2.1. Structures of Porphyrin and Its Derivatives
In brief, the predicted ground state geometry and selected bond angles and dihedral (torsional) angles of the porphyrin and its derivatives in water, used as a solvent, at B3LYP/6-311G(d,p) level of the DFT are provided in
Figure 1 and
Table 1. the results of the calculations indicated that while the
meso-substitution of porphyrin with tetraphenyl or tetrasulfonatophenyl cause slightly out-of-plane distortion from the planar structure of the macrocycle (within 3° to 5°) for TPP and TSPP; the protonation of the porphyrin core leads to a significant distortion from its planarity within 10° to 20° for H
2TPyr, H
2TPP, H
2TSPP and H
6TSPP. Furthermore, with respect to the average plane of porphryin to macrocycle as seen in
Figure 1 and
Table 1, the peripheral phenyl and sulfonatophenyl substituents are oriented at a tilt angle of about 72° for the TPP and TSPP (unprotonated structures) and about 48° for the H
2TPP, H
2TSPP and H
6TSPP (protonated structures).
Figure 1.
Molecular structures of unsubstituted porphyrin (TPyr), meso-tetraphenylporphyrin (TPP), and anionic meso-tetrakis(p-sulfonatophenyl)porphyrin (TSPP) as well as their protonated structures (H2TPyr, H2TPP, H2TSPP and H6TSPP) in water at the B3LYP/6-311G(d,p) level of the DFT.
Figure 1.
Molecular structures of unsubstituted porphyrin (TPyr), meso-tetraphenylporphyrin (TPP), and anionic meso-tetrakis(p-sulfonatophenyl)porphyrin (TSPP) as well as their protonated structures (H2TPyr, H2TPP, H2TSPP and H6TSPP) in water at the B3LYP/6-311G(d,p) level of the DFT.
Table 1.
Selected dihedral angles (D) and bond angles (A) of free-base porphyrin (TPyr), meso-tetraphenylporphyrin (TPP), and the anionic meso-tetrakis(p-sulfonatophenyl)porphyrin (TSPP) as well as their protonated structures (H2TPyr, H2TPP, H2TSPP and H6TSPP). The calculations were done in water at the B3LYP/6-311G(d,p) level of the DFT.
Table 1.
Selected dihedral angles (D) and bond angles (A) of free-base porphyrin (TPyr), meso-tetraphenylporphyrin (TPP), and the anionic meso-tetrakis(p-sulfonatophenyl)porphyrin (TSPP) as well as their protonated structures (H2TPyr, H2TPP, H2TSPP and H6TSPP). The calculations were done in water at the B3LYP/6-311G(d,p) level of the DFT.
| | TPyr | TPP | TSPP | H2TPyr | H2TPP | H2TSPP | H6TSPP |
|---|
| D(Cβ, Cα, Cm, Cϕ) | N.A | 3.6 | 4.0 | N.A | 19.4 | 19.6 | 19.0 |
| D(Cβ, Cα, Cm, Cα′) | 180.0 | −176.5 | −176.4 | −169.5 | −166.6 | −160.5 | −161.6 |
| D(Cα, Cm, Cα’, N) | 0.0 | 2.3 | 2.4 | 10.3 | 20.6 | 20.7 | 20.0 |
| D(Cα, Cm, Cϕ, C) | N.A. | 72.1 | 71.0 | N.A | 47.8 | 47.2 | 49.6 |
| D(Cα, Cm, Cα′, Cβ′) | 180.0 | −177.2 | −176.9 | −169.5 | −160.6 | −160.5 | −160.8 |
| D(Cm, Cα′, Cβ′, Cβ″) | 180.0 | 179.8 | 179.7 | −178.8 | −176.4 | −174.4 | −176.9 |
| D(Cα′, Cβ′, Cβ″, Cα″) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| D(Cβ′, Cβ″, Cα″, Cm′) | 180.0 | −179.8 | −179.7 | 178.8 | 176.4 | 176.4 | 176.9 |
| D(Cβ″, Cα″, Cm′, Cϕ′) | N.A | −2.6 | −2.6 | N.A | −19.4 | −19.4 | −18.9 |
| D(Cα″, Cm′, Cϕ′, C) | N.A | −72.1 | −70.9 | N.A | −47.8 | −47.5 | −49.9 |
| D(Cα′, N, Cα″, Cβ″) | 0.0 | 0.4 | 0.4 | 2.2 | 4.2 | 4.2 | 4.1 |
| A(Cβ, Cα, Cm) | 127.9 | 123.0 | 123.1 | 127.7 | 128.0 | 128.0 | 128.0 |
| A(Cα, Cm, Cϕ) | N.A | 118.2 | 118.2 | N.A | 118.3 | 118.3 | 118.2 |
| A(Cϕ, Cm, Cα′) | N.A | 116.6 | 116.5 | N.A | 118.3 | 118.4 | 118.4 |
| A(Cα, Cm, Cα′) | 127.0 | 125.2 | 125.3 | 127.4 | 123.4 | 123.4 | 123.9 |
| A(Cm, Cα′, Cβ′) | 123.4 | 126.9 | 126.9 | 127.7 | 128.0 | 128.0 | 127.9 |
| A(N, Cα, Cm) | 125.6 | 126.3 | 126.2 | 125.5 | 125.4 | 125.3 | 125.2 |
| A(Cm, Cα′, N) | 125.7 | 126.6 | 126.6 | 125.5 | 125.4 | 125.3 | 125.3 |
The calculated bond lengths are in agreement with X-ray measurements within ca. ±0.01 Å. As result, the calculations indicated clearly indicated that the protonation of the porphyrin core does not only destroy planarity of the macrocycle, but also, has an effect on the tilt angle of the phenyl and p-sulfonatophenyl substitution groups.
2.2. Raman Spectra of Porphyrin and Derivatives
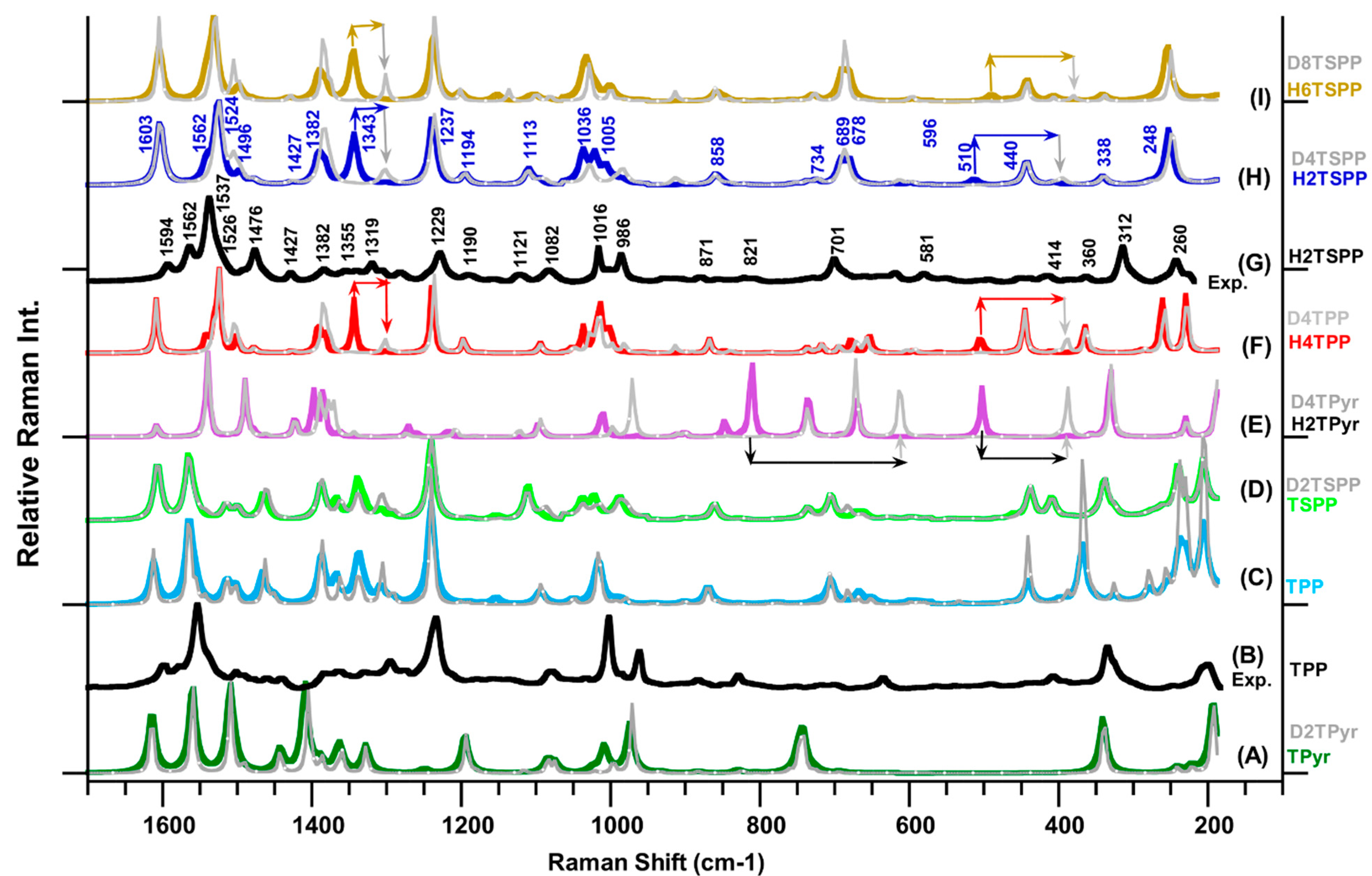
Figure 2 provides the measured Raman spectra of the TPP (
Figure 2B, from our previous work [
23] and H
2TSPP (
Figure 2G), which reveal many Raman peaks with strong and medium intensity as well as many weak or very weak features dispersed through the spectrum. When comparing the observed Raman spectrum of the H
2TSPP with the TPP, the Raman patterns in spectra of both molecules are very similar, but some of the Raman peaks positions are significantly shifted in frequency. For instance, while the most intense peak at 1564 cm
−1 and relatively weak peaks at 1595, 1577, 1438, 1327, 1234 and 334 cm
−1 in the spectrum of TPP are red-shifted to 1537 cm
−1 (the strongest one), 1494, 1562, 1427, 1339, 1229 and 312 cm
−1, respectively. The peaks at 1002, 962, 334 and 201 cm
−1 are respectively blue-shifted to 1014, 983, 314 and 236 cm
−1 in the spectrum of H
2TSPP. Also, the Raman bands at 1476 and 701 cm
−1 Raman spectrum of the H
2TSPP are significantly enhanced reference to corresponding peaks in the TPP spectrum.
Figure 2 provide the calculated Raman spectra of the TPyr/D
2TPyr, H
2TPyr/ D
4TPyr, TPP/D
2TPP, H
2TPP/(D
4TPP), TSPP/D
2TSPP, H
2TSPP/D
4TSPP, and H
6TSPP/D
8TSPP in water (used as a solvent), with the observed Raman spectra of the TPP and H
2TSPP for comparison. (It is worthy to note that where D indicates that deuterium atoms covalently bound to the N atoms at porphyrin core; for dicationic D
6TSPP, four of eight D atoms covalently bound to O atoms within sulfonato group, -SO
3−, other four covalently bound to the N atoms at the core). While only the data for the Raman features of TPP, TSPP and H
2TSPP are presented in
Table 2; the observed Raman spectrum of the H
2TSPP is assigned in connection with the calculated Raman spectra of the compounds studied here and observed Raman spectrum of TPP (from our previous work [
23]). Our conclusion may be summarized as follows:
(1) the Raman peak at 1593 cm
−1: as seen in
Figure 2, the calculations produced an active Raman band at about 1600 cm
−1 for all of the porphyrin derivatives. However, the nuclear motion of the molecules showed that this calculated Raman band is due to C-C bond stretching ν(C-C) within phenyl rings and rocking of their H ρ(CH), no any contribution comes from macrocycle and sulfonato groups (-SO
3−). The spectra of the free base porphyrin (TPyr) and protonated TPyr (H
2TPyr) produced a peak around 1600 cm
−1, which results from asymmetric stretching of the C
α-C
m-C
α (ν
a(C-C
m-C)) and bending deformation of the C-N(H)-C/C-N-C bonds, θ(C-N(H)-C/C-N-C), and rocking of H on C
m atoms (ρ(C
mH)), as seen in
Figure 3. Therefore, the vibrational motion of the phenyl substitution is responsible for the observed Raman band at 1593 cm
−1 in the observed spectrum of protonated TSPP (H
2TSPP). Furthermore, in our previous work [
23], the measured and calculated Raman spectrum of the TPP revealed the same result and also these results are shown in
Table 2 and
Figure 2.
Figure 2.
Calculated Raman spectra of porphyrin derivatives in water (used as solvent in the calculations) at the B3LYP/6-311G(d,p) level of the DFT: (A) free-base porphyrin (TPyr) and deuterated-TPyr (D
2TPyr); (C)
meso-tetraphenylporphyrin (TPP) and (D
2TPP); (D) anionic
meso-tetrakis(
p-sulfonatophenyl)porphyrin (TSPP) and deuterated-TSPP (D
2TSPP), and (E) protonated-TPyr (H
2TPyr) and deuterated-H
2TPyr (D
4TPyr); (F) protonated-TPP (H
2TPP) and deuterated-H
2TPP (D
4TPP); (H) protonated TSPP (H
2TSPP) and deuterated-H
2TSPP (D
4TSPP); and (I) dicationic TSPP (H
6TSPP) and deuterated-H
6TSPP (D
8TSPP). The measured Raman spectra of: (B) TPP (taken from ref. [
23]) and (G) H
2TSPP. It should be noted that the plot Raman spectra in the gray color belong to the deuterated molecules, and the line arrows show the frequency shift in the deuterated molecule.
Figure 2.
Calculated Raman spectra of porphyrin derivatives in water (used as solvent in the calculations) at the B3LYP/6-311G(d,p) level of the DFT: (A) free-base porphyrin (TPyr) and deuterated-TPyr (D
2TPyr); (C)
meso-tetraphenylporphyrin (TPP) and (D
2TPP); (D) anionic
meso-tetrakis(
p-sulfonatophenyl)porphyrin (TSPP) and deuterated-TSPP (D
2TSPP), and (E) protonated-TPyr (H
2TPyr) and deuterated-H
2TPyr (D
4TPyr); (F) protonated-TPP (H
2TPP) and deuterated-H
2TPP (D
4TPP); (H) protonated TSPP (H
2TSPP) and deuterated-H
2TSPP (D
4TSPP); and (I) dicationic TSPP (H
6TSPP) and deuterated-H
6TSPP (D
8TSPP). The measured Raman spectra of: (B) TPP (taken from ref. [
23]) and (G) H
2TSPP. It should be noted that the plot Raman spectra in the gray color belong to the deuterated molecules, and the line arrows show the frequency shift in the deuterated molecule.
(2) The observed peak at 1563 cm
−1: while the measured Raman spectrum of the H
2TSPP displayed a relatively weak bands at 1563 cm
−1, its calculated Raman spectrum displayed only an extremely very weak peak at 1568 cm
−1 in this region (as a result of C-C bond stretching within phenyl rings and rocking of their H, accompanied by relatively weak asymmetric stretching of C
α-C
m-C
β bond stretching, no any contribution comes from sulfonato groups). When we examined this peak caused by the same vibrational motion for the TPP, TSPP, H
2TPP and H
6TSPP, their calculated Raman spectra again exhibited an extremely very weak peak at 1588, 1574, 1585 and 1578 cm
−1, respectively. Actually, the calculated spectrum of the TSPP produced the most intense Raman band at 1,564 cm
−1, but its vibrational motions indicated that this mode of frequency shifted to 1524 cm
−1 in the spectrum of H
2TSPP. This problem is not only observed for H
2TSPP, but also, we found the same problem for the TPP (seen
Figure 2 and
Table 2). Therefore, we believe that the DFT calculations might underestimate the intensity of this peak.
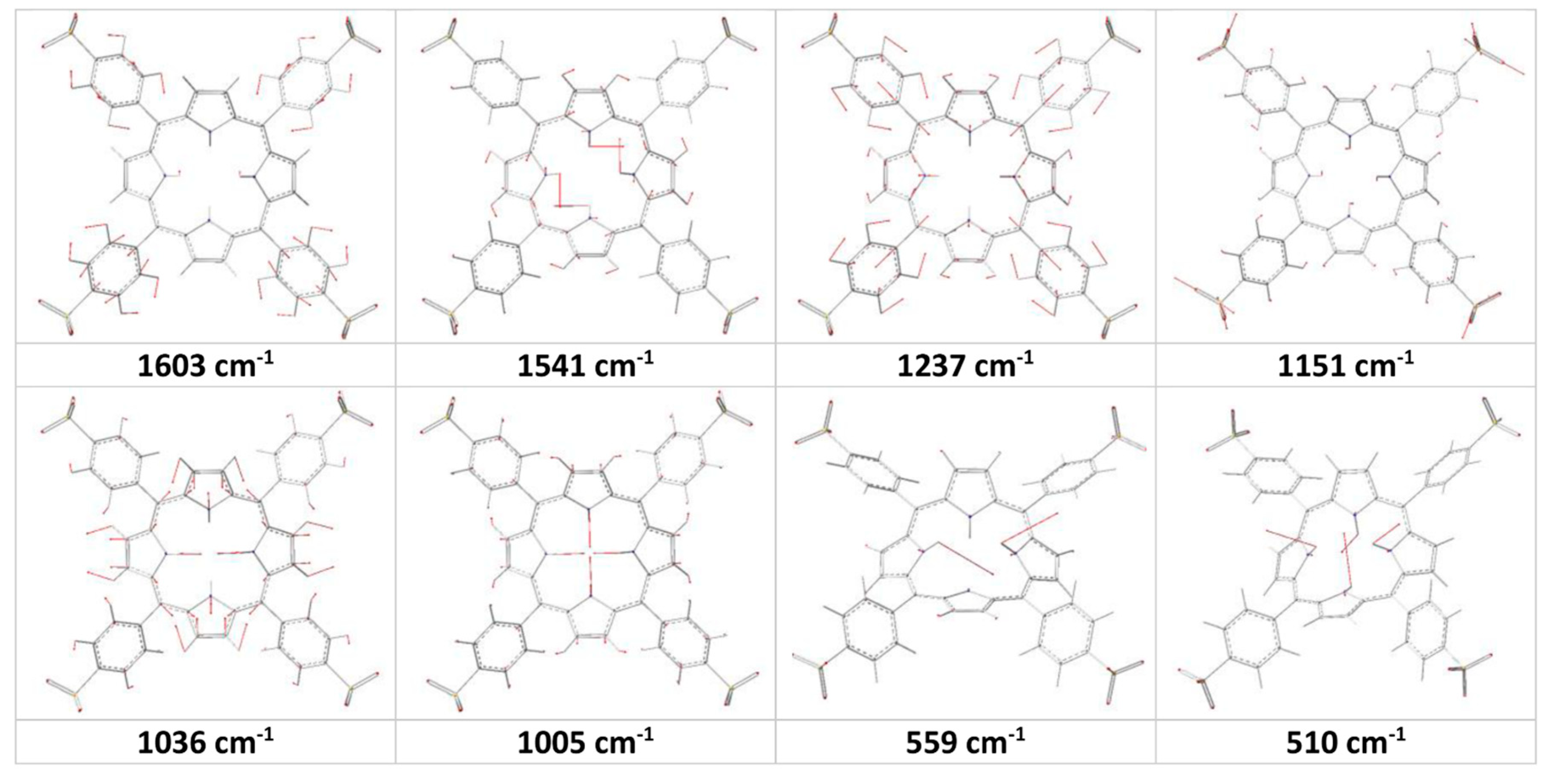
Figure 3.
Calculated molecular motions for some vibrational bands of the H2TSPP.
Figure 3.
Calculated molecular motions for some vibrational bands of the H2TSPP.
(3) The observed Raman spectra of TPP and H2TSPP exhibited a strong band at 1553 cm−1 (with a shoulder ca. at 1540 cm−1) and at 1537 cm−1 (with a shoulder at ca. 1528 cm−1), respectively. Their calculated spectra displayed the strongest Raman band at 1564 and 1524 cm−1, respectively, as a results of the same vibrational motion: Cβ-Cβ bond stretching, ν(Cβ-Cβ), symmetric stretching of Cα-Cm-Cα bonds, νs(Cα-Cm-Cα), which lead to bending deformation of the C-N-C bonds, θ(C-N(H)-C). The observed shoulders at 1540 cm−1 (TPP) and at 1528 cm−1 (H2TSPP) correspond to the calculated peaks at 1559/1540 cm−1 (in the TPP/H2TSPP and caused by νs(Cα-Cm-Cα)/ρ(C-N(H)-C)/ρ(NH)) and 1555/1529 cm−1 (in the TPP/H2TSPP, owing to ν(Cβ-Cβ)/ρ(CβH) and relatively weak θ(C-N(H)-C)). Both of the experimental and calculated Raman spectra showed that the strongest band in the TPP spectrum is significantly red shifted in the H2TSPP spectrum. This large red-shift in this peak position (about exp. 14 cm−1 and ca. 40 cm−1) is mainly caused by the out-of-plane distortion of the macrocycle as a result of protonation of the N atoms at porphyrin core, not due to the substitution effect, which is responsible from the observed Raman band at 1563 cm−1 in the spectrum of H2TSPP. Also, the frequency shift of this peak in the calculated spectra of the TPyr, TPP and TSPP is found in the calculated spectrum of the H2TPyr, H2TPP and H6TSPP.
Table 2.
Observed and calculated Raman active modes of frequencies (in cm
−1) of the H
2TSPP (C
2v) with the TPP (C
2v point group) and TSPP (C
2v) for comparison. The calculations were carried out in water used as solvent at B3LYP/6-311G(d,p) level of the DFT. Where ∆ν
sc stands for the scaled vibrational frequencies (∆ν
sc. = 0.96∆ν
calc +40) and S
R and I
R indicate the calculated Raman scattering activity and intesity, respectively;
∆ν
exp and I
R/exp stand for the observed Raman frequency and Intensity, respectively. The experimental values of Raman spectrum of the TPP are taken from our previous work [
23].
Table 2.
Observed and calculated Raman active modes of frequencies (in cm−1) of the H2TSPP (C2v) with the TPP (C2v point group) and TSPP (C2v) for comparison. The calculations were carried out in water used as solvent at B3LYP/6-311G(d,p) level of the DFT. Where ∆νsc stands for the scaled vibrational frequencies (∆νsc. = 0.96∆νcalc +40) and SR and IR indicate the calculated Raman scattering activity and intesity, respectively; ∆νexp and IR/exp stand for the observed Raman frequency and Intensity, respectively. The experimental values of Raman spectrum of the TPP are taken from our previous work [23].
| TPP | TSPP | H2TSPP | |
|---|
| | ∆νsc. | SR | IR | | | H2 | ∆νsc. | SR | IR | H4 | ∆νsc. | SR | IR | ∆νexp | IR/exp |
|---|
| A2 | 1612 | 25 | 24 | 1595 | 29 | A2 | 1607 | 44 | 42 | A2 | 1603 | 45 | 40 | 1593 | 21 | C-C bond stretching within phenyl rings and rocking of their H, no any contribution comes from macrocycle and sulfonato groups (-). |
| A1 | 1612 | 29 | 27 | A1 | 1607 | 53 | 50 | A1 | 1603 | 55 | 49 |
| A1 | 1588 | <1 | <1 | 1577 | 27 | A1 | 1574 | 1 | 1 | A1 | 1568 | 2 | 1 | 1563 | 42 | C-C bond stretching within phenyl rings and rocking of their H, accompanied by relatively weak asymmetric stretching of Cα-Cm-Cβ bond stretching, no any contribution comes from sulfonato groups. |
| A1 | 1564 | 100 | 100 | 1553
1540 | 100
40 | A1 | 1564 | 100 | 100 | A1 | 1524 | 100 | 100 | 1537 | 100 | Cβ-Cβ bond stretching, ν(Cβ-Cβ), symmetric stretching of Cα-Cm-Cα bonds, νs(Cα-Cm-Cα) that leads to bending deformation of the C-N-C bonds, θ(C-N(H)-C). |
| A2 | 1559 | 8 | 8 | A2 | 1558 | 9 | 9 | A2 | 1540 | 29 | 28 | 1528 | 42 | νs( Cα-Cm-Cα)/rocking of C-N(H)-C and H on N atoms, ρ(C-N(H)-C)/ρ(NH) |
| A1 | 1555 | 15 | 15 | A1 | 1554 | 8 | 8 | A1 | 1529 | 26 | 25 | Asymmetric stretching of Cα-Cm-Cα) bonds νa(Cα-Cm-Cα)/θ(C-N(H)-C). |
| A1 | 1514 | 24 | 25 | 1502 | 21 | A1 | 1515 | 21 | 23 | A1 | 1477 | 5 | 5 | 1476 | 38 | ν(Cβ-Cβ) and rocking of the H on C atoms within macrocycle (not on the phenyl groups), ρ(CβH), and relatively weak θ(C-N(H)-C) |
| A2 | 1502 | 14 | 15 | 1491 | 16 | A2 | 1499 | 14 | 15 | A1 | 1496 | 7 | 7 | 1489 | 15 | ρ(CH within phenyl groups only) |
| A1 | 1501 | 4 | 4 | A1 | 1498 | 3 | 4 | A2 | 1495 | 8 | 8 |
| A2 | 1466 | 31 | 36 | 1461 | 13 | A2 | 1464 | 37 | 44 | | | | | ν(Cm-Cα)/ρ(CβH), and relatively weak νa(C-N(H)-C) |
| A1 | 1454 | 3 | 3 | 1438 | 12 | A1 | 1454 | 2 | 2 | A1 | 1426 | 3 | 3 | 1428 | 11 | νs(Cα-Cm-Cα)/ρ(C-N(H)-C)/ρ(CH) |
| A2 | 1387 | 46 | 61 | 1378 | 20 | A2 | 1387 | 44 | 58 | A2 | 1391 | 31 | 38 | 1384 | 15 | νa(Cβ-Cα-N(H))/ρ(CβH-CβH) |
| A1 | 1367 | 22 | 30 | | | A1 | 1367 | 22 | 29 | A1 | 1382 | 20 | 25 | 1354 | 14 | ν(Cβ-Cα)/θ(C-Cm-C)/θ(C-N(H)-C), which leading to macrocycle getting a square shape) |
| A2 | 1339 | 19 | 26 | 1327 | 20 | A2 | 1338 | 42 | 60 | A2 | 1343 | 52 | 70 | 1340 | 14 | ν(Cϕ-Cm)/ρ(CβH)/ρ(NH), and relatively weak νa(C-N(H)-C) |
| A2 | 1335 | 26 | 38 | | | A2 | 1329 | 13 | 19 | | | | | 1319 | 22 |
| A1 | 1306 | 13 | 19 | | | A1 | 1305 | 7 | 10 | A1 | 1321 | 1 | 1 | 1304 | 14 | νa(C-C-C) within phenyl groups/θ(C-N(H)-C)/ρ(CH). |
| A1 | 1291 | 4 | 7 | | | A1 | 1290 | 4 | 6 | A1 | 1300 | 1 | 2 | 1283 | 12 |
| A1 | 1239 | 75 | 127 | 1234 | 84 | A1 | 1239 | 77 | 131 | A1 | 1237 | 60 | 97 | 1229 | 34 | ν(Cϕ-Cm) (primarily)/νs(C-N(H)-C)/ρ(CH)/ν(Cβ-Cβ) (relatively weak) |
| A2 | 1189 | 0. | 0. | | | A2 | 1188 | 1 | 1 | A2 | 1194 | 3 | 6 | 1190 | 8 | ρ(CH) within phenyl groups. |
| A1 | 1189 | 1 | 2 | | | A1 | 1189 | 1 | 1 | A1 | 1194 | 5 | 9 |
| A2 | 1152 | 4 | 8 | 1137 | 10 | A2 | 1153 | 3 | 5 | | | | | | | ρ(NH)/ρ(CβH) and relatively weak structural deformation |
| | | | | | | A2 | 1146 | <1 | <1 | A2 | 1151 | <1 | <1 | 1122 | 9 | νa(O-S-O) within sulfonato groups |
| A1 | 1097 | 1 | 2 | 1080 | 21 | A1 | 1109 | 19 | 42 | A1 | 1108 | 9 | 18 | 1082 | 14 | ν(S-O)/θ(C-C(S)-C) within sulfonato groups. |
| A1 | 1091 | 5 | 11 | A1 | 1092 | 3 | 6 | A1 | 1093 | 2 | 4 | ρ(CβH) |
| A1 | 1048 | 2 | 6 | | | A1 | 1036 | 1 | 3 | A1 | 1033 | <1 | 1 | | | θ(C-C-C) within the phenyl groups |
| A2 | 1013 | 6 | 15 | | | A2 | 1037 | 1 | 4 | A2 | 1035 | 1 | 3 | | |
| A1 | 1020 | 4 | 10 | 1002 | 85 | A1 | 1020 | 2 | 5 | A1 | 1036 | 3 | 7 | 1016 | 40 | Expansion of the pyrrole/pyrroline groups along N(H)…N(H) direction due to ν(Cα-Cβ), leading to macrocycle getting rectangular shape instead of square shape. |
| A1 | 983 | 2 | 4 | 962 | 44 | A1 | 986 | 2 | 5 | A1 | 1005 | 2 | 5 | 1002 | 15 | Expansion of the pyrrole/pyrroline groups along N(H)…N(H) direction in the same phase like macrocycle getting square shape or similar to breathing of the macrocycle |
| A2 | 990 | 1 | 2 | | | A2 | 988 | 1 | 2 | A2 | 992 | 0. | 1 | | | Out of plane wagging of the H on the phenyl rings, w(CH) |
| A1 | 899 | 1 | 3 | | | A1 | 899 | 1 | 3 | A1 | 904 | <1 | 1 | 986 | 32 | Bending deformation inside entire molecule. |
| A1 | 868 | 3 | 13 | | | A1 | 859 | 4 | 14 | A1 | 858 | 2 | 8 | 879 | 6 | w(CH on the macrocycle and phenyl rings) |
| A1 | 815 | <1 | <1 | | | A1 | 814 | <1 | <1 | A1 | 820 | <1 | 1 | 821 | 5 |
| A1 | 768 | <1 | <1 | | | A1 | 749 | <1 | 1 | A1 | 751 | 1 | 2 | | | Out of plane bending deformation of whole molecule including w(CH/NH) |
| A1 | 727 | <1 | <1 | | | A1 | 734 | 3 | 15 | A1 | 734 | 1 | 5 | 728 | 5 | ν(S-O) and expansion of the phenyl rings along S…Cm direction including w(CH/NH) |
| | | | | | | | | | | A2 | 689 | 5 | 31 | 701 | 27 | Out of plane twisting of the macrocycle |
| A1 | 665 | 2 | 12 | 636 | 13 | A1 | 669 | 1.33 | 9 | A1 | 678 | 5 | 33 |
| A1 | 584 | <1 | 2 | | | A1 | 589 | <1 | 1 | A1 | 596 | <1 | 3 | 580 | 10 | w(NH and CH on the macrocycle and phenyl rings) and wagging of the macrocycle. |
| A1 | 531 | 0. | 3 | | | A1 | 574 | 0. | 1 | A1 | 566 | 0. | 1 | 548 | 5 | Wagging of entire molecule |
| | | | | | | | | | | A1 | 510 | 0. | 2 | 494 | 3 | w(NH) |
| A2 | 468 | <1 | 1 | | | A2 | 457 | 1 | 13 | A2 | 468 | <1 | 1 | | | In-plane wagging of macrocycle and translational motion of phenyl rings. |
| A2 | 437 | 2 | 33 | 408 | 15 | A2 | 435 | 2 | 25 | A2 | 440 | 2 | 25 | 439 | 5 | Out of plane bending of the phenyl rings. |
| | | | | | | A1 | 407 | 2 | 47 | A1 | 404 | <1 | 6 | 414 | 8 | Breathing macrocycle and translational motion of phenyl rings in opposite phase. |
| A1 | 365 | 6 | 160 | 334 | 50 | A1 | 336 | 3 | 111 | A1 | 338 | 1 | 24 | 314 | 41 | Breathing of whole molecule. |
| A2 | 322 | <1 | 17 | | | | | | | | | | | 363 | 7 | Out of plane wagging of macrocycle. |
| A1 | 235 | 2 | 127 | 201 | 29 | A1 | 237 | 2 | 152 | A1 | 248 | 2 | 144 | 242 | 26 | Out of plane wagging of macrocycle. |
| A1 | 252 | <1 | 28 | | | A1 | 257 | <1 | 18 | A1 | 252 | <1 | 12 | | | Out of plane wagging of phenyl rings and relatively weak out of plane wagging macrocycle. |
(4) The calculated Raman spectra of the TPP, H
2TPP, TSPP, H
2TSPP and H
6TSPP revealed a relatively strong peak around 1238 cm
−1, arise from the ν(C
ϕ-C
m) (predominantly), ν
s(C-N(H)-C), ρ(CH) and ν(C
β-C
β) (relatively weak). This vibrational mode of frequency is assigned to the observed Raman bands at 1229 cm
−1 (H
2TSPP) and 1234 cm
−1 (TPP). It is clear that this mode is mainly due to bond stretching between the
meso-position C atom (C
m within macrocycle) and the adjacent C atom (within the
meso-substituted phenyl or
p-sulfonatophenyl groups). For the unsubstituted free base porphyrin molecules, this peak is red-shifted to 1194 cm
−1 and 1217 cm
−1 in the calculated spectra of the TPyr and H
2TPyr, respectively, which mainly result from rocking of the H atom (covalently bound to
meso-carbon atom), ρ(C
mH), including vibrational bond stretching within the macrocycle (see
Figure 3). The question here is that why this peak is not shifted in the
meso-substituted phenyl/sulfonatophenyl porphyrin molecules, but it is shifted in the TPyr and H
2TPyr? This may be explained by the existence of the electrostatic repulsion interactions between the H atoms on the C
m atoms and H atoms on the β-C atoms (C
β) within the pyrrole/pyrroline rings in the case of the TPyr and H
2TPyr. For instance, this frequency shift in the TPyr (1194 cm
−1) is larger than in the H
2TPyr (214 cm
−1) due to the out of plane distortion from planarity of the protonated porphyrin (H
2TPyr, see
Figure 1) that leads to decreasing electrostatic repulsion interaction between the H atoms on the C
β and C
m atoms resulting from increasing distance between them.
In the case of
meso-phenyl/sulfonatophenyl-substituted porphyrin molecules, this electrostatic repulsion interaction between the H atoms on the C
β atoms and H atoms on the
meso-phenyl substituent lead to rotation of the
meso-phenyl/sulfonatophenyl groups about C
m-C
ϕ bond, up to the tilt angle of 71° for unprotonated structures and about 48° for the protonated porphyrin molecules (relative to macrocycle, see
Figure 1 and
Table 1). Therefore, due to decreasing in these electrostatic repulsion interactions, the calculations did not produced a significant frequency shift in this peak position (~1238 cm
−1) in the Raman spectra of the substituted porphyrin molecules.
(5) In the range of 1050–950 cm
−1, there are two well-known Raman band that are influenced by the protonation and deuteration of the macrocycle. While the observed Raman spectrum of the TPP (excited at 488 nm) exhibited two bands at 1002 and 962 cm
−1, which are respectively blue shifted to 1016 and 986 cm
−1 in the H
2TSPP spectrum (excited at 514 nm). These large shift in the peak positions may be due to protonation of the porphyrin core, which leads to saddle-type distortions of the porphyrin core (in the other words, leading to increasing the degree of the freedom of the rocking of the N-H bonds as a result of decreasing electrostatic repulsion between these H atoms). Furthermore, the Gaussview animation software showed that the band at 1036 cm
−1 (H
4TSPP) is due to expansion of the pyrrole/pyrroline groups along N(H)…N(H) direction in opposite direction (
Figure 3), as a result of the ν(C
α-C
β), leading to macrocycle getting rectangular shape instead of square shape, and the bands at 1005 cm
−1 (H
4TSPP) is due to expansion of the pyrrole/pyrroline groups along N(H)…N(H) direction in the same phase like macrocycle getting square shape or similar to breathing of the macrocycle or the breathing of the pyrrole rings as assigned by Rich and McHale [
29].
(6) Two other important peaks in low frequency region are predicted at 248 and 338 cm
−1 for H
2TSPP and 235 and 365 cm
−1 for TPP are due to out of plane twisting of the macrocycle only and breathing of entire molecule, respectively, which are consistent with their experimental values (242 and 338 cm
−1 for the H
2TSPP; 235 and 334 cm
−1 for the TPP). These observed and calculated Raman bands of TPP are red and blue shifted as a result of the protonation of the macrocycle, not due to sulfonato-substituted groups. The rest of the assignments of the observed Raman bands are given in
Table 2.
Isotopic Effect on the Raman Spectrum
The results of the calculations showed that the deuteration of the N atoms at the porphyrin core also produce a significant red shift in their frequencies. There is a strong evidence on this observation comes from the polarized resonance Raman scattering (RRS) spectra of the aggregated H
2TSPP (protonated TSPP) and D
2TSPP (deuterated TSPP) by Rich and McHale [
29]. The authors reported that the RRS spectra of the aggregated H
2TSPP and D
2TSPP (excited at 488 nm) exhibits frequency shifts of some of the well-known Raman modes besides changes in the relative intensities of the Raman modes upon deuteration. They found the most notable frequency shifts observed for the Raman bands at 983 cm
−1 and 1013 cm
−1 in the Raman spectrum of protonated TSPP (H
2TSPP) aggregate that shift to 957 and 1004 cm
−1 in the spectrum of aggregated D
2TSPP. They also concluded that these two modes are pyrrole breathing modes and thus the red shifts can be attributed to the substitution of deuterium ions with the labile protons in the porphyrin core [
29]. Our measured Raman spectra of the TPP and H
2TSPP, and protonated and deuterated spectra of aggregated H
2TSPP and D
4TSPP clearly showed that while the protonation of the porphyrin core leads to blue shift in the frequency, the deuteration leads to red shift in the spectral position of these two Raman bands.
By comparing the spectral positions of these two peaks in the calculated Raman spectra of protonated and deuterated porphyrin core with their corresponding unprotonated ones (see
Table 3), we can see that while the protonation of the free base and meso-substituted porphyrin core caused a blue shift in frequency of these two bands, the deuteration caused a red shift. For instance, while these two Raman band at 1020 and 985 cm
−1 in the spectrum of TSPP are blue shifted to 1036 and 1005 cm
−1 in the protonated TSPP (H
2TSPP), these peaks are red shifted to 1012 and 980 cm
−1 in the spectrum of deuterated TSPP (D
2TSPP). When the four N atoms at the core are deuterated (D
4TSPP), then, these two peaks are shifted from 1036 and 1005 cm
−1 (in the H
4TSPP) to 1026 and 983 cm
−1 in the D
4TSPP.
Table 3.
The predicted Raman active bands of frequencies (for the protonated (see
Table 2 for TPP and H
2TSPP) and deuterated (Ref. [
29]) porphyrin derivatives) that experimentally exhibited significant frequency shift in the range of 1040–950 cm
−1.
Table 3.
The predicted Raman active bands of frequencies (for the protonated (see Table 2 for TPP and H2TSPP) and deuterated (Ref. [29]) porphyrin derivatives) that experimentally exhibited significant frequency shift in the range of 1040–950 cm−1.
| TPyr | H2TPyr | TPP | Exp. | H2TPP | TSPP | H2TSPP | Exp. | H6TSPP |
|---|
| 1007 | 1013 | 1020 | 1002 | 1036 | 1020 | 1036 | 1016 | 1036 |
| 972 | 1010 | 983 | 962 | 1002 | 985 | 1005 | 983 | 1001 |
| D2TPyr | D4TPyr | D2TPP | | D4TPP | D2TSPP | D4TSPP | Exp. [29] | D6TSPP |
| 996 | 995 | | | 1026 | 1012 | 1026 | 1004 | 1026 |
| 968 | 968 | | | 980 | 977 | 983 | 957 | 979 |
The effect of the deuteration of the N atoms on the frequency shift in the rest of intense Raman bands in the calculated spectrum are less than 5 cm
−1, which is consistent with the experimental observation [
29], but there are many predicted Raman bands with weak intensities which exhibited large shift in the frequency (see
Figure 2). The other
meso-substituted and free base porphyrin derivatives showed the similar results, which are in accordance with experimental observations as discussed above. Furthermore, the theoretical calculations indicated that the
meso-substituted groups do not any significant effect on the spectral position of these two Raman bands. The blue shift of these two peaks for the protonation of the porphyrin core is not so surprising when taking into account of the electrostatic repulsion effect between H atoms (covalent bonded to N atoms) at the porphyrin core. This effect can be minimized by out of plane distortion (or deviation from the planarity) of the macrocycle as discussed above. The red shift also can be expected due to isotopic effect since the vibrational frequency is inversely proportional to the square root of mass of atoms that contribute to the vibrational mode. Furthermore, the deuteration of the porphyrin core and one of oxygen atoms within the sulfonato (SO
3D) groups exhibited new Raman peaks in the range of 2630–2720 cm
−1.
2.3. IR Spectra of Porphyrin and Derivatives
We also calculated the IR spectra of TPyr, TPP, TSPP, H
2TPyr, H
2TPP, H
2TSPP and H
6TSPP as well as their deuterated structures (D
2TPyr, D
2TPP, D
2TSPP, D
4TPyr, D
4TPP, D
4TSPP and D
6TSPP, where D
2 and D
4 indicate the number of D atoms at porphyrin core and D
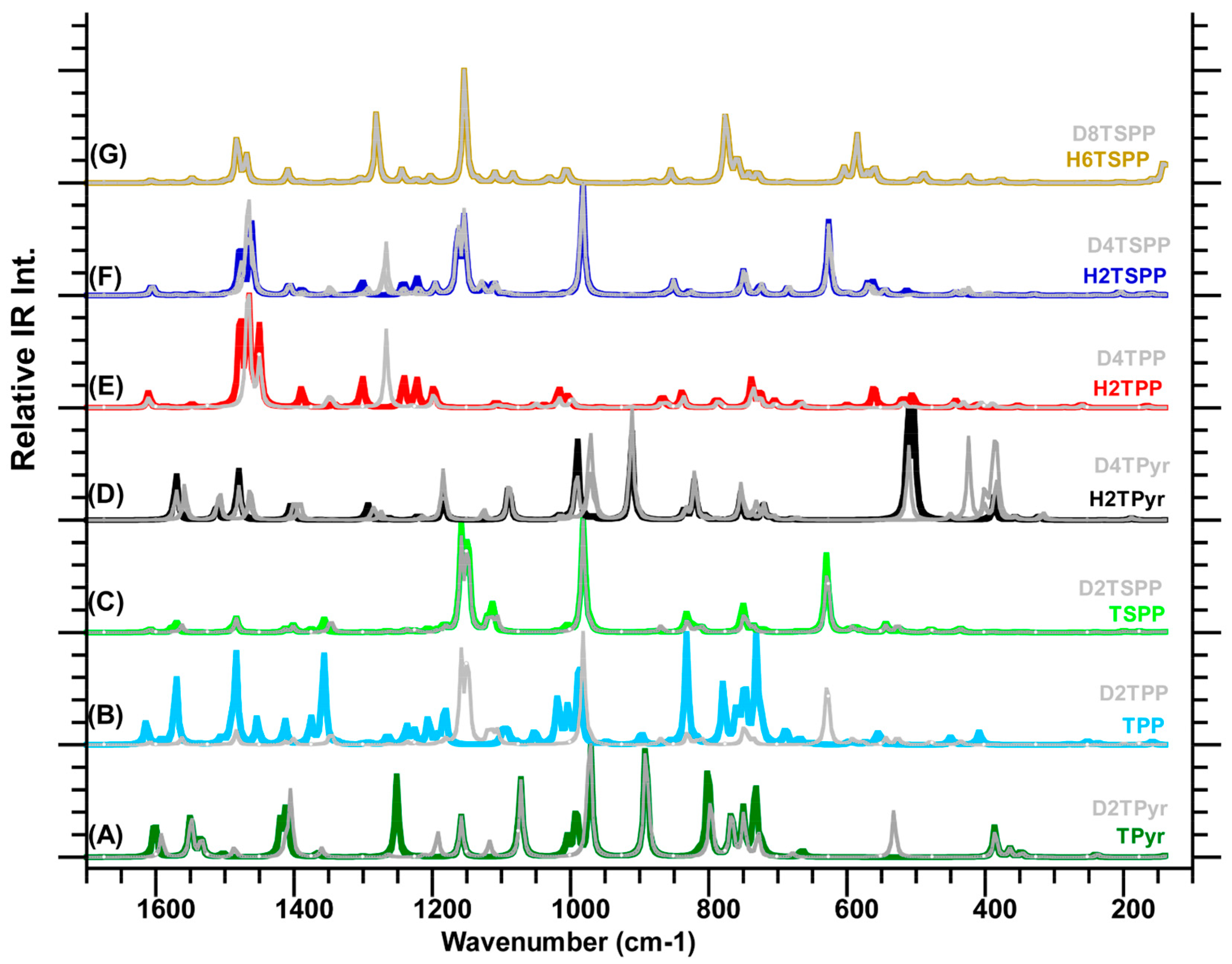
6 stands for the deuteration of the porphyrin core and sulfonato groups) at the same level of the DFT. Their spectra exhibited many IR features with medium and relatively weak intense in addition intense IR bands, which are dispersed throughout the full spectral range as seen in
Figure 4. The calculations coupled with the animated motions indicated that these calculated IR vibrational modes are principally associated with: (a) symmetric and asymmetric skeletal deformations of the macrocycle and phenyl rings; (b) out-of-plane deformation of the macrocycle and the phenyl rings; and (c) rocking and wagging of the CH and NH bonds. The assignments of selected IR features for these molecules are provided in
Table 4. In order to test the reliability of the calculated IR spectra of the molecules investigated here, we also compared their IR peaks in the calculated spectra of the TPP and H
2TSPP (protonated TSPP) with its experimentally measured IR spectra of TPP [
30] and H
2TSPP [
31]. As seen in
Table 4, the calculated and measured IR frequencies are well correlated, which indicates that the calculated IR spectra of the molecules investigated in this work are reasonable. We should point out that two types of scaling factors used for the IR spectrum of TPP: one (0.96 × ω
cal + 40 in cm
−1) that we used through paper for the Raman and IR spectra discussed here, but a scaling factor of 0.976 provided best fit to experimentally measured IR data (not for Raman) of TPP only. Only the latter one is given in
Table 4.
The key conclusions on the calculated IR spectra as following: (1) when carefully examination the predicted IR features for the H2TSPP in connection with the IR spectra of the TPyr/H2TPyr, TPP/H2TPP and TSPP/H6TSPP by taking into account of their vibrational motions of the modes of frequencies, the peaks at 439, 974, 1125, 1193, 1405, 1566 and 1603 cm−1 in the IR spectrum of the H4TSPP are caused by skeletal deformation of the meso-phenyl substitution rings only such as rocking or wagging of its H and vibrational bond stretching, no any contribution comes from the sulfonato groups -SO3); (2) the predicted IR peaks are caused by the vibrational motion of the -SO3 groups: the strongest IR peak at 980 cm−1 in the H2TSPP spectrum (which results from the symmetric stretching of O-S-O bonds, νs(O-S-O); a medium intense one at 1151 cm−1 (due to asymmetric stretching of O-S-O, νa(O-S-O)); and at a relatively strong peak at 624 cm−1 is due to bending deformation of the SO3 groups like closing and opening umbrella shape; (3) a relatively strong IR peak at 1160 cm−1 in the spectrum of the H2TSPP in water used as solvent that is caused by νa(O-S-O) and rocking of CH in phenyl rings, ρ(CH on phenyl); (4) the bond stretching, bending deformation and rocking of CH bonds within sulfonatophenyl substitutes; ν(S-C)), θ(C-C(S)-C) and ρ(CH on phenyl only) produced a very weak IR peak at about 1108 cm−1 in spectra of the porphyrins substituted with sulfonatophenyl groups.
The frequency shift predicted is that of 1108 cm
−1 mode in the sulfonatophenyl-substituted porphyrin which shift to 1049 cm
−1 in the spectra of phenyl substituted porphyrin; (5) the predicted IR peaks result from due to ν(S-C)/θ(phenyl) and relatively weak w(CH and NH) and out of plane bending (or twisting) deformation of macrocycle (at 748 cm
−1) and out of plane twisting of the entire molecule, including θ(O-S-O)/w(CH and NH) (at 566 cm
−1) have very weak intensity, where the θ and w represent the bending deformation and wagging vibration, respectively; (6) the vibrational mode of frequency only result from the H atoms at porphyrin core (NH) caused two IR peaks at low frequency region of 510 and 559 cm
−1 (due to w(NH)), with very weak intensity; (7) the IR band caused by only rocking of the CβH produced a very weak peak at 1089 cm
‒1, which is appeared almost at same position for the other systems; (8) the vibrational mode of frequencies caused by the bond stretching (ν), rocking (ρ), wagging (w) and bending deformation (θ) of the C and H atoms within macrocycle are predicted at 751, 825, 848, 1036, 1240, 1299 and 1386 cm
−1 with a very weak or very weak intensity. These predicted IR features of frequencies are somewhat shifted in spectra of the protonated porphyrins, which are in agreement with experimental observation as provided in
Table 4 from refs. [
30,
31]. It is worth noting that each assignment made in
Table 4 was carried out based on the nuclear motion of the atoms within molecule using the Gaussview
visualization program; (9) the bonding stretching between the
meso-C atoms (C
m within the macrocycle) and C
ϕ atom within the phenyl substitution involved in the mode of frequencies (due to ν
s/a(C
ϕ-C
m-C
α)/θ(C
ϕ-C
m-C
α)/ν
a(C
β-C
β-C
α)/θ(C
α-N-C
α)/ρ(CH) are found at 1220 and 1348 cm
−1. The peaks caused by symmetric/asymmetric bond stretching, bending deformation or wagging/rocking vibrational motion of the atoms within free base porphyrin (macrocycle) are predicted at 1004, 1460 and 1545 cm
−1; (10) while in-plane rotational motion of the pyrroline rings, including relatively weak out-of-plane twisting deformation of the phenyl rings produced a weak peak at 439 cm
−1, a weak peak at 427 cm
−1 (rocking of phenyl rings (ρ(phenyl) and wagging of macrocycle w(macrocycle and another weak peak at 484 cm
−1 (caused by twisting of phenyl (phenyl) and w(macrocycle)) are found in the calculated spectrum of H
2TSSP. Detailed descriptions for each band are given in
Table 4.
2.3.1. Isotopic (or Deuteration) Effect on the IR Spectrum
The calculated IR spectra of the molecules studied here clearly indicated that there are not some shifted in the vibrational frequency, but also, the deuterated porphyrin core or sulfonato groups exhibit relatively intense IR peaks in the range of 2630–2660 cm−1 (unscaled values) due to N-D bond stretching and around 2711 cm−1 (unscaled value) resulting from the O-D bond stretching. The most momentous frequency shifts predicted are those of 540 and 490 cm−1 (H2TSPP, where all N atoms at porphyrin core are hydrogenated or protonated) shifted to 396 and 366 cm−1 in the D4HTSPP (indicates that all N atoms at porphyrin core were deuterated) in the range of low frequency region (below 700 cm−1) due to wagging of the N-D bond, w(ND).
Table 4.
Assignments of the IR active modes of frequencies (in cm
−1) of the meso substituted porphyrin derivatives: TPP (C
2v point group), TSPP (C
2v), H
2TSPP (C
2v) and H
6TSPP (C
2v) for comparison. The calculations were carried out in water used as solvent at B3LYP/6-311G(d,p) Level of the DFT. Where ∆ν
sc stands for scaled vibrational frequencies ((a) ∆ν
sc = 0.96∆ν
calc +40 as used for the Raman spectra for all molecules studied here) and I
IR indicate the calculated IR intensity. In the assignments, the signs ν, θ, ρ and w indicates the bonding stretching, bending deformation, rocking and wagging, respectively. It should be noted that two different scaling factor used for the TPP: (a) ∆ν
sc = 0.96∆ν
calc +40) and (b) ∆ν
sc = 0.976∆ν
calc. The latter one (b) yields best fitting to observed IR spectrum (from refs. [
30,
31]) of the TPP only, not for others; however, the scaling factor of ∆ν
sc = 0.96∆ν
calc +40 yields the best fitting to observed IR spectrum of H
2TSPP (from ref. [
31]) and Raman spectra of the TPP (our previous work, ref. [
23]) and H
2TSPP (present work).
Table 4.
Assignments of the IR active modes of frequencies (in cm−1) of the meso substituted porphyrin derivatives: TPP (C2v point group), TSPP (C2v), H2TSPP (C2v) and H6TSPP (C2v) for comparison. The calculations were carried out in water used as solvent at B3LYP/6-311G(d,p) Level of the DFT. Where ∆νsc stands for scaled vibrational frequencies ((a) ∆νsc = 0.96∆νcalc +40 as used for the Raman spectra for all molecules studied here) and IIR indicate the calculated IR intensity. In the assignments, the signs ν, θ, ρ and w indicates the bonding stretching, bending deformation, rocking and wagging, respectively. It should be noted that two different scaling factor used for the TPP: (a) ∆νsc = 0.96∆νcalc +40) and (b) ∆νsc = 0.976∆νcalc. The latter one (b) yields best fitting to observed IR spectrum (from refs. [30,31]) of the TPP only, not for others; however, the scaling factor of ∆νsc = 0.96∆νcalc +40 yields the best fitting to observed IR spectrum of H2TSPP (from ref. [31]) and Raman spectra of the TPP (our previous work, ref. [23]) and H2TSPP (present work).
| TPP | TSPP | H2TSPP | H6TSPP | Assignments |
|---|
| Sym | ∆νsc.a | ∆νsc.b | IIR | ∆νexp[30] | ∆νexp[31] | Sym | ∆νsc.a | IIR | Sym | ∆νsc.a | IIR | ∆νexp[31] | Sym | ∆νsc.a | IIR |
|---|
| B2 | 447 | 414 | 6 | 409 | 406 | B2 | 431 | 1 | B2 | 439 | 3 | 415 | B2 | 439 | 1 | In-plane rotational motion of the pyrroline rings, including relatively weak out-of-plane twisting deformation of the phenyl rings, but no contributions come from the pyrroline rings |
| | | | | | | B2 | 438 | 13 | B2 | 427 | 5 | 445 | | | | Rocking of phenyl rings (ρ(phenyl) and wagging of macrocycle w(macrocycle). |
| B2 | 447 | 414 | 6 | | | B2 | 475 | 59 | B2 | 439 | 3 | 457 | B2 | 469 | 1 | Out-of-plane bending of phenyl groups only. |
| B1 | 553 | 521 | 10 | | 516 | B1 | 523 | 2 | A1 | 484 | 8 | | A1 | 484 | 8 | Twisting of phenyl τ(phenyl) and w(macrocycle) |
| A1 | 584 | 553 | 2 | | | | | | A1 | 510 | 8 | | A1 | 487 | 4 | w(NH only) |
| B2 | 570 | 539 | 2 | 559 | 558 | | | | B1 | 559 | 10 | | B1 | 567 | 10 |
| | | | | | | A1 | 540 | 8 | A1 | 566 | 14 | 560 | | | | Out of plane twisting of the molecule and θ(O-S-O)/w(CH and NH) |
| | | | | | | B1 | 541 | 1 | B1 | 567 | 6 | 580 | | | |
| | | | | | | B2 | 627 | 45 | B2 | 624 | 63 | 637 | B2 | 582 | 39 | Due to bending deformation of the SO3− groups like closing and opening umbrella shape. |
| | 647 | 618 | 2 | 619 | 618 | A1 | 657 | 0.6 | | | | | | | | In plane bending deformation of phenyl rings, including w(NH and CβH only) and out of plane deformation of the macrocycle. |
| A1 | 666 | 636 | 4 | 638 | 636 | A1 | 668 | 1.0 | | | | | | | |
| B2 | 688 | 659 | 6 | 658 | 657 | | | | | | | | | | |
| B2 | 728 | 700 | 43 | 699 | 701 | | | | | | | | | | | w(CH on phenyl) and relatively weak out of plane deformation of the phenyl rings. |
| | | | | | | B2 | 732 | 6 | B2 | 722 | 12 | 715 | B2 | 725 | 5 | w(CH on phenyl) and out of plane deformation of the phenyl rings and macrocycle. |
| | | | | | | B2 | 747 | 16 | B2 | 748 | 19 | 741 | B2 | 739 | 6 | Primarily due to ν(S-C)/θ(phenyl) and relatively weak w(CH an NH) and out of plane bending (or twisting) deformation of macrocycle. |
| A1 | 745 | 716 | 61 | | | A1 | 749 | 10 | A1 | 751 | 6 | A1 | 753 | 19 | Primarily due to w(CβHs an NH) and out of plane bending (or twisting) deformation of macrocycle, relatively weak out of plane deformation of the phenyl. |
| B2 | 757 | 729 | 32 | 727 | 728 | | | | | | | | | | | w(CβH an NH) and out of plane bending (or twisting) deformation of macrocycle, relatively weak out of plane deformation of the phenyl. |
| B1 | 776 | 749 | 37 | | | B1 | 757 | | | | | | | | | w(CH in phenyl and macrocycle) and out of plane bending (or twisting) deformation of phenyl rings the macrocycle. |
| B2 | 776 | 749 | 21 | 746 | 748 | B2 | 767 | 1 | | | | | | | |
| | | | | | | | | | | | | | A1 | 769 | 32 | Mainly due to ν(S-O(H)), including w(CβHs an NH) and out of plane bending (or twisting) deformation of macrocycle, relatively weak out of plane deformation of the phenyl. |
| | | | | | | | | | | | | | B2 | 773 | 66 |
| A1 | 815 | 788 | 3 | 785 | 788 | B1 | 823 | 7 | B1 | 825 | 2 | 800 | B1 | 826 | 3 | W(CβHs and NH) and out of plane bending deformation of macrocycle |
| A1 | 829 | 802 | 100 | 798 | 799 | A1 | 829 | 20 | A1 | 848 | 23 | 854 | A1 | 852 | 15 |
| B1 | 894 | 869 | 10 | 875 | 871 | B1 | 896 | 1 | | | | | | | | θ(N-Cα-Cβ and N-Cα’-Cβ’ in the same phase)/θ(Cm-Cα-N)/θ(Cα-Cm-Cα)/θ(phenyl)/ρ(CβH) |
| B1 | 985 | 960 | 91 | 964 | 962 | B1 | 968 | 5 | B1 | 974 | 1 | 966 | B1 | 998 | 2 | W(CH on phenyl) |
| | | | | | | B2 | 980 | 63 | B2 | 980 | 100 | 984 | | | | νs(O-S-O) |
| B2 | 1001 | 977 | 34 | | | B2 | 1002 | 5 | B2 | 1004 | 2 | 1012 | B2 | 1004 | 9 | θ(N-Cα-Cβ and N-Cα’-Cβ’ in the same phase)/θ(Cm-Cα-N)/θ(Cα-Cm-Cα)/θ(phenyl)/ρ(CβH) |
| A1 | 1020 | 996 | 0. | 979 | 980 | A1 | 1020 | 0. | A1 | 1036 | 2 | 1039 | A1 | 1035 | 1 | Expansion of the pyrrole/pyrroline groups along N(H)…N(H) direction due to ν(Cα-Cβ), leading to macrocycle getting rectangular shape instead of square shape. |
| B1 | 1014 | 991 | 11 | | | | | | | | | | | | | θ(C-C-C in phenyl) |
| B2 | 1016 | 993 | 33 | 999 | 1002 | B2 | 1016 | 1 | | | | | | | | ρ(CβH)/θ(C-C-C in phenyl)/θ(Cm-Cα-N) |
| B1 | 1049 | 1026 | 9 | 1031 | 1032 | | | | B1 | 1033 | <1 | | | | | ρ(CH in phenyl)/θ(C-C-C in phenyl) |
| B1 | 1087 | 1065 | 9 | 1069 | 1072 | | | | B1 | 1089 | 1 | | | | | ρ(CβH) |
| A1 | 1094 | 1072 | 10 | | | | | | | | | | | ρ(CβH and CH on phenyl). |
| | | | | | | B1 | 1109 | 21 | B1 | 1108 | 11 | | B1 | 1107 | 3 | ν(C-S)/θ(C-C(S)-C)/ρ(CH on phenyl only) |
| | | | | | | A1 | 1116 | 14 | A1 | 1125 | 6 | 1125 | A1 | 1133 | 1 | ρ(CH on phenyl only) |
| | | | | | | B2 | 1146 | 64 | B1 | 1151 | 30 | | | | | νa(O-S-O) |
| | | | | | | A1/B1 | 1156 | 100 | A1/B1 | 1160 | 35 | 1188a | A1/B1 | 1151 | 100 | νa(O-S-O)/ρ(CH on phenyl) |
| B2 | 1180 | 1159 | 38 | | 1155 | B2 | 1180 | 8 | | | | | | | | νa(C-N-C)/θ(C-N-C)/ ρ(CβH) |
| B2 | 1189 | 1168 | 4 | 1174 | 1176 | B2 | 1188 | 0. | B2 | 1193 | 9 | | B2 | 1200 | 3 | ρ(CH on phenyl only) |
| B1 | 1204 | 1183 | 25 | 1187 | 1187 | B1 | 1204 | 4 | | | | | | | | ρ(NH) and relatively weak θ(whole molecule) |
| B2 | 1224 | 1203 | 13 | 1211 | 1211 | B2 | 1224 | 2 | B2 | 1220 | 14 | 1218 | B2 | 1221 | 2 | νs(C-N-C)/νs(Cϕ-Cm-Cα)/ρ(NH and CH) |
| B1 | 1235 | 1215 | 17 | 1220 | 1219 | B1 | 1235 | 3 | B1 | 1240 | 12 | | B1 | 1241 | 7 | νa(C-N-C)/ρ(NH and CβH) |
| B1 | 1262 | 1242 | 9 | 1247 | 1251 | B1 | 1262 | 2 | B1 | 1299 | 11 | | B1 | 1301 | 2 | ν(Cβ-Cα)/θ(C-N-C)/ρ(CβH and NH). |
| B1 | 1355 | 1337 | 47 | 1348 | 1351 | B1 | 1354 | 8 | B1 | 1348 | 1 | 1350 | B1 | 1387 | 1 | νa(Cβ-Cβ-Cα)/ θ(Cα-N-Cα)/νa(Cϕ-Cm-Cα)/θ(Cϕ-Cm-Cα)/ρ(CH). |
| B1 | 1373 | 1356 | 24 | 1359 | 1358 | B1 | 1373 | 4 | B1 | 1386 | 5 | 1384 | B1 | 1387 | 1 | νa(C-N-C)/ρ(CβH and NH). |
| B2 | 1411 | 1394 | 22 | 1400 | 1400 | B2 | 1411 | 4 | | | | | | | | ν(Cβ-Cα)/ν(Cα-Cm) which also leading to νa(Cβ-Cα-Cm), including ρ(CβH). |
| A1 | 1451 | 1435 | 12 | 1459 | 1437 | B2 | 1399 | 2 | B1 | 1405 | 9 | 1395 | B1 | 1407 | 6 | ρ(CH on phenyl), including relatively weak νs(C-C-Cϕ) |
| B2 | 1482 | 1466 | 82 | 1471 | 1471 | B2 | 1482 | 16 | B2 | 1460 | 62 | 1472 | B2 | 1467 | 17 | Ν(Cβ-Cβ)/ν(Cα-Cm) that leading to θ(C-N-C) |
| B1 | 1492 | 1486 | 7 | 1488 | 1493 | | | | | | | | | | | ν(Cα-Cm)/ν(Cβ-Cβ’)/νa(Cα-NH-Cβ), including ρ(CH) on the macrocycle only. |
| B1 | 1568 | 1554 | 67 | | 1555 | B1 | 1567 | 10 | B1 | 1545 | 1 | 1554 | B1 | 1545 | 3 |
| B1 | 1588 | 1574 | 1 | 1573 | 1575 | | | | B2 | 1566 | 1 | 15581564 | | | | νa( C-C-C) within phenyl rings and ρ(H on phenyl) |
| B2 | 1612 | 1598 | 30 | 1595 | 1595 | B2 | 1607 | 2 | B1 | 1603 | 8 | 15921597 | B2 | 1604 | 1 | ν(C-C)/ρ(CH) within phenyl rings, including θ(C-C-C in phenyl). |
Figure 4.
Calculated IR spectra of porphyrin derivatives calculated in water used as solvent at the B3LYP/6-311G(d,p) level of the DFT: (A) free-base porphyrin (TPyr) and deuterated-TPyr (D2TPyr); (B) meso-tetraphenylporphyrin (TPP) and (D2TPP); (C) anionic meso-tetrakis(p-sulfonatophenyl)porphyrin (TSPP) and deuterated-TSPP (D2TSPP), and (D) protonated-TPyr (H2TPyr) and deuterated-H2TPyr (D4TPyr); (E) protonated-TPP (H2TPP) and deuterated-H2TPP (D4TPP); (F) protonated TSPP (H2TSPP) and deuterated-H2TSPP (D4TSPP); and (G) dicationic TSPP (H6TSPP) and deuterated-H6TSPP (D8TSPP). It should be noted that the plot IR spectra in the gray color belong to the deuterated molecules.
Figure 4.
Calculated IR spectra of porphyrin derivatives calculated in water used as solvent at the B3LYP/6-311G(d,p) level of the DFT: (A) free-base porphyrin (TPyr) and deuterated-TPyr (D2TPyr); (B) meso-tetraphenylporphyrin (TPP) and (D2TPP); (C) anionic meso-tetrakis(p-sulfonatophenyl)porphyrin (TSPP) and deuterated-TSPP (D2TSPP), and (D) protonated-TPyr (H2TPyr) and deuterated-H2TPyr (D4TPyr); (E) protonated-TPP (H2TPP) and deuterated-H2TPP (D4TPP); (F) protonated TSPP (H2TSPP) and deuterated-H2TSPP (D4TSPP); and (G) dicationic TSPP (H6TSPP) and deuterated-H6TSPP (D8TSPP). It should be noted that the plot IR spectra in the gray color belong to the deuterated molecules.
In the high or mid frequency region, when the deuterium atom(s) is involved in the vibrational mode of frequency that lead to a red shift in the frequency as much as up to 10 cm
−1 for the D
2TSPP spectrum. The deuteration also have an effect on the relative intensity of the IR peaks in some cases, see
Figure 4. These shift in the high frequency shift for the D
2TPyr, D
4TPyr and D
4TPP are more significant than these for D
2TSPP and D
4TSPP. This observation indicates that above the low frequency region, the frequency shift due to deuteration decreases with increasing size of the substituent group.
2.3.2. Solvent Effect on the IR Spectrum
Also, the solvent effect on the IR spectrum of the H2TSPP was investigated. We used dimethylsulfoxide (DMSO) and toluene instead of water as solvent beside vacuum (gas-phase) in the calculations. The calculations indicated that, below 1100 cm−1, there is no any significant frequency shift in the peak positions, but there are above 1100 cm−1. For instance, while the IR peak centered at around 1200 cm−1 in gas phase is shifted to 1188 cm−1 (toluene), 1166 cm−1 (DMSO), and 1160 cm−1 (water), the IR peaks centered around 1477 and 1453 cm−1 in gas phase spectrum that are shifted to 1490 and 1468 cm−1 (toluene), 1497 and 1480 cm−1 (DMSO), and 1499 and 1481 cm−1 (water). These results indicate that the IR features, in high energy region, of the porphyrin and its derivatives, at least for H2TSPP, are sensitive to its environment. For that reason, the experimentally measured IR spectrum may exhibits frequency shifts in the spectral location of peaks in the different environments; accordingly we need to be careful about the assignment of the IR features.
2.4. Calculated Electronic Spectra of Porphyrin and Its Derivatives
Excited states are not only important to processes such as for electronics, photosynthesis, but they also play a crucial role in the fields of renewable energy, material design and medicine. Therefore, we investigated vertical electronic transitions of the porphyrin derivatives studied here for both singlet triplet states (up to 24 singlet and 24 triplet states) that are provided in
Figure 5 and
Table 5.
Figure 5.
Calculated dipole allowed electronic transitions of porphyrin derivatives in water used as a solvent in the calculations at the TD-B3LYP/6-31G(d,p) level of the TD-DFT: free-base porphyrin (TPyr), meso-tetraphenylporphyrin (TPP), dianionic meso-tetrakis(p-sulfonatophenyl)porphyrin (TSPP), and protonated-TPyr (H2TPyr), protonated-TPP (H2TPP), protonated TSPP (H2TSPP) and dicationic TSPP (H6TSPP).
Figure 5.
Calculated dipole allowed electronic transitions of porphyrin derivatives in water used as a solvent in the calculations at the TD-B3LYP/6-31G(d,p) level of the TD-DFT: free-base porphyrin (TPyr), meso-tetraphenylporphyrin (TPP), dianionic meso-tetrakis(p-sulfonatophenyl)porphyrin (TSPP), and protonated-TPyr (H2TPyr), protonated-TPP (H2TPP), protonated TSPP (H2TSPP) and dicationic TSPP (H6TSPP).
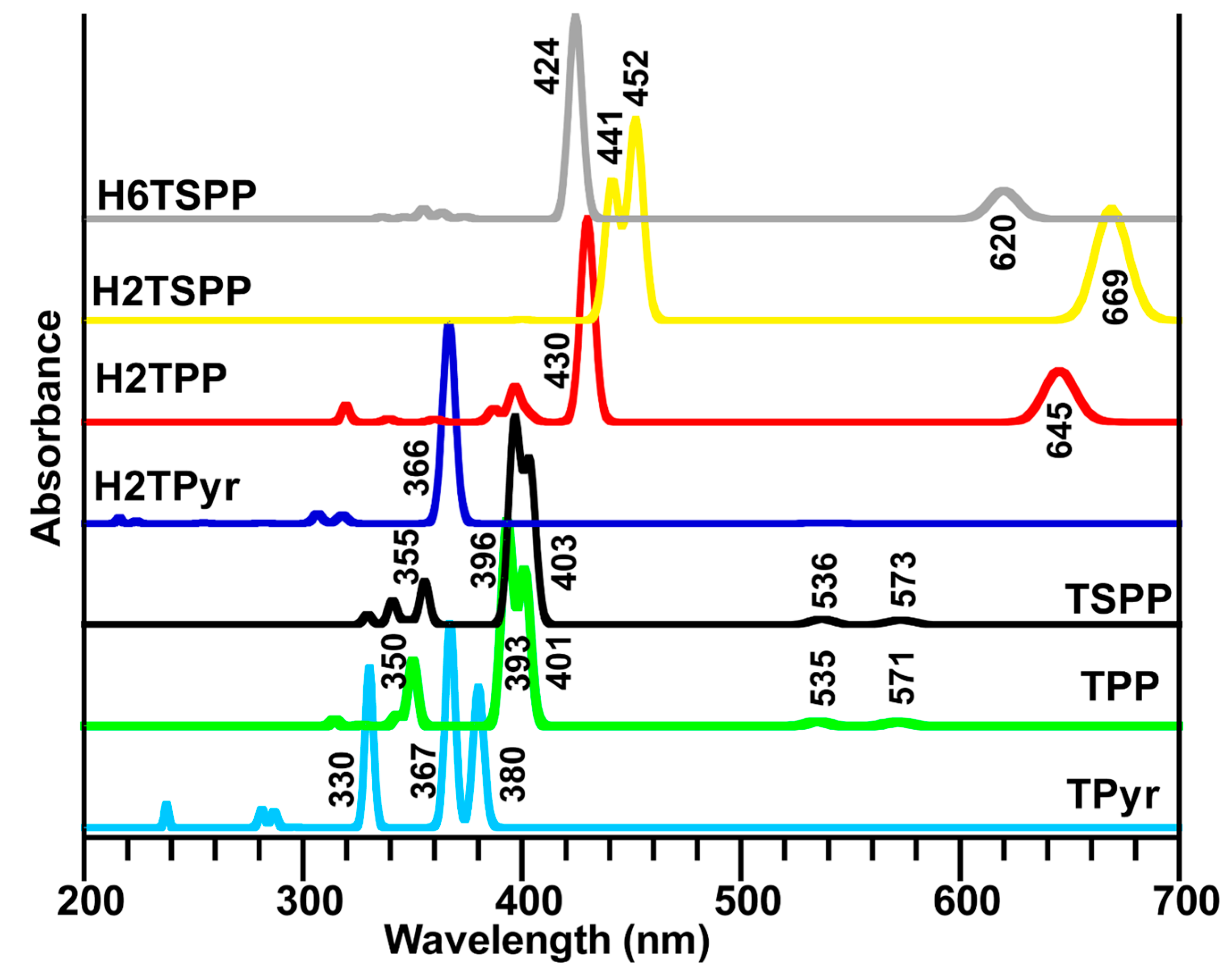
The calculations mainly produced a strong electronic absorption band in the range of 450–360 nm and a few weak or very weak electronic transition below and above the strong band. The strongest band is known as Soret band (B-band, in the range of about 400–450 nm) and weaker bands (in longer wavelength region, about 450–750 nm) are known as Q-bands. The electronic spectra of porphyrin and its derivatives studied here discussed below. It is worthy to note that the percentage given in parenthesis indicates the contributions from the different HOMO (H)→LUMO (L) transitions to a desired electronic transitions. The minor contributions are not shown here.
Table 5.
The selected values of the calculated singlet-singlet (S0→Sn) and singlet-triplet (S0→Tn) vertical electronic transitions for the TPyr, H2TPyr, TPP, H2TPP, TSPP, H2TSPP, and H6TSPP (in water used as a solvent) at TD-B3LYP/6-31G(d,p) level of the TD-DFT. The percentages in the parenthesis indicate the contributions from the different HOMO(H)→LUMO(L) transitions to a desired electronic transitions and the minor contributions are not provided here.
Table 5.
The selected values of the calculated singlet-singlet (S0→Sn) and singlet-triplet (S0→Tn) vertical electronic transitions for the TPyr, H2TPyr, TPP, H2TPP, TSPP, H2TSPP, and H6TSPP (in water used as a solvent) at TD-B3LYP/6-31G(d,p) level of the TD-DFT. The percentages in the parenthesis indicate the contributions from the different HOMO(H)→LUMO(L) transitions to a desired electronic transitions and the minor contributions are not provided here.
| TPyr/S0→Sn | S0→Tn |
| Sn | (eV) | (nm) | F | Sym | Major Contribs | Tn | (eV) | (nm) | Sym | Major Contribs |
| 1 | 2.30 | 540 | 0.0005 | B1U | H−1− > L+1 (40%), | 1 | 1.51 | 822 | B2U | H−1− > L (21%), |
| H− > L (59%) | H− > L+1 (79%) |
| 2 | 2.45 | 506 | 0.0003 | B2U | H−1− > L (47%), | 2 | 1.82 | 682 | B1U | H− > L (94%) |
| H− > L+1 (53%) |
| 3 | 3.26 | 380 | 0.8144 | B1U | H−3− > L (22%), | 3 | 2.04 | 608 | B2U | H−1− > L (78%), |
| H−1− > L+1 (48%), |
| H− > L (29%) | H− > L+1 (22%) |
| 4 | 3.38 | 367 | 1.1911 | B2U | H−1− > L (50%), | 4 | 2.07 | 598 | B1U | H−1− > L+1 (94%) |
| H− > L+1 (47%) |
| 5 | 3.44 | 360 | | B3G | H−2− > L (98%) | 7 | 2.90 | 428 | B3G | H−2− > L (88%) |
| 6 | 3.66 | 339 | | AG | H−2− > L+1 (99%) | 8 | 2.96 | 419 | B1U | H−3− > L (86%) |
| 7 | 3.76 | 330 | 0.6934 | B1U | H−3− > L (76%), | 9 | 3.15 | 393 | AG | H−2− > L+1 (93%) |
| H−1− > L+1 (12%), |
| H− > L (12%) |
| 8 | 3.76 | 330 | 0.2479 | B2U | H−3− > L+1 (93%) | 11 | 3.33 | 373 | B3G | H−8− > L+1 (16%), |
| H− > L+2 (72%) |
| 16 | 4.33 | 287 | 0.0914 | B2U | H−5− > L+1 (97%) | 13 | 3.39 | 366 | B2U | H−3− > L+1 (96%) |
| 18 | 4.41 | 281 | 0.1037 | B1U | H−5− > L (99%) | 15 | 3.61 | 343 | AG | H−8− > L (26%), |
| H−1− > L+2 (68%) |
| 23 | 5.22 | 237 | 0.1338 | B1U | H−2− > L+2 (98%) | 16 | 3.64 | 340 | B3G | H−4− > L+1 (79%) |
| H2TPyr/S0→Sn | S0→Tn |
| Sn | (eV) | (nm) | F | Sym | Major contribs | Tn | (eV) | (nm) | Sym | Major contribs |
| 1 | 2.31 | 538 | 0.0007 | E | H−1− > L+1 (48%), | 1 | 1.63 | 763 | E | H−1− > L+1 (30%), |
| H− > L (52%) | H− > L (70%) |
| 2 | 2.31 | 538 | 0.0007 | E | H−1− > L (48%), | 2 | 1.63 | 763 | E | H−1− > L (30%), |
| H− > L+1 (52%) | H− > L+1 (70%) |
| 3 | 3.39 | 366 | 1.4554 | E | H−1− > L+1 (52%), | 3 | 1.96 | 632 | E | H−1− > L+1 (69%), |
| H− > L (48%) | H− > L (31%) |
| 4 | 3.39 | 366 | 1.4554 | E | H−1− > L (52%), | 4 | 1.96 | 632 | E | H−1− > L (69%), |
| H− > L+1 (48%) | H− > L+1 (31%) |
| 7 | 3.90 | 318 | 0.0597 | E | H−5− > L+1 (45%), | 7 | 3.23 | 384 | B1 | H−3− > L+1 (28%), |
| H−2− > L (28%), |
| H−4− > L+1 (53%) | H− > L+2 (31%) |
| 8 | 3.90 | 318 | 0.0597 | E | H−5− > L (45%), | 8 | 3.32 | 374 | E | H−3− > L+1 (44%), |
| H−4− > L (53%) | H−2− > L (44%) |
| 11 | 4.05 | 306 | 0.0695 | E | H−5− > L+1 (54%), | 9 | 3.37 | 367 | E | H−5− > L+1 (42%), |
| H−4− > L+1 (45%) | H−4− > L+1 (48%) |
| 12 | 4.05 | 306 | 0.0695 | E | H−5− > L (54%), | 10 | 3.37 | 367 | E | H−5− > L (42%), |
| H−4− > L (45%) | H−4− > L (48%) |
| TPP/S0→Sn | S0→Tn |
| Sn | (eV) | (nm) | F | Sym | Major contribs | Tn | (eV) | (nm) | Sym | Major contribs |
| 1 | 2.17 | 571 | 0.0337 | B2 | H−1− > L+1 (32%), | 1 | 1.40 | 884 | B1 | H−1− > L (16%), |
| H− > L (67%) | H− > L+1 (84%) |
| 2 | 2.32 | 535 | 0.0359 | B1 | H−1− > L (37%), | 2 | 1.66 | 745 | B2 | H− > L (98%) |
| H− > L+1 (63%) |
| 3 | 3.09 | 401 | 1.2834 | B2 | H−3− > L (10%), | 3 | 1.99 | 623 | B1 | H−1− > L (84%), |
| H−1− > L+1 (62%), |
| H− > L (27%) | H− > L+1 (15%) |
| 4 | 3.16 | 393 | 1.6972 | B1 | H−1− > L (62%), | 4 | 2.06 | 602 | B2 | H−1− > L+1 (97%) |
| H− > L+1 (37%) |
| 6 | 3.54 | 350 | 0.5462 | B2 | H−3− > L (87%) | 5 | 2.84 | 436 | A2 | H−2− > L (88%) |
| 8 | 3.62 | 343 | 0.0909 | B1 | H−3− > L+1 (98%) | 6 | 2.90 | 428 | B2 | H−3− > L (82%) |
| 19 | 3.95 | 314 | 0.0267 | B1 | H−10− > L (39%), | 7 | 3.08 | 403 | A1 | H−2− > L+1 (91%) |
| H−8− > L+1 (57%) |
| 20 | 3.95 | 314 | 0.0216 | B2 | H−14− > L (15%), | 8 | 3.15 | 393 | A2 | H−16− > L+1 (10%),
H− > L+2 (74%) |
| H−11− > L (56%), |
| H−10− > L+1 (14%), |
| H−8− > L (10%) |
| H2TPP/S0→Sn | S0→Tn |
| Sn | (eV) | (nm) | F | Sym | Major contribs | Tn | (eV) | (nm) | Sym | Major contribs |
| 1 | 1.92 | 645 | 0.304 | A' | H−1− > L+1 (16%), | 1 | 1.22 | 1020 | A" | H− > L+1 (98%) |
| H− > L (84%) |
| 2 | 1.92 | 645 | 0.3039 | A" | H−1− > L (16%), | 2 | 1.22 | 1020 | A' | H− > L (98%) |
| H− > L+1 (84%) |
| 3 | 2.89 | 430 | 1.2029 | A' | H−1− > L+1 (74%), | 3 | 2.02 | 615 | A" | H−1− > L (95%) |
| H− > L (14%) |
| 4 | 2.89 | 430 | 1.2026 | A" | H−1− > L (74%), | 4 | 2.02 | 615 | A' | H−1− > L+1 (95%) |
| H− > L+1 (14%) |
| 10 | 3.13 | 396 | 0.2053 | A" | H−5− > L (89%) | 5 | 2.67 | 464 | A" | H−3− > L (13%), |
| H−2− > L+1 (13%), |
| H− > L+2 (56%) |
| 11 | 3.13 | 396 | 0.2056 | A' | H−5− > L+1 (89%) | 6 | 2.76 | 449 | A' | H−7− > L+1 (16%), |
| H−6− > L (16%), |
| H−3− > L+1 (29%), |
| H−2− > L (29%) |
| 15 | 3.20 | 387 | 0.078 | A" | H−8− > L (80%) | 7 | 2.88 | 431 | A' | H−3− > L+1 (46%), |
| H−2− > L (46%) |
| 16 | 3.20 | 387 | 0.0778 | A' | H−8− > L+1 (80%) | 8 | 2.88 | 430 | A" | H−3− > L (46%), |
| H−2− > L+1 (46%) |
| 21 | 3.45 | 360 | 0.0351 | A' | H−10− > L (12%), | 9 | 2.93 | 424 | A' | H−5− > L+1 (82%) |
| H−9− > L (80%) |
| 22 | 3.66 | 339 | 0.039 | A" | H−10− > L+1 (13%), | 10 | 2.93 | 424 | A" | H−5− > L (82%) |
| H−9− > L+1 (80%) |
| TSPP/S0→Sn | S0→Tn |
| Sn | (eV) | (nm) | F | Sym | Major contribs | Tn | (eV) | (nm) | Sym | Major contribs |
| 1 | 3.88 | 319 | 0.1998 | A' | H−10− > L (78%),
H−9− > L (11%) | 11 | 2.94 | 421 | A" | H−7− > L (17%), |
| H−6− > L+1 (17%), |
| H−3− > L (13%), |
| H−2− > L+1 (13%), |
| H− > L+2 (31%) |
| 5 | 2.16 | 573 | 0.0419 | B2 | H−1− > L+1 (32%), | 1 | 1.40 | 884 | B1 | H−1− > L (16%), |
| H− > L (67%) | H− > L+1 (84%) |
| 6 | 2.31 | 536 | 0.0506 | B1 | H−1− > L (36%), | 2 | 1.67 | 744 | B2 | H− > L (97%) |
| H− > L+1 (64%) |
| 10 | 3.07 | 403 | 1.4382 | B2 | H−1− > L+1 (62%), | 3 | 1.99 | 624 | B1 | H−1− > L (84%), |
| H− > L (28%) | H− > L+1 (15%) |
| 11 | 3.13 | 396 | 1.8378 | B1 | H−1− > L (62%), | 4 | 2.05 | 604 | B2 | H−1− > L+1 (97%) |
| H− > L+1 (36%) |
| 36 | 3.49 | 355 | 0.3924 | B2 | H−10− > L (83%) | 5 | 2.84 | 437 | A2 | H−9− > L (49%), |
| H−7− > L (39%) |
| 38 | 3.56 | 348 | 0.0392 | B1 | H−10− > L+1 (28%), | 6 | 2.89 | 429 | B2 | H−11− > L (36%), |
| H−8− > L (69%) | H−10− > L (48%) |
| 47 | 3.64 | 341 | 0.2178 | B2 | H−11− > L (83%) | 7 | 3.07 | 404 | A1 | H−9− > L+1 (39%), |
| H−7− > L+1 (51%) |
| 48 | 3.77 | 329 | 0.0936 | B1 | H−11− > L+1 (82%), | 8 | 3.14 | 395 | A2 | H− > L+2 (70%) |
| H−10− > L+1 (11%) |
| H2TSPP/S0→Sn | S0→Tn |
| Sn | (eV) | (nm) | F | Sym | Major contribs | Tn | (eV) | (nm) | Sym | Major contribs |
| 1 | 1.85 | 669 | 0.4223 | B2 | H−5− > L+1 (13%), | 1 | 1.17 | 1056 | B2 | H− > L (97%) |
| H− > L (87%) |
| 2 | 1.85 | 669 | 0.4138 | B1 | H−5− > L (13%), | 3 | 2.34 | 530 | A1 | H−3− > L+1 (47%), |
| H− > L+1 (87%) | H−2− > L (52%) |
| 4 | 2.35 | 528 | 0.0001 | A1 | H−3− > L+1 (47%), | 5 | 2.34 | 529 | B1 | H−4− > L+1 (47%), |
| H−2− > L (53%) | H−1− > L (52%) |
| 5 | 2.35 | 528 | 0.0005 | B2 | H−4− > L (53%), | 7 | 2.01 | 618 | B1 | H−5− > L (95%) |
| H−1− > L+1 (47%) |
| 10 | 2.40 | 518 | 0.0001 | B2 | H−4− > L (47%), | 9 | 2.39 | 518 | A1 | H−3− > L+1 (53%), |
| H−1− > L+1 (53%) | H−2− > L (47%) |
| 12 | 2.74 | 452 | 0.7601 | B1 | H−6− > L (79%), | 13 | 2.51 | 494 | A2 | H−8− > L+1 (34%), |
| H−7− > L (35%), |
| H−5− > L (15%) | H− > L+2 (23%) |
| 13 | 2.75 | 451 | 0.7369 | B2 | H−6− > L+1 (83%), | 14 | 2.62 | 474 | B1 | H−6− > L (90%) |
| H−5− > L+1 (12%) |
| 16 | 2.81 | 441 | 0.5315 | B2 | H−9− > L (17%), | 17 | 2.65 | 467 | A1 | H−8− > L (46%),
H−7− > L+1 (46%) |
| H−6− > L+1 (16%), |
| H−5− > L+1 (57%) |
| 18 | 2.81 | 441 | 0.5073 | B1 | H−9− > L+1 (16%), | 18 | 2.69 | 462 | A2 | H−8− > L+1 (47%),
H−7− > L (47%) |
| H−6− > L (19%), |
| H−5− > L (54%) |
| 19 | 2.97 | 417 | 0.0013 | B1 | H−13− > L (14%), | 19 | 2.72 | 455 | A2 | H−8− > L+1 (13%), |
| H−7− > L (12%), |
| H−10− > L+1 (74%) | H− > L+2 (63%) |
| 22 | 3.10 | 400 | 0.0081 | B2 | H−13− > L+1 (12%), | 20 | 2.96 | 419 | A2 | H−12− > L+1 (43%), |
| H−10− > L (82%) | H−11− > L (47%) |
| H6TSPP/S0→Sn | S0→Tn |
| Sn | (eV) | (nm) | F | Sym | Major contribs | Tn | (eV) | (nm) | Sym | Major contribs |
| 1 | 2.00 | 620 | 0.2467 | B2 | H−1− > L+1 (23%), | 1 | 1.31 | 946 | B2 | H− > L (96%) |
| H− > L (77%) |
| 2 | 2.00 | 619 | 0.2377 | B1 | H−1− > L (23%), | 2 | 1.31 | 945 | B1 | H− > L+1 (96%) |
| H− > L+1 (77%) |
| 3 | 2.92 | 424 | 1.7153 | B1 | H−1− > L (74%), | 3 | 1.94 | 639 | B1 | H−1− > L (94%) |
| H− > L+1 (23%) |
| 4 | 2.92 | 424 | 1.7145 | B2 | H−1− > L+1 (75%), | 4 | 1.94 | 638 | B2 | H−1− > L+1 (94%) |
| H− > L (22%) |
| 7 | 3.32 | 374 | 0.0199 | B2 | H−4− > L (92%) | 5 | 2.74 | 452 | A2 | H− > L+2 (72%) |
| 8 | 3.32 | 373 | 0.0240 | B1 | H−4− > L+1 (92%) | 6 | 3.00 | 413 | A1 | H−7− > L (26%), |
| H−6− > L+1 (26%), |
| H−3− > L (13%), |
| H−2− > L+1 (18%) |
| 11 | 3.41 | 363 | 0.0560 | B1 | H−5− > L (88%) | 7 | 3.08 | 403 | A2 | H−3− > L+1 (37%), |
| H−2− > L (40%) |
| 12 | 3.42 | 363 | 0.0614 | B2 | H−5− > L+1 (88%) | 8 | 3.09 | 401 | A1 | H−3− > L (36%), |
| H−2− > L+1 (34%), |
| H−1− > L+2 (14%) |
| 15 | 3.50 | 355 | 0.0822 | B1 | H−8− > L (94%) | 9 | 3.16 | 393 | B2 | H−5− > L+1 (16%), |
| H−4− > L (66%) |
| 16 | 3.50 | 355 | 0.0918 | B2 | H−8− > L+1 (94%) | 10 | 3.16 | 393 | B1 | H−5− > L (17%), |
| H−4− > L+1 (65%) |
| 20 | 3.59 | 346 | 0.0365 | B2 | H−10− > L (10%),
H−9− > L (84%) | 11 | 3.20 | 387 | A2 | H−12− > L+1 (11%), |
| H−11− > L (12%), |
| H−7− > L+1 (29%), |
| H−6− > L (30%) |
| 21 | 3.69 | 336 | 0.0363 | B1 | H−10− > L+1 12%), | 12 | 3.23 | 384 | B1 | H−8− > L (66%) |
| H−9− > L+1 (81%) |
2.4.1. The Electronic Spectra of the TPyr and Protonated-TPyr (H2TPyr)
The electronic spectrum of the TPyr molecule exhibited two weaker electronic transitions below the Soret band (S): S
0→S
1 (B
1u), H−1→L+1 (40%) and H→L (59%), at 540 nm (with oscillator strength, f = 0.0005) and S
0→S
2 (B
2u), H−1→L+1 (47%) and H→L+1 (53%), at 506 nm (f = 0.0003); two strong electronic transitions in the range of Soret band (B-band): S
0→S
4 (B
2u), results from H−1→L (50%), and H→L+1 (47%), at 367 nm (f = 1.1911), and S
0→S
3 (B
1u), originates from H−3→L (23%), H−1→L+1 (49%) and H→L (30%), at 380 nm (f = 0.8144) beside a few weaker bands: S
0→S
7 (B
1u), H−3→L (76%), H−1→L+1 (12%) and H→L (12%), at 330 nm (f = 0.6934); S
0→S
8 (B
2u), H−3→L+1 (93%), at 330 nm (f = 0.2479); S
0→S
13 (B
3u), H−7→L+1 (99%), at 296 nm (f = 0.0019); S
0→S
16 (B
2u), H−5→L+1 (97%), at 287 nm (f = 0.0914); S
0→S
18 (B
1u), H−5→L (99%), at 281 nm (f = 0.1037); and S
0→S
23 (B
1u), H−2→L+2 (98%), at 237 nm (f = 0.1338). Where H−n and L+n molecular orbitals are not only pure π/σ and π*/σ* molecular orbitals (MOs) as seen in
Figure 6, they also include nonbinding atomic orbitals (AOs) in particular cases such as: H is a symbol of the π(C
β-C
β/C
m-C
α)+n(N); similarly, H−1: π(C
β-C
α), H−2: π(C
α-N-C
α/C
β-C
β); H−3: π(C
α-N-C
α/C
β-C
β)+n(minor, N/C
m); H−4/H+5: π(C
β-C
β)+n(N); H−6/H−7: n(N)+σ(minor; C
β-C
α); L/L+1: π*(C
β-C
β/C
β-C
α/C
m-C
α)+n(minor; N); L+2: π*(C
β-C
α)+n(C
m); L+3: π*(C
β-C
β/C
m-C
α)+n(N/C
α); L+4/L+5: π*(C
β-C
β/C
m-C
α)+n(N/C
α/C
β); L+6/L+7: σ*(H-H/macrocycle)+n(N); and L+8: n*(C).
Figure 6.
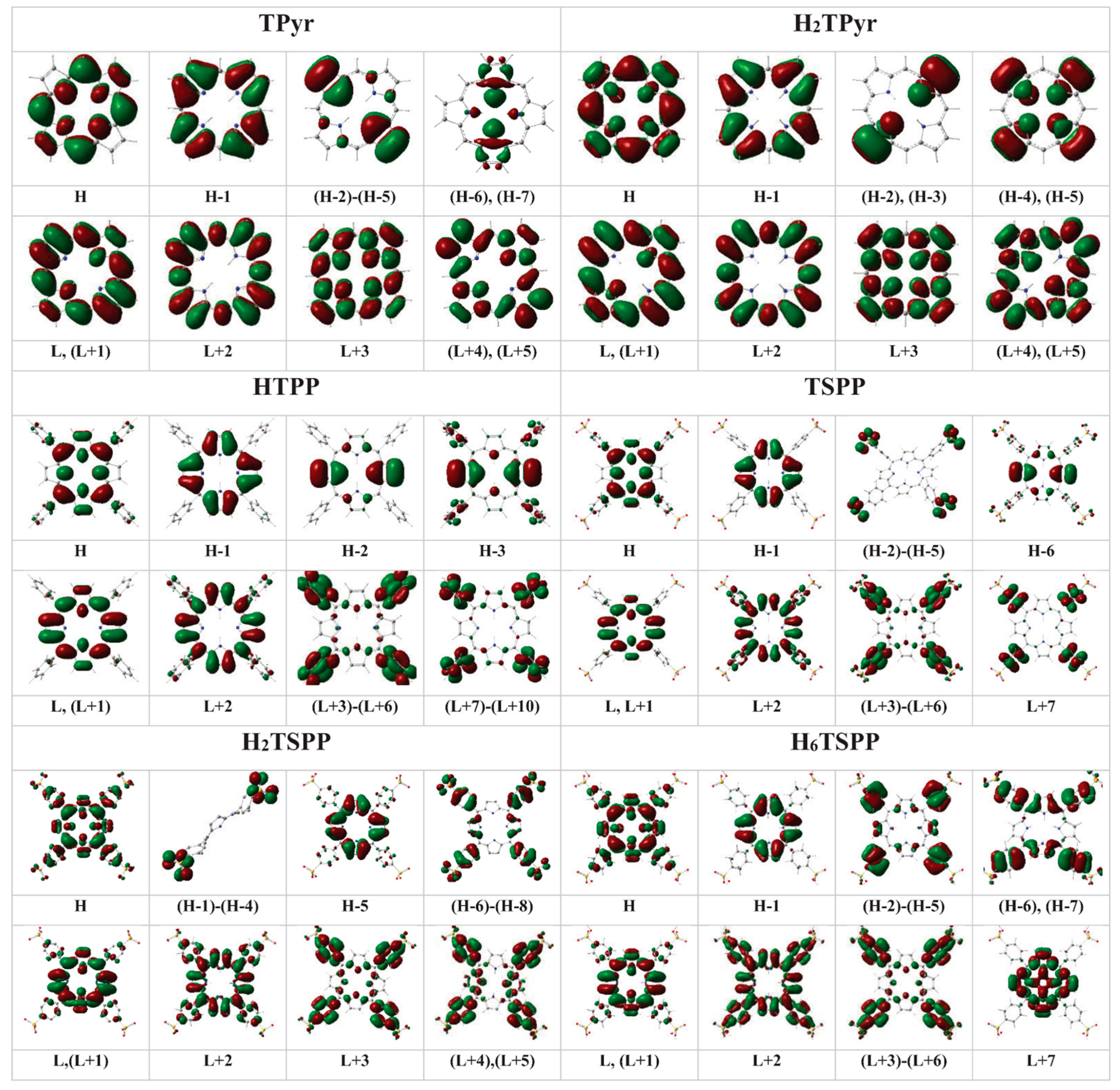
The plotted electron densities in the HOMOs (H) and LUMO (Ls) of: free-base porphyrin (TPyr), meso-tetraphenylporphyrin (TPP), dianionic meso-tetrakis(p-sulfonatophenyl)porphyrin (TSPP), and protonated-TPyr (H2TPyr), protonated-TPP (H2TPP), protonated TSPP (H2TSPP) and dicationic TSPP (H6TSPP) molecules.
Figure 6.
The plotted electron densities in the HOMOs (H) and LUMO (Ls) of: free-base porphyrin (TPyr), meso-tetraphenylporphyrin (TPP), dianionic meso-tetrakis(p-sulfonatophenyl)porphyrin (TSPP), and protonated-TPyr (H2TPyr), protonated-TPP (H2TPP), protonated TSPP (H2TSPP) and dicationic TSPP (H6TSPP) molecules.
The electronic spectrum of the protonated TPyr (H
2TPyr) exhibited Q- and B-bands originate from the S
0→S
1/2 (E), H−1→L (48%) and H→L+1(52%), at 538 nm (f = 0.0007); S
0→S
3/4 (E), H−1/→L+1 (53%) and H→L (49%), at 366 nm (f = 1.4554); S
0→S
22/23 (E), H−5→L+1 (45%) and H−4→L+1 (53%), at 318 nm (f = 0.0597); S
0→S
11/12 (E), H−5→L+1 (54%) and H−4→L+1 (45%), at 306 nm (f = 0.0695); S
0→S
15 (B
2), H−1→L+2 (78%), at 282 nm (f = 0.0049); S
0→S
18 (B
2), H−7→L (10%), H−6→L+1 (10%), H−1→L+2 (11%) and H→L+3 (66%), at 224 nm (f = 0.0337); S
0→S
22/23 (E), H−3→L+2 (95%), at 216 nm (f = 0.0407). Where the H: π(C
β-C
β and C
α-C
m-C
α)+n(N); H−1: π(C
α-C
β); H−2/H−3: π(C
α-C
β)+n(N); H−4/H−5: π(C
β-C
β)+n(N); H−6/H−7: π(C
β-C
α-C
m)+n(minor, N); H−8: π(C
α-C
m-C
α); L/L+1: π(C
β-C
α)+n(minor, N); L+2: π(C
β-C
α) + n(C
m); L+3: π(C
β-C
β)+n(N/C
α/C
m); L+4: π(C
β-C
β and C
α-C
m)+n(N/C
α/C
m). Comparing the electronic spectrum of TPyr with that of H
2TPyr, while the TPyr exhibit three strong bands at 380, 360 and 330 nm (see
Figure 5 and
Table 5), H
2TPyr showed a strong band at 366 nm only in the Soret band region. In the Q-band region, the electronic band positions are shifted from 540 and 506 nm in TPyr to 539 nm (doubly degenerated) in H
2TPyr.
Furthermore, the calculations produced twenty-four triplet states in the range of 822–249 nm for the TPyr molecule (in water used as solvent). There are two triplet states at 373 nm with symmetry B3g, T8(B3g), and at 366 nm (T9(B2u)) that are almost overlap with the strongly dipole allowed singlet electronic state at 367 nm (S4(B2u)). This finding indicate that there is no only possibility of the internal-conversion (IC) process between the S4(B2u at 376 nm) and S1(B1u at 540 nm) and S2(B2u at 506 nm), but also, the possibility of the inter-system-crossing (ISC) process through potential energy surface (PES) touching or strong vibrational coupling between S4 and T8/9 electronic states in the excited state.
For the protonated TPyr (H
2TPyr) molecules exhibited similar futures such as: the IC between the S
3/4 (the strongest bands or B-band) at 366 nm (with the symmetry E) and the S
1 (at 538 nm with symmetry E); the ISC process between the S
1 (at 538 nm with symmetry E) and T
7/8 (triplet states at 367 nm with the symmetry E), see
Table 5.
2.4.2. The Electronic Spectra of TPP and H2TPP
While the TPP molecule exhibited two weak peaks at 571 nm (S
0→S
1, f = 0.00337) and 535 nm (S
0→S
2, f = 0.0359) in Q-band region, the H
2TPP produced only a double degenerated peak that is red shifted to 645 nm (S
0→S
1/2, f = 0.3040). In the B-band region, two strong peaks at 401 and 393 nm (S
0→S
3/4, f = 1.2834/1.6972) in the TPP absorption spectrum and a doubly degenerated band at 430 nm (S
0→S
3/4, f = 1.209) in the H
2TPP. Both spectra exhibited a few weak and very weak allowed electronic transitions in the high energy region as seen in
Table 5 and
Figure 5. Jiang
et al. [
32] measured absorption and EPR spectra of some porphyrins (TPP and derivatives) and metalloporphyrins compounds. The measured absorption spectrum of the TPP produced an intense electronic transition at 417 nm (B-band) and several Q-bands with weak intensity at 514, 550, 593 and 646 nm. As seen in
Table 5 and
Figure 5, while these observed electronic bands are consistent with the calculated values as mentioned above, the electronic transition at 646 nm is not. The calculations did not even produce any dipole forbidden singlet-singlet electronic transition with wavelength longer than 571 nm, but, a weak singlet-singlet transition was predicated at 645 nm for the protonated TPP (H
2TPP) molecule. This observation suggest that the H
2TPP might be formed in the TPP solution with a low concentration and high pH value (pH > 7) or there might be singlet-triplet transition (predicted at 623 nm) through the intensity borrowing process. The authors have also reported that both steric hindrance and electron effects of the functional groups influenced the UV-vis absorption indicate that the TPP of Soret bands of the studied
para-substituted
meso-tetraphenylporphine derivatives moved slightly toward short wavelength (3–5 nm). This experimental observation is consistent with our calculations, for instance, when molecular system going from the TPyr, TPP to TSPP or from the H
2TPyr, H
2TPP to H
2TSPP (see
Figure 5 or
Table 5).
Additionally, the results of the calculations of the TPP (in solution/water) indicated the possibility of the IC process between the B-bands (S3 at 401 nm (with symmetry B2), S4 at 393 nm (B1) and S6 at 350 nm (B2)) and S1 (at 571 nm (B2)/S2 (at 535 nm (B1); the ISC process between the B-bands (S3 at 401 nm (B2) and T7 (at 403 nm (A1))/T8 (at 393 nm (A2)/T11 (at 348 nm (A2) through surface touching since the these singlet and triplet states are almost overlapping, based on the calculations.
For the protonated TPP molecule (H2TPP), the IC process between the B-bands (S3/4 at 430 nm (A' and A")) and S1/2 (at 645 nm (A' and A")), the ISC process between the doubly degenerated Q-band (S1/2 at 645 nm (A' and A")) and T6 (at 449 nm with the symmetry A') and between the degenerated B-band (S3/4 at 430 nm (A' and A")) and T7/8 (at 431 and 430 nm (A' and A")).
2.4.3. Calculated Electronic Spectra of the TSPP, H2TSPP and H6TSPP
The experimentally observed absorption spectra of the free base TSPP exhibited an intense band (S-band, also known as B-band) at ~410 nm and several weak bands (known as Q-bands) at about 515, 550, 580, 640 nm by Akins
et al. [
21] and Zhang
et al. [
31]. Also, the authors have reported that the absorption spectrum of the dianionic TSPP (here we referred as H
2TSPP) displayed the S-band at 432 nm and Q-bands at 540, 580 and 642 nm. Also, Akins and coworkers have measured fluorescence spectra of free-base TSPP (pH = 12) and monomeric H
2TSPP (pH = 4.5), and aggregate TSPP. Their measured fluorescence spectra at excitation 412, 432, and 488 nm (the B-bands for the TSPP, H2TSPP and the aggregated-TSPP, respectively) showed that emission arises from the lowest excited electronic singlet state (S
1) associated with the Q-bands, while excitation might be to the S
2 state, the higher excited state associated with the B-band. (The fluorescence spectrum at excitation 412 nm exhibited a broadened and red degraded peak at 642 nm and a weak one at 702 nm; at the excitation 432 nm, they observed an intense broadened peak with a shoulder at 655 nm. These experimental clearly indicate an internal conversion (CI) from the B-band to the Q-band [
21].
In the Q-band region, while the calculated exhibited two weak transitions at 573 nm (S
0→S
1 with symmetry B
2 and f = 0.0419) and at 536 nm (S
0→S
2 with symmetry B
1, f = 0.0506) in the TSPP spectrum; and at 669 nm (S
0→S
1/2 with symmetries B
2 and B
1, f = 0.4223/0.4138) and 528 nm (S
0→S
4/5 with symmetries A
1 and B
2, f = 0.0001/0.0005), and at 518 nm (S
0→S
10, B
2 symmetry and f = 0.0001) in the H
2TSPP; however, the H
6TSPP (or dicationic TSPP) exhibited only one doubly degenerated weak electronic band at 620/619 nm (S
0→S
1/2 with symmetries B
2 and B
1, f = 0.2467/0.2377). In the B-band (Soret band) region, the calculations revealed a strong band at 403 (S
0→S
3 with B
2 symmetry and f = 1.4382), and at 396 nm (S
0→S
4 with symmetry B
1 and f = 1.8378) for the TSPP; a strong band at 452/451 nm (S
0→S
12/13, B
1 and B
2 symmetries, f = 0.7601/0.7369) and a medium intense band at 441 nm (S
0→S
16, B
2 symmetry and f = 0.5315) for the H
2TSPP; and a strong transition at 424 nm (S
0→S
3/4, B
1 and B
2 symmetries and f = 1.7153/1.7145) for H
6TSPP. Also, many weak electronic transitions are found above these strong bands (see
Table 5). The predicted B-bands and Q-bands for these molecules are compatible with the experimental data [
21,
31].
The predicted possible IC and ISC processes: for the TSPP, the IC (internal conversion) can happen from S
3 (at 403 nm) to S
1 (at 573 nm) and S
2 (at 536 nm). The ISC (intersystem crossing) process may take place from the S
3 (at 403 nm)/S
4 (at 396 nm) to the T
7 (at 404 nm)/T
8 (at 395 nm), but not, take place between the Q-bands and any triplet states, based on the calculations. For the H
2TSPP (or dianionic TSPP): the IC process may take place from the B-bands, S
12/13(at 452/451 nm)/S
16 (at 441 nm), to Q-bands S
10 (at 518 nm)/S
4/5(at 528 nm)/S
1/2(at 669 nm), which is consistent with experimental observation (at 665 nm) [
21]; the ISC process may occur from the Q-bands to the triplet states such as S
4/5(at 528 nm) to T
3,4,5,6(at 530 and 529 nm), and S
10 (at 518 nm) to T
15,16,17,18(at 518 nm). In the high energy region, the ISC may originate from B-bands to triplet states: S
12/13(at 452/451 nm) to the T
19 (at 455 nm). Likewise, for the H
6TSPP dicationic molecule: the IC process may happen between the B-bands (S
3/4(at 424 nm)) and Q-bands (S
1/2(at 620/619 nm)); the ISC process between the Q-bands and triplet states such as from S
1/2(at 620/619 nm) to T
3/4(at 639 and 638 nm). In order to the ISC process happens between the B-band and the closest triplet state(s) (T) that is expected very strong vibrational coupling in the excited state since the energy separation between the S
3/4(B
1/B
2) at 424 nm and the closest triplet states at 452 nm (T
5(A
2) and at 413 nm (T
6(A
1) are 28 nm (−0.18 eV or −1652 cm
−1) and −13 nm (0.08 eV or 645 cm
−1), respectively.
The examination of the results of the calculated absorption spectra for the porphyrin molecules studied here (
Table 5) revealed two important points that are: (1) the
meso-substitutions of the porphyrin with the phenyl/sulfonatophenyl and the protonation of the N atoms at porphyrin core bring about significant red shift in the spectral position of the B-bands and Q-bands; (2) even though the ISC process between the B-band(s) and triplet states occurs for the all porphyrin derivatives, but, there would be an ISC process between the Q-bands (singlet) and triplet state(s) if both of the protonation and meso substitution of the porphyrin with the phenyl/sulfonatophenyl take place.
Figure 6 provides the electron densities in the HOMOs and LUMOs molecular orbitals and nonbonding atomic orbitals (n) involved in the electronic transitions (see
Table 5). The plotted electron densities indicated that the HOMO (Hs) and LUMOs (Ls) are as a resluts of binding/antibinding (π/π*) and nonbinding (n) atomic orbitals beween the atoms within the macrocycle and phenhyl ring such as H: π(C
α-C
m-C
α/C
β-C
β/C-C in phenyl)+n(N); (H-1) to (H-4): generates from the nobonding orbitals of oxygen atom, n(O); H-5: π(C
α-C
β); (H-6)-(H-8): π(C-C-C in phenyl)+ minor n(O and C
β); L and (L+1): π*(C
α-C
m/C
β-C
β)+n*(N); L+2: π*(C
ϕ-C
m/C
β-C
α ) and minor n*(C in phenyl); and (L+3)-(L+5): π*(C-C in phenyl)+n*(C(S) and C
ϕ in phenyl) for the H
2TSPP. For the H
6TSPP, H: π(C
α-C
m-C
α/C
β-C
β)+n(N and C
ϕ/C(S) in phenyl) H-1: π(C
α-C
β); (H-2)-(H-5): π(C-C-C in phenyl) + n(C
α and C
β, minor); (H-6) and (H-7): π(C(S)-C/C-C
ϕ in phenyl and C
m-C
α-C
β); L and (L+1): π*(C
α-C
m/C
β-C
β)+n*(N); L+2: π*(C
ϕ-C
m/C
β-C
β and C-C/C-S in phenyl); (L+3)-(L+6): π*(C-C/C-S in phenyl) and n*( C
ϕ)/n*(C
α and N, minor); and L+7: π*(C
β-C
β)+n*(N and C
α).
2.5. Relaxed Potential Energy Surfaces (PESs) Scan of TSPP Molecule
The ground state (S
0) relaxed PES scan of the TSPP molecule was calculated in water (used as a solvent) by rotating one of four dihedral angles θ (C
α-C
m-C
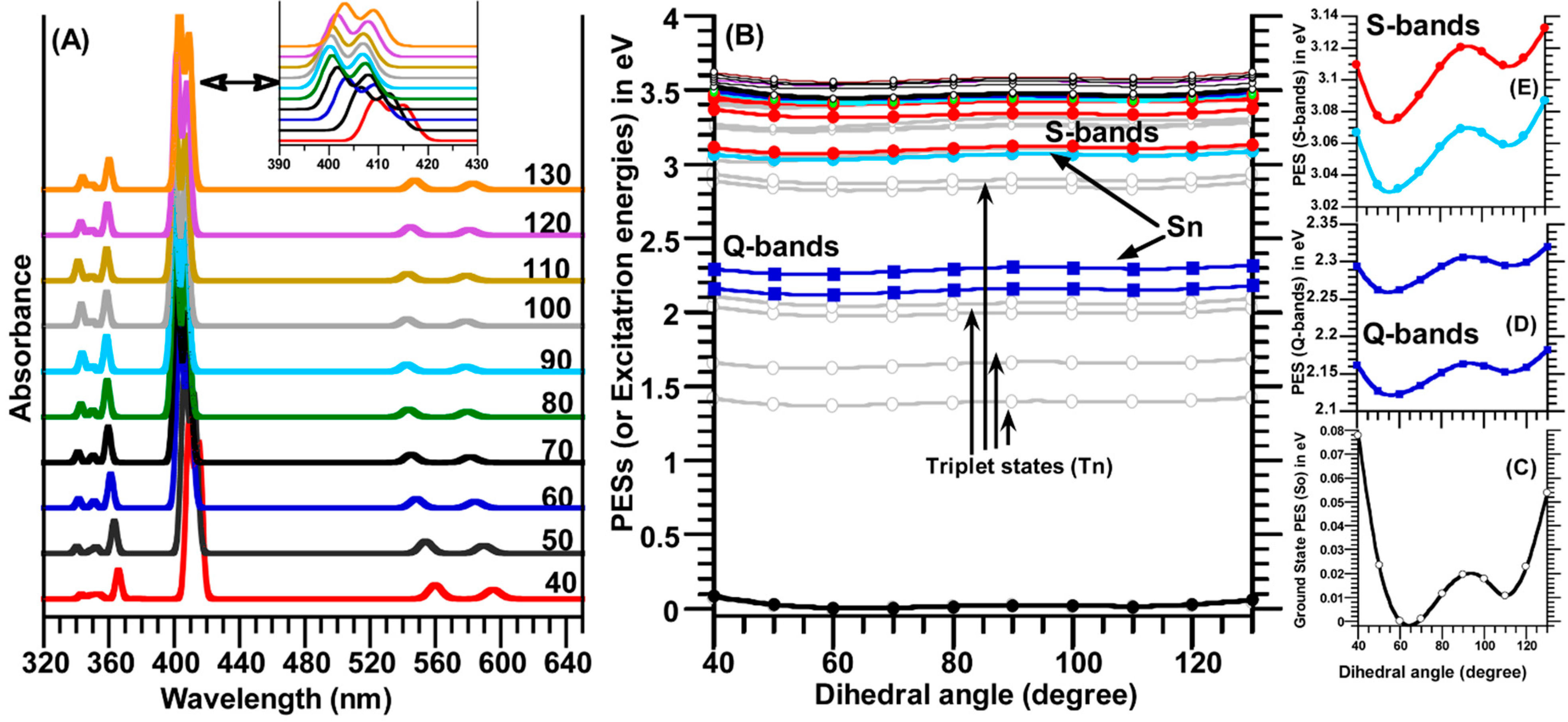
ϕ-C) in the region of 40° to 130° with a step size of 10°. As seen in
Figure 7B,C, the calculated relaxed PES(S
0) curve exhibited two minima at dihedral angles of ~66° and 110°.
Figure 7.
(
A) The calculated electronic spectra (S
0→S
n,
n = 1–24) and (
B) relaxed potential energy surfaces (PESs) scan for the ground state S
0 and upper states S
n and T
n (
n = 1–24) of the TSPP molecule as function of the dihedral angle (C
α-C
m-C
ϕ-C(ph)) rotation in the range of 40° to 130° with a step size of 10°; (
C–
E) display the change in the PES for the S
0 (the ground state), Soret bands (B-bands) and Q-bands in low scale for a better view. It should be noted that only one of the four meso-sulfonatophenyl groups rotated around C
m-C
ϕ bond. Note that the S-bands in
Figure 7E indicates the Soret bands (or B-bands).
Figure 7.
(
A) The calculated electronic spectra (S
0→S
n,
n = 1–24) and (
B) relaxed potential energy surfaces (PESs) scan for the ground state S
0 and upper states S
n and T
n (
n = 1–24) of the TSPP molecule as function of the dihedral angle (C
α-C
m-C
ϕ-C(ph)) rotation in the range of 40° to 130° with a step size of 10°; (
C–
E) display the change in the PES for the S
0 (the ground state), Soret bands (B-bands) and Q-bands in low scale for a better view. It should be noted that only one of the four meso-sulfonatophenyl groups rotated around C
m-C
ϕ bond. Note that the S-bands in
Figure 7E indicates the Soret bands (or B-bands).
These two minima on the relaxed PES curve of the ground state (S
0), at 65° and 110°, correspond to the lowest ground state (with symmetry of C
2v) and an energetically more stable local state (with symmetry of C
2), respectively. The local minima (at 110° ) lies only 0.0132 eV (or 106 cm
−1) above the lowest ground state at ~66°) for the TSPP molecule (
Figure 7C). The highest potential energy barrier is only 0.0219 eV (177 cm
−1) at the dihedral angle of 90° when the molecule going from the lowest ground state to this local state. This finding implies that the meso-sulfonatophenyl substitution groups can be easily rotated around C
m-C
ϕ bond at room temperature since the thermal energy (kT) at 298 K is 207.2 cm
−1. Therefore, the self-assembly or arrangement of the TSPP molecules in any environment might be very easily formed since the predicted potential energy barrier is low as much as 0.0132 eV/106 cm
−1 (at the dihedral angle of 90°). It is should be point out that the calculated ground state relaxed PES scan was obtained for only one of four meso-sulfonatophenyl groups rotation within the TSPP molecule. If the relaxed PES scan is obtained for the rotations of all four meso-substitutional groups, there would be several different local minimum with a different potential energy barriers on the ground state PES. In this case, these small changes in the potential energy barrier distribution of the TSPP molecules may be used such as scanning nanocalorimetry measurement purpose or other electronic purposes that responding to very small changes in energy.
The PES scan of the singlet (S
n) and triplet (T
n) excited states were obtained by calculating the vertical electronic transitions S
0→S
n and S
0→T
n (
n = 1–24) energies for each optimized ground state geometry of the TSPP molecule at the dihedral angle θ(C
α-C
m-C
ϕ-C(ph)) and taking into account of the SCF energy corrections, using the following equation:
where E
0 and E(θ) correspond to the calculated global (total SCF) energies at the lowest ground state and the relaxed potential energy at the dihedral angle θ(C
α-C
m-C
ϕ-C
1), respectively; and (S
0→S
n/S
0→T
n; θ) indicates the vertical electronic transition energy between S
0 and excited energy levels S
0/T
n at the rotated dihedral angle rotated θ. As seen in
Figure 7C–E, change in the electronic energy levels of the singlet and triplet states as function of the dihedral angle rotation, PESs (for S
n and T
n states), are similar to change in the PES(S
0) curve. The calculated dipole allowed electronic transitions at each rotated dihedral angle are given in
Figure 7A. The results of the calculations displayed that the red shift in spectral position of Soret bands (B-band) increases with the increasing rotational dihedral angle in both right-handed and left-handed rotational direction around its equilibrium dihedral angle of ~66° in the ground state.

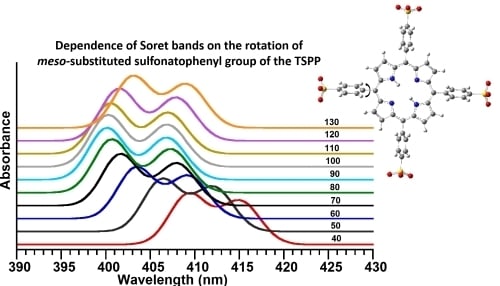
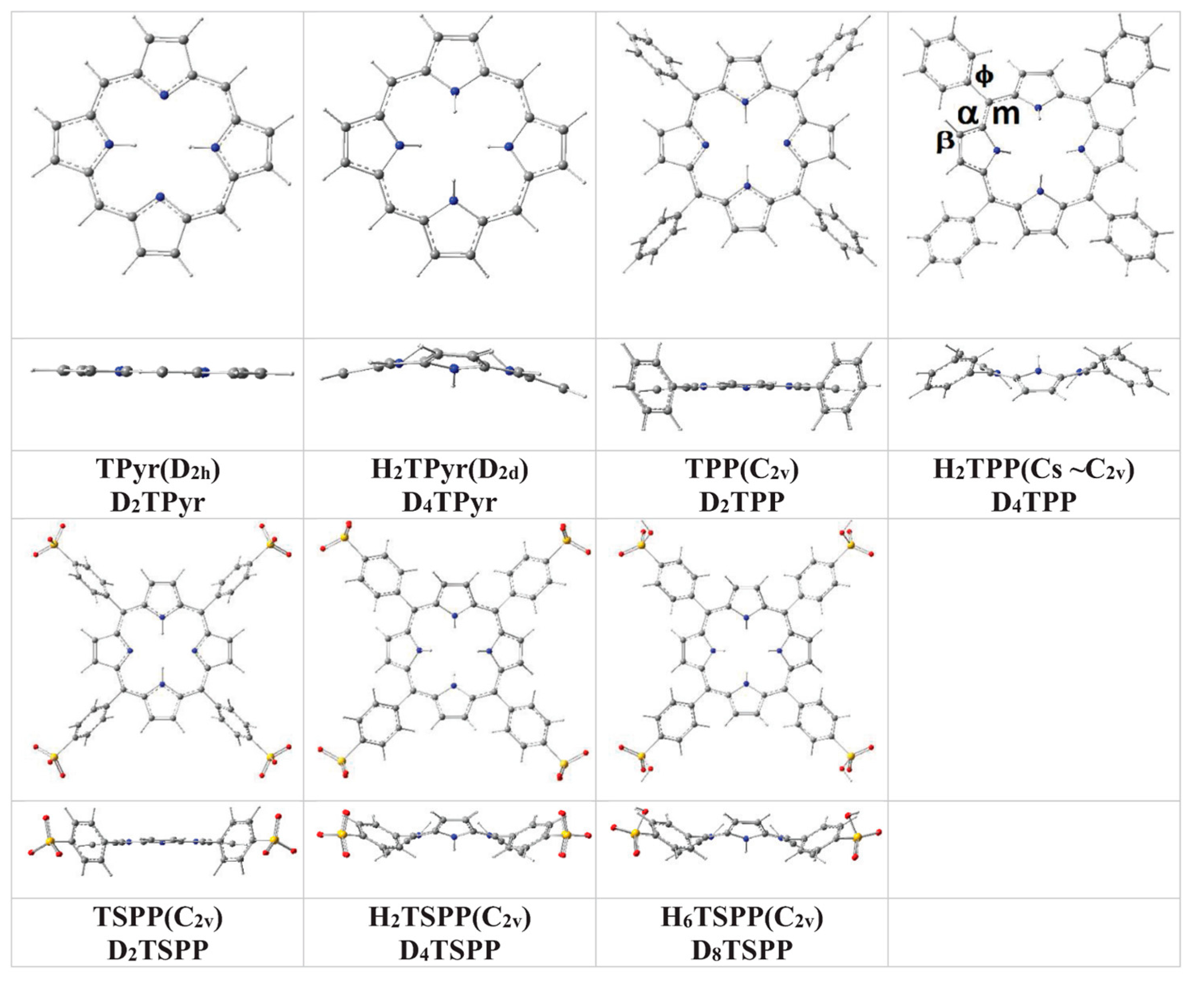

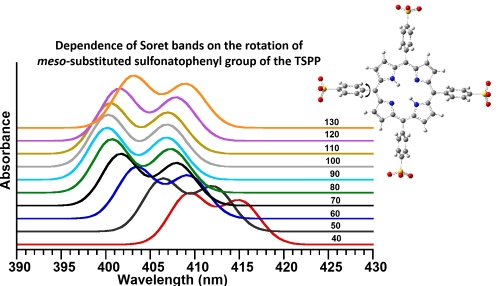{kind=link}
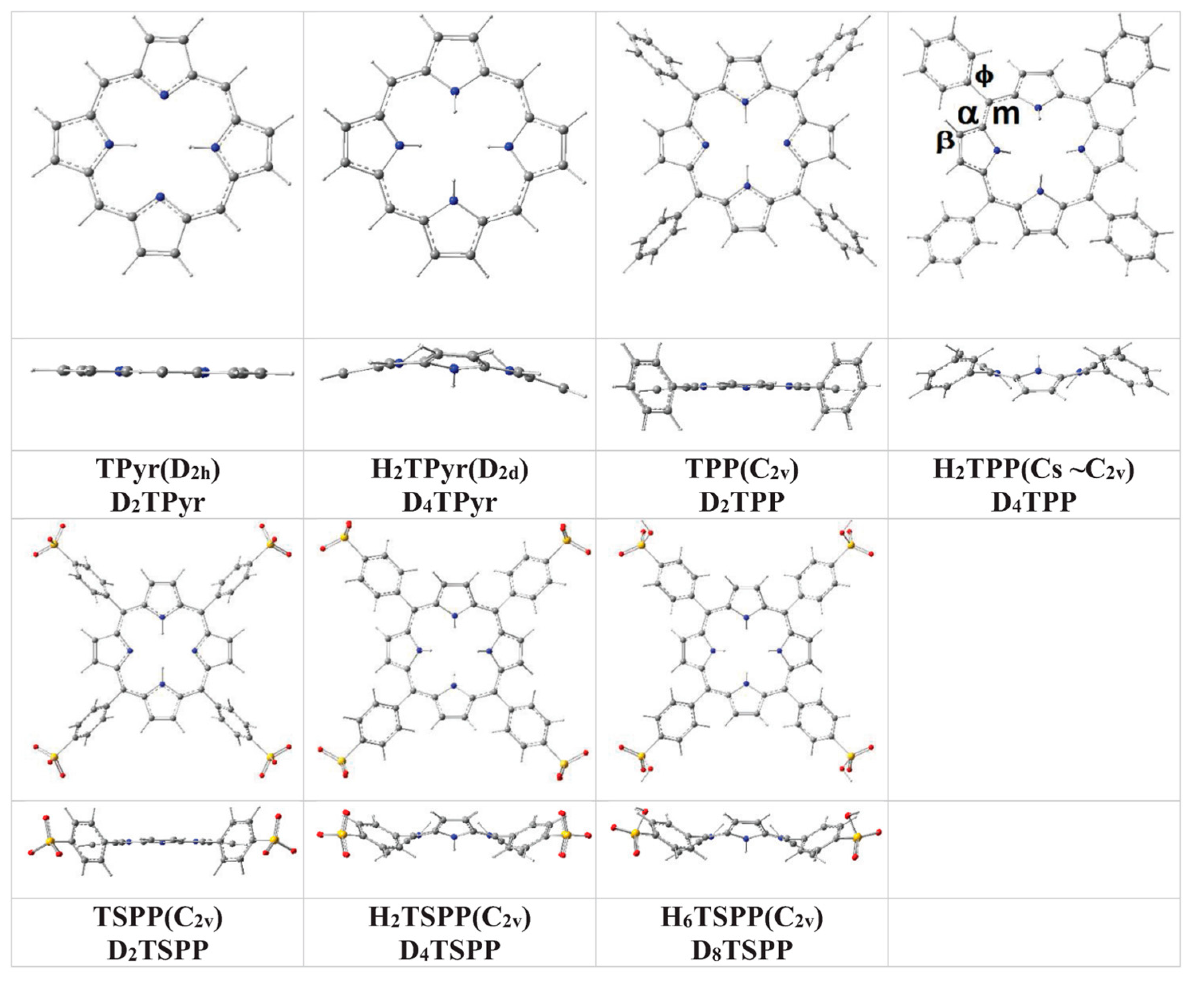{kind=link}
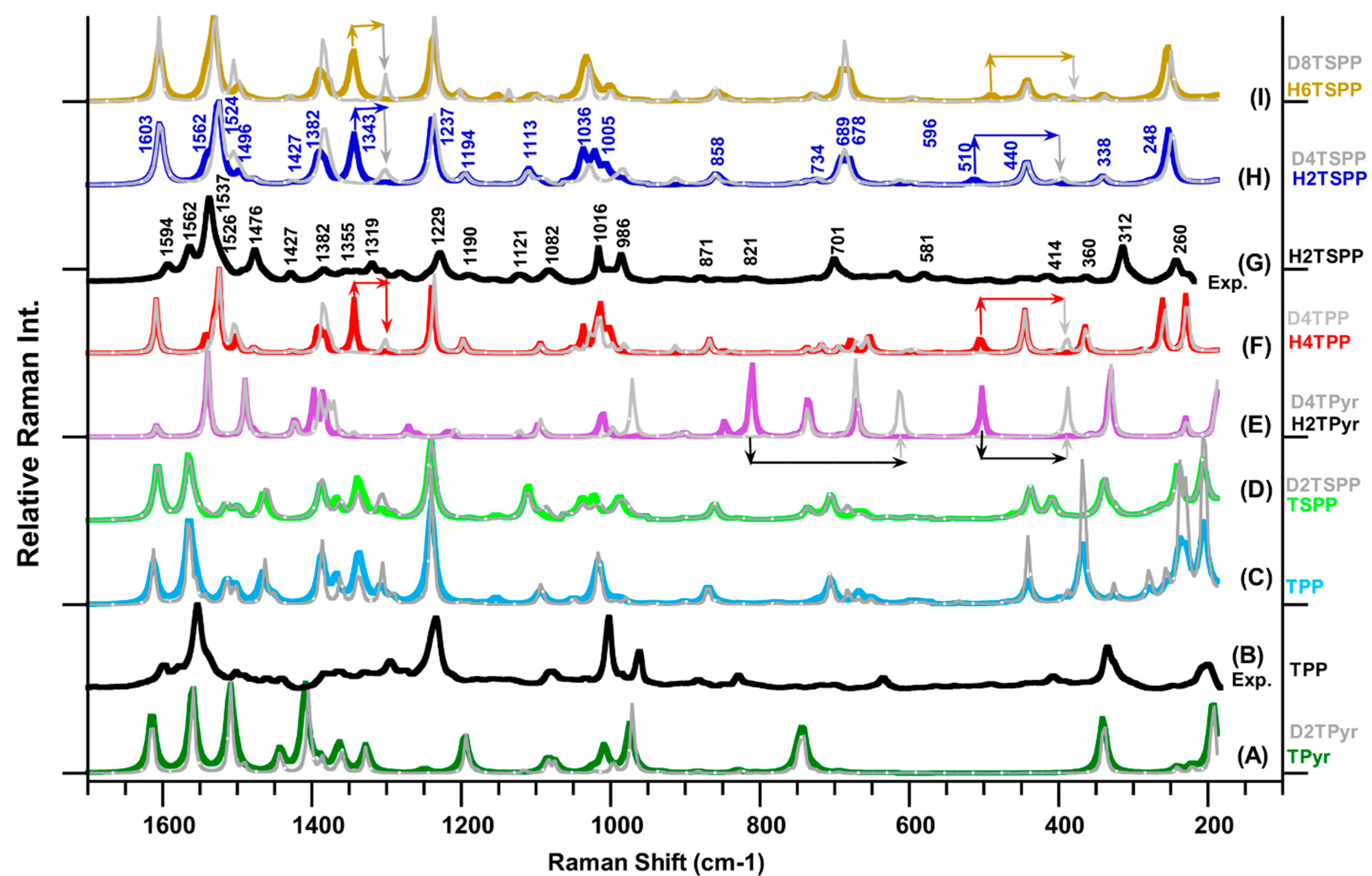{kind=link}
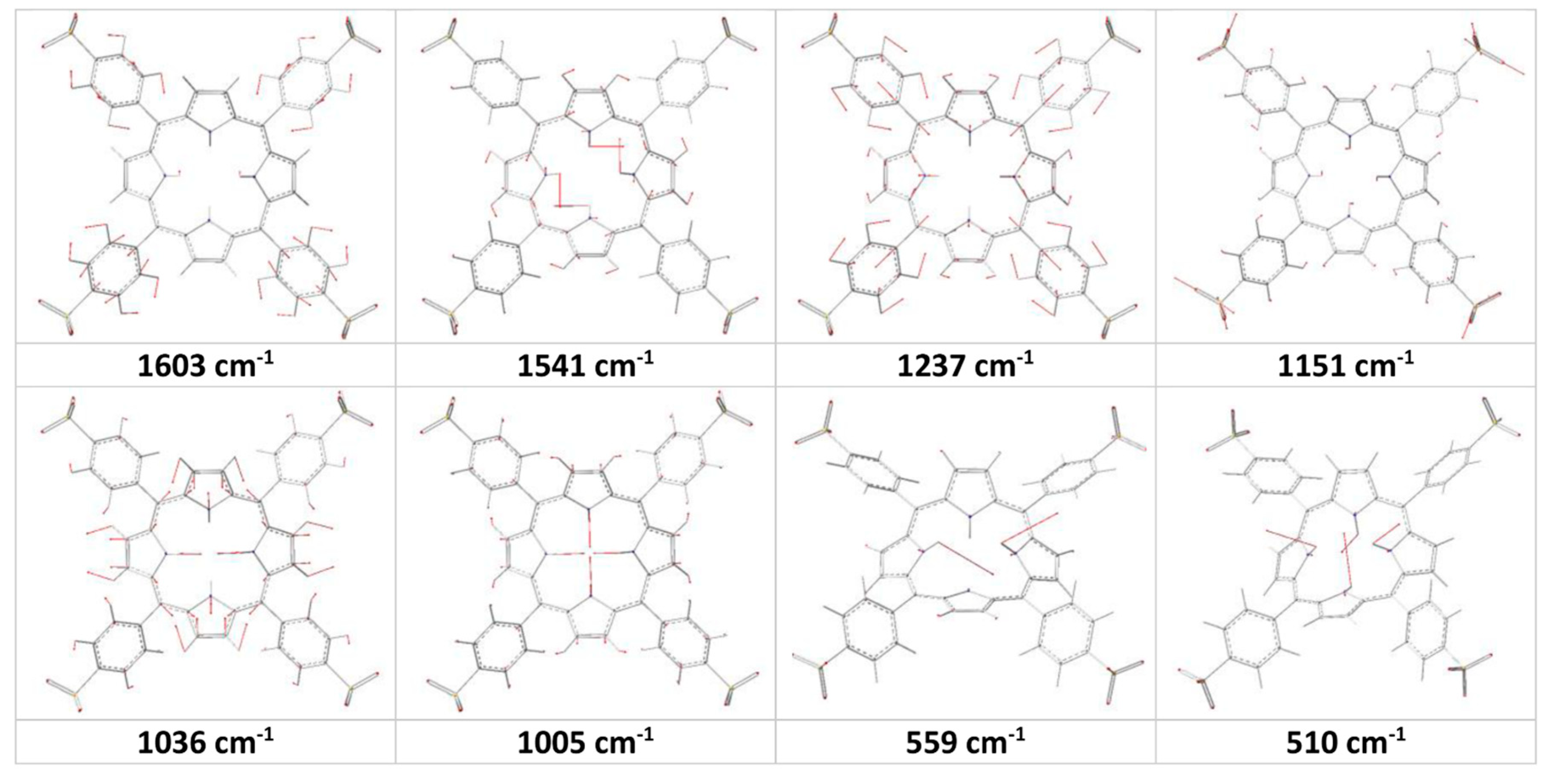{kind=link}
{kind=link}
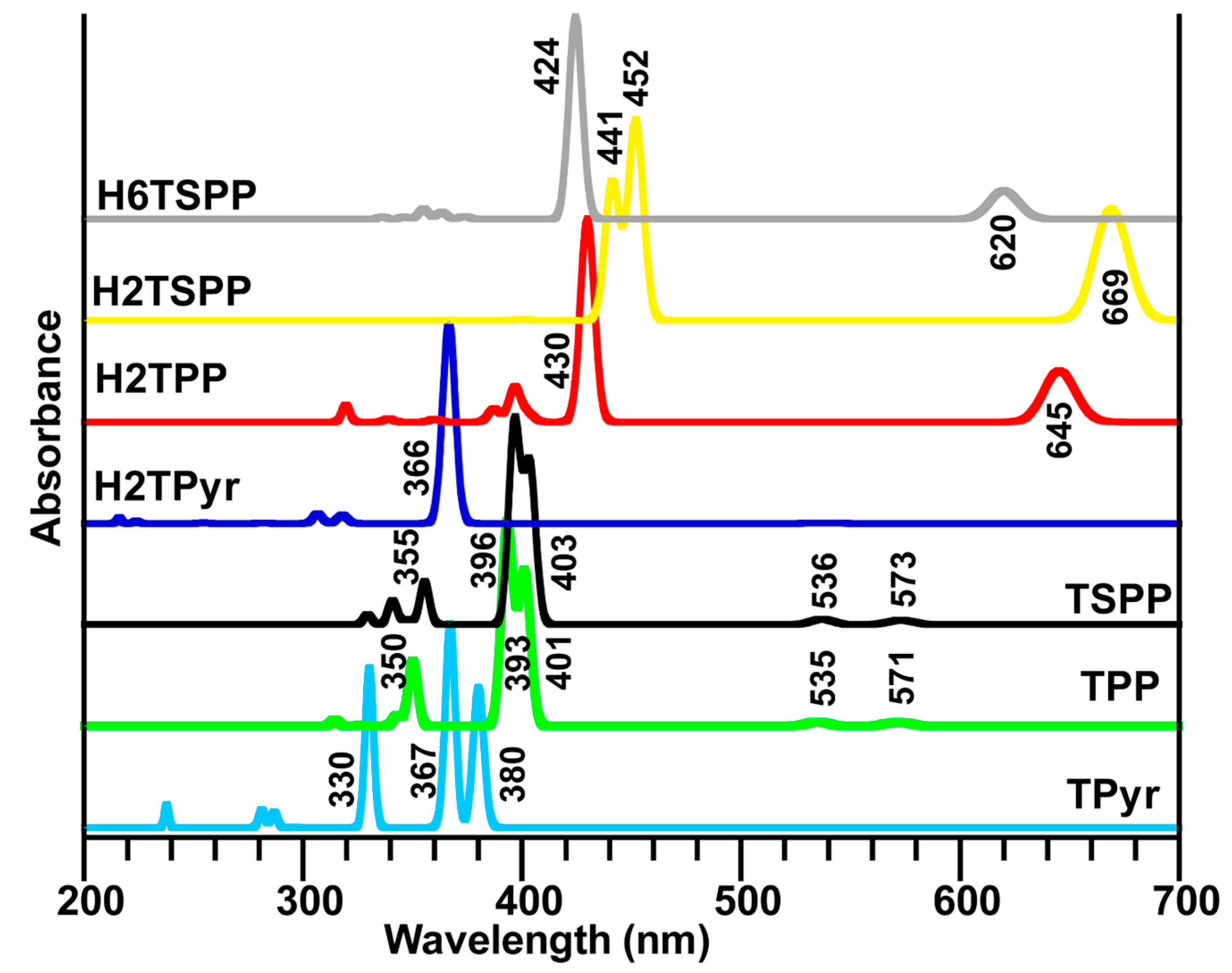{kind=link}
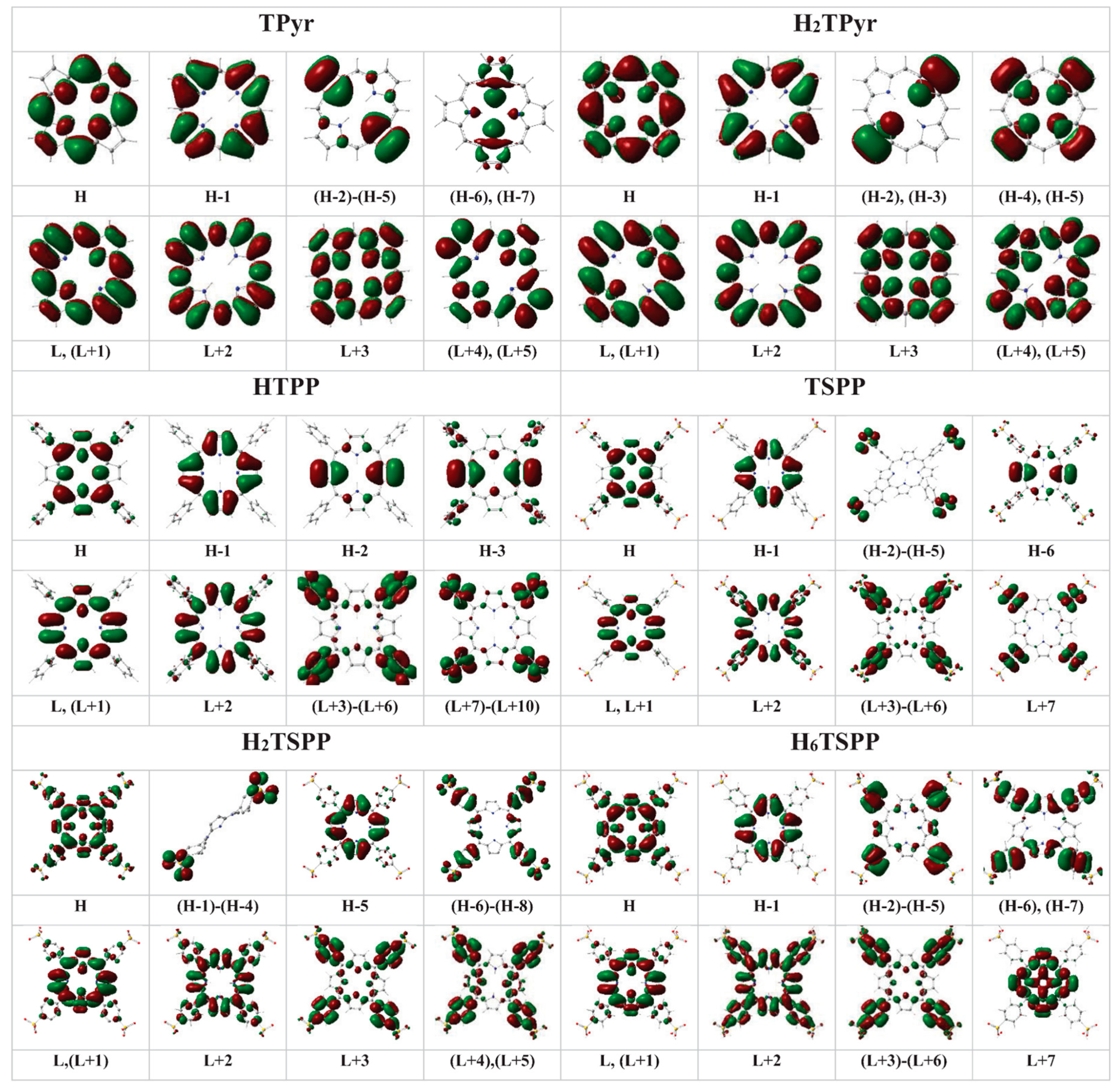{kind=link}
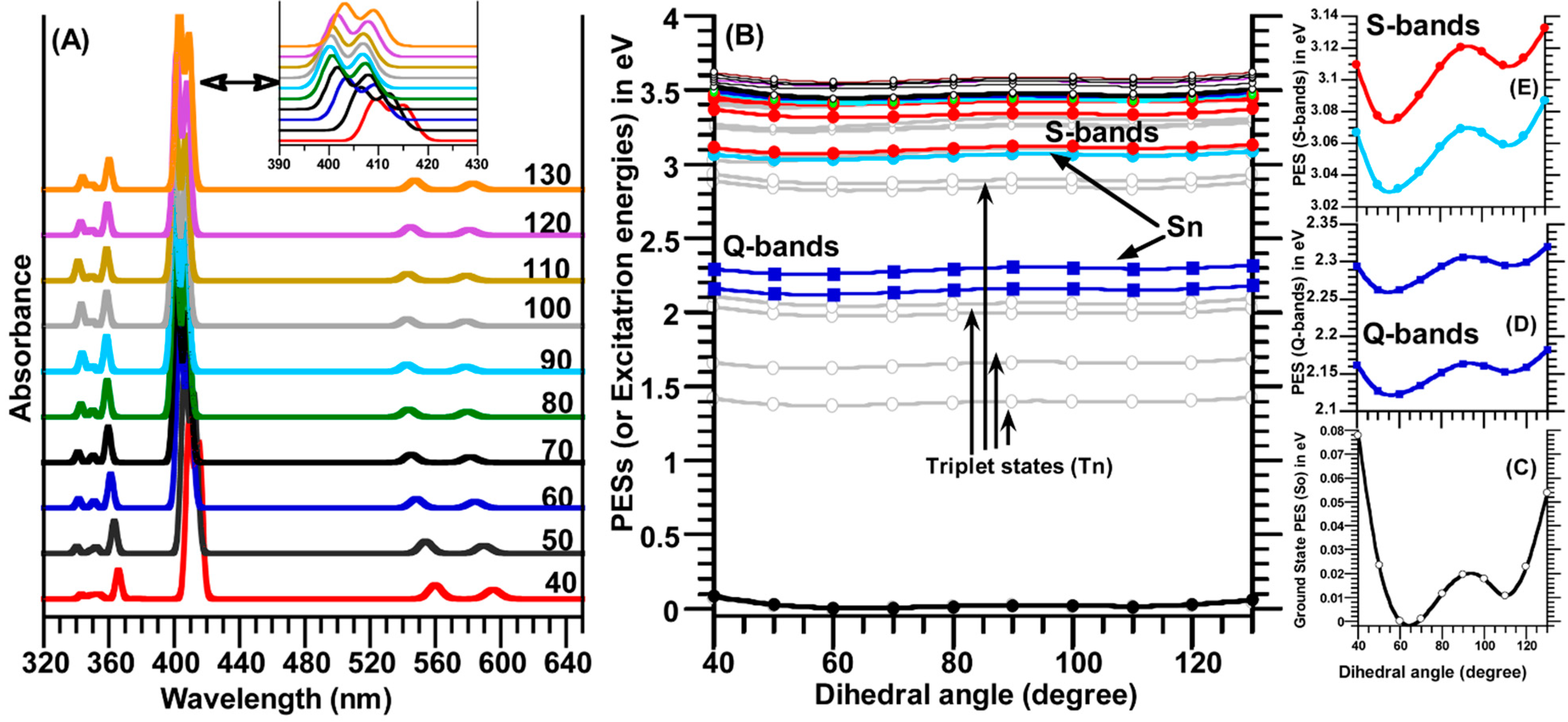{kind=link}