General Information
All reactions were carried out under an argon atmosphere unless otherwise noted. When necessary, solvents were dried prior to use. Dry THF, dry Et2O and dry CH2Cl2 were purchased from Kanto Chemical Co., Inc. (Tokyo, Japan). Optical rotations were measured on a JASCO P-2200 digital polarimeter with a sodium (D line) lamp. IR spectra were recorded on a JASCO Model A-202 spectrophotometer. 1H-NMR spectra and 13C-NMR spectra were obtained on JEOL JNM-GX400, JNM-α400, JNM-AL400 or JNM-ECX400 spectrometers in deuterated solvent using tetramethylsilane as an internal standard. Deuteriochloroform was used as a solvent, unless otherwise stated. High-resolution mass spectra were obtained on a Waters LCT Premier XE (ESI) or JEOL JMS-700 (FAB). Preparative and analytical TLC were carried out on silica gel plate (Kieselgel 60 F254, E. Merck AG., Darmstadt, Germany) using UV light, 1 M aq. sulfuric acid, and/or 5% molybdophosphoric acid in ethanol for detection. Kanto silica 60N (spherical, neutral, 105–210 μm) was used for column chromatography. All anodic oxidation was conducted using HA-151A (Hokuto Denko, Tokyo, Japan) as a potentiostat and galvanostat, using glassy carbon plate as anodes, and platinum plate or wire as cathode.
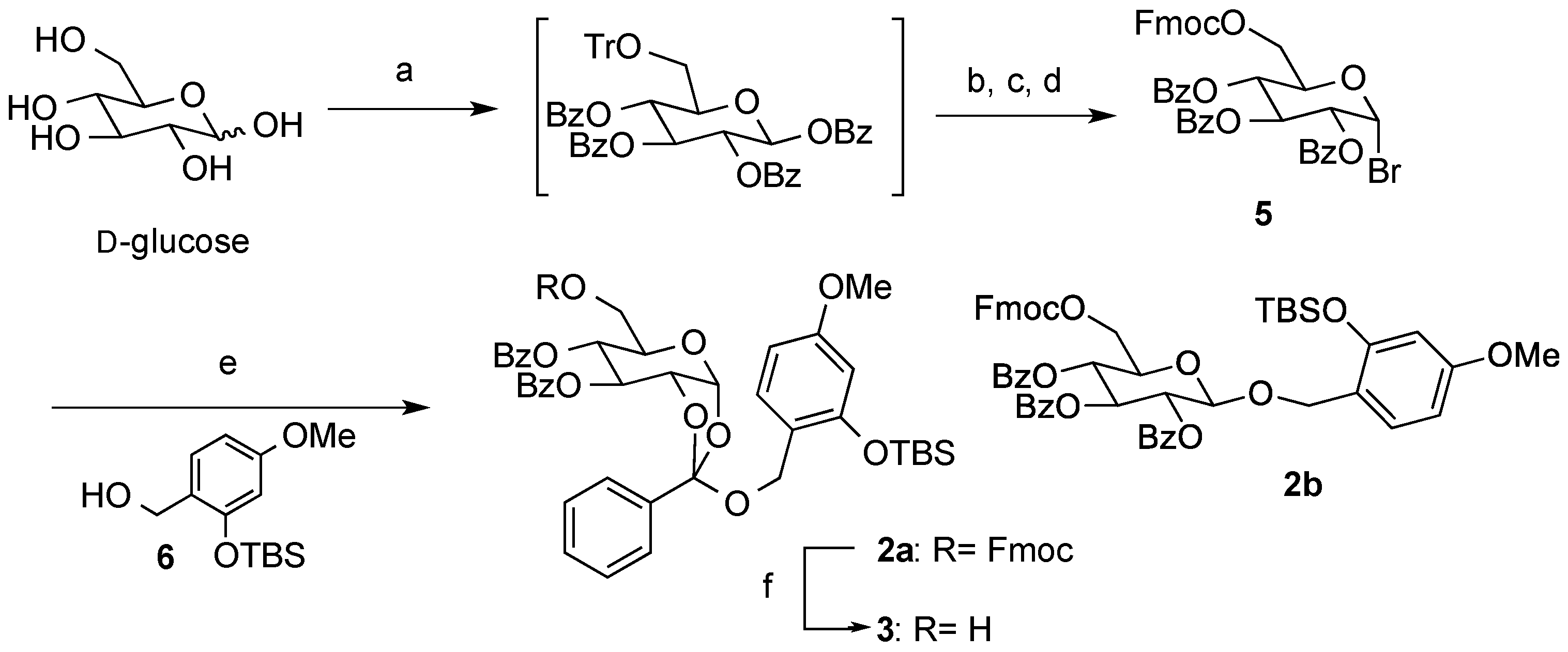
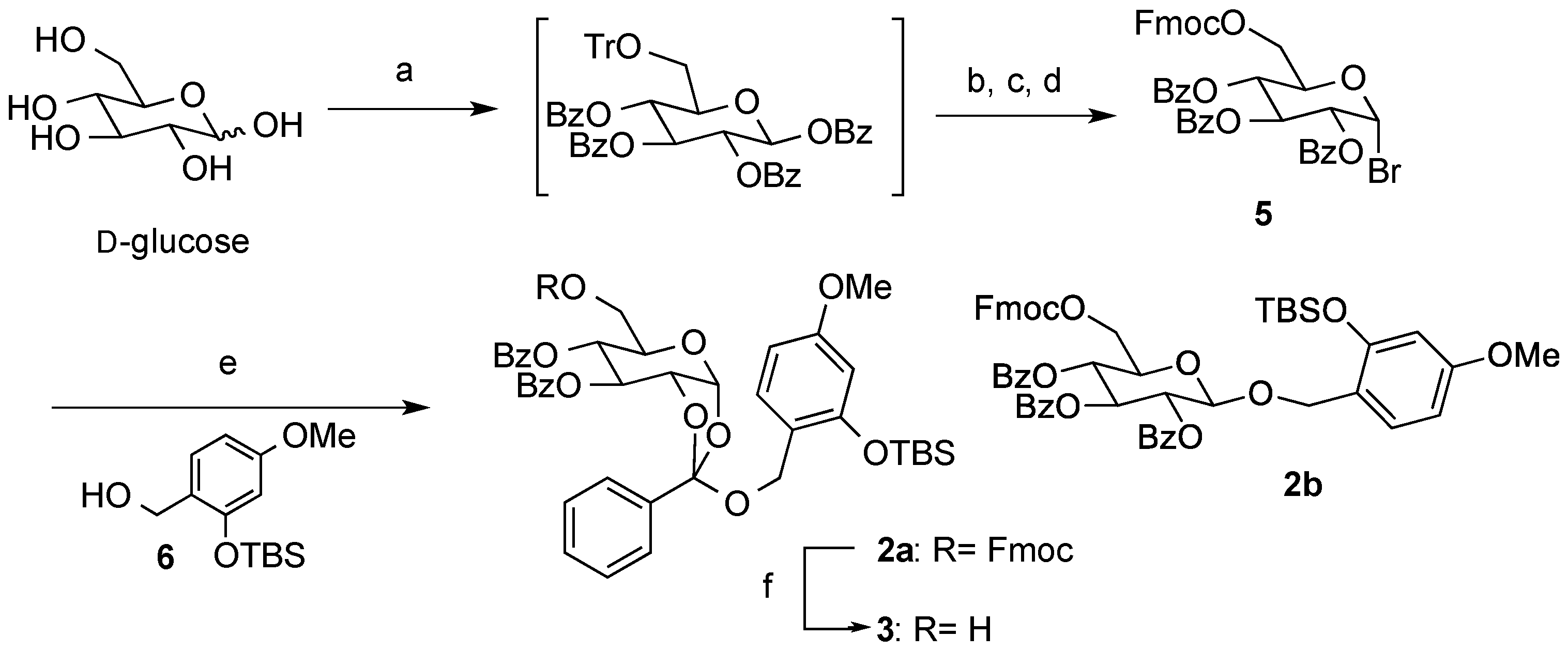
2,3,4-Tri-O-benzoyl-6-O-9-fluorenylmethyloxycarboxyl-α-D-glucopyranosyl bromide (5). To a suspension of d-glucose (4.55 g, 0.025 mol) in pyridine (50 mL) were added TrCl (10.6 g, 0.038 mol) and Et3N (18 mL, 0.13 mol), and the mixture was stirred at room temperature overnight. After the addition of BzCl (23 mL, 0.20 mol) at the same temperature, stirring was continued overnight. The reaction was quenched with sat. aq. NaHCO3, and the mixture was extracted with CHCl3. The organic layer was dried (Na2SO4), and concentrated in vacuo. After removal of the high polarity byproducts by silica gel short column (hexane/EtOAc = 5:1), a crude was used in the next step without further purification. The crude product was solved in MeOH/EtOAc (1:1, 100 mL). After the addition of 5% Pd-C (cat.), the mixture was stirred overnight under H2 atmosphere. The mixture was filtered and the filtrate was concentrated in vacuo. After silica gel short column chromatography (hexane/EtOAc = 3:1), a crude was used in the next step without further purification.
A mixture of the crude and FmocCl (9.80 g, 0.038 mol) in pyridine (100 mL) was stirred overnight. The reaction was quenched by sat. aq. NaHCO3, and the mixture was extracted with CHCl3. The organic layer was dried (Na2SO4), and concentrated in vacuo. After purification by silica gel short column chromatography (hexane/EtOAc = 10:1), a crude was used in the next step without further purification. A mixture of the crude, HBr-AcOH (250 mL), AcOH (60 mL), and Ac2O (60 mL) was stirred overnight. The mixture was extracted with CHCl3. The organic layer was dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 5:1) to give 5 as a colorless oil (8.43 g, 43% in 4 steps): [α]23D +76.4 (c 1.00, CHCl3); IR (film) 1732, 1601, 1261 cm−1; 1H-NMR (400 MHz) δ 4.40 (5H, m, H-6a,6b, Fmoc), 4.67 (1H, m, H-5), 5.36 (1H, dd, J = 9.8, 4.0 Hz, H-2), 5.78 (1H, t, J = 9.8 Hz, H-4), 6.28 (1H, t, J = 9.8 Hz, H-3), 6.89 (1H, d, J = 4.0 Hz, H-4), 7.40 (11H, m, Ar), 7.54 (2H, m, Ar), 7.64 (2H, m, Ar), 7.78 (2H, m, Ar), 7.89 (2H, m, Ar), 8.00 (4H, m, Ar); 13C-NMR (100 MHz) δ 46.5 (Fmoc), 64.9 (C-6), 67.7 (C-4), 70.4 (C-3), 70.5 (C-2), 71.3 (C-5), 72.5 (FmocCH2), 86.7 (C-1), 120.0 (Ar), 125.2 (Ar), 125.3 (Ar), 127.2 (Ar), 127.9 (Ar), 128.3 (Ar), 128.4 (Ar), 128.5 (Ar), 128.7 (Ar), 129.7 (Ar), 129.9 (Ar), 130.0 (Ar), 133.3 (Ar), 133.7 (Ar), 133.85 (Ar), 141.2 (Ar), 141.3 (Ar), 143.2 (Ar), 143.4 (Ar), 154.7 (C=O), 165.1 (C=O), 165.2 (C=O), 165.5 (C=O). ESI-MS: calcd for C42H33O10NaBr 799.1155 (M+Na)+, found m/z 799.1151.
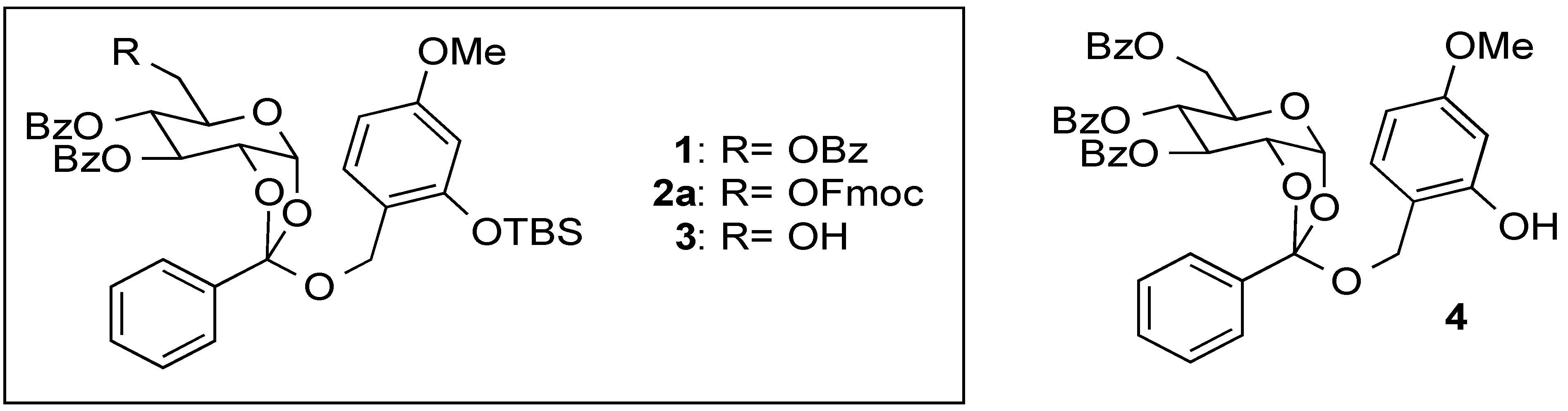
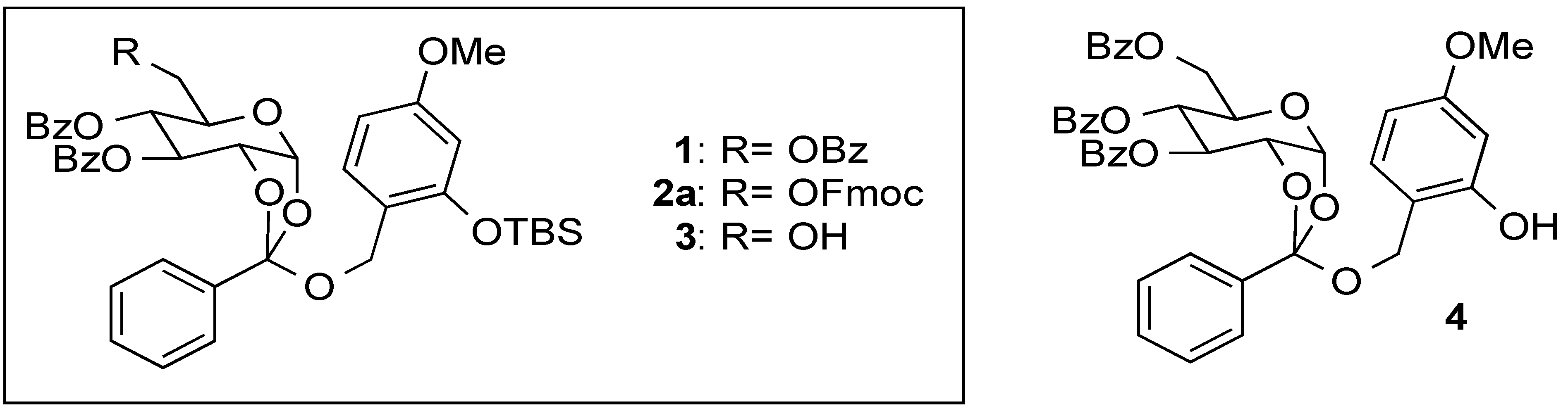
(2R,5R,6R,7S)-5-(((((9H-Fluoren-9-yl)methoxy)carbonyl)oxy)methyl)-2-((2-((tert-butyldimethylsilyl)oxy)-4-methoxybenzyl)oxy)-2-phenyltetrahydro-3aH-[1,3]dioxolo[4,5-b]pyran-6,7-diyl dibenzoate (2a) and 2-tert-Buthyldimethylsyloxy-4-methoxyphenylmethyl2,3,4-tetra-O-benzoyl-6-O-9-fluorenylmethyloxycarboxyl-β-d-glucopyranoside (2b). To a solution of 5 (71 mg, 0.092 mmol) in anhydrous toluene (0.9 mL) were added 6 (74 mg, 0.27 mmol), Hg(CN)2 (46 mg, 0.18 mmol), and MS 4A at 100 °C, and the mixture was stirred overnight. The mixture was filtered through a Celite pad, and the filtrate was washed with sat. aq. NaHCO3. The organic extracts were dried (Na2SO4), and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/acetone = 7:1) to give 2a (45 mg, 50%) and 2b (26 mg, 30%) as oil: 2a: [α]23D +1.2 (c 1.00, CHCl3); IR (film) 1728, 1613, 1257 cm−1; 1H-NMR (400 MHz) δ 0.15 (6H, s, SiCH3), 0.93 (9H, s, tBu), 3.74 (3H, s, OCH3), 4.16 (2H, m, Fmoc), 4.29 (6H, m, H-5,6a,6b, ArCH2, Fmoc), 4.78 (1H, m, H-2), 5.37 (1H, bd, H-4), 5.76 (1H, br, H-3), 6.04 (1H, bd, H-1), 6.31 (1H, d, J = 2.2 Hz, Ar), 6.48 (1H, dd, J = 8.3, 2.2 Hz, Ar), 7.18 (1H, d, J = 8.3 Hz, Ar), 7.41 (11H, m, Ar), 7.57 (4H, m, Ar), 7.75 (2H, m, Ar), 7.82 (2H, m, Ar), 7.97 (2H, m, Ar), 8.11 (2H, m, Ar); 13C-NMR (100 MHz) δ −4.3 (SiCH3), 18.1 (tBu), 25.6 (tBu), 46.6 (Fmoc), 55.2 (OCH3), 61.3 (ArCH2), 67.3 (C-6), 68.3 (C-2), 69.2 (C-3), 70.1 (C-4), 72.3 (FmocCH2), 77.2 (C-5), 97.4 (C-1), 105.3 (Ar), 105.9 (Ar), 119.9 (Ar), 120.5 (Ar), 121.4 (Ar), 125.3 (orthoester-C), 126.5 (Ar), 127.1 (Ar), 127.2 (Ar), 127.8 (Ar), 128.3 (Ar), 1283.4 (Ar), 128.5 (Ar), 129.1 (Ar), 129.6 (Ar), 129.9 (Ar), 130.0 (Ar), 133.5 (Ar), 133.6 (Ar), 135.3 (Ar), 141.2 (Ar), 143.2 (Ar), 154.0 (C=O), 154.9 (C=O), 159.9 (C=O), 165.2 (C=O). ESI-MS: calcd for C56H56O13SiNa 987.3388 (M+Na)+, found m/z 987.3373. 2b: [α]23D −14.9 (c 1.00, CHCl3); IR (film) 1735, 1610, 1262 cm−1; 1H-NMR (400 MHz) δ 0.10 (3H, s, SiCH3), 0.16 (3H, s, SiCH3), 0.94 (9H, s, tBu), 3.72 (3H, s, OCH3), 4.06 (1H, ddd, J = 9.8, 5.3, 3.2 Hz, H-5), 4.23 (1H, t, J = 7.6 Hz, Fmoc), 4.40 (4H, m, H-6a,6b, Fmoc), 4.76 (1H, d, J = 12.4 Hz, ArCH2), 4.83 (1H, d, J = 12.4 Hz, ArCH2), 4.86 (1H, d, J = 8.0 Hz, H-1), 5.58 (1H, dd, J = 9.8, 8.0 Hz, H-2), 5.60 (1H, t, J = 9.8 Hz,H-4), 5.83 (1H, t, J = 9.8 Hz, H-3), 6.27 (1H, dd, J = 8.7, 2.5 Hz, Ar), 6.30 (1H, d, J = 2.5 Hz, Ar), 7.12 (1H, d, J = 8.7 Hz, Ar), 7.35 (14H, m, Ar), 7.50 (2H, m, Ar), 7.62 (2H, m, Ar), 7.76 (2H, m, Ar), 7.83 (4H, m, Ar), 7.92 (1H, m, Ar); 13C-NMR (100 MHz) δ −4.4 (SiCH3), −4.3 (SiCH3), 18.1 (tBu), 25.6 (tBu), 29.7 (tBu), 46.6 (Fmoc), 55.2 OCH3), 65.6 (ArCH2), 66.3 (C-6), 69.6 (C-4), 70.2 (C-3), 71.6 (C-2), 72.0 (C-5), 72.9 (FmocCH2), 99.1 (C-1), 105.4 (Ar), 105.9 (Ar), 119.6 (Ar), 119.9 (Ar), 125.2 (Ar), 125.3 (Ar), 127.2 (Ar), 127.8 (Ar), 128.1 (Ar), 128.2 (Ar), 128.4 (Ar), 128.8 (Ar), 129.3 (Ar), 129.7 (Ar), 129.8 (Ar), 129.9 (Ar), 131.2 (Ar), 133.0 (Ar), 133.2 (Ar), 133.4 (Ar), 141.2 (Ar), 143.2 (Ar), 143.4 (Ar), 154.7 (C=O), 154.9 (C=O), 160.2 (C=O), 164.9 (C=O), 165.2 (C=O), 165.8 (C=O). ESI-MS: calcd for C56H56O13SiNa 987.3388 (M+Na)+, found m/z 987.3381.
(2R,5R,6R,7S)-2-((2-((tert-Butyldimethylsilyl)oxy)-4-methoxybenzyl)oxy)-5-(hydroxymethyl)-2-phenyl-tetrahydro-3aH-[1,3]dioxolo[4,5-b]pyran-6,7-diyl dibenzoate (3). To a solution of 2a (30 mg, 0.032 mmol) in pyridine (0.5 mL) was added Et3N (13 µL, 0.095 mmol) at room temperature. After being stirred overnight, the reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/acetone = 2:1) to give 3 as a clear oil (19 mg, 81%): [α]23D +9.5 (c 1.00, CHCl3); IR (film) 3527, 1725, 1613, 1261 cm−1; 1H-NMR (400 MHz) δ 0.15 (6H, s, SiCH3), 0.90 (9H, s, tBu), 3.65 (1H, m, H-6), 3.75 (3H, s, OCH3), 3.78 (1H, m, H-6'), 3.89 (1H, m, H-5),4.29 (1H, d, J = 11.2 Hz, ArCH2), 4.36 (1H, d, J = 11.2 Hz, ArCH2), 4.75 (1H, ddd, J = 5.2, 2.9, 0.8 Hz, H-2), 5.37 (1H, m, H-4), 5.74 (1H, m, H-3), 6.03 (1H, bd, H-1), 6.31 (1H, d, J = 2.5 Hz, Ar), 6.48 (1H, dd, J = 8.5, 2.5 Hz, Ar), 7.18 (1H, d, J = 8.5 Hz, Ar), 7.44 (7H, m, Ar), 7.60 (2H, m, Ar), 7.81 (2H, m, Ar), 7.98 (2H, m, Ar), 8.05 (2H, m, Ar); 13C-NMR (100 MHz) δ −4.3 (SiCH3), 18.1 (tBu), 25.6 (tBu), 55.2 (OCH3), 61.2 (ArCH2), 62.4 (C-6), 68.3 (C-2), 69.4 (C-3), 69.8 (C-4), 72.6 (C-5), 97.5 (C-1), 105.3 (Ar), 105.9 (Ar), 120.5 (Ar), 121.1 (Ar), 126.4 (orthoester-C), 128.3 (Ar), 128.4 (Ar), 128.5 (Ar), 129.0 (Ar), 129.1 (Ar), 129.6 (Ar), 129.9 (Ar), 130.0 (Ar), 133.5 (Ar), 133.6 (Ar), 135.3 (Ar), 154.0 (C=O), 159.9 (C=O), 164.6 (C=O), 165.6 (C=O). ESI-MS: calcd for C41H46O11NaSi 765.2707 (M+Na)+, found m/z 765.2710.
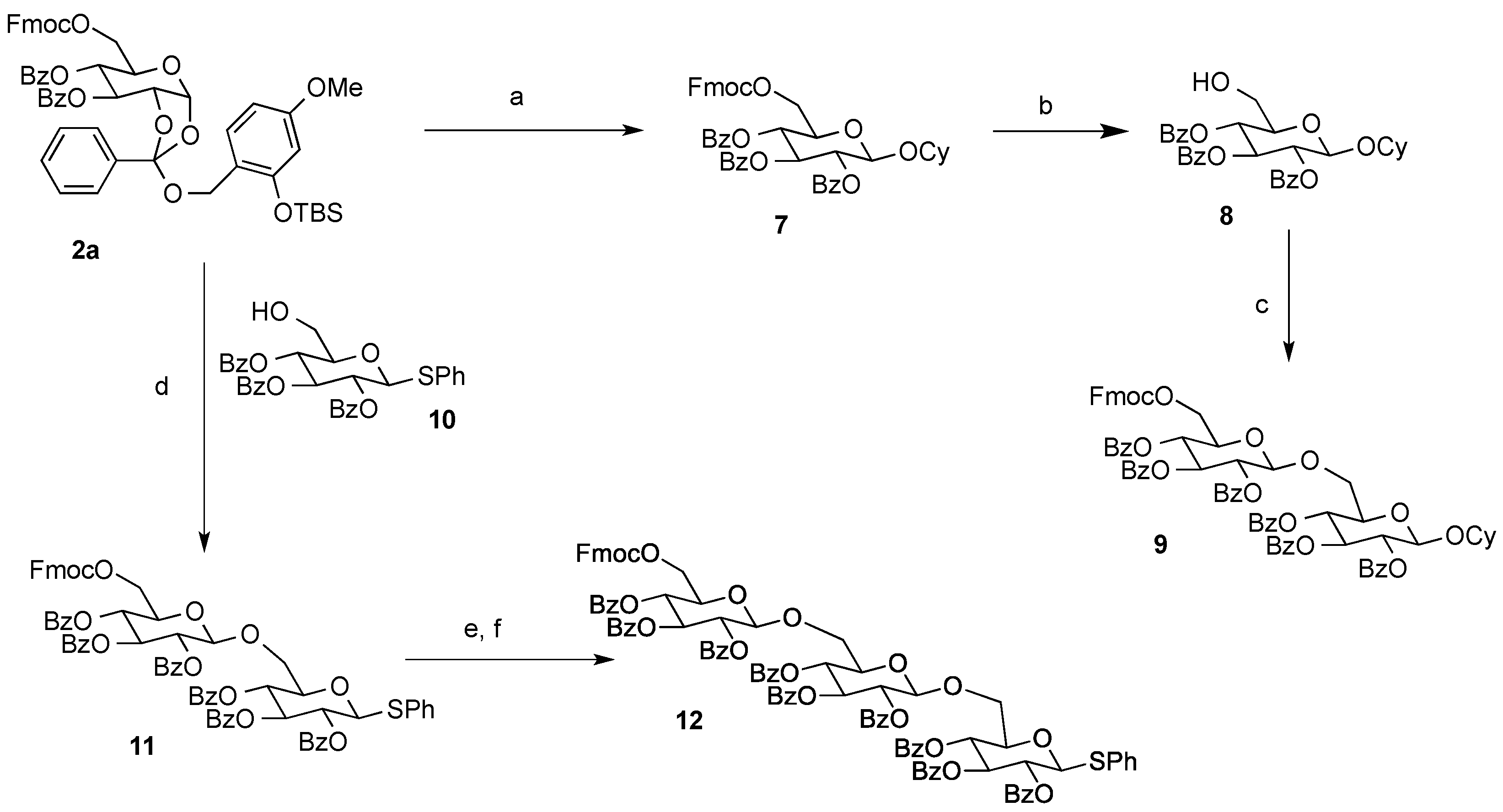
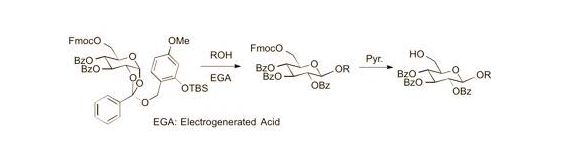
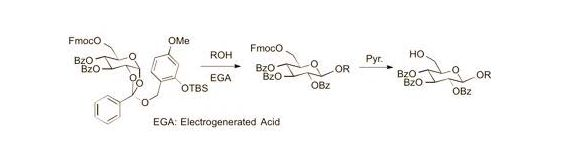
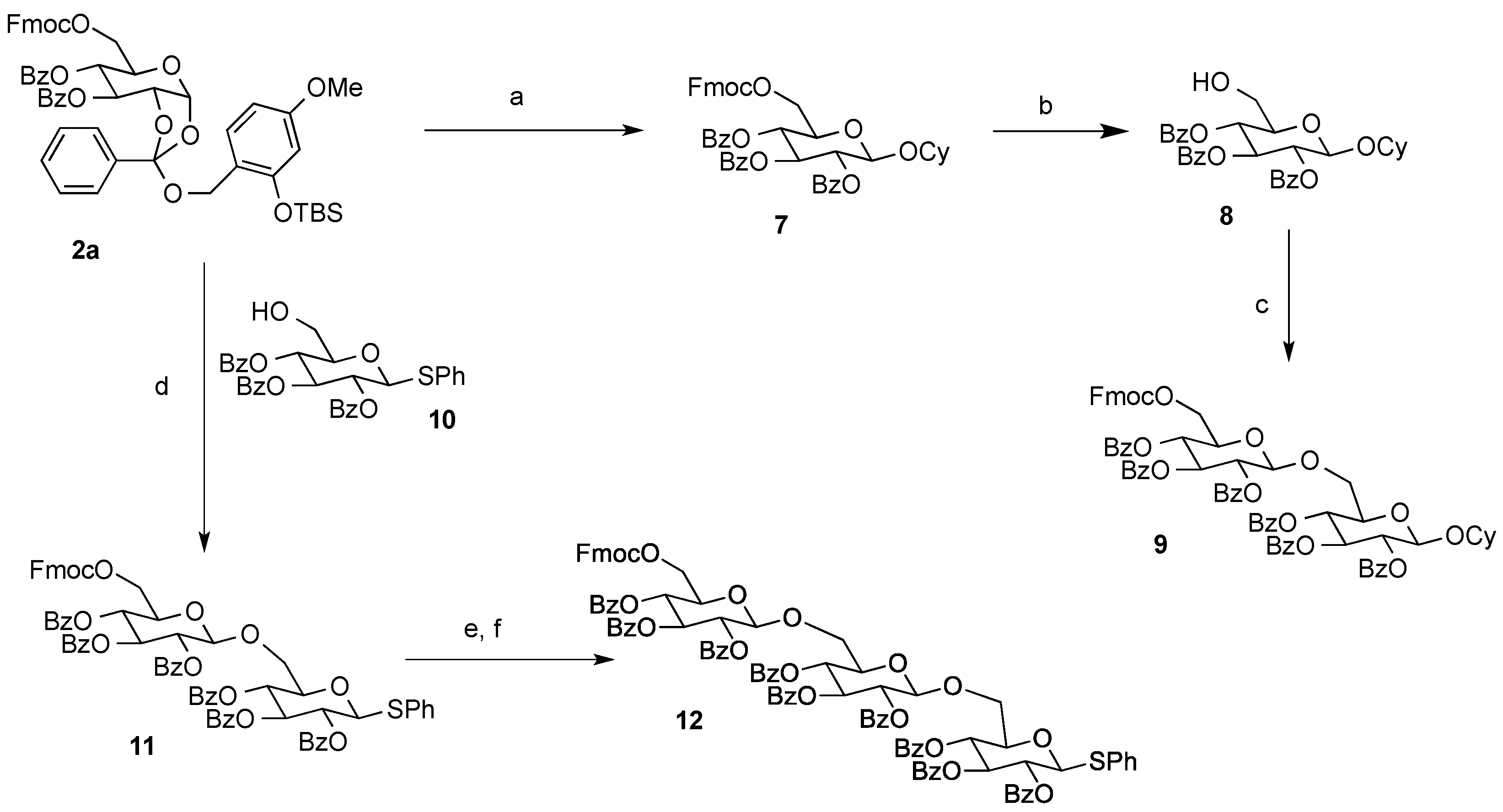
Cyclohexyl 2,3,4-tri-O-benzoyl-6-O-9-fluorenylmethyloxycarboxyl-β-d-glucopyranoside (7). A 0.1 M solution of cyclohexanol in 1,2-DCE (10 mL) containing 4Å MS and Bu4NClO4 (3.42 g, 1 M) as a supporting salt, was electrolyzed by constant current electrolysis (C.C.E., 6 mA/cm2) at 40 °C, using a glassy carbon plate (1.5 cm × 1.5 cm) as an anode and a Pt plate (1.8 cm × 1.8 cm) as a cathode. After the electrolysis, the reaction mixture (3.4 mL, 0.1 M EGA solution, 3 equiv.) was added to a solution of 2a (0.11 g, 0.11 mmol), cyclohexanol (0.4 mL, 0.34 mmol), and 4Å MS in 1,2-DCE (1.1 mL). After being stirred 20 min, the mixture was filtered through a Celite pad and the filtrate was washed with sat. aq. NaHCO3. The organic extracts were dried (Na2SO4), and concentrated in vacuo. The residue was purified by preparative TLC (hexane/acetone = 5:1) to give 7 as an oil (0.056 g, 62%): [α]23D −4.10 (c 1.00, CHCl3); IR (film) 1732, 1601, 1262 cm−1; 1H-NMR (400 MHz) δ 1.22 (5H, m, cyclohexyl), 1.46 (2H, m, cyclohexyl), 1.70 (2H, m, cyclohexyl), 1.88 (1H, m, cyclohexyl), 3.72 (1H, m, cyclohexyl), 4.09 (1H, m, Fmoc), 4.22 (1H, m, Fmoc), 4.36 (3H, m, H-5,6a, Fmoc), 4.46 (1H, d, J = 11.4, 6.5 Hz, H-6b), 4.93 (1H, d, J = 8.0 Hz, H-1), 5.51 (1H, dd, J = 9.6, 8.0 Hz, H-2), 5.55 (1H, t, J = 9.6 Hz, H-4), 5.90 (1H, t, J = 9.6 Hz, H-3), 7.30 (11H, m, Ar), 7.51 (2H, m, Ar), 7.60 (2H, m, Ar), 7.77 (2H, m, Ar), 7.84 (2H, m, Ar), 7.95 (4H, m, Ar); 13C-NMR (100 MHz) δ 23.4 (cyclohexyl), 23.6 (cyclohexyl), 25.3 (cyclohexyl), 31.5 (cyclohexyl), 33.1 (cyclohexyl), 46.6 (Fmoc), 66.5 (C-6), 69.8 (C-4), 70.2 (C-2,3), 72.0 (FmocCH2), 72.9 (C-5), 78.2 (cyclohexyl), 99.7 (C-1), 120.0 (Ar), 125.2 (Ar), 127.2 (Ar), 127.9 (Ar), 128.3 (Ar), 128.4 (Ar), 128.7 (Ar), 128.8 (Ar), 129.5 (Ar), 129.7 (Ar), 129.8 (Ar), 133.1 (Ar), 133.2 (Ar), 133.5 (Ar), 141.2 (Ar), 143.2 (Ar), 123.3 (Ar), 154.8 (C=O), 164.9 (C=O), 165.3 (C=O), 165.8 (C=O). ESI-MS: calcd for C48H45O117997.2962 (M+H)+, found m/z 797.2936.
Cyclohexyl 2,3,4-tri-O-benzoyl-β-d-glucopyranoside (8). To a solution of 7 (54 mg, 0.068 mmol) in pyridine (1 mL) was added Et3N (19 µL, 0.14 mmol) at room temperature, and the mixture was stirred overnight. The mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/acetone = 2:1) to give 8 as an oil (37 mg, 95%): [α]23D −11.5 (c 1.00, CHCl3); IR (film) 3523, 1731, 1601, 1263 cm−1; 1H-NMR (400 MHz) δ 1.05 (3H, m, cyclohexyl), 1.48 (6H, m, cyclohexyl), 1.77 (1H, m, cyclohexyl), 2.41 (1H, bs, OH), 3.68 (4H, m, cyclohexyl, H-5,6a,6b), 4.38 (1H, d, J = 8.0 Hz, H-1), 5.37 (2H, bt, H-2,4), 5.82 (1H, t, J = 9.6 Hz, H-3), 7.18 (3H, m, Ar), 7.31 (4H, m, Ar), 7.41 (2H, m, Ar), 7.74 (2H, m, Ar), 7.85 (4H, m, Ar); 13C-NMR (100 MHz) δ 23.4 (cyclohexyl), 23.6 (cyclohexyl), 25.4 (cyclohexyl), 31.5 (cyclohexyl), 33.2 (cyclohexyl), 61.5 (C-6), 69.7 (C-4,4',6'), 72.1 (C-2,3,3'), 72.9 (C-2',5'), 74.5 (C-5), 78.1 (cyclohexyl), 99.6 (C-1,1'), 128.2 (Ar), 128.3 (Ar), 128.5 (Ar), 128.6 (Ar), 128.9 (Ar), 129.5 (Ar), 129.6 (Ar), 129.7 (Ar), 129.9 (Ar), 133.1 (Ar), 133.2 (Ar), 133.6 (Ar), 164.9 (C=O), 165.8 (C=O), 165.9 (C=O). ESI-MS: calcd for C33H34O9Na 597.2101 (M+Na)+, found m/z 597.2112.
Cyclohexyl 2,3,4-tri-O-benzoyl-β-d-glucopyranosyl-(1→6)-2,3,4-tri-O-benzoyl-6-O-9-fluorenyl-methyloxycarboxyl-β-d-glucopyranoside (9). A solution of cyclohexanol in 1,2-DCE (10 mL, 0.1 M) containing 4Å MS and Bu4NClO4 (3.42 g, 1 M) as a supporting salt, was electrolyzed by constant current electrolysis (C.C.E., 6 mA/cm2) at 40 °C, using a glassy carbon plate (1.5 cm × 1.5 cm) as an anode and a Pt plate (1.8 cm × 1.8 cm) as a cathode. The reaction mixture (0.6 mL, 0.1 M EGA solution, 3 equiv.) was added to a solution of 2a (31 mg, 0.032 mmol) and 8 (37 mg, 0.064 mmol), and MS 4A in 1,2-DCE (0.5 mL). After being stirred 20 min, the mixture was filtered through a Celite pad, and the filtrate was washed with sat. aq. NaHCO3. The organic extract was dried (Na2SO4), and concentrated in vacuo. The residue was purified by preparative TLC (hexane/acetone = 2:1) to give 9 as an oil (16 mg, 38%): [α]23D −9.8 (c 1.00, CHCl3); IR (film) 1734, 1601, 1261 cm−1; 1H-NMR (400 MHz) δ 1.41 (10H, m, cyclohexyl), 3.62 (1H, m, cyclohexyl), 4.00 (4H, m, Fmoc, H-5,5'), 4.29 (5H, m, H-6a,6b,6a',6b'), 4.78 (1H, d, J = 7.8 Hz, H-1'), 5.04 (1H, d, J = 7.8 Hz, H-1), 5.34 (1H, t, J = 9.8 Hz, H-4'), 5.41 (1H, dd, J = 9.8, 7.8 Hz, H-2'), 5.48 (1H, dd, J = 9.8, 7.8 Hz, H-2), 5.52 (1H, t, J = 9.8 Hz, H-4), 5.80 (1H, t, J = 9.8 Hz, H-3'), 5.83 (1H, t, J = 9.8 Hz, H-3), 7.41 (22H, m, Ar), 7.62 (2H, m, Ar), 7.78 (6H, m, Ar), 7.92 (8H, m, Ar); 13C-NMR (100 MHz) δ 22.9 (cyclohexyl), 23.2 (cyclohexyl), 25.4 (cyclohexyl), 31.2 (cyclohexyl), 32.9 (cyclohexyl), 46.6 (Fmoc), 66.0 (C-6), 68.3 (C-6'), 69.3(C-4), 69.9 (C-4'), 70.3 (C-3), 71.7 (C-2,3'), 71.9 (C-2'), 72.1 C-5'), 72.8 (C-5), 72.9 (cyclohexyl), 74.1 (FmocCH2), 99.4 (C-1'), 100.8 (C-1), 119.9 (Ar), 125.3 (Ar), 125.4 (Ar), 127.2 (Ar), 127.8 (Ar), 128.2 (Ar), 128.3 (Ar), 128.4 (Ar), 128.7 (Ar), 128.9 (Ar), 129.3 (Ar), 129.5 (Ar), 129.6 (Ar), 129.7 (Ar), 129.8 (Ar), 133.0 (Ar), 133.1 (Ar), 133.2 (Ar), 133.4 (Ar), 133.5 (Ar), 141.2 (Ar), 143.3 (Ar), 143.4 (Ar), 154.8 (C=O), 164.9 (C=O), 165.1 (C=O), 165.2 (C=O), 165.4 (C=O), 165.6 (C=O), 165.7 (C=O). ESI-MS: calcd for C75H66O19Na 1293.4096 (M+Na)+, found m/z 1293.4078.
Phenyl 2,3,4-tri-O-benzoyl-6-O-9-fluorenylmethyloxycarboxyl-β-d-glucopyranosyl-(1→6)-2,3,4-tri-O-benzoyl-1-thio-β-d-glucopyranoside (11). A solution of cyclohexanol in 1,2-DCE (10 mL, 0.1 M) containing 4Å MS and Bu4NClO4 (3.42 g, 1 M) as a supporting salt was electrolyzed by constant current electrolysis (C.C.E., 6 mA/cm2) at 40 °C, using a glassy carbon plate (1.5 cm × 1.5 cm) as an anode and a Pt plate (1.8 cm × 1.8 cm) as a cathode. After the electrolysis, the reaction mixture (1.3 mL, 0.1 M EGA solution, 3 equiv.) was added to a solution of 2a (34 mg, 0.036 mmol), 10 (42 mg, 0.072 mmol), m and 4Å MS in 1,2-DCE (0.5 mL). After being stirred for 20 min, the reaction mixture was filtered through a Celite pad, and the filtrate was washed with sat. aq. NaHCO3. The organic extracts were dried (Na2SO4), and concentrated in vacuo. The residue was purified by preparative TLC (hexane/acetone = 2:1) to give 11 as an oil (29 mg, 63%): [α]23D +8.9 (c 1.00, CHCl3); IR (film) 1733, 1601, 1261 cm−1; 1H-NMR (400 MHz) δ 3.99 (4H, m, Fmoc, H-5,5'), 4.24 (1H, m, Fmoc), 4.38 (4H, m, H-6a,6b,6a',6b'), 4.92 (1H, d, J = 9.8 Hz, H-1'), 4.97 (1H, d, J = 8.0 Hz, H-1), 5.31 (1H, t, J = 9.8 Hz, H-4'), 5.34 (1H, t, J = 9.8 Hz, H-3'), 5.51 (2H, m, H-2,4), 5.81 (1H, t, J = 9.8 Hz, H-3'), 5.85 (1H, t, J = 9.8 Hz, H-3), 7.23 (2H, m, Ar), 7.39 (25H, m, Ar), 7.61 (2H, m, Ar), 7.74 (4H, m, Ar), 7.85 (4H, m, Ar), 7.93 (6H, m, Ar); 13C-NMR (100 MHz) δ 46.6 (Fmoc), 65.9 (C-6), 68.4 (C-6'), 69.4 (C-4'), 69.5 (C-4'), 70.2 (C-3'), 70.4 (C-3), 71.6 (C-2), 72.0 (C-2'), 72.8 (C-5'),74.0 (FmocCH2), 78.3 (C-5), 85.9 (C-1'), 101.0 (C-1), 119.9 (Ar), 125.3 (Ar), 125.4 (Ar), 127.2 (Ar), 128.2 (Ar), 128.3 (Ar), 128.4 (Ar), 128.5 (Ar), 128.6 (Ar), 128.7 (Ar), 128.8 (Ar), 129.1 (Ar), 129.2 (Ar), 129.7 (Ar), 129.8 (Ar), 131.7 (Ar), 133.2 (Ar), 133.4 (Ar), 133.5 (Ar), 141.2 (Ar), 141.3 (Ar), 143.3 (Ar), 143.4 (Ar), 154.8 (C=O), 164.9 (C=O), 165.1 (C=O), 165.2 (C=O), 165.3 (C=O), 165.6 (C=O), 165.7 (C=O). ESI-MS: calcd for C75H60O18NaS 1303.3398 (M+Na)+, found m/z 1303.3385.
Phenyl 2,3,4-tri-O-benzoyl-6-O-9-fluorenylmethyloxycarboxyl-β-d-glucopyranosyl-(1→6)-2,3,4-tri-O-benzoyl-β-d-glucopyranosyl-(1→6)-2,3,4-tri-O-benzoyl-1-thio-β-d-glucopyranoside (12). To a solution of 11 (29 mg, 0.022 mmol) in pyridine (0.5 mL) was added Et3N (6.3 µL, 0.045 mmol) at room temperature. After being stirred overnight, the mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/acetone = 2:1) to give an alcohol as an oil (19 mg, 82%): [α]23D −2.1 (c 1.00, CHCl3); IR (film) 3062, 1731, 1601, 1262 cm−1; 1H-NMR (400 MHz) δ 2.81 (1H, dd, J = 8.0, 5.6 Hz, OH), 3.60 (1H, m, H-5), 3.74 (2H, m, H-5',6a'), 3.95 (1H, dd, J = 10.5, 5.6 Hz, H-6b'), 4.05 (2H, m, H-6a,6b), 4.92 (2H, bt, H-1,1'), 5.23 (1H, t, J = 9.6 Hz, H-4'), 5.40 (3H, m, H-2,2'), 5.81 (1H, t, J = 9.6 Hz, H-3'), 5.85 (1H, t, J = 9.6 Hz, H-3), 7.38 (19H, m, Ar), 7.53 (4H, m, Ar), 7.76 (2H, m, Ar), 7.81 (2H, m, Ar), 7.93 (8H, m, Ar); 13C-NMR (100 MHz) δ 61.2 (C-6), 67.9 (C-6'), 69.4 (C-4'), 69.9 (C-4), 70.4 (C-3'), 71.5 (C-3), 72.8 (C-2), 73.9 (C-2'), 74.6 (C-5'), 77.8 (C-5), 86.0 (C-1'), 100.5 (C-1), 128.2 (Ar), 128.3 (Ar), 128.4 (Ar), 128.5 (Ar), 128.7 (Ar), 128.8 (Ar), 129.1 (Ar), 129.2 (Ar), 129.7 (Ar), 129.8 (Ar), 129.9 (Ar), 131.5 (Ar), 133.2 (Ar), 133.3 (Ar), 133.5 (Ar), 133.6 (Ar), 164.9 (C=O), 165.1 (C=O), 165.6 (C=O), 165.7 (C=O), 165.9 (C=O). ESI-MS: calcd for C60H50O16NaS 1081.2717 (M+Na)+, found m/z 1081.2732.
A solution of cyclohexanol in 1,2-DCE (10 mL, 0.1 M) containing 4Å MS and Bu4NClO4 (3.42 g, 1 M) as a supporting salt, was electrolyzed by constant current electrolysis (C.C.E., 6 mA/cm2) at 40 °C, using a glassy carbon plate (1.5 cm × 1.5 cm) as an anode and a Pt plate (1.8 cm × 1.8 cm) as a cathode. The reaction mixture (0.6 mL, 0.1 M EGA solution, 3 equiv.) was added to a solution of 2a (37 mg, 0.038 mmol), the alcohol (20 mg, 0.019 mmol), and 4Å MS in 1,2-DCE (0.5 mL). After being stirred at 40 °C for 20 min, the reaction mixture was filtered through a Celite pad, and the filtrate was washed with saturated aqueous NaHCO3. The organic extracts were dried (Na2SO4), and concentrated in vacuo. The residue was purified by preparative TLC (Et2O/hexane = 2:1) to give 12 as a clear oil (4.1 mg, 12%): [α]23D −5.4 (c 1.00, CHCl3); IR (film) 1733, 1601, 1261 cm−1; 1H-NMR (400 MHz) δ 3.62 (1H, dd, J = 11.2, 5.0 Hz, H-6a), 3.84 (3H, m, Fmoc, H-5',6), 4.01 (2H, m, H-5,5''), 4.31 (6H, m, H-6a,6b,6a',6b'), 4.61 (1H, d, J = 8.0 Hz, H-1'), 4.99 (1H, d, J = 10.0 Hz, H-1''), 5.08 (1H, d, J = 8.0 Hz, H-1), 5.10 (1H, t, J = 10.0 Hz, H-4), 5.21 (1H, dd, J = 9.8, 8.0 Hz, H-2''), 5.51 (4H, m, H-2,2',4',4''), 5.68 (1H, t, J = 9.8 Hz, H-3'), 5.84 (1H, t, J = 9.8 Hz, H-3''), 6.06 (1H, t, J = 9.8 Hz, H-3), 7.18 (2H, m, Ar), 7.36 (34H, m, Ar), 7.58 (2H, m, Ar), 7.76 (2H, m, Ar), 7.92 (14H, m, Ar); 13C-NMR (100 MHz) δ 46.6 (Fmoc), 66.2 (C-6), 68.3 (C-6''), 69.4 (C-6'), 69.7 (C-4''), 70.1 (C-4), 70.2 (C-4'), 70.5 (C-3''), 71.7 (C-3), 71.9 (C-2,2',3'), 72.0 (C-2''), 72.6 (C-5'), 72.7 (C-5''), 73.9 (C-5), 74.1 ((FmocCH2), 86.3 (C-1''), 100.5 (C-1), 101.2 (C-1'), 119.9 (Ar), 120.0 (Ar), 125.3 (Ar), 125.4 (Ar), 127.2 (Ar), 127.8 (Ar), 128.1 (Ar), 128.2 (Ar), 128.3 (Ar), 128.4 (Ar), 128.5 (Ar), 128.6 (Ar), 128.7 (Ar), 128.8 (Ar), 129.0 (Ar), 129.1 (Ar), 129.3 (Ar), 129.4 (Ar), 129.7 (Ar), 129.8 (Ar), 130.0 (Ar), 132.1 (Ar), 132.8 (Ar), 133.0 (Ar), 133.2 (Ar), 133.4 (Ar), 141.2 (Ar), 143.3 (Ar), 143.4 (Ar), 154.8 (C=O), 164.9 (C=O), 165.2 (C=O), 165.3 (C=O), 165.7 (C=O). ESI-MS: calcd for C102H82O26NaS 1777.4713 (M+Na)+, found m/z 1777.4736.

{kind=link}
{kind=link}
{kind=link}
{kind=link}