RNA/aTNA Chimeras: RNAi Effects and Nucleases Resistance of Single and Double Stranded RNAs



Abstract
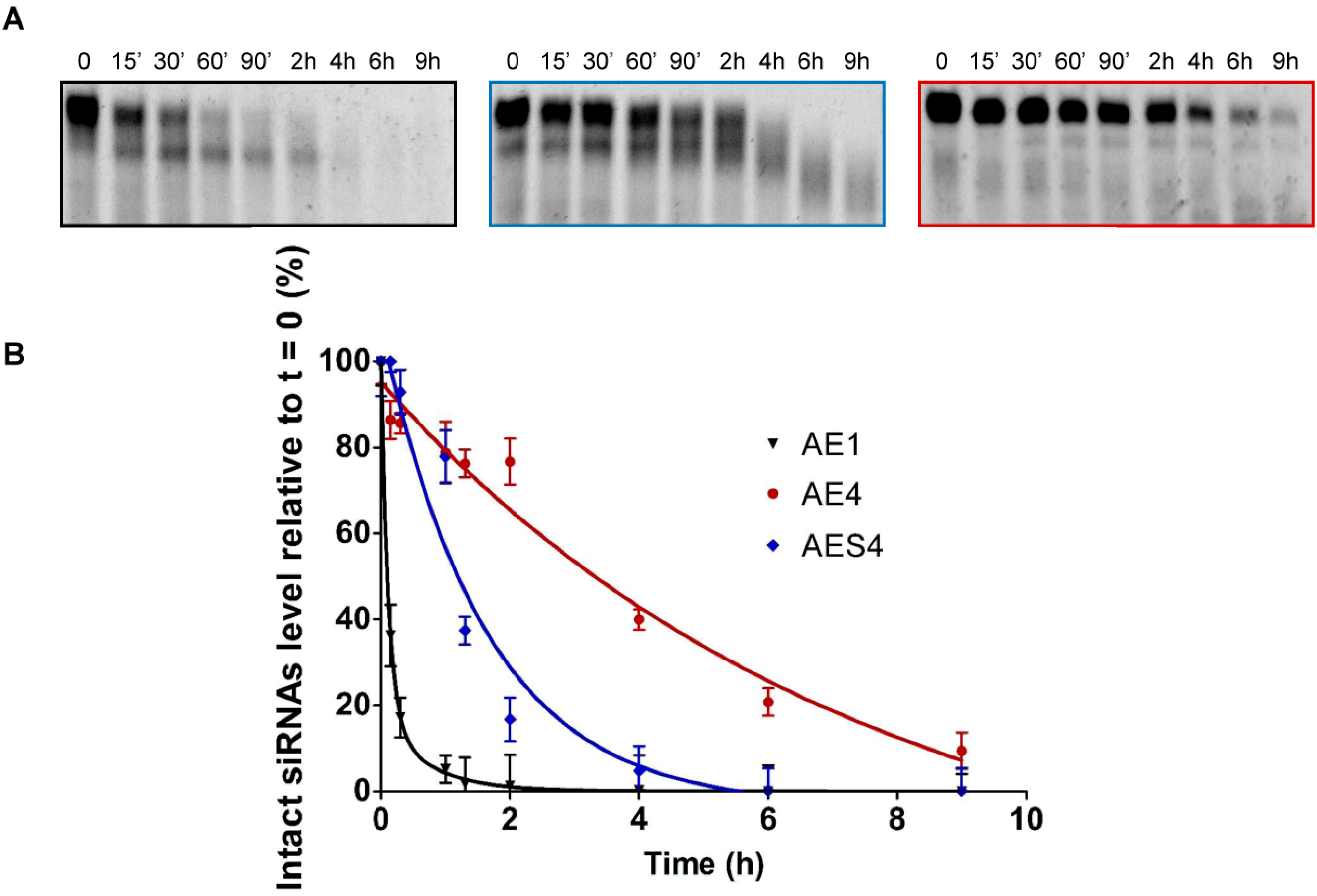
:
1. Introduction
2. Results and Discussion
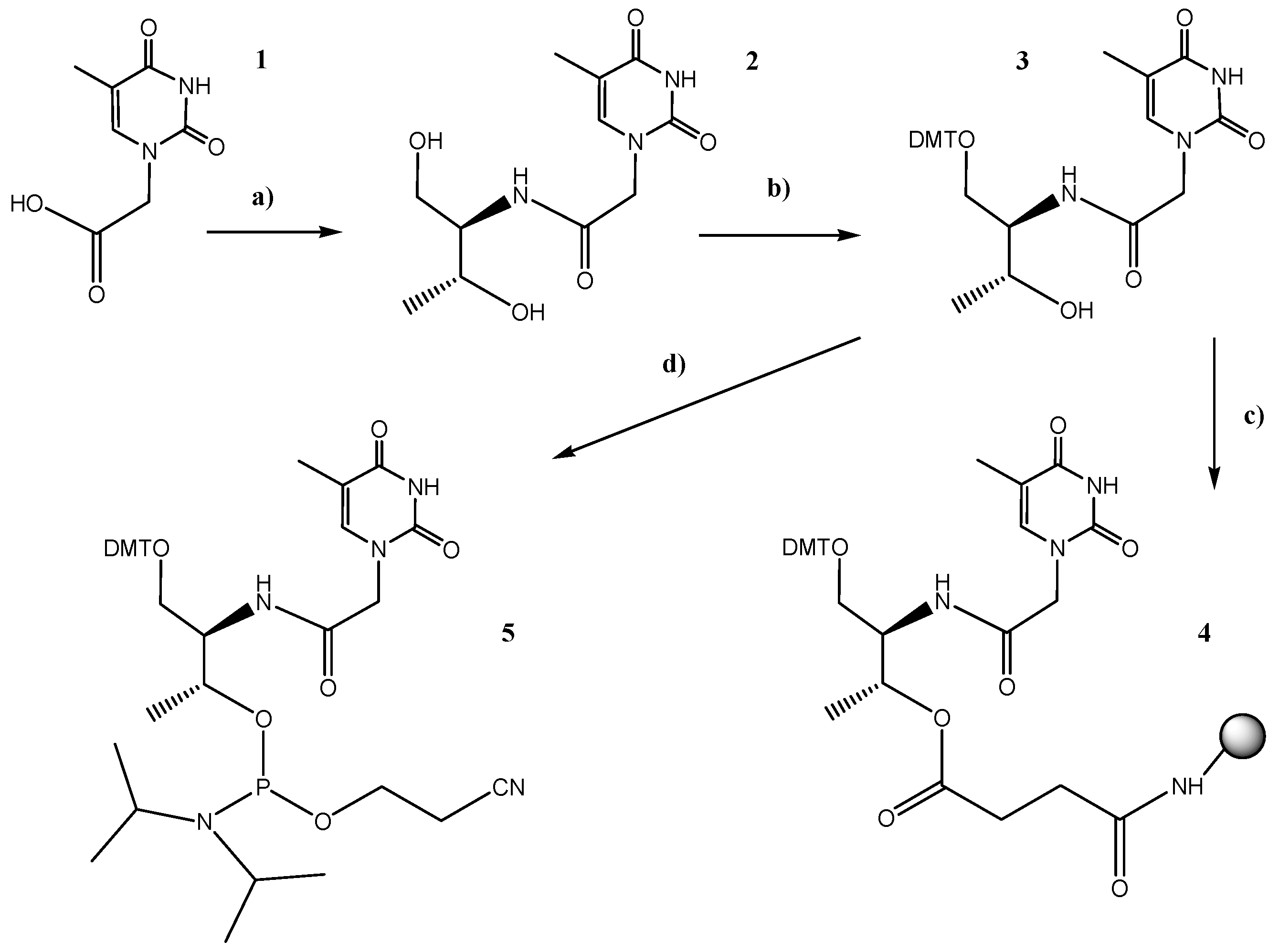
2.1. Synthesis of the l-threoninol-thymine Building Block

2.2. RNA Synthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
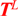 : l-threoninol-thymine monomer,
: l-threoninol-thymine monomer,  : thymidine monomer with phosphorothioate linkage, T: thymidine.
: thymidine monomer with phosphorothioate linkage, T: thymidine.
| ON | Sequence | MW Calculated | MW Found |
|---|---|---|---|
| AS1 | 5′-UUUUUCUCCUUCUUCAGAUTT | 6439 | 6434 |
| SS1 | 5′-AUCUGAAGAAGGAGAAAAATT | 6829 (+Na) | 6829 (+Na) |
| AS2 | 5′-UUUUUCUCCUUCUUCAGAU | 6497 | 6492 |
| SS2 | 5′-AUCUGAAGAAGGAGAAAAA | 6864 | 6859 |
| AS3 | 5′-UCCUUUCUUUCUUUCGAUATT | 6439 | 6433 |
| SS3 | 5′-UAUCGAAAGAAAGAAAGGATT | 6806 | 6800 |
| SS4 | 5′- A CUGAAGAAGGAGAAAAATT | 6833 | 6827 |
| AS5 | 5′-UUUCUUGUUCUGAAUGUCCTT | 6742 | 6736 |
| SS5 | 5′-GGACAUUCAGAACAAGAAATT | 6518 | 6512 |
| AS6 | 5′-UUUCUUGUUCUGAAUGUCC | 6800 | 6795 |
| SS6 | 5′-GGACAUUCAGAACAAGAAA | 6576 | 6570 |
| ASP | 5′-UUUUUCUCCUUCUUCAGAU  | -- | -- |
| SSP | 5′- AUCUGAAGAAGGAGAAAAA | -- | -- |
: L-threoninol-thymine monomer; : thymidine monomer with phosphorothioate linkage; T: thymidine.
| siRNA | ON | Sequence | Tm [°C] | IC50 [pM] |
|---|---|---|---|---|
| AE1 | SS1 | TTAAAAAGAGGAAGAAGUCUA-5′ | 67.8 | 9.8 ± 0.2 |
| AS1 | 5′-UUUUUCUCCUUCUUCAGAUTT | |||
| AE2 | SS1 | TTAAAAAGAGGAAGAAGUCUA-5′ | N.D. | 6.3 ± 0.5 |
| AS2 | 5′-UUUUUCUCCUUCUUCAGAU | |||
| AE3 | SS2 | AAAAAGAGGAAGAAGUCUA-5′ | N.D. | 14.3 ± 0.3 |
| AS1 | 5′-UUUUUCUCCUUCUUCAGAUTT | |||
| AE4 | SS2 | AAAAAGAGGAAGAAGUCUA-5′ | 67.4 | 7.2 ± 0.4 |
| AS2 | 5′-UUUUUCUCCUUCUUCAGAU | |||
| AES2 | SS1 | TTAAAAAGAGGAAGAAGUCUA-5′ | N.D. | 6.5 ± 0.2 |
| ASP | 5′- UUUUUCUCCUUCUUCAGAU | |||
| AES3 | SSP | AAAAAGAGGAAGAAGUCUA-5′ | N.D. | 10.5 ± 0.4 |
| AS1 | 5′-UUUUUCUCCUUCUUCAGAUTT | |||
| AES4 | SSP | AAAAAGAGGAAGAAGUCUA-5′ | 67.8 | 8.3 ± 0.3 |
| ASP | 5′-UUUUUCUCCUUCUUCAGAU | |||
| SCR | SS3 | TTAGGAAAGAAAGAAAGCUAU-5′ | N.D. | Not active |
| AS3 | 5′-UCCUUUCUUUCUUUCGAUATT |
: l-threoninol-thymine monomer, T: thymidine.
| siRNA | ON | Sequence |
|---|---|---|
| APO1 | SS5 | TTAAAGAACAAGACUUACAGG-5′ |
| AS5 | 5′-UUUCUUGUUCUGAAUGUCCTT | |
| APO6 | SS6 | AAAGAACAAGACUUACAGG-5′ |
| AS6 | 5′-UUUCUUGUUCUGAAUGUCC |
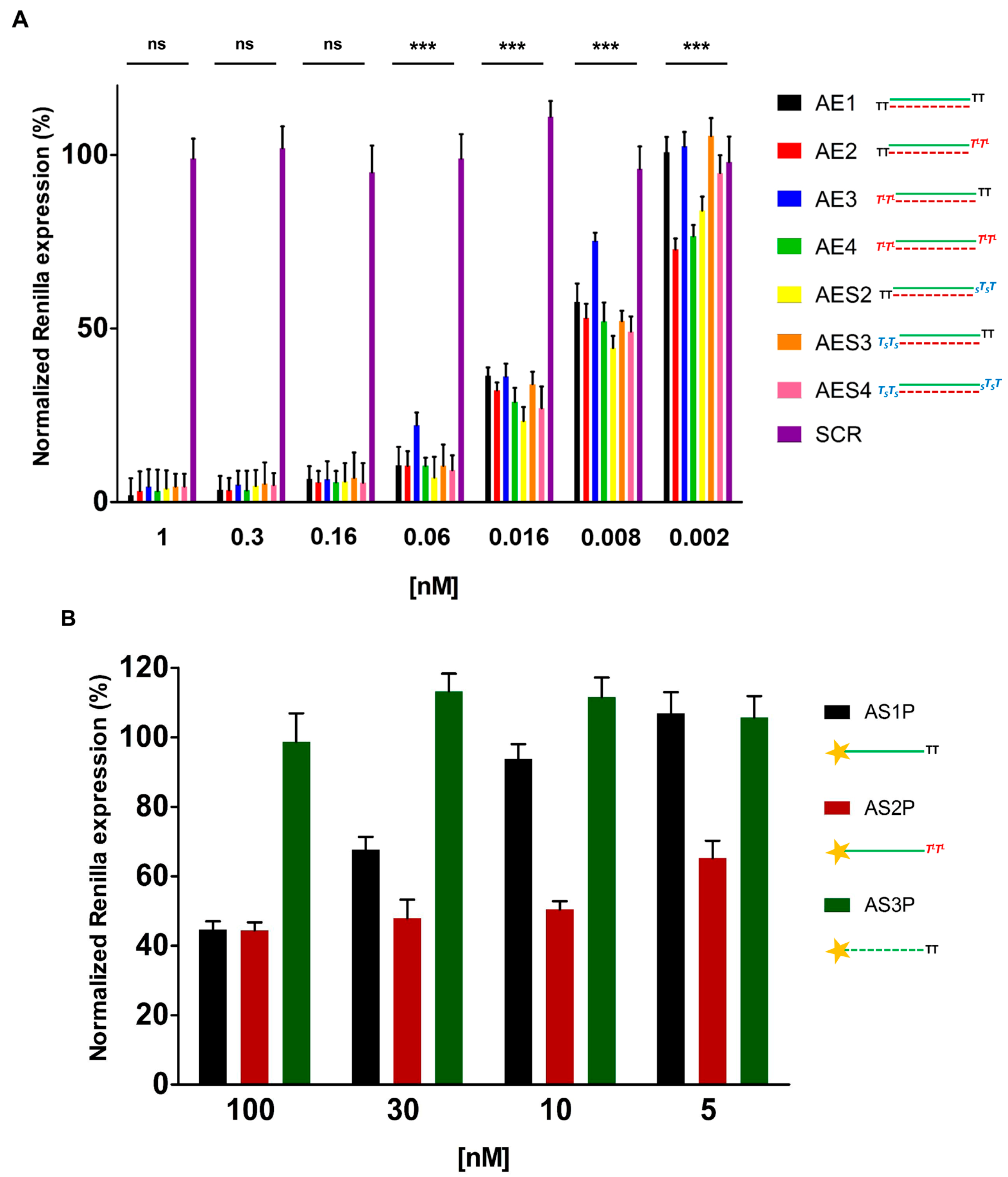
2.3. In Vitro Evaluation of Double-Stranded and Single-Stranded Antisense siRNAs Potency
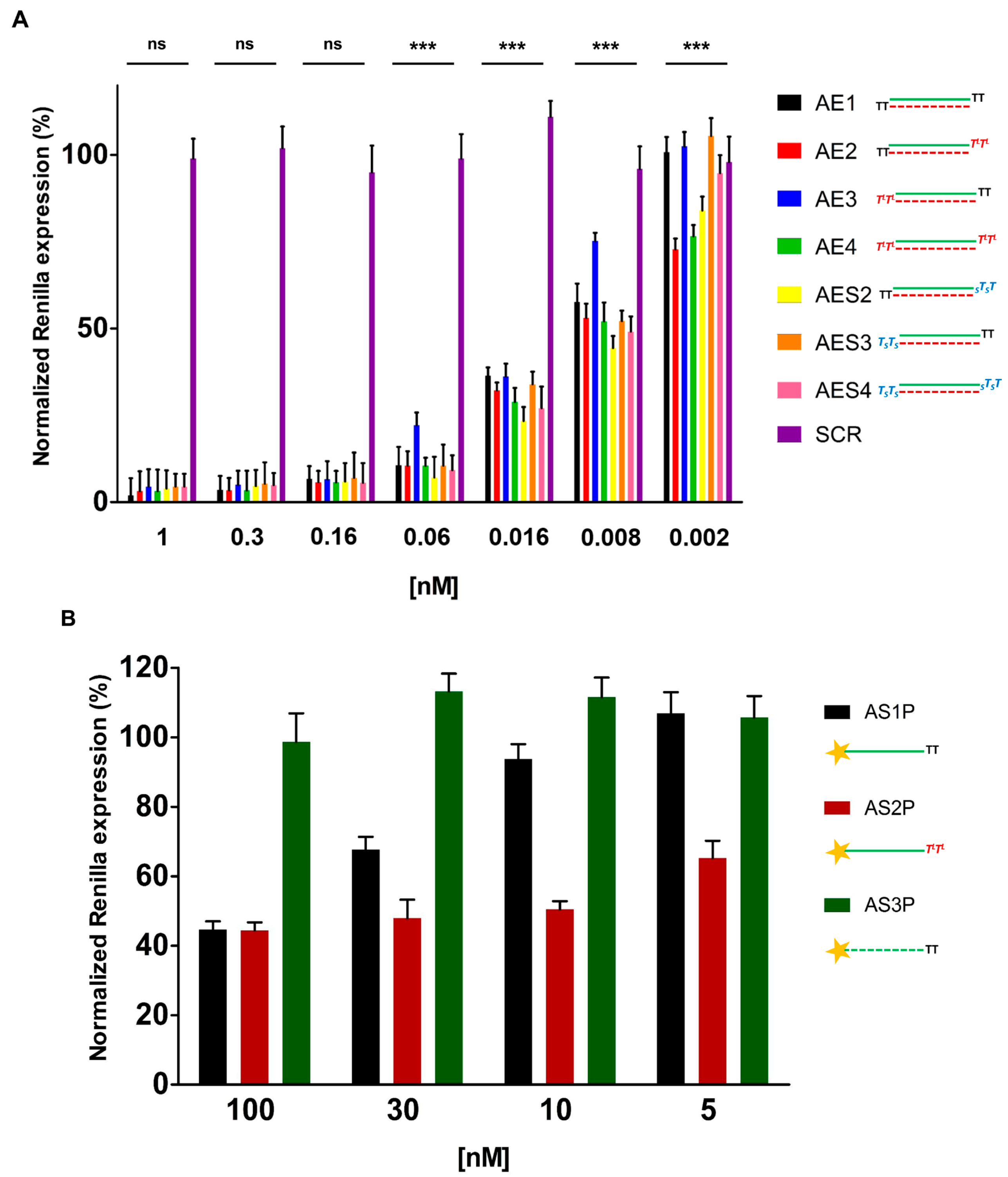
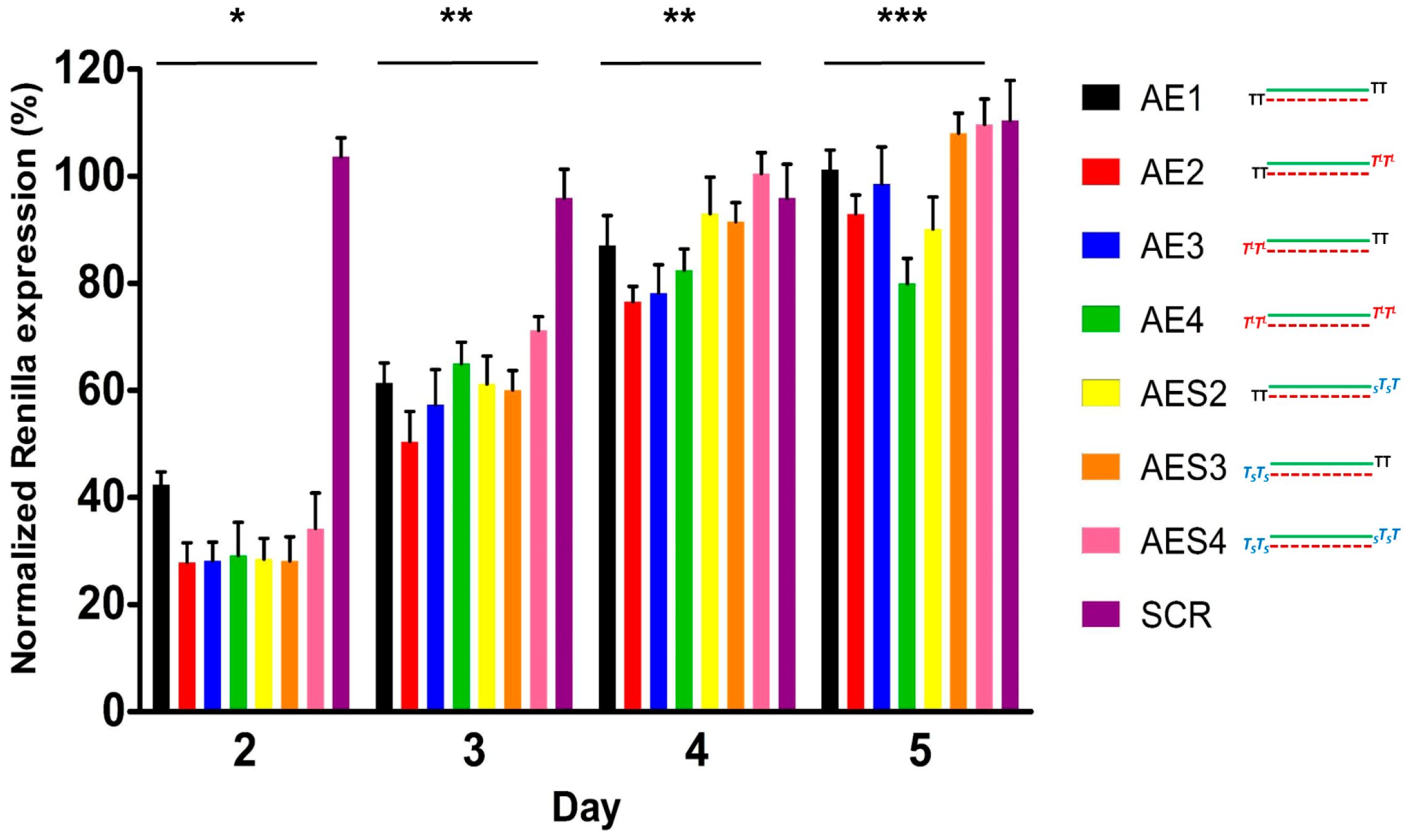
2.4. Over Time Silencing Activity Comparison of siRNAs Targeting Renilla
2.5. l-threoninol Modified siRNA Silencing Depends on Ago2-Mediated Mechanism
2.6. Effect of l-threoninol Modified siRNA on the HeLa Cell Survival
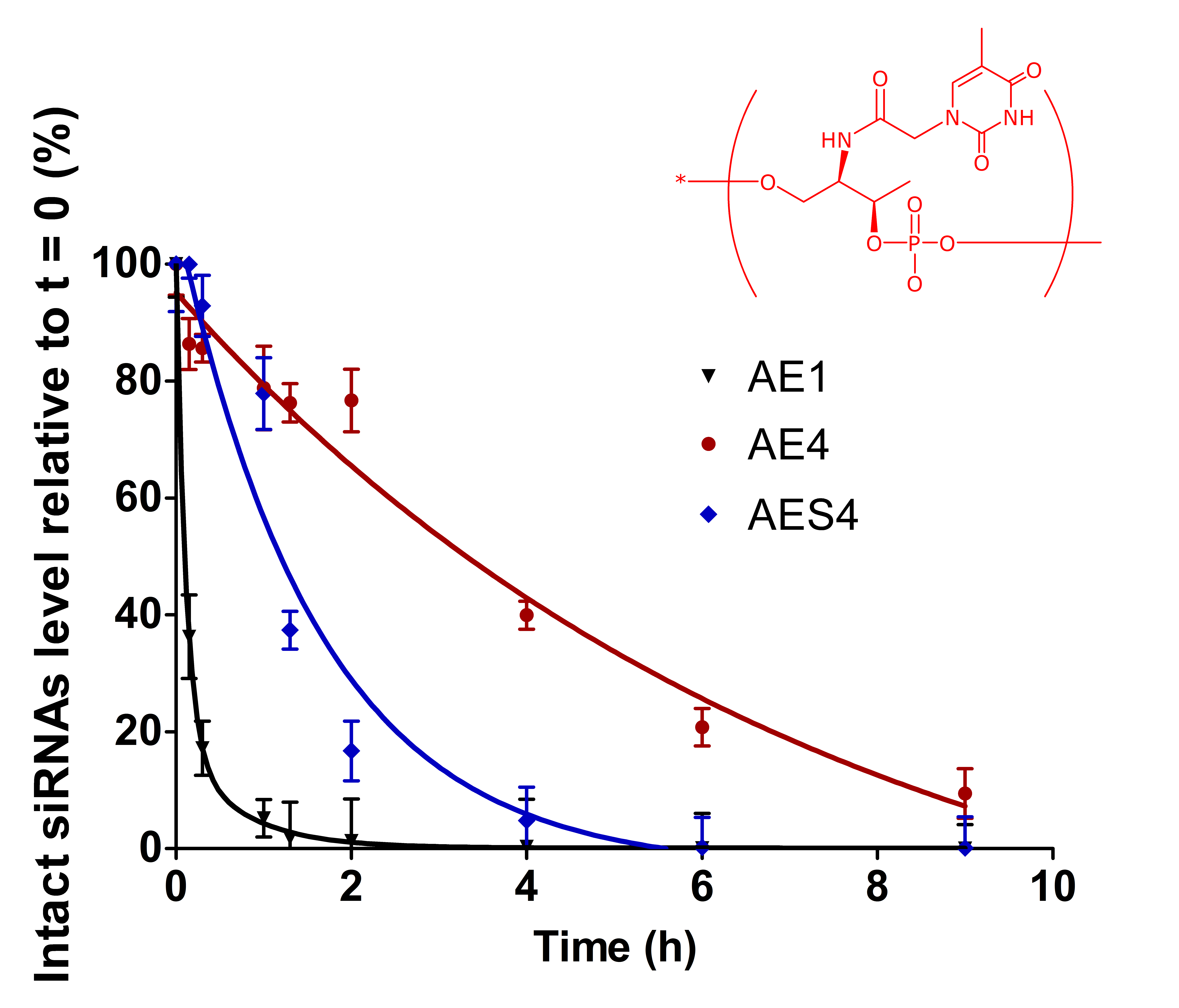
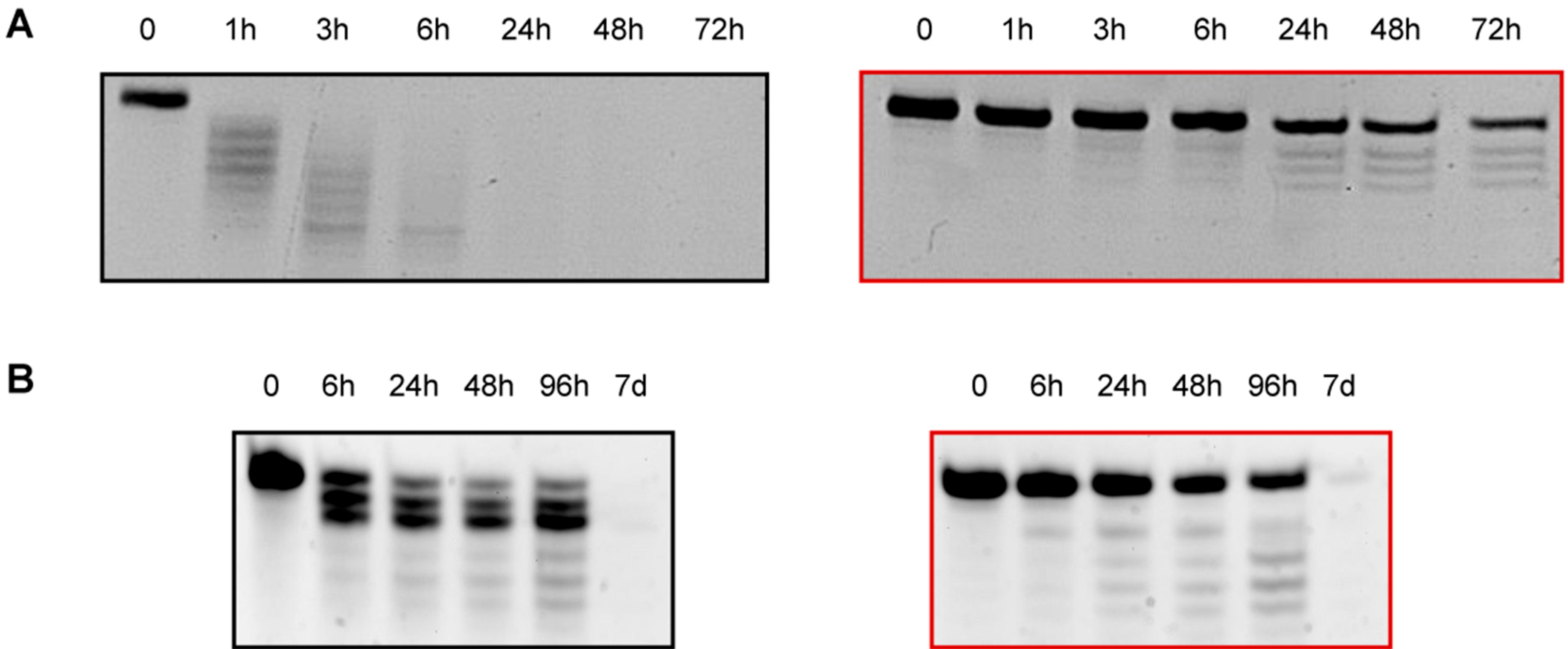
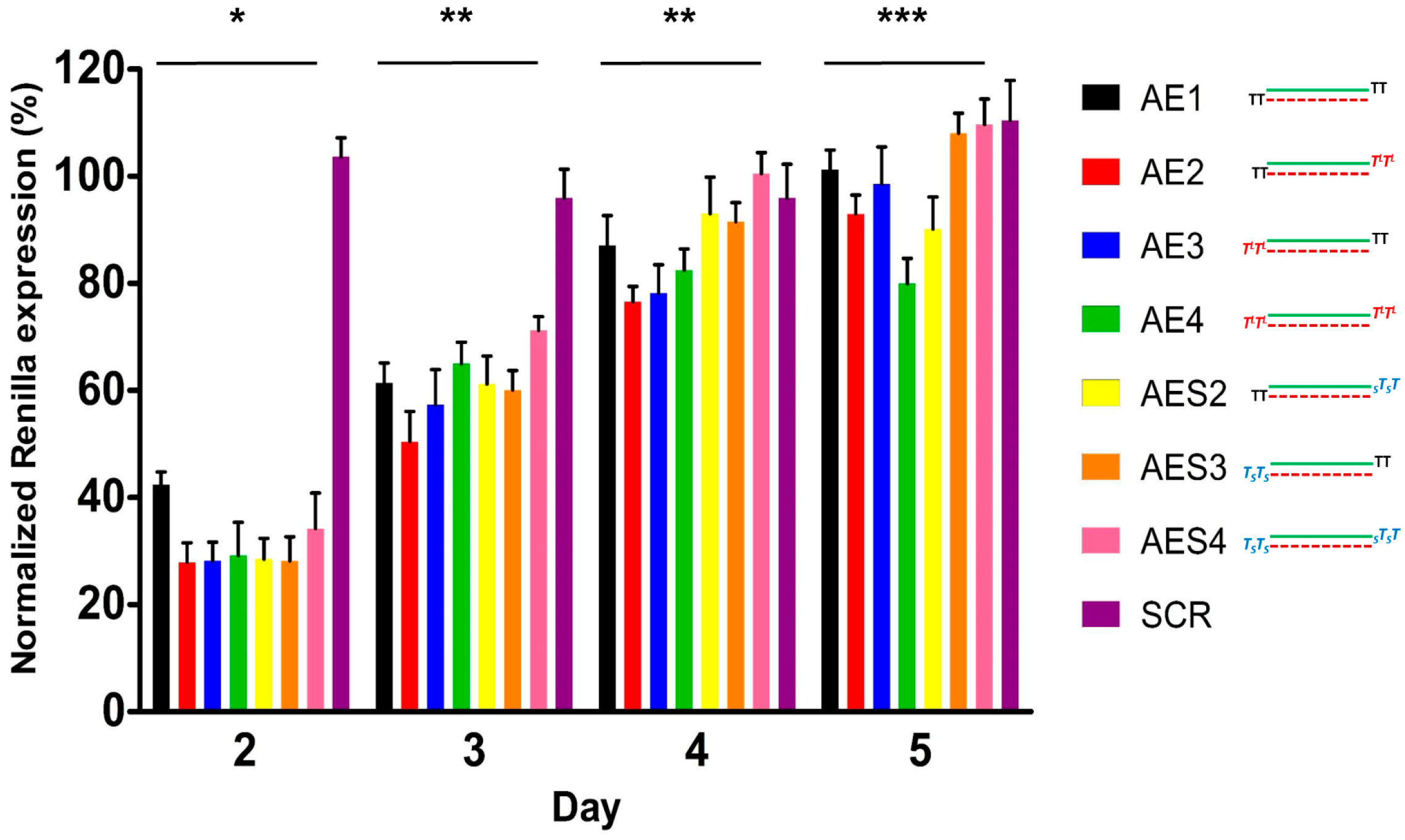
2.7. Human Serum Nucleases Stability of Chemically Modified siRNAs
2.8. 3′-/5′-Exonuclease Resistance Studies of Modified ssRNAs
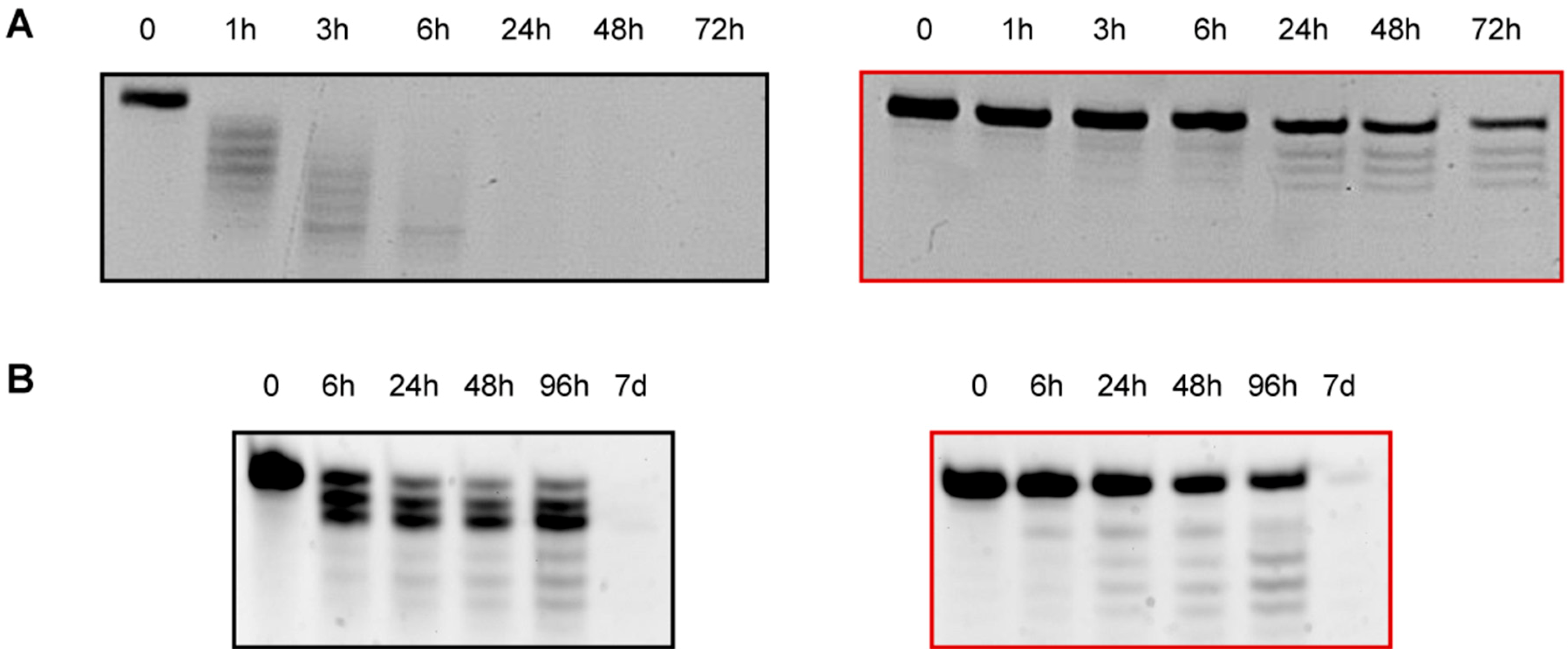
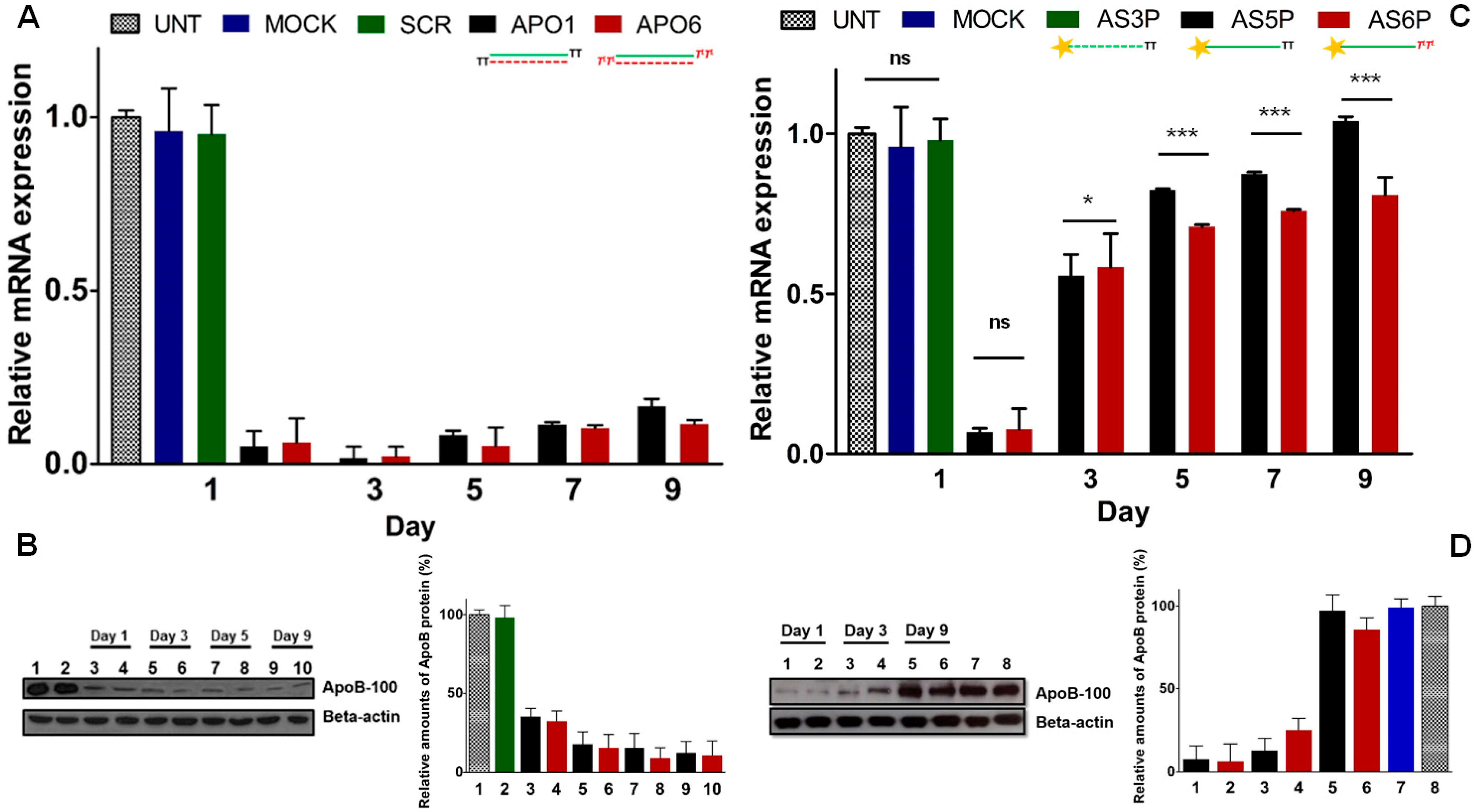
2.9. Keeping the Silencing: Evaluation of Long-Term RNAi Activity on ApoB Gene

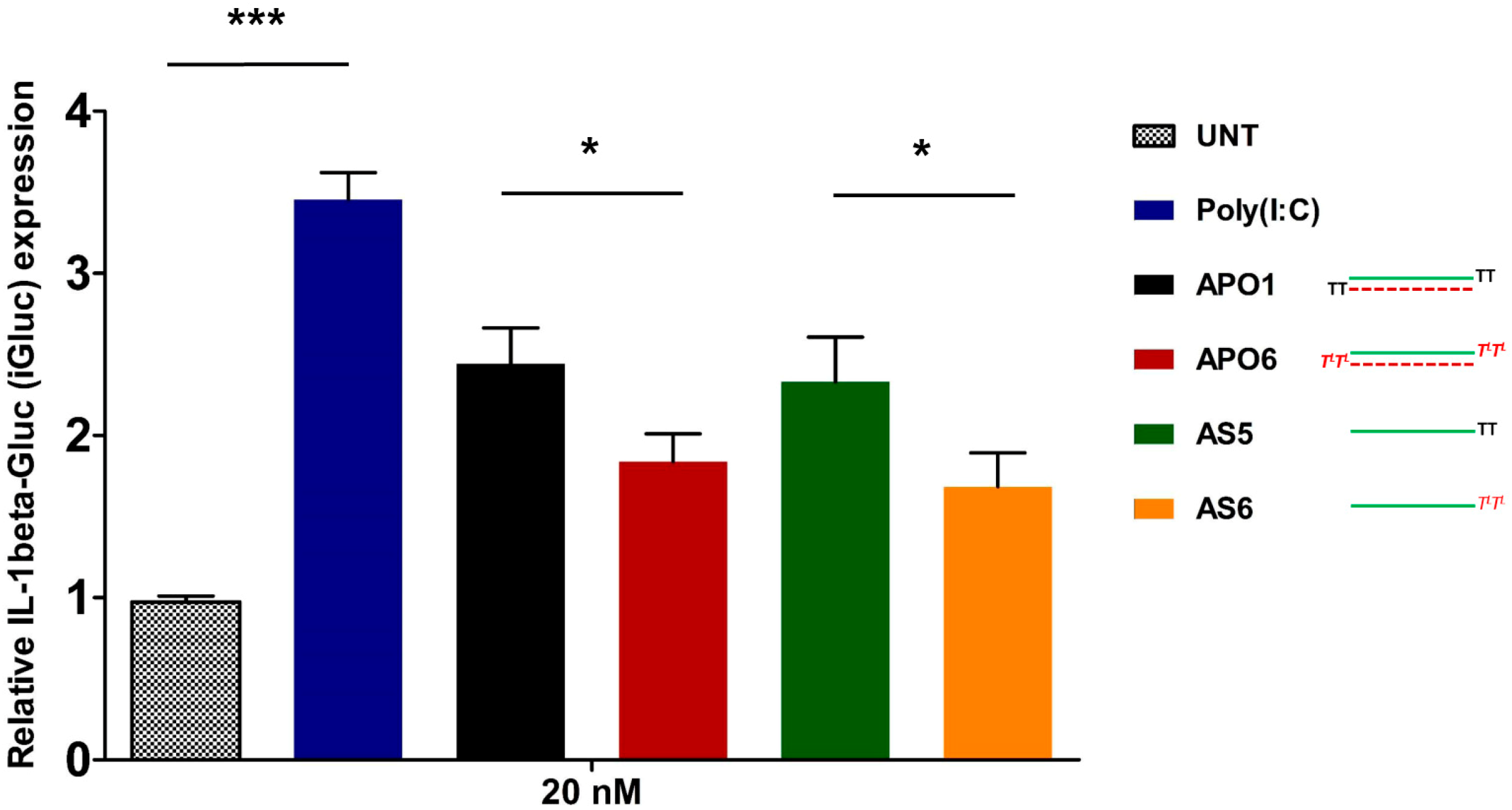
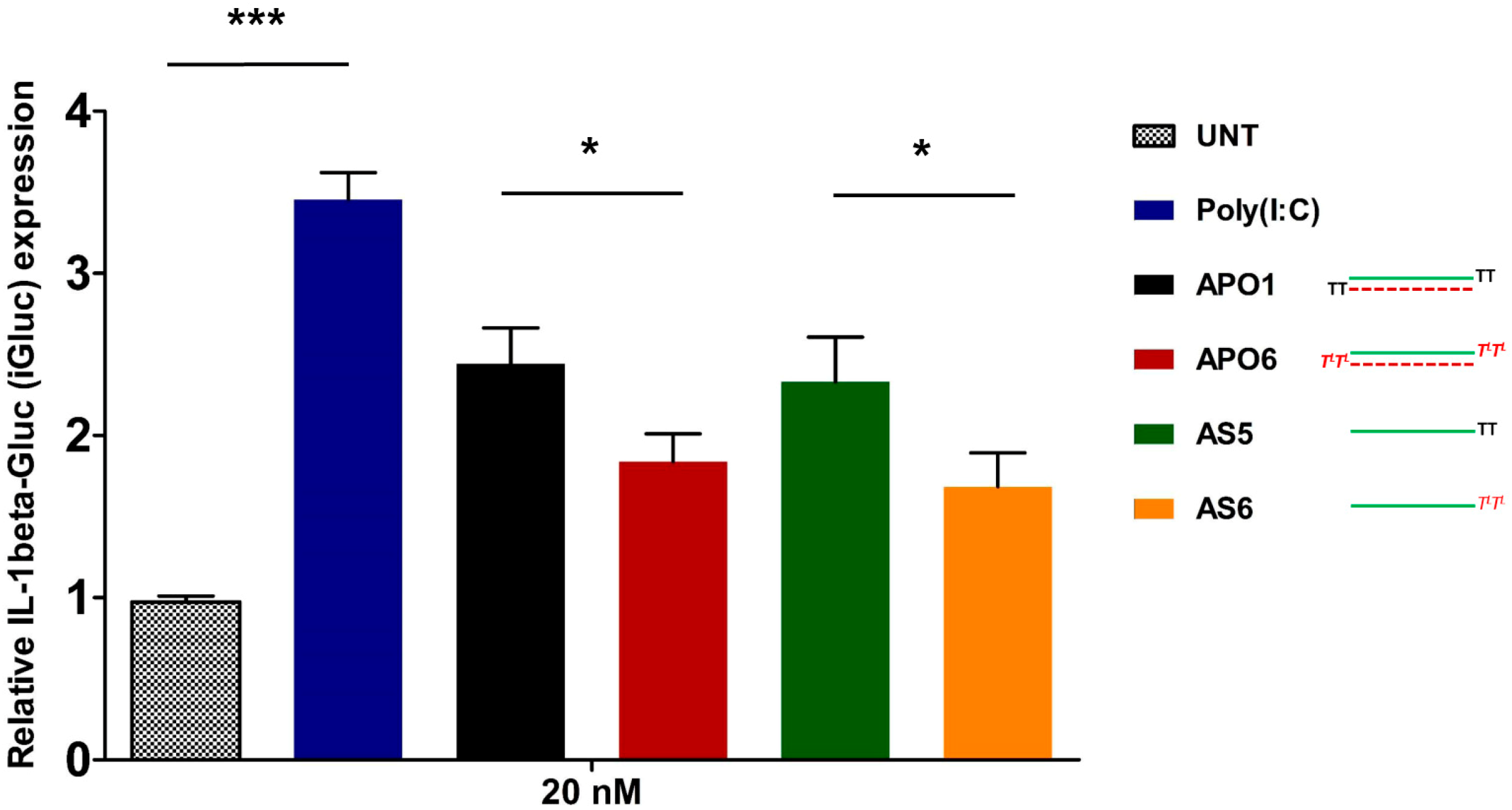
2.10. siRNA-Mediated Innate Immune System Activation
3. Experimental Section
3.1. Abbreviations and Acronyms
3.2. General Experimental Methods
3.3. Synthesis of Building Blocks
3.3.1. l-threoninol-thymine
3.3.2. DMT-Protected l-threoninol-thymine
3.3.3. Solid Support Functionalization
3.3.4. Synthesis of the Phosphoramidite Derivative
3.4. RNA Synthesis
3.5. Deprotection and Purification of Unmodified and Modified RNA Oligonucleotide
3.6. Thermal Denaturation Studies
3.7. Evaluation of Stability of RNAs to Exonucleases
3.8. Serum Nucleases Stability Assay
3.9. Cells
3.10. Luciferase Assay
3.11. Ago2-Mediated Silencing Assay
3.12. MTT Assay
3.13. THP-1 Interferon Assay
3.14. Single-Stranded Antisense siRNA 5′-End Phosphorylation
3.15. HepG2 Transfection
3.16. THP-1 iGluc C1
3.17. Isolation of RNA and RT-qPCR
3.18. Western Blot Analysis
3.19. Statistical Analysis
4. Conclusions
Acknowledgments
Supplementary Materials
Supplementary Files
Supplementary File 1Author Contributions
Conflicts of Interest
References
- Paterson, B.M.; Roberts, B.E.; Kuff, E.L. Structural gene identification and mapping by DNA-mRNA hybrid-arrested cell-free translation. Proc. Natl. Acad. Sci. USA 1977, 74, 4370–4374. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Rana, T.M. Illuminating the silence: Understanding the structure and function of small RNAs. Nat. Rev. Mol. Cell Biol. 2007, 8, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.; Doudna, J.A. Molecular Mechanisms of RNA Interference. Ann. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, C.C.; Choy, K.W.; Du, Q.; Chen, J.; Wang, Q.; Li, L.; Chung, T.K.; Tang, T. Therapeutic potentials of gene silencing by RNA interference: Principles, challenges, and new strategies. Gene 2014, 538, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Sledz, C.A.; Holko, M.; de Veer, M.J.; Silverman, R.H.; Williams, B.R. Activation of the interferon system by short-interfering RNAs. Nat. Cell Biol. 2003, 5, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Judge, A.D.; Sood, V.; Shaw, J.R.; Fang, D.; McClintock, K.; MacLachlan, I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 2005, 23, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Deleavey, G.F.; Damha, M.J. Designing chemically modified oligonucleotides for targeted gene silencing. Chem. Biol. 2012, 8, 937–954. [Google Scholar] [CrossRef]
- Martinez, J.; Patkaniowska, A.; Urlaub, H.; Lührmann, R.; Tuschl, T. Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell 2002, 110, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Holen, T.; Amarzguioui, M.; Babaie, E.; Prydz, H. Similar behaviour of single-strand and double-strand siRNAs suggests they act through a common RNAi pathway. Nucl. Acids Res. 2003, 31, 2401–2407. [Google Scholar] [CrossRef] [PubMed]
- Haringsma, H.J.; Li, J.J.; Soriano, F.; Kenski, D.M.; Flanagan, W.M.; Willingham, A.T. mRNA knockdown by single strand RNA is improved by chemical modifications. Nucl. Acids Res. 2012, 40, 4125–4136. [Google Scholar] [CrossRef] [PubMed]
- Lima, W.F.; Prakash, T.P.; Murray, H.M.; Kinberger, G.A.; Li, W.; Chappell, A.E.; Li, C.S.; Murray, S.F.; Gaus, H.; Seth, P.P.; et al. Single-stranded siRNAs activate RNAi in animals. Cell 2012, 150, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, H.; Toda, T.; Murayama, K.; Liang, X.; Kashida, H. Unexpectedly stable artificial duplex from flexible acyclic threoninol. J. Am. Chem. Soc. 2010, 132, 14702–14703. [Google Scholar] [CrossRef] [PubMed]
- Murayama, K.; Tanaka, Y.; Toda, T.; Kashida, H.; Asanuma, H. Highly stable duplex formation by artificial nucleic acids acyclic threoninol nucleic acid (aTNA) and serinol nucleic acid (SNA) with acyclic scaffolds. Chem. Eur. J. 2013, 19, 14151–14158. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Meares, C.F. Synthesis, metal chelate stability studies, and enzyme digestion of a peptide-linked DOTA derivative and its corresponding radiolabeled immunoconjugates. Bioconjug. Chem. 1993, 4, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Bramsen, J.B.; Kjems, J. Development of therapeutic-grade small interfering RNAs by chemical engineering. Front. Genet. 2012, 3, 154. [Google Scholar] [CrossRef]
- Lima, W.F.; Wu, H.; Nichols, J.G.; Sun, H.; Murray, H.M.; Crooke, S.T. Binding and cleavage specificities of human Argonaute2. J. Biol. Chem. 2009, 284, 26017–26028. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Huang, Y.; Li, J.; Yi, F.; Zheng, J.; Huang, H.; Wei, N.; Shan, Y.; An, M.; Zhang, H.; et al. Comprehensive analysis of sequence-specific stability of siRNA. FASEB J. 2010, 24, 4844–4855. [Google Scholar] [CrossRef] [PubMed]
- Choung, S.; Kim, Y.J.; Kim, S.; Park, H.O.; Choi, Y.C. Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem. Biophys. Res. Commun. 2006, 342, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, M.; Cieslak, M.; Stec, W.J.; Goding, J.W.; Koziolkiewicz, M. Nucleotide pyrophosphatase/Phosphodiesterase 1 is responsible for degradation of antisense phosphorothioate oligonucleotides. Oligonucleotides 2007, 17, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Gijsbers, R.; Aoki, J.; Arai, H.; Bollen, M. The hydrolysis of lysophospholipids and nucleotides by autotaxin (NPP2) involves a single catalytic site. FEBS Lett. 2003, 538, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Linn, S.M.; Lloyd, R.S.; Roberts, R.J. Nucleases; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1993. [Google Scholar]
- Volkov, A.A.; Kruglova, N.S.; Meschaninova, M.I.; Venyaminova, A.G.; Zenkova, M.A.; Vlassov, V.V.; Chernolovskaya, E.L. Selective protection of nuclease-sensitive sites in siRNA prolongs silencing effect. Oligonucleotides 2009, 19, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.W.; Davis, M.E. Effect of siRNA nuclease stability on the in vitro and in vivo kinetics of siRNA-mediated gene silencing. Biotechnol. Bioeng. 2007, 97, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, Y.; Ito, A.; Ito, H.; Urushihara, M.; Takai, J.; Fujii, T.; Liang, X.; Kashida, H.; Asanuma, H. Selective labeling of mature RISC using a siRNA carrying fluophore-quencher pair. Chem. Sci. 2013, 4, 4016–4021. [Google Scholar] [CrossRef]
- Braasch, D.A.; Jensen, S.; Liu, Y.; Kaur, K.; Arar, K.; White, M.A.; Corey, D.R. RNA interference in mammalian cells by chemically-modified RNA. Biochemistry 2003, 42, 7967–7975. [Google Scholar] [CrossRef] [PubMed]
- Hoerter, J.A.; Krishnan, V.; Lionberger, T.A.; Walter, N.G. SiRNA-Like double-stranded RNAs are specifically protected against degradation in human cell extract. PLoS One 2011, 6, e20359. [Google Scholar] [CrossRef] [PubMed]
- Raemdonck, K.; Remaut, K.; Lucas, B.; Sanders, N.N.; Demeester, J.; de Smedt, S.C. In situ analysis of single-stranded and duplex siRNA integrity in living cells. Biochemistry 2006, 45, 10614–10623. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, A.J.; Barrett, P.H.; van Bockxmeer, F.M.; Burnett, J.R. Lipid disorders and mutations in the APOB gene. Clin. Chem. 2004, 50, 1725–1732. [Google Scholar] [CrossRef] [PubMed]
- Pullinger, C.R.; North, J.D.; Teng, B.B.; Rifici, V.A.; Ronhild de Brito, A.E.; Scott, J. The apolipoprotein B gene is constitutively expressed in HepG2 cells: Regulation of secretion by oleic acid, albumin, and insulin, and measurement of the mRNA half-life. J. Lipid Res. 1989, 7, 1065–1077. [Google Scholar]
- Aston, N.S.; Watt, N.; Morton, I.E.; Tanner, M.S.; Evans, G.S. Copper toxicity affects proliferation and viability of human hepatoma cells (HepG2 line). Hum. Exp. Toxicol. 2000, 19, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.A.; Dahlman, J.E.; Langer, R.S.; Anderson, D.G. Silencing or stimulation? SiRNA delivery and the immune system. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Robbins, M.; Judge, A.; MacLachlan, I. SiRNA and innate immunity. Oligonucleotides 2009, 19, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M. Induction of inflammatory cytokines and interferon responses by double-stranded and single-stranded siRNAs is sequence-dependent and requires endosomal localization. J. Mol. Biol. 2005, 348, 1079–1090. [Google Scholar] [CrossRef]
- Reyes-Darias, J.A.; Berzal-Herranz, A. Detection of immune response activation by exogenous nucleic acids by a multiplex RT-PCR method. Mol. Cell. Probes 2014, 28, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M. Single-stranded small interfering RNA are more immunostimulatory than their double-stranded counterparts: A central role for 2′-hydroxyl uridines in immune responses. Eur. J. Immunol. 2006, 36, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Schroecksnadel, S.; Jenny, M.; Fuchs, D. Myelomonocytic THP-1 cells for in vitro testing of immunomodulatory properties of nanoparticles. J. Biomed. Nanotechnol. 2011, 7, 209–210. [Google Scholar] [CrossRef] [PubMed]
- Bartok, E.; Bauernfeind, F.; Khaminets, M.G.; Jakobs, C.; Monks, B.; Fitzgerald, K.A.; Latz, E.; Hornung, V. iGLuc: A luciferase-based inflammasome and protease activity reporter. Nat. Methods 2013, 10, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, X.; Rodríguez, L.; Prévot, J.; Oleaga, C.; Ciudad, C.J.; Noé, V. Stability and immunogenicity properties of the gene-silencing polypurine reverse Hoogsteen hairpins. Mol. Pharm. 2014, 11, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Terrazas, M.; Alagia, A.; Faustino, I.; Orozco, M.; Eritja, R. Functionalization of the 3’-ends of DNA and RNA strands with N-ethyl-N-coupled nucleosides: A promising approach to avoid 3'-exonuclease-catalyzed hydrolysis of therapeutic oligonucleotides. ChemBioChem 2013, 14, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Vaish, N.; Chen, F.; Seth, S.; Fosnaugh, K.; Liu, Y.; Adami, R.; Brown, T.; Chen, Y.; Harvie, P.; Johns, R.; et al. Improved specificity of gene silencing by siRNAs containing unlocked nucleobase analogs. Nucl. Acids Res. 2011, 39, 1823–1832. [Google Scholar] [PubMed]
- O’Carroll, D.; Mecklenbrauker, I.; Das, P.P.; Santana, A.; Koenig, U.; Enright, A.J.; Miska, E.A.; Tarakhovsky, A. A Slicer-independent role for Argonaute 2 in hematopoiesis and the microRNA pathway. Genes Dev. 2007, 21, 1999–2004. [Google Scholar] [PubMed]
- Waaler, J.; Machon, O.; von Kries, J.P.; Wilson, S.R.; Lundenes, E.; Wedlich, D.; Gradl, D.; Paulsen, J.E.; Machonova, O.; Dembinski, J.L.; et al. Novel synthetic antagonists of canonical Wnt signaling inhibit colorectal cancer cell growth. Cancer Res. 2011, 71, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Grijalvo, S.; Alagia, A.; Puras, G.; Zárate, J.; Pedraz, J.L.; Eritja, R. Cationic vesicles based on non-ionic surfactant and synthetic aminolipids mediate delivery of antisense oligonucleotides into mammalian cells. Colloids Surf. B 2014, 119, 30–37. [Google Scholar] [CrossRef]
- Rajan, J.V.; Warren, S.E.; Miao, E.A.; Aderem, A. Activation of the NLRP3 inflammasome by intracellular poly I:C. FEBS Lett. 2010, 584, 4627–4632. [Google Scholar] [CrossRef] [PubMed]
- NIH/NCBI/Primer-BLAST. Available online: http://www.ncbi.nlm.nih.gov/tools/primer-blast/ (accessed on 3 November 2014).
- Somoza, A.; Terrazas, M.; Eritja, R. Modified siRNAs for the study of the paz domain. Chem. Commun. 2010, 46, 4270–4272. [Google Scholar]
- Potenza, N.; Moggio, L.; Milano, G.; Salvatore, V.; di Blasio, B.; Russo, A.; Messere, A. RNA interference in mammalia cells by RNA-3’-PNA chimeras. Int. J. Mol. Sci. 2008, 9, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Ittig, D.; Luisier, S.; Weiler, J.; Schümperli, D.; Leumann, C.J. Improving gene silencing of siRNAs via tricyclo-DNA modification. Artif. DNA PNA XNA 2010, 1, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Ueno, Y.; Watanabe, Y.; Shibata, A.; Yoshikawa, K.; Takano, T.; Kohara, M.; Kitade, Y. Synthesis of nuclease-resistant siRNAs possessing universal overhangs. Bioorg. Med. Chem. 2009, 17, 1974–1981. [Google Scholar] [CrossRef]
- Gaglione, M.; Potenza, N.; di Fabio, G.; Romanucci, V.; Mosca, N.; Russo, A.; Novellino, E.; Cosconati, S.; Messere, A. Tuning RNA interference by enhancing siRNA/PAZ recognition. ACS Med. Chem. Lett. 2013, 4, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Ueno, Y.; Inoue, T.; Yoshida, M.; Yoshikawa, K.; Shibata, A.; Kitamura, Y.; Kitade, Y. Synthesis of nuclease-resistant siRNAs possessing benzene-phosphate backbones in their 3'-overhang regions. Bioorg. Med. Chem. Lett. 2008, 18, 5194–5196. [Google Scholar] [CrossRef] [PubMed]
- Lingel, A.; Simon, B.; Izaurralde, E.; Sattler, M. Nucleic acid 3'-end recognition by the Argonaute2 PAZ domain. Nat. Struct. Mol. Biol. 2004, 11, 576–577. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Juranek, S.; Li, H.; Sheng, G.; Wardle, G.S.; Tuschl, T.; Patel, D.J. Nucleation, propagation and cleavage of target RNAs in Ago silencing complexes. Nature 2009, 461, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 1–4 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alagia, A.; Terrazas, M.; Eritja, R. RNA/aTNA Chimeras: RNAi Effects and Nucleases Resistance of Single and Double Stranded RNAs. Molecules 2014, 19, 17872-17896. https://doi.org/10.3390/molecules191117872
Alagia A, Terrazas M, Eritja R. RNA/aTNA Chimeras: RNAi Effects and Nucleases Resistance of Single and Double Stranded RNAs. Molecules. 2014; 19(11):17872-17896. https://doi.org/10.3390/molecules191117872
Chicago/Turabian StyleAlagia, Adele, Montserrat Terrazas, and Ramon Eritja. 2014. "RNA/aTNA Chimeras: RNAi Effects and Nucleases Resistance of Single and Double Stranded RNAs" Molecules 19, no. 11: 17872-17896. https://doi.org/10.3390/molecules191117872
APA StyleAlagia, A., Terrazas, M., & Eritja, R. (2014). RNA/aTNA Chimeras: RNAi Effects and Nucleases Resistance of Single and Double Stranded RNAs. Molecules, 19(11), 17872-17896. https://doi.org/10.3390/molecules191117872