH), 5.04 (br s, 2H, NH2), 3.84–3.74 (m, 1H, CHH), 3.74–3.66 (m, 1H, CHH), 2.94–2.82 (m, 1H, CHH), 2.63–2.57 (m, 1H, CHH), 2.43–2.34 (m, 1H, CHH), 2.14–2.09 (m, 1H, CHH); 13C-NMR (150 MHz, CDCl3) δ 166.9, 164.8, 162.6, 161.0, 160.5, 156.6, 155.0, 153.3, 148.6, 147.8, 146.7, 137.9, 136.2, 127.8, 123.4, 116.7, 116.6, 116.2, 109.6, 109.4, 102.6, 102.1, 91.8, 64.2, 52.7, 33.8, 19.5; MS (ESI pos ion) m/z: calcd for C23H18Cl2F2N4O3 506.1, found 507.1 [M + H]+; HR-MS (ESI) Calcd for C23H19Cl2F2N4O3 507.0802 [M + H]+, found 507.0816.
3.2.5. Preparation of 19a and 19b
Compound 18a or 18b (40 mg, 0.07 mmol) was dissolved in TFA and heated to reflux for 6 h. The solvent was removed and the residue was purified by column chromatography, giving the title compound 19a or 19b as a pale yellow solid.
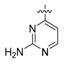
N-(4-((2-aminopyrimidin-4-yl)oxy)-3-fluorophenyl)-1-(4-fluorophenyl)-2-oxopiperidine-3-carbox-amide (19a). 71% yield; 1H-NMR (600 MHz, CDCl3) δ 10.20 (s, 1 H, NH), 7.95 (s, 1H, ArH), 7.75 (d, 1H, J = 11.8 Hz, ArH), 7.21 (dd, 2H, J = 8.7, 4.8 Hz, ArH), 7.18 (d, 1H, J = 8.6 Hz, ArH), 7.13 (t, 2H, J = 8.2 Hz, ArH), 7.07 (t, 1H, J = 8.4 Hz, ArH), 6.49 (s, 1H, ArH), 5.77 (s, 1H, NH), 3.67 (q, 2H, J = 6.0 Hz, NCH2), 3.57 (s, 1H, CH), 2.57–2.46 (m, 1H, CHH), 2.25–2.16 (m, 1H, CHH), 2.13–1.95 (m, 2H, CH2); 13C-NMR (150 MHz, CDCl3) δ 166.3, 162.5, 160.9, 154.4, 152.8, 138.4, 137.8, 134.1, 128.2, 128.1, 123.1, 116.7, 116.5, 115.6, 108.9, 108.7, 52.7, 47.7, 29.8, 23.0, 21.7; MS (ESI pos ion) m/z: calcd for C22H19F2N5O3 439.1, found 440.1 [M + H]+; HR-MS (ESI) Calcd for C22H20F2N5O3 440.1534 [M + H]+, found 440.1528.
N-(4-((2-aminopyrimidin-4-yl)oxy)-3-fluorophenyl)-3-chloro-1-(4-fluorophenyl)-2-oxopiperidine-3-carboxamide (19b). 87% yield; 1H-NMR (600 MHz, CDCl3) δ 10.04 (s, 1H, NH), 7.96 (d, 1H, J = 6.7 Hz, ArH), 7.72 (dd, 1H, J = 11.8, 2.5 Hz, ArH), 7.25–7.18 (m, 3H, ArH), 7.16–7.06 (m, 3H, ArH), 6.47 (d, 1H, J = 6.7 Hz, ArH), 3.79 (ddd, 1H, J = 12.4, 10.1, 4.7 Hz, CHH), 3.69 (dt, 1H, J = 11.3, 4.5 Hz, CHH), 2.88 (ddd, 1H, J = 14.9, 11.6, 3.1 Hz, CHH), 2.63–2.49 (m, 1H, CHH), 2.44–2.31 (m, 1H, CHH), 2.15–2.05 (m, 1H, CHH); 13C-NMR (150 MHz, CDCl3) δ 171.2, 166.8, 164.9, 162.6, 161.0, 154.5, 152.8, 137.9, 137.9, 137.2, 137.1, 134.8, 134.7, 127.9, 127.9, 127.8, 123.3, 116.7, 116.6, 116.0, 109.3, 109.2, 109.1, 109.1, 100.0, 99.0, 64.4, 52.7, 33.8, 19.4; MS (ESI pos ion) m/z: calcd for C22H18ClF2N5O3 473.1, found 474.0 [M + H]+; HR-MS (ESI) Calcd for C22H19ClF2N5O3 474.1145 [M + H]+, found 474.1151.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}