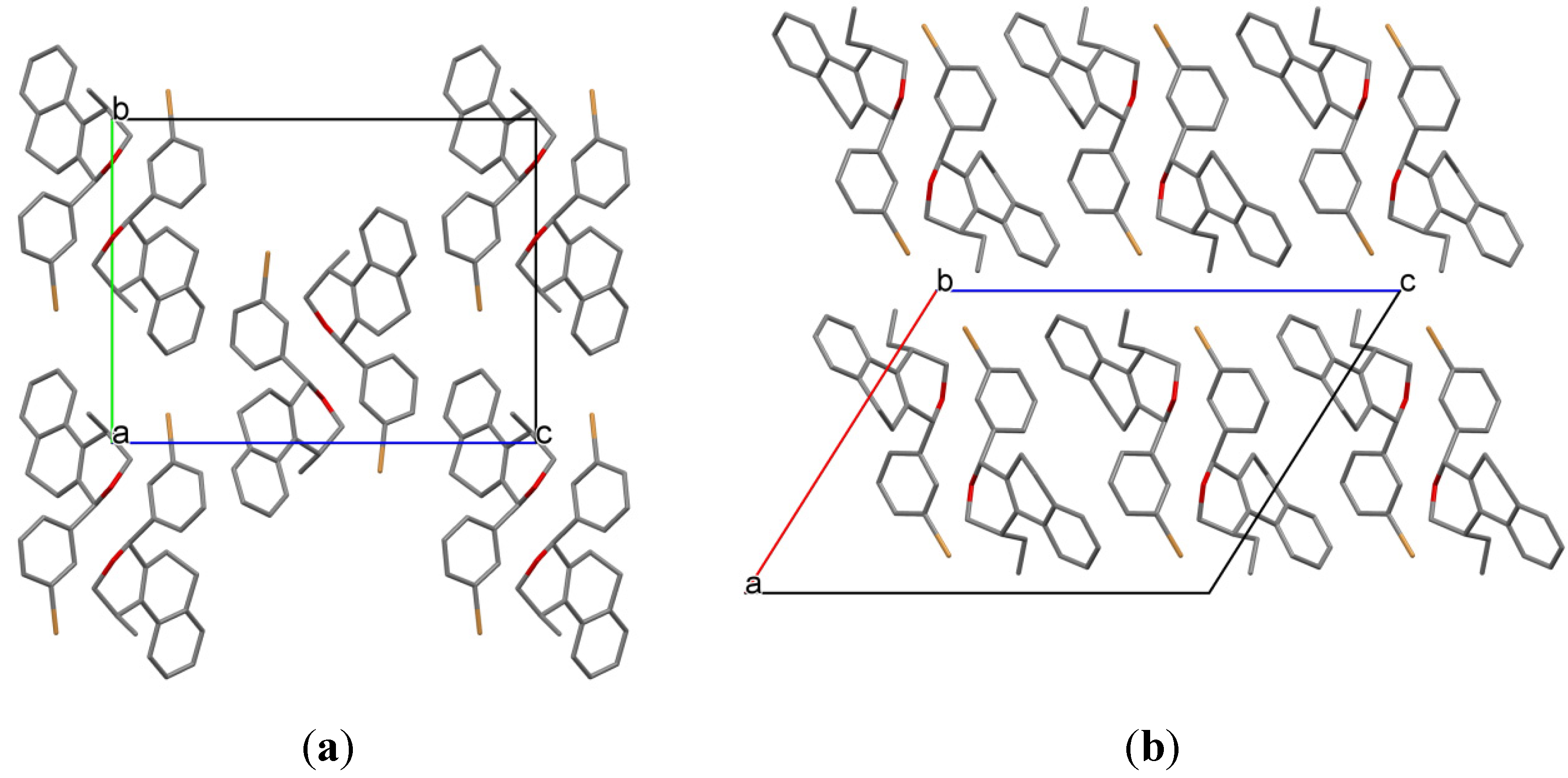
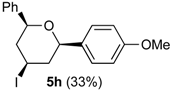
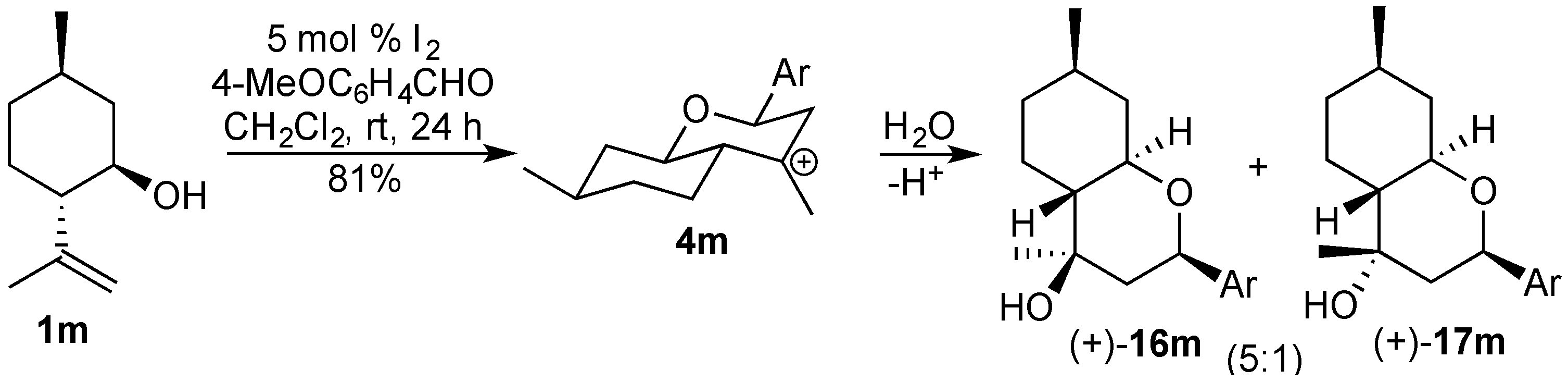
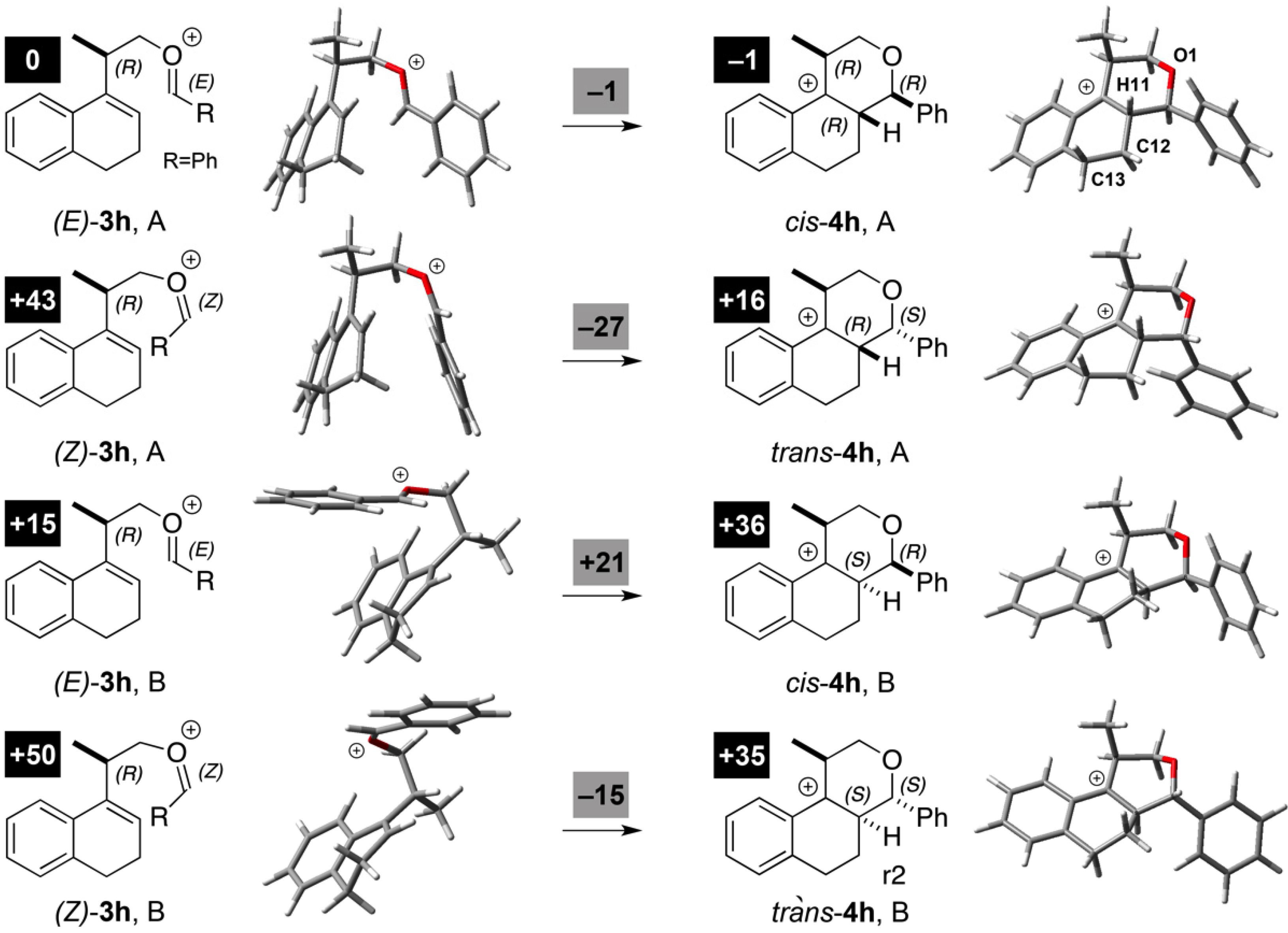
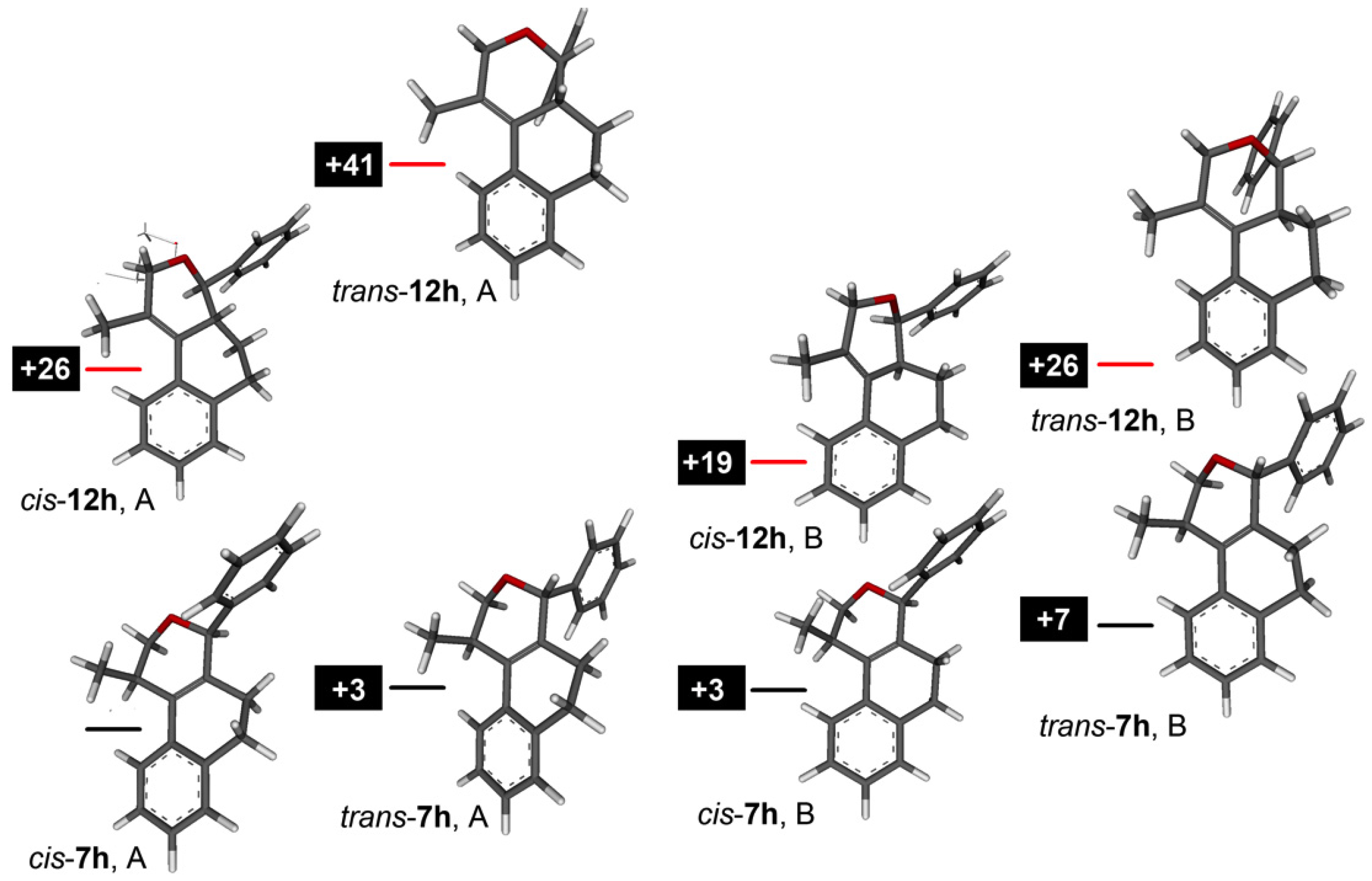
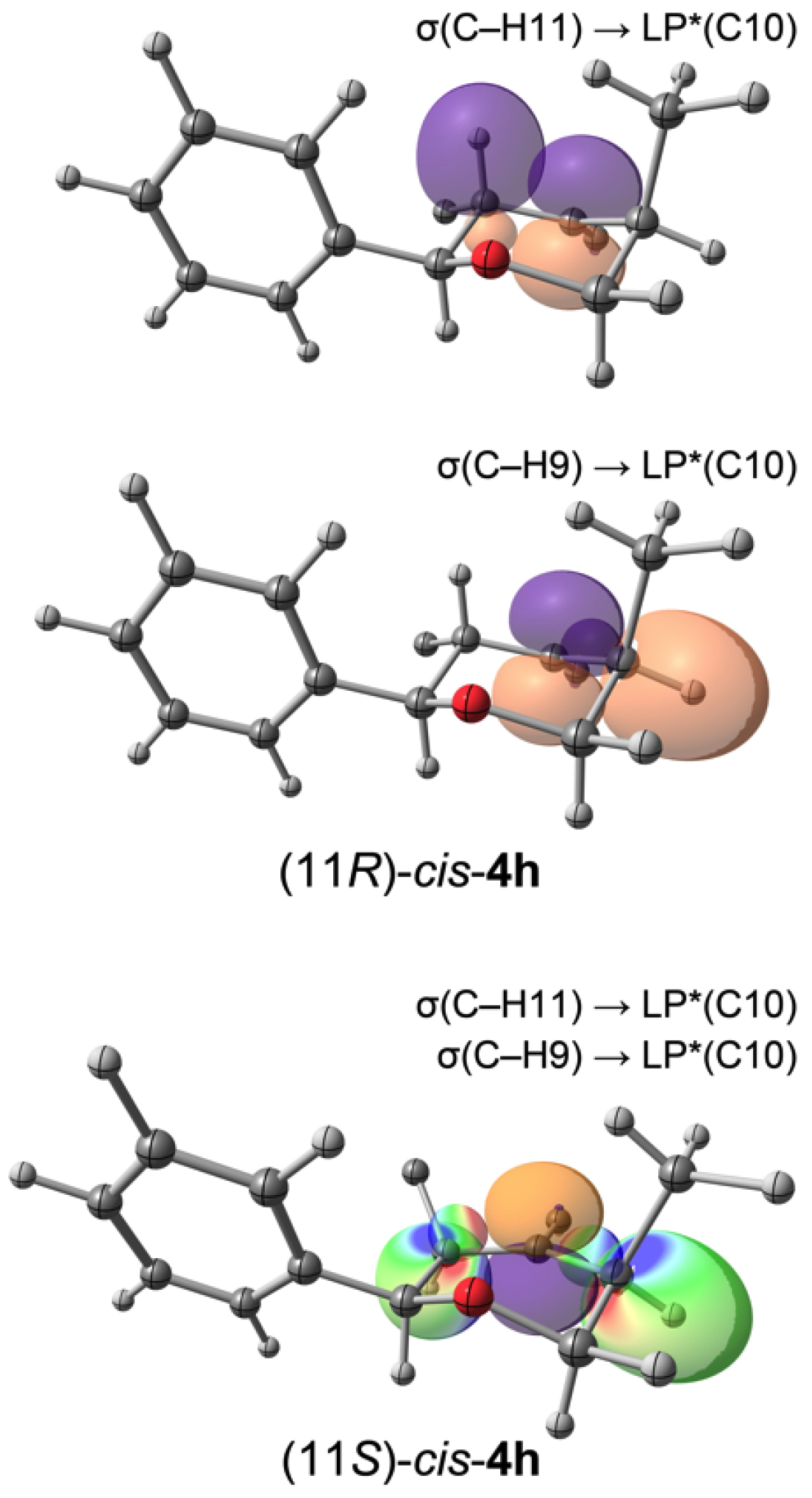
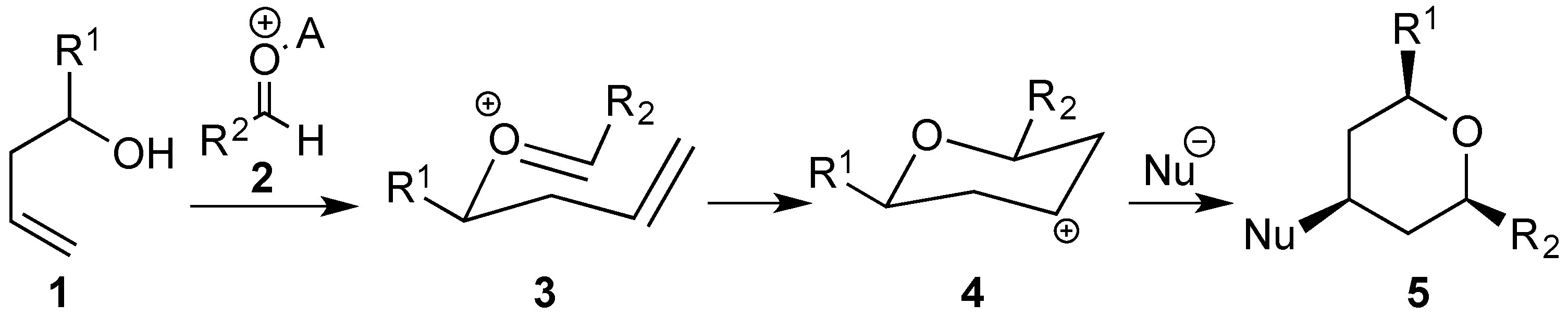
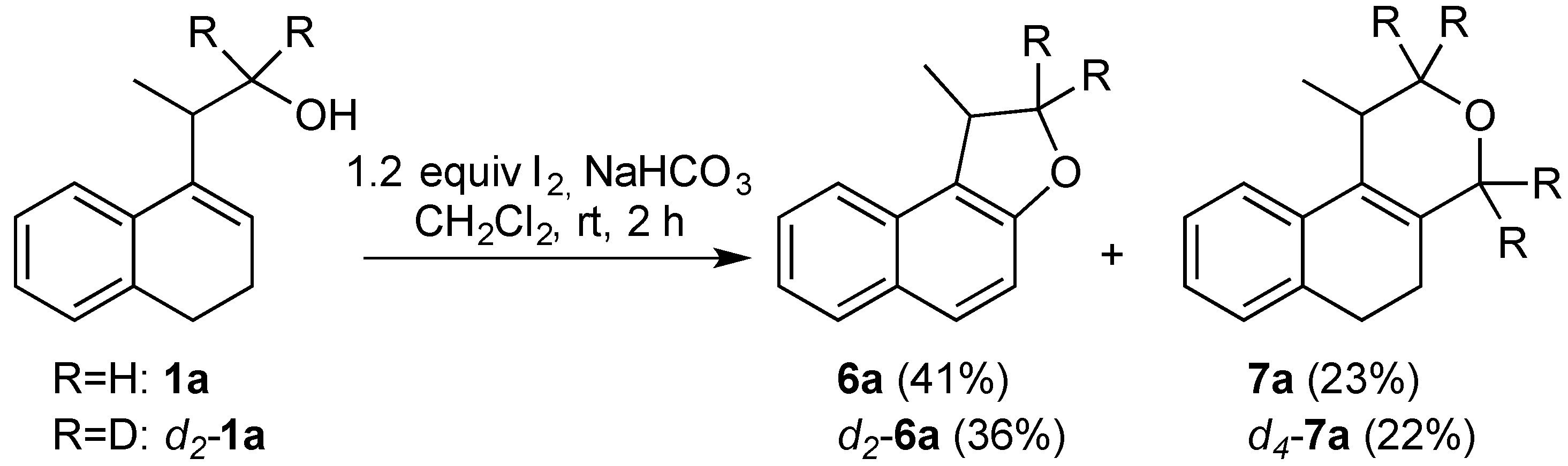
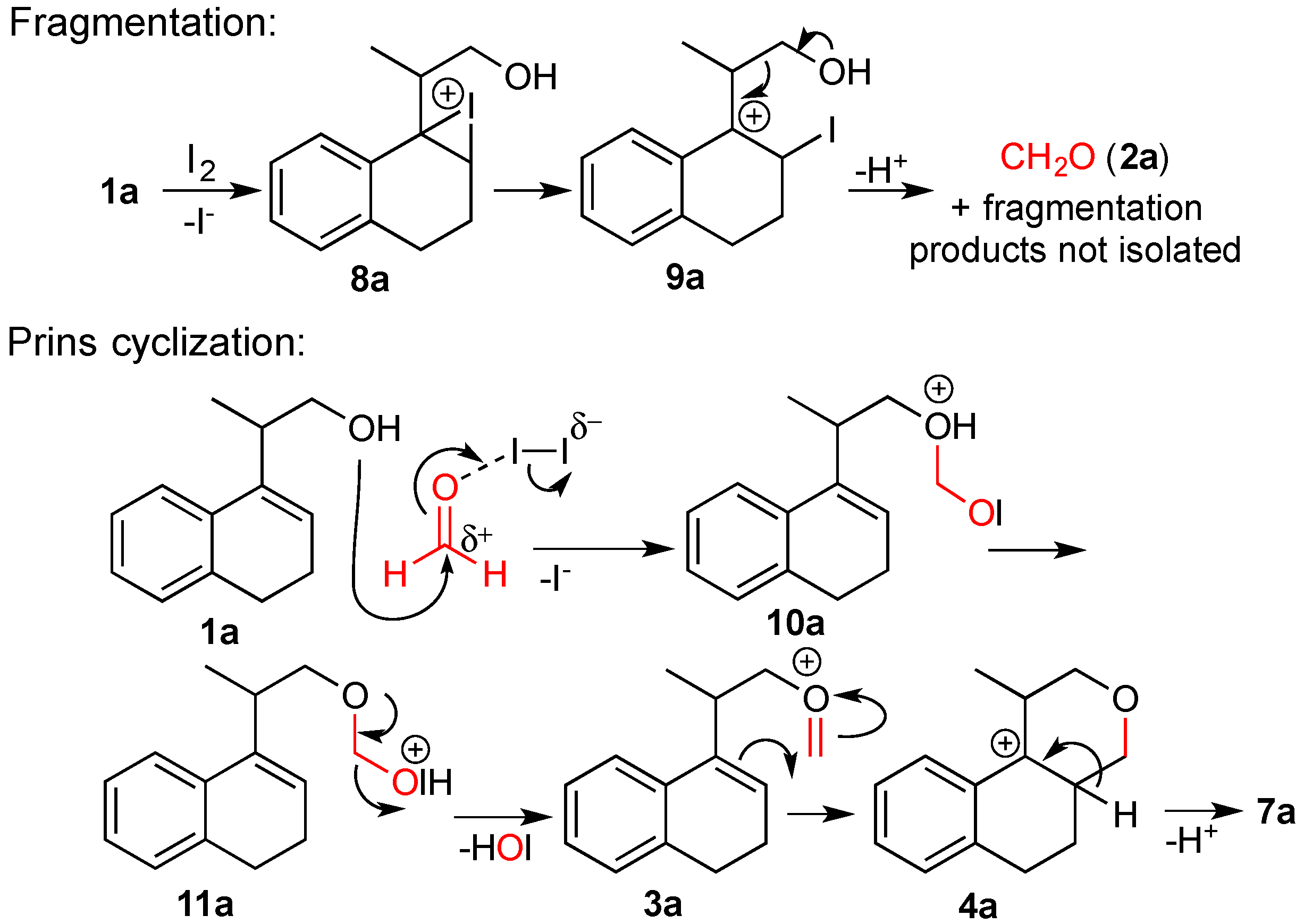
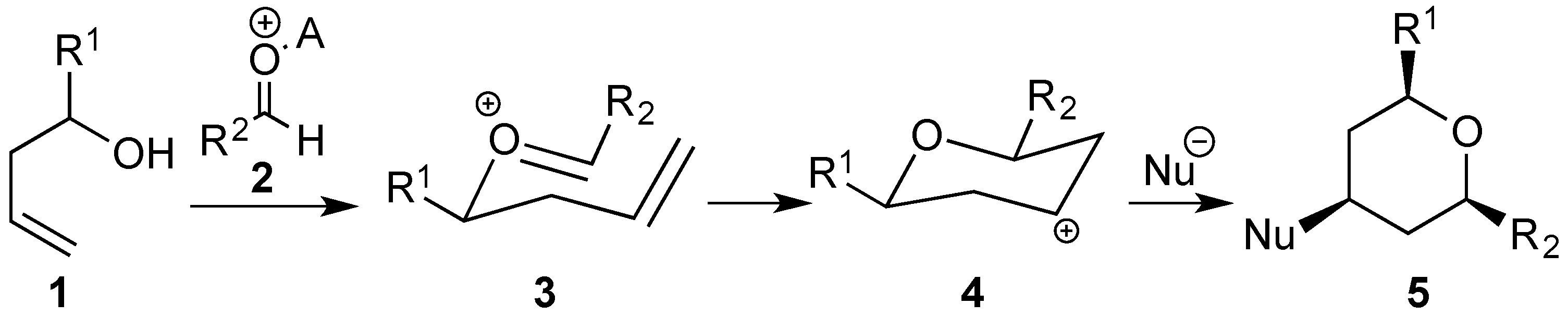
3.2. Prins Cyclizations
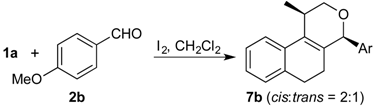
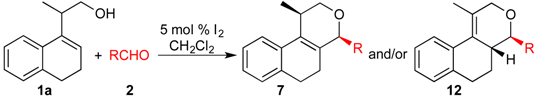
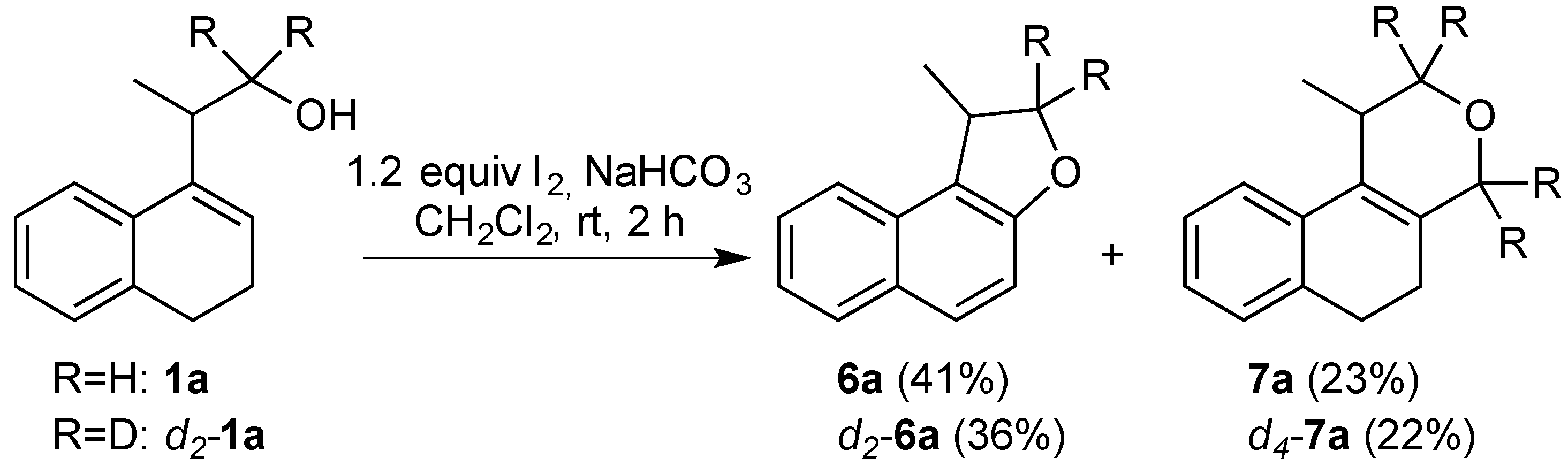
Prins cyclization of 1a and 2c. General Procedure for Iodine-Catalyzed Prins cyclization. To a stirred solution of 1a (0.078 g, 0.414 mmol) and 2c (0.050 mL, 0.41 mmol) in CH2Cl2 (5 mL), was added I2 (0.0052, 0.021 mmol). The mixture was refluxed for 3 h. Na2SO3 (0.0030 g, 0.021 mmol) and H2O (10 mL) were added. The aqueous phase was extracted with AcOEt (3 × 5 mL). The combined organic was washed with brine (5 mL) and dried over anhydrous MgSO4. The solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (5% AcOEt in hexane), affording 7c (cis:trans = 1.3:1, 0.11 g, 0.35 mmol, 84%) as colorless viscous oil. The relative configuration was assigned by NMR analysis, including NOESY experiments of enriched samples of cis-7c and trans-7c that were obtained after successive purifications of the product by flash column chromatography (1% AcOEt in hexanes).
(±)-cis-2,4,5,6-Tetrahydro-4-(2-methoxyphenyl)-1-methyl-1H-benzo[f]isochromene (cis-7c). IR (film): 3063, 2960, 2936, 2836, 1599, 1491, 1462, 756, 736 cm−1. 1H-NMR (300 MHz, CDCl3) δ: 1.41 (d, J = 6.9 Hz, 3H), 1.77–1.87 (m, 1H), 1.94–2.05 (m, 1H), 2.67 (t, J = 7.9 Hz, 2H), 2.76–2.78 (m, 1H), 3.83–3.87 (m, 1H), 3.87 (s, 3H), 3.98 (dd, J = 10.8, 3.3 Hz, 1H), 5.70 (s, 1H), 6.91–6.97 (m, 2H), 7.08–7.16 (m, 2H), 7.21–7.27 (m, 1H), 7.29–7.38 (m, 3H). 13C-NMR (75 MHz, CDCl3) δ: 18.5, 24.4, 28.0, 28.8, 55.6, 70.2, 73.0, 110.9, 120.7, 122.1, 126.4, 127.6, 128.5, 129.3, 131.8, 133.7, 134.0, 135.8, 157.9. LRMS m/z (rel. int.): 306 (M+∙, 25), 264 (17), 245 (13), 231 (10), 199 (11), 141 (17), 135 (100). HRMS [ESI(+)] calcd. for [C21H22O2+Na+] 329.1517, found 329.1494.
(±)-trans-2,4,5,6-Tetrahydro-4-(2-methoxyphenyl)-1-methyl-1H-benzo[f]-isochromene (trans-7c). IR (film): 3060, 3018, 2959, 2929, 2835, 1599, 1587, 1489, 1462, 756, 736 cm−1. 1H-NMR (300 MHz, CDCl3) δ: 1.26 (d, J = 7.0 Hz, 3H), 1.81–2.04 (m, 2H), 2.62–2.81 (m, 3H), 3.54 (dd, J = 11.2, 2.6 Hz, 1H), 3.81–3.95 (m, 1H), 3.90 (s, 3H), 5.75 (s, 1H), 6.86–6.96 (m, 2H), 7.11–7.39 (m, 6H). 13C-NMR (75 MHz, CDCl3) δ: 18.2, 25.8, 28.1, 28.3, 55.6, 66.1, 71.5, 110.8, 119.7, 122.3, 126.3, 126.7, 127.6, 129.3, 129.6, 132.3, 132.8, 133.7, 136.3, 158.3. LRMS m/z (rel. int.): 306 (M+∙, 32), 264 (18), 245 (16), 231 (8), 199 (11), 141 (19), 135 (100). HRMS [ESI(+)] calcd. for [C21H22O2+Na+] 329.1517, found 329.1511.
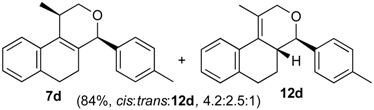
Prins cyclization of 1a and 2d. The reaction was performed following the general procedure, but using 1a (0.096 g, 0.51 mmol), 2d (0.060 mL, 0.41 mmol), CH2Cl2 (5 mL), I2 (0.0065, 0.025 mmol). A mixture of 7d and 12d (cis:trans:12d = 4.2:2.5:1, 0.12 g, 0.43 mmol, 84%) was obtained as a colorless viscous oil. This mixture was subjected to another flash column chromatography (1% AcOEt in hexanes). Partially pure samples could be obtained for characterization separately.
(±)-cis-2,4,5,6-Tetrahydro-1-methyl-4-p-tolyl-1H-benzo[f]isochromene (cis-7d). IR (film): 3058, 3024, 2954, 2919, 2849, 1272, 1178, 1109, 766, 754 cm−1. 1H-NMR (200 MHz, CDCl3) δ: 1.43 (d, J = 6.8 Hz, 3H), 1.68–2.05 (m, 2H), 2.35 (s, 3H), 2.67 (t, J = 7.6 Hz, 2H), 2.73–2.78 (m, 1H), 3.88 (dd, J = 10.9, 1.8 Hz, 1H), 3.97 (dd, J = 10.8, 2.8 Hz, 1H), 5.09 (s, 3H), 7.08–7.19 (m, 3H), 7.21–7.29 (m, 3H), 7.30–7.47 (m, 2H). 13C-NMR (50 MHz, CDCl3) δ: 18.7, 21.2, 24.5 28.0, 28.9, 70.0, 80.5, 122.1, 126.5, 126.6, 127.7, 128.6, 129.2, 131.9, 133.1, 133.8, 135.7, 137.7, 138.0. LRMS m/z (rel. int.): 290 (M+∙, 18), 276 (14), 275 (72), 257 (8), 247 (25), 229 (10), 215 (8), 203 (13), 171 (9), 155 (11), 128 (29), 127 (22), 119 (93), 91 (88), 65 (51), 43 (100). HRMS [ESI(+)] calcd. for [C21H22O+Na]+ 313.1563, found 313.1535.
(±)-(4R,4aR)-4,4a,5,6-Tetrahydro-1-methyl-4-p-tolyl-2H-benzo[f]isochromene (12d) and (±)-trans-2,4,5,6-tetrahydro-1-methyl-4-p-tolyl-1H-benzo[f]isochromene (trans-7d). IR (film): 3059, 3020, 2925, 2873, 1716, 1452, 1273, 1103, 1038, 816, 760 cm−1. 1H-NMR (200 MHz, CDCl3) δ: 12d: 1.21–1.81 (m, 2H), 1.92 (s, 3H), 2.53 (s, 3H), 2.60–2.87 (m , 3H), 4.10 (d, J = 9.6 Hz, 1H), 4.24 (d, J = 17.2 Hz, 1H), 4.35 (d, J = 16.6 Hz, 1H), 7.11–7.41 (m, 7H), 7.44 (d, J = 3.0 Hz, 1H), trans-7d: 1.22 (d, J = 6.8 Hz, 3H), 2.34 (s, 3H), 3.50 (dd, J = 11.3, 3.3 Hz, 1H), 3.99 (d, J = 11.3, 3.7 Hz, 1H), 5.17 (s, 1H). Other signals overlap with the major diastereomer. 13C-NMR (75 MHz, CDCl3) δ: 12d: 16.6, 21.2, 26.1, 28.3, 40.7, 71.3, 83.3, 125.0, 126.3, 126.5, 126.6, 127.6, 129.1, 128.4, 128.9, 129.1, 132.6, 136.2, 137.7, 137.9, 138.0, 138.1. trans-7d: 17.9, 21.2, 26.0, 28.2, 28.2, 66.7, 78.8, 122.6, 127.0, 127.3, 127.6, 129.2, 129.6, 129.7, 130.2, 132.6, 133.5, 133.7, 134.8, 136.2, 136.3. LRMS m/z (rel. int.): 290 (M+∙, 17), 275 (72), 247 (25), 229 (10), 215 (8), 203 (13), 155 (11), 128 (29), 127 (21), 119 (93), 91 (88), 65 (51), 43 (100). HRMS [ESI(+)] calcd. for [C21H22O+Na]+ 313.1563, found 313.1532.
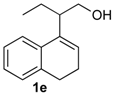
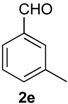
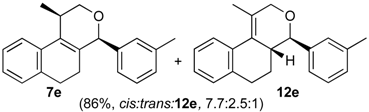
Prins cyclization of 1a and 2e. The reaction was performed following the general procedure, but using 1a (0.080 g, 0.42 mmol), 2e (0.050 mL, 0.42 mmol), CH2Cl2 (5 mL), I2 (0.0054, 0.021 mmol). A mixture of 7e and 12e (cis:trans:12e = 7.7:2.5:1, 0.11 g, 0.37 mmol, 86%) was obtained as colorless viscous oil. This mixture was subjected to another flash column chromatography (1% AcOEt in hexanes) for characterization.
(±)-2,4,5,6-Tetrahydro-1-methyl-4-m-tolyl-1H-benzo[f]isochromene (7e). IR (film): 3060, 3022, 2963, 2930, 1718, 1488, 1459, 1451, 1278, 1200, 1115, 766, 746 cm−1. 1H-NMR (200 MHz, CDCl3) δ: cis-7e: 1.45 (d, J = 6.8 Hz, 3H), 1.69–2.06 (m, 2H), 2.34 (s, 3H), 2.67 (t, J = 8.0 Hz, 2H), 2.74–2.78 (m, 1H), 3.97 (dd, J = 10.8, 2.8 Hz, 1H), 3.88 (dd, J = 10.9, 1.8 Hz, 1H), 5.09 (s, 3H), 7.08–7.19 (m, 3H), 7.21–7.29 (m, 3H), 7.30–7.47 (m, 2H). trans-7e: 1.22 (d, J = 6.8 Hz, 3H), 2.37 (s, 3H), 3.51 (dd, J = 11.2, 4 Hz, 1H), 4.41 (dd, J = 10.5, 2.8 Hz, 1H). Other signals overlap with the major diastereomer. 13C-NMR (50 MHz, CDCl3) δ: cis-7e: 18.8, 21.4, 24.4, 28.0, 28.9, 70.4, 80.8, 122.1, 125.3, 125.8, 126.5, 126.6, 127.7, 128.4, 128.5, 129.0, 129.3, 131.9, 133.0, 133.8, 135.7, 138.2, 140.5. LRMS m/z (rel. int.): 290 (M+∙, 37), 249 (11), 248 (69), 247 (39), 233 (25), 215 (11), 155 (17), 141 (15), 129 (33), 119 (100). HRMS [ESI(+)] calcd. for [C21H22O+Na]+ 313.1563, found 313.1541.
(±)-(4R,4aR)-4,4a,5,6-Tetrahydro-1-methyl-4-m-tolyl-2H-benzo[f]isochromene (12e). IR (film): 3094, 2925, 2869, 1648, 1484, 1445, 1101, 913, 760, 745 cm−1. 1H-NMR (500 MHz, CDCl3) δ: 1.48–1.56 (m, 1H), 1.75–1.81 (m, 1H), 1.92 (t, J = 1.0 Hz, 3H), 2.36 (s, 3H), 2.59–2.64 (m, 1H), 2.67–2.73 (m, 1H), 2.76–2.82 (m, 1H), 4.10 (d, J = 9.5 Hz, 1H), 4.26 (ddd, J = 16.5, 3.0, 1.0 Hz, 1H), 4.35 (dd, J = 16.5, 1.0 Hz, 1H), 7.08–7.10 (m, 1H), 7.12–7.15 (m, 2H), 7.16–7.18 (m, 1H), 7.19–7.20 (m, 1H), 7.24–7.27 (m, 2H), 7.43 (dd, J = 7.5, 1.5, 1H). 13C-NMR (75 MHz, CDCl3) δ: 16.6, 21.4, 26.1, 28.3, 40.7, 71.3, 83.5, 124.8, 125.0, 126.7, 126.9, 128.2, 128.3, 128.4, 128.8, 128.9, 129.7, 134.8, 137.9, 138.1, 141.0. LRMS m/z (rel. int.): 290 (M+∙, 1.4), 233 (9.8), 215 (2.5), 170 (100). HRMS [ESI(+)] calcd. for [C21H22O+Na]+ 313.1563, found 313.1555.
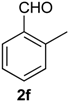
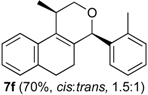
Prins cyclization of 1a and 2f. The reaction was performed following the general procedure, but using 1a (0.081 g, 0.43 mmol), 2f (0.050 mL, 0.41 mmol), CH2Cl2 (5 mL), I2 (0.0055, 0.022 mmol). Compound 3f (cis:trans = 1.5:1, 0.089 g, 0.30 mmol, 70%) was obtained as colorless viscous oil. This mixture was subjected to another flash column chromatography (1% AcOEt in hexanes). Pure samples could be obtained for characterization.
(±)-cis-2,4,5,6-Tetrahydro-1-methyl-4-o-tolyl-1H-benzo[f]isochromene (cis-7f). White solid, m.p. 155–157 °C. IR (film): 3060, 3017, 2954, 2919, 2850, 1486, 1458, 1122, 761, 734 cm−1. 1H-NMR (300 MHz, CDCl3) δ: 1.38 (d, J = 6.9 Hz, 3H), 1.79–2.04 (m, 2H), 2.50 (s, 3H), 2.66–2.72 (m, 2H), 2.79–2.81 (m, 1H), 3.83 (dd, J = 11.1, 2.4 Hz, 1H), 3.96 (dd, J = 11.1, 3.3 Hz, 1H), 5.40 (s, 1H), 7.10–7.15 (m, 2H), 7.17–7.21 (m, 3H), 7.25–7.27 (m, 1H), 7.31–7.35 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ: 18.3, 19.3, 24.6, 27.9, 28.8, 70.2, 77.2, 122.1, 126.1, 126.5, 127.7, 128.0, 130.6, 1334, 133.8, 135.8, 137.4, 138.2. LRMS m/z (rel. int.): 290 (M+∙, 73), 248 (70), 247 (55), 234 (17), 233 (100). HRMS [ESI(+)] calcd. for [C21H22O+Na]+ 313.1563, found 313.1584.
(±)-trans-2,4,5,6-Tetrahydro-1-methyl-4-o-tolyl-1H-benzo[f]isochromene (trans-7f). Viscous oil. IR (film): 3062, 3017, 2962, 2918, 2897, 1485, 1457, 1119, 1036, 765, 749, 739 cm−1. 1H-NMR (500 MHz, CDCl3) δ: 1.25 (d, J = 6.5 Hz, 3H), 1.85 (ddd, J = 15.5, 5.1, 5.0 Hz, 1H), 2.00–2.04 (m, 1H), 2.50 (s, 3H), 2.64–2.85 (m, 3H), 3.54 (dd, J = 11.5, 3.0 Hz, 1H), 3.92–3.94 (m, 1H), 5.45 (s, 1H), 7.09–7.17 (m, 4H), 7.19–7.23 (m, 2H), 7.29–7.32 (2H). 13C-NMR (75 MHz, CDCl3) δ: 18.2, 19.2, 26.2, 28.2, 28.3, 29.7, 77.2, 122.5, 125.2, 126.4, 126.5, 127.6, 128.1, 129.0, 130.9, 132.9, 133.7, 136.3, 136.5, 138.4. LRMS m/z (rel. int.): 290 (M+∙, 48), 248 (47), 247 (35), 233 (65), 215 (13), 199 (11), 128 (26), 119 (100). HRMS [ESI(+)] calcd. for [C21H22O+Na]+ 313.1563, found 313.1573.
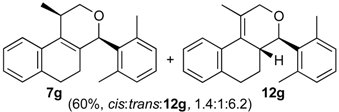
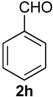
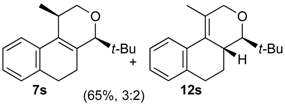
Prins cyclization of 1a and 2g. The reaction was performed following the general procedure, but using 1a (0.17 g, 0.89 mmol), 2g (0.12 mL, 0.89 mmol), CH2Cl2 (5 mL), I2 (0.011 g, 0.045 mmol). A mixture of 7g and 12g (cis:trans:12g = 1.4:1:6.2, 0.16 g, 0.53 mmol, 60%) was obtained as colorless viscous oil. This mixture was subjected to another flash column chromatography (1% AcOEt in hexanes). Partially pure samples could be obtained for characterization.
(±)-(4R,4aR)-4,4a,5,6-Tetrahydro-1-methyl-4-(2,6-dimethylphenyl)-2H-benzo[f]isochromene (12g). Solid, m.p: 148–150 °C. IR (film): 3066, 3015, 2919, 2857, 2797, 1443, 1374, 1106, 1095, 762, 735 cm−1. 1H-NMR (200 MHz, CDCl3) δ: 1.43–1.64 (m, 1H), 1.69–1.84 (m, 1H), 1.95 (s, 3H), 2.42 (s, 3H), 2.51 (s, 3H), 2.73–2.97 (m, 3H), 4.19 (d, J = 16.6 Hz, 1H), 4.35 (d, J = 16.4 Hz, 1H), 4.67 (d, J = 10.0 Hz, 1H), 7.04–7.22 (m, 6H), 7.43–7.47 (m, 1H). 13C-NMR (75 MHz, CDCl3) δ: 16.8, 27.4, 28.4, 38.9, 71.3, 79.2, 124.9, 126.7, 127.3, 127.5, 128.8, 129.1, 129.8, 134.7, 136.6, 137.4. LRMS m/z (rel. int.): 304 (M+∙, 0.34), 247 (2.5), 232 (1.0), 171 (9.7), 170 (100). HRMS [ESI(+)] calcd. for [C22H24O+Na] 327.1725, found 327.1732.
(±)-2,4,5,6-Tetrahydro-1-methyl-4-(2,6-dimethylphenyl)-1H-benzo[f]isochromene (7g). IR (film): 3095, 3062, 3021, 2917, 2855, 2731, 1486, 1377, 1126, 1101, 776, 729 cm−1. 1H-NMR (500 MHz, CDCl3) δ: cis-7g: 1.44 (d, J = 7.0 Hz, 1H), 1.74–1.82 (m, 2H), 2.19 (s, 3H), 2.42 (s, 3H), 2.51–2.93 (m, 3H), 4.08 (d, J = 11.2, 1.5 Hz, 1H), 4.05 (d, J = 11.5, 2.7 Hz, 1H), 5.59 (s, 1H), 6.98–7.24 (m, 7H), 7.30 (d, J = 7.0 Hz, 1H), trans-7g: 1.05 (d, J = 7.0 Hz, 1H), 1.50–1.71 (m, 2H), 2.48 (s, 3H), 2.49 (s, 3H), 3.16–3.22 (m, 1H), 3.52 (d, J = 11.5, 10.5Hz, 1H), 4.32 (d, J = 11.2, 6.0 Hz, 1H), 5.72 (s, 1H), 6.88 (d, J = 7.0 Hz, 1H). Other signals overlap with the major diastereomer. 13C-NMR (50 MHz, CDCl3) δ: cis-7g: 17.9, 21.0, 21.2, 23.7, 28.1, 28.6, 72.3, 77.6, 122.0, 123.3, 125.9, 126.39, 126.45, 127.6, 127.8, 128.2, 129.9, 132.3, 133.9, 134.0, 135.7, 137.5, 138.4. trans-7g: 16.1, 19.6, 21.0, 25.1, 27.8, 28.3, 72.3, 76.2, 125.3, 127.2, 127.3, 127.7, 128.0, 129.9, 134.0, 134.7, 135.7, 136.2, 137.0, 138.5. LRMS m/z (rel. int.): cis-7g: 304 (M+∙, 50), 262 (31), 261 (22), 248 (11), 247 (71), 229 (17), 152 (14), 141 (20), 133 (100), trans-7g: 304 (67), 262 (45), 261 (30), 248 (15), 247 (100). HRMS [ESI(+)] calcd. for [C22H24O+Na] 327.1725, found 327.1717.
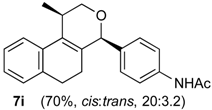
Prins cyclization of 1a and 2i. The reaction was performed following the general procedure, but using 1a (0.075 g, 0.40 mmol), 2i (0.065 g, 0.40 mmol), CH2Cl2 (5 mL), I2 (0.0051 g, 0.020 mmol). A mixture of 7i (cis:trans = 20:3.2, 0.933 g, 0.28 mmol, 70%) was obtained as colorless viscous oil.
(±)-N-(4-(2,4,5,6-tetrahydro-1-methyl-1H-benzo[f]isochromen-4-yl)phenyl)acetamide (7i). IR (film): 3308, 3197, 3123, 3059, 2964, 2929, 2871, 1684, 1671, 1601, 1540, 1411, 1372, 1318, 1267, 767 736 cm−1. 1H-NMR (200 MHz, CDCl3) δ: cis-7i: 1.43 (d, J = 6.8 Hz, 3H), 1.64–1.98 (m, 2H), 2.15 (s, 3H), 2.60–2.68 (m, 2H), 2.73–2.78 (m, 1H), 3.87 (d, J = 10.9, 2.0 Hz, 1H), 3.96 (d, J = 11.0, 3.0 Hz, 1H), 5.09 (s, 1H), 7.06–7.20 (m, 3H), 7.23–7.51 (m, 5H).
trans-7i: 1.21 (d, J = 6.8 Hz, 3H), 2.08 (s, 3H), 3.50 (dd, J = 11.4, 3.8 Hz, 1H), 5.16 (s, 1H). Other signals overlap with the major compound. 13C-NMR (75 MHz, CDCl3) δ: cis-7i: 18.8, 24.4, 24.6, 27.9, 28.8, 70.3, 80.2, 119.7, 122.1, 126.5 126.7, 127.7, 129.3, 132.0, 133.7, 135.7, 137.8, 168.3. trans-7i: 17.9, 24.6, 28.1, 28.2, 77.2, 78.6, 119.7, 122.6, 125.3, 126.3, 126.5, 127.6, 129.8, 132.9, 136.6, 137.1. LRMS m/z (rel. int.): 333 (M+∙, 28), 292 (13), 291 (69), 290 (45), 288 (20), 249 (18), 233 (13), 162 (55), 155 (12), 141 (13), 129 (15), 128 (23), 115 (16), 43 (100). HRMS [ESI(+)] calcd. for [C22H23O2+Na] 356.1626, found 356.1633.
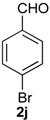
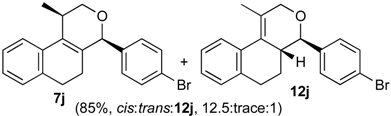
Prins cyclization of 1a and 2j. The reaction was performed following the general procedure, but using 1a (0.056 g, 0.30 mmol), 2j (0.055 g, 0.30 mmol), CH2Cl2 (5 mL), I2 (0.0038 g, 0.015 mmol). A mixture of 7j and 12j (cis:trans:12j = 12.5:0.01:1, 0.091 g, 0.26 mmol, 85%) was obtained as a colorless viscous oil. Aldehyde 2j (12%) was also recovered. This mixture was subjected to another flash column chromatography (1% AcOEt in hexanes). Partially pure samples could be obtained for characterization.
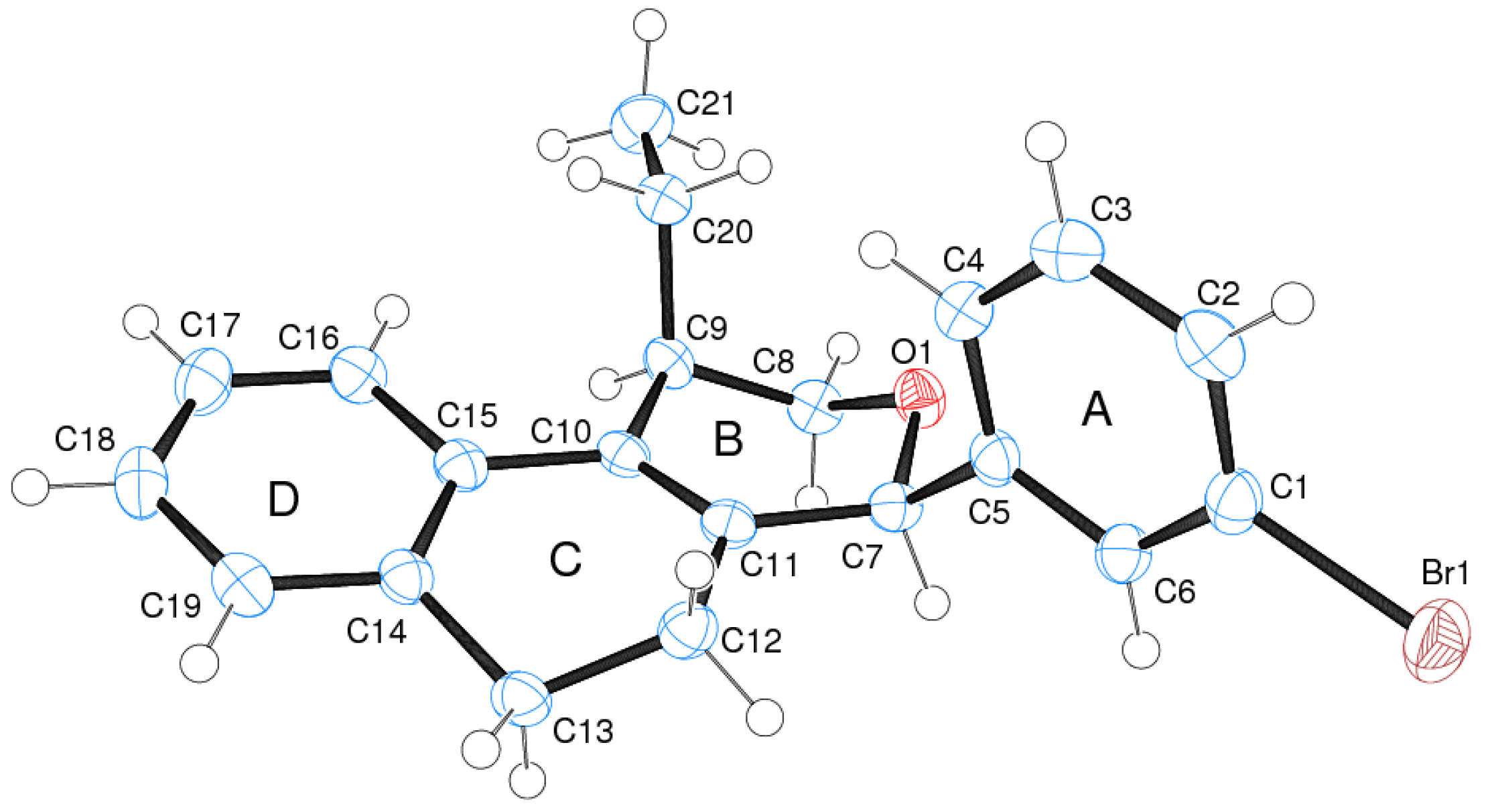
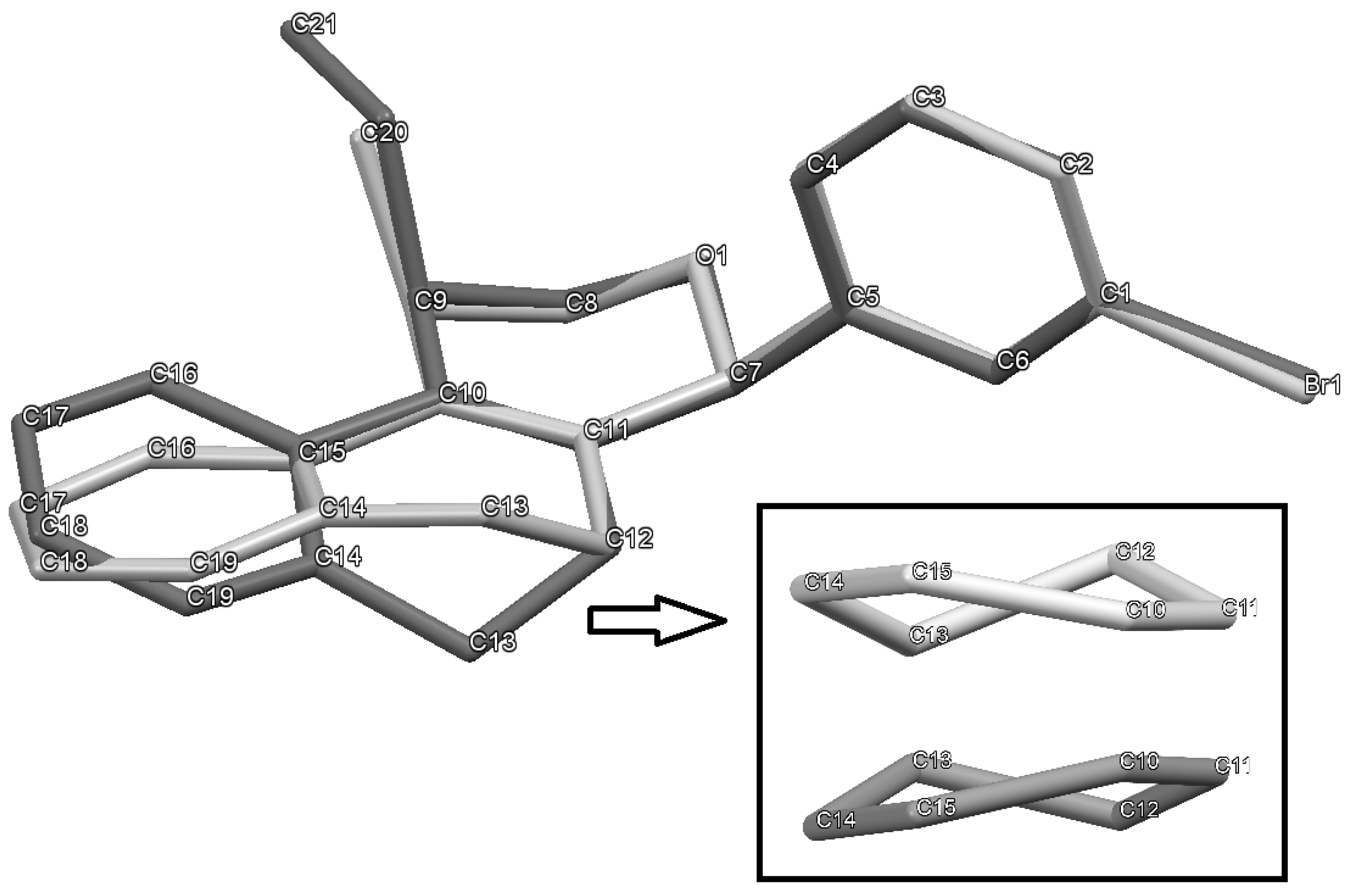
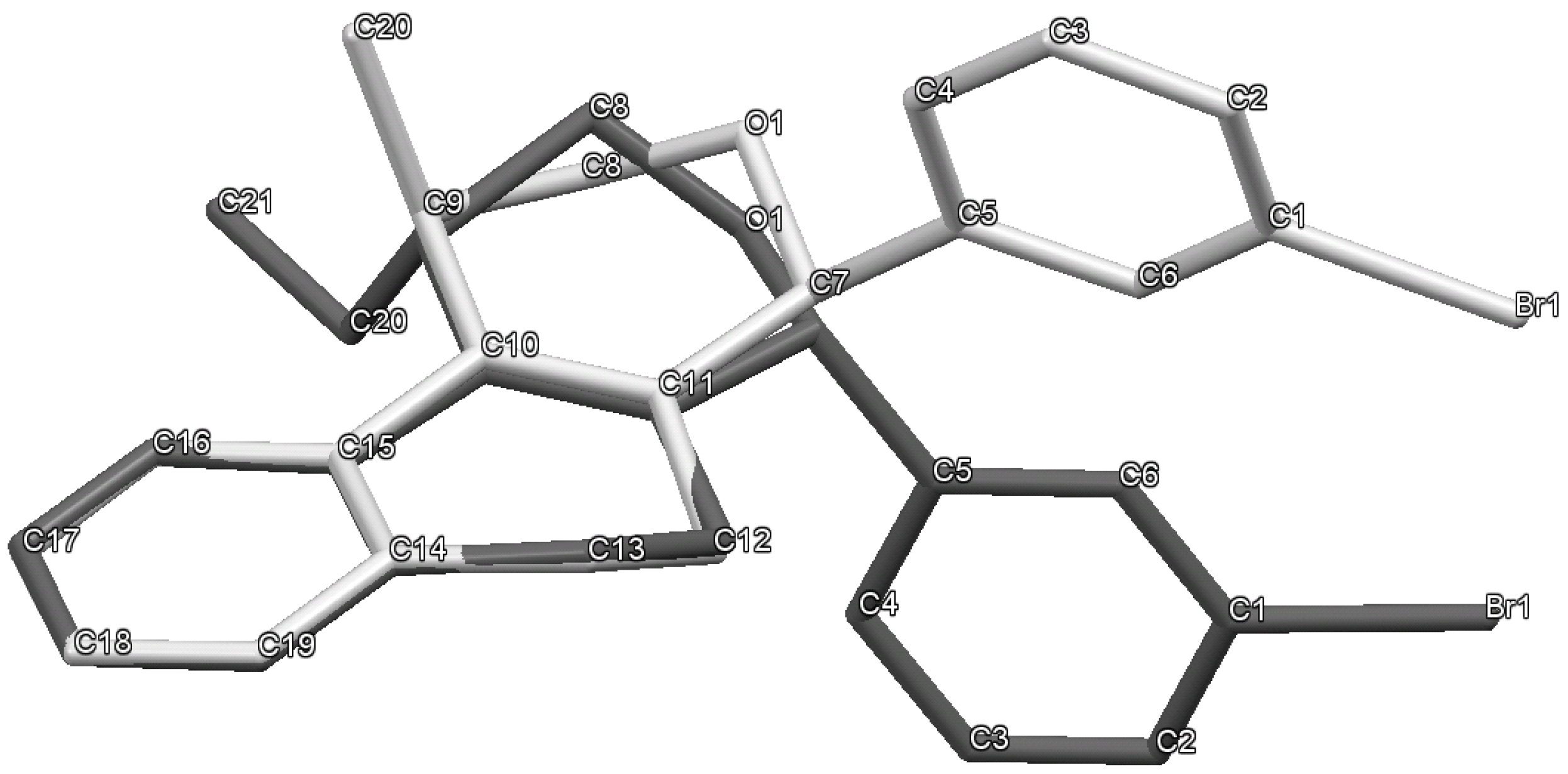
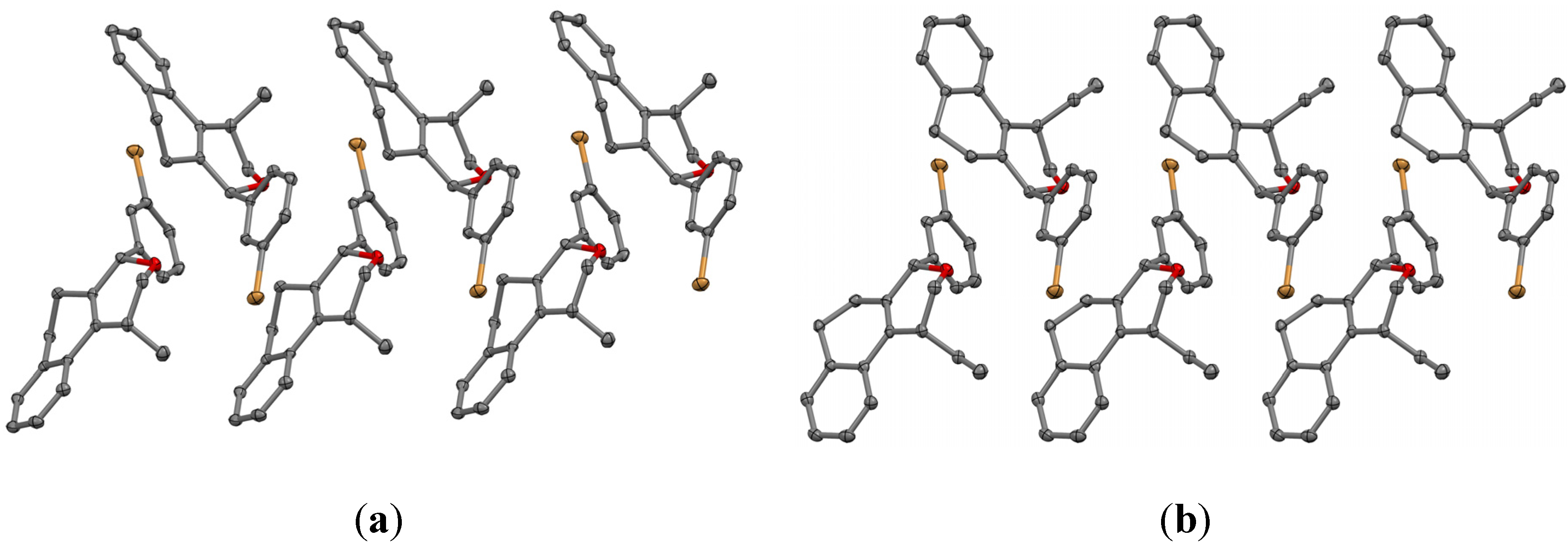
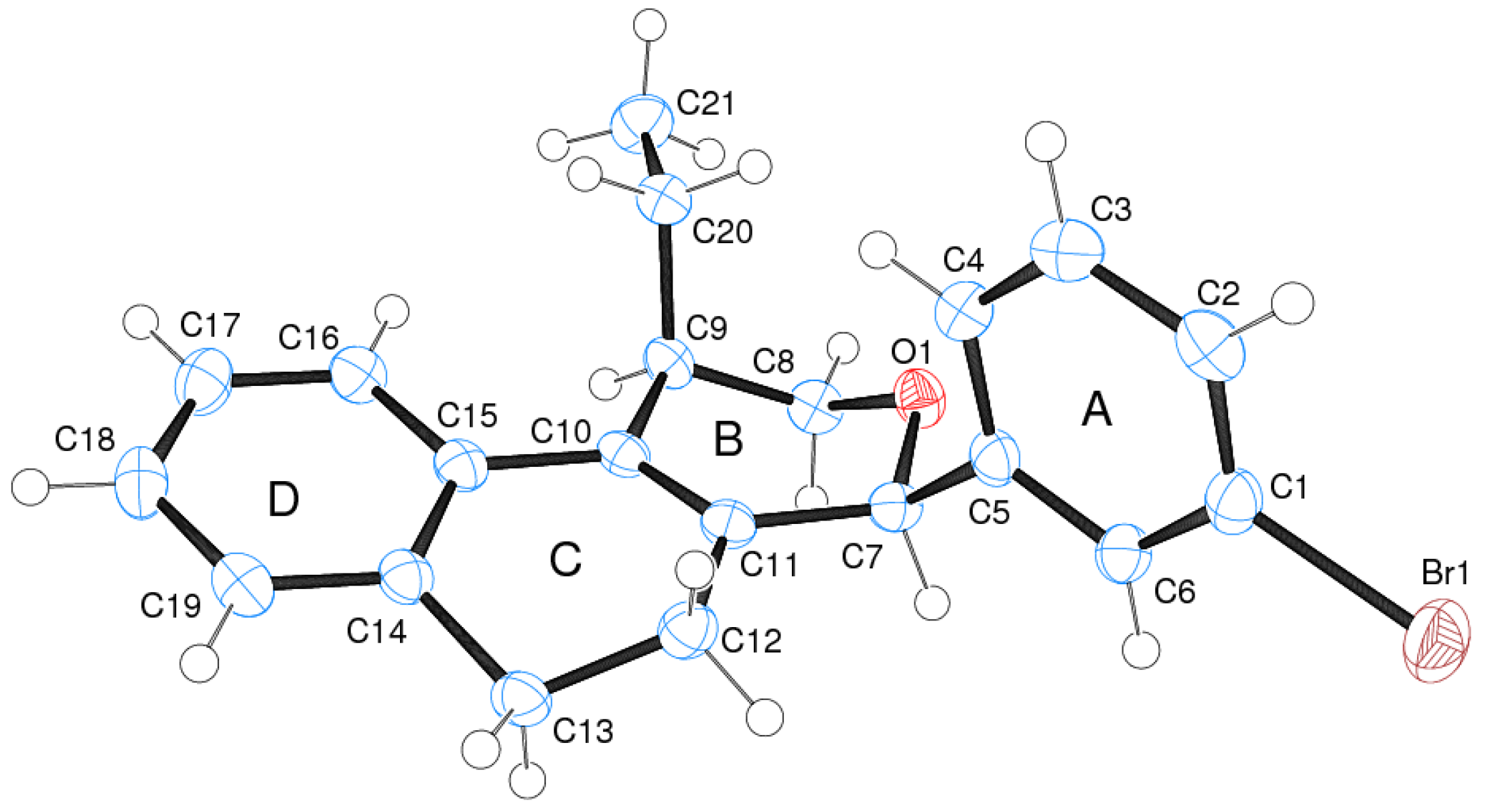
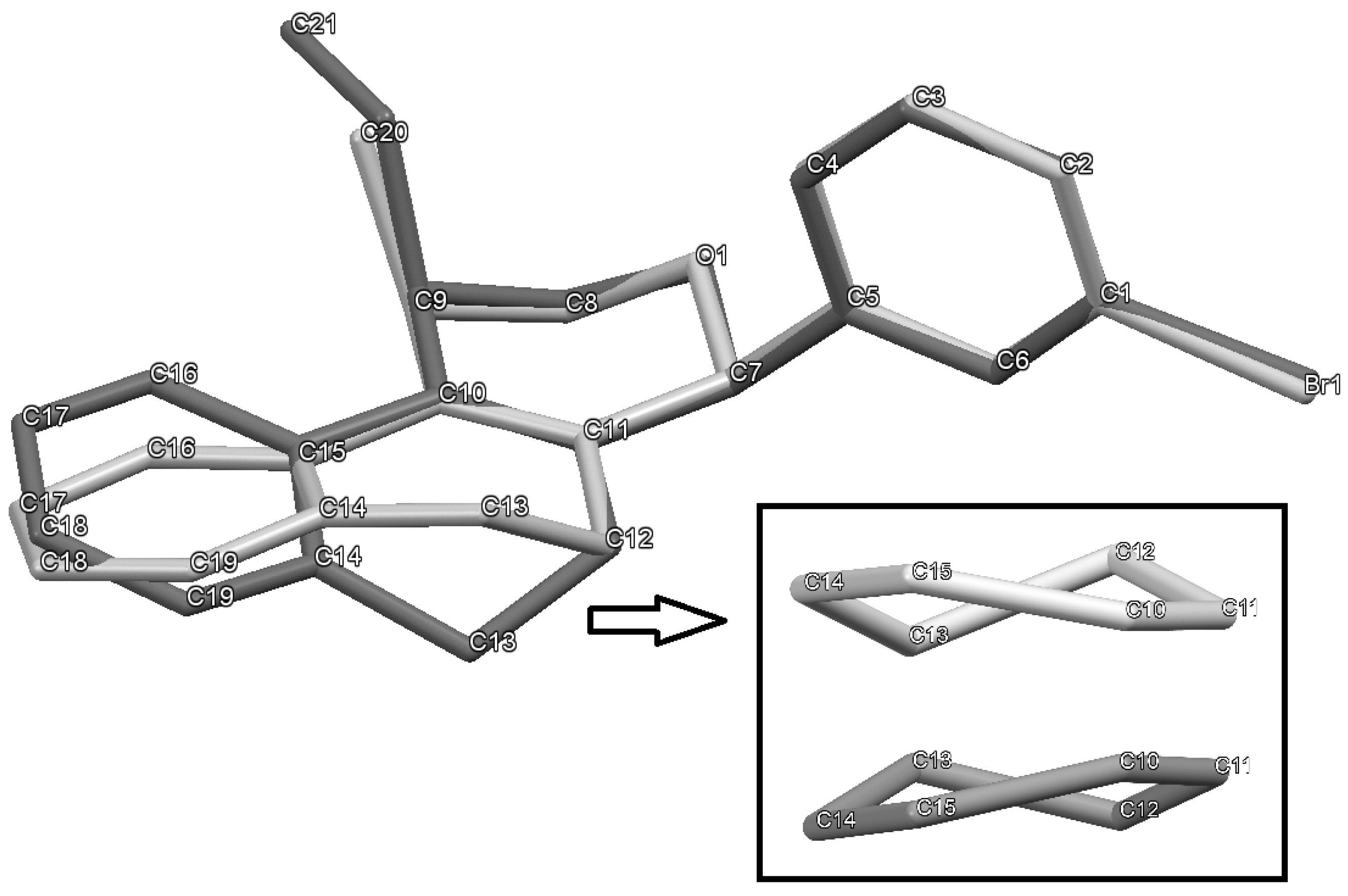
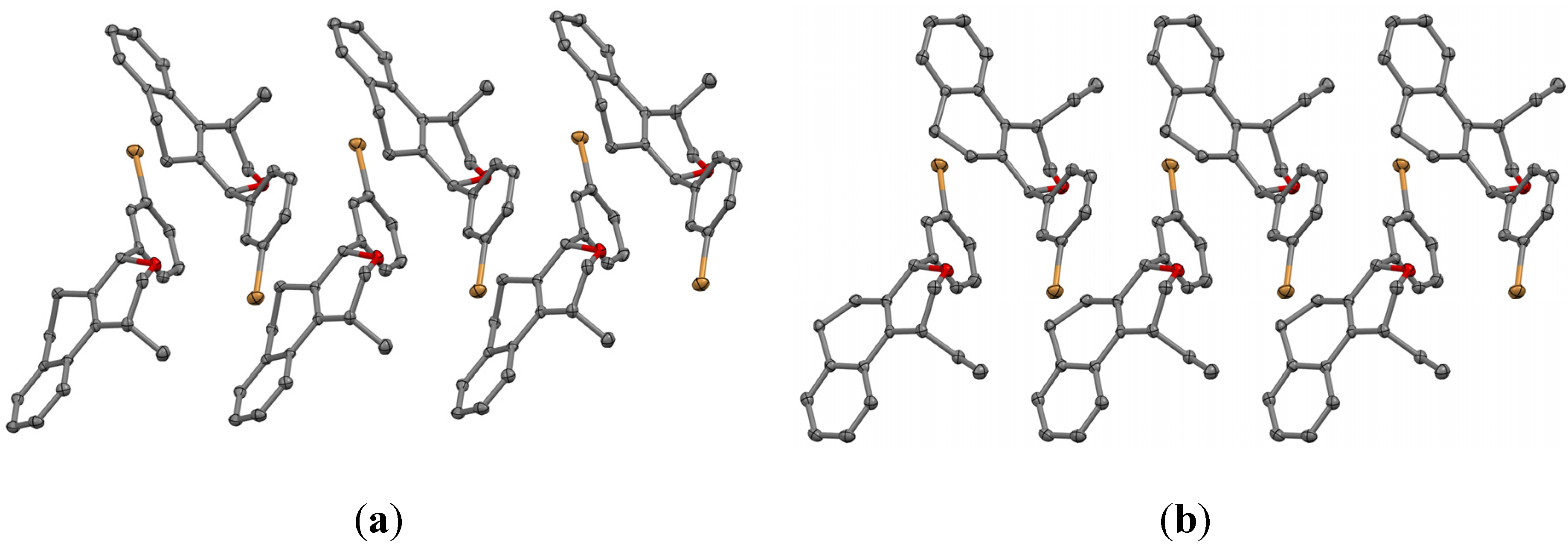
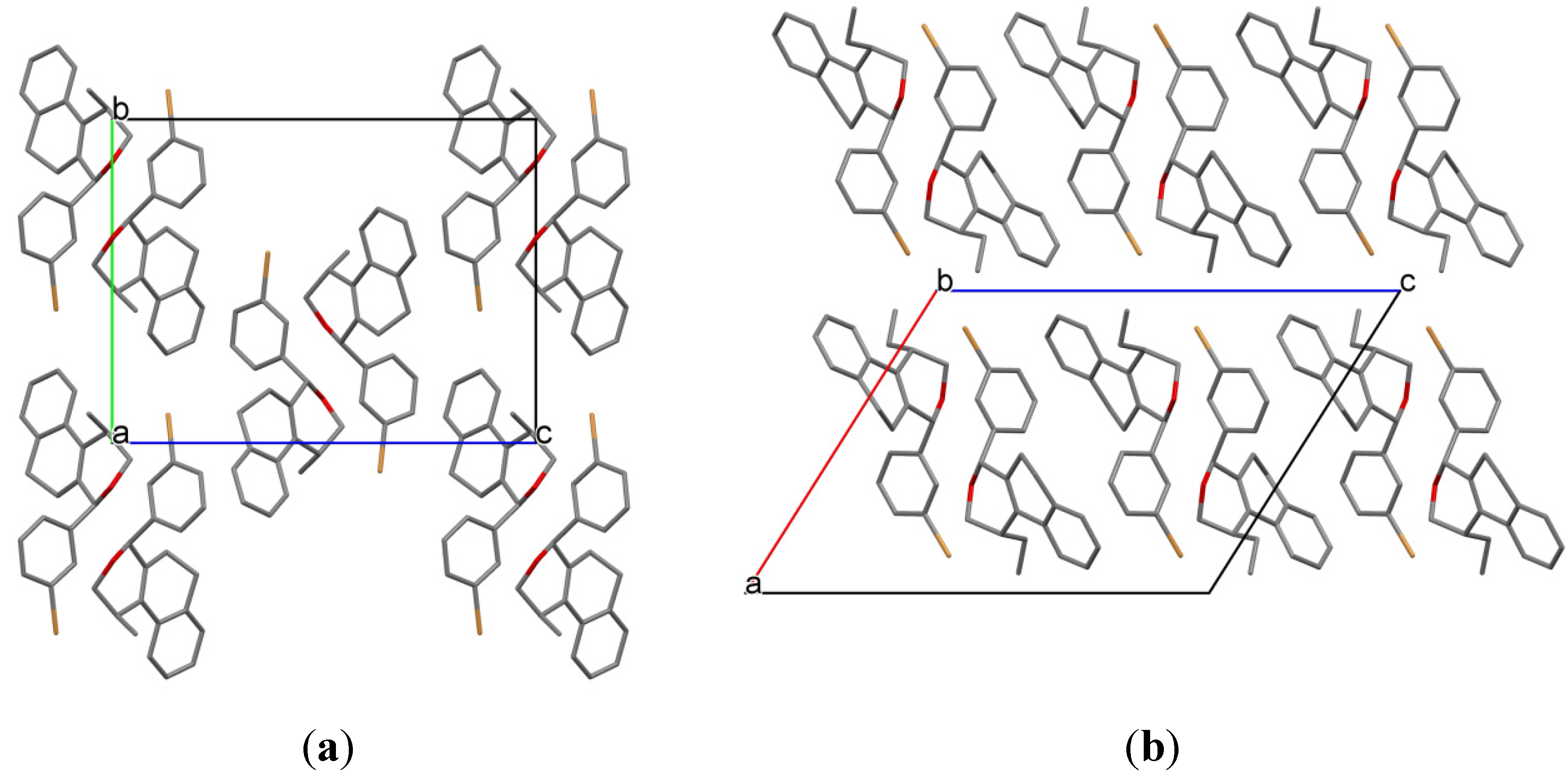
(±)-cis-4-(4-Bromophenyl)-2,4,5,6-tetrahydro-1-methyl-1H-benzo[f]isochromene (cis-7j). IR (film): 3065, 2962, 2927, 1712, 1487, 1462, 1453, 1276, 768, 735 cm−1. 1H-NMR (200 MHz, CDCl3) δ: 1.43 (d, J = 7.0 Hz, 3H), 1.67–2.05 (m, 2H), 2.57–2.80 (m, 3H), 3.87 (dd, J = 11.0, 2.0 Hz, 1H), 3.95 (dd, J = 11.0, 3.0 Hz, 1H), 5.08 (s, 1H), 7.07–7.12 (m, 1H), 7.15–7.21 (m, 1H), 7.24–7.35 (m, 4H), 7.46 (t, J = 2.0 Hz, 1H), 7.51 (t, J = 1.8 Hz, 1H). 13C-NMR (50 MHz, CDCl3) δ: 18.8, 24.3, 27.9, 28.8, 70.4, 80.1, 122, 126.5, 126.8, 127.7, 130.3, 131.7, 132.2, 132.3, 133.5, 135.6, 139.8. LRMS m/z (rel. int.): 356 (M+∙+2, 20), 354 (M+∙, 20), 314 (47), 312 (47), 233 (46), 215 (32), 185 (60), 183 (60), 129 (100). HRMS [ESI(+)] calcd. for [C20H19BrO+H] 355.0698, found 355.0549.
(±)-(4R,4aR)-4-(4-bromophenyl)-4,4a,5,6-tetrahydro-1-methyl-2H-benzo[f]isochromene (12j) and (±)-trans-4-(4-bromophenyl)-2,4,5,6-tetrahydro-1-methyl-1H-benzo[f]isochromene (trans-7j). IR (film): 3067, 2956, 2926, 1719, 1590, 1484, 1454, 1271, 757, 733 cm−1. 1H-NMR (200 MHz, CDCl3) δ: 12j: 1.37–2.05 (m, 2H), 1.92 (s, 3H), 2.60–2.79 (m, 3H), 4.10 (d, J = 9.6 Hz, 3H), 4.23 (dd, J = 16.5, 1.4 Hz, 1H), 4.35 (d, J = 15.4 Hz, 1H), 7.07–7.52 (m, 7H), 7.66–7.78 (m, 1H). trans-7j: 1.21 (d, J = 6.8 Hz, 3H), 3.50 (dd, J = 11.4, 4.0 Hz, 1H), 5.16 (s, 1H). Other signals overlap with the major diastereomer. 13C-NMR (50 MHz, CDCl3) δ: 12j: 16.5, 26.0, 28.2, 40.8, 67.0, 78.4, 125.1, 125.3, 126.7, 126.9, 128.4, 128.9, 129.3, 130.8, 131.0, 131.5, 131.8, 132.4, 134.6, 137.7. trans-7j: 17.8, 25.9, 28.1, 28.2, 71.2, 82.3, 121.9, 122.3, 122.7, 126.8, 127.6, 129.4, 129.8, 133.1, 133.5, 135.1, 136.2, 138.2, 140.2. LRMS m/z (rel. int.): 356 (M+∙+2, 20), 354 (M+∙, 20), 314 (41), 312 (47), 233 (40), 215 (27), 185 (47), 183 (53), 157 (17), 155 (29), 129 (100). HRMS [ESI(+)] calcd. for [C20H19BrO+H] 355.0698, 357.0677, found 355.0319, 357.0301.
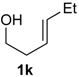
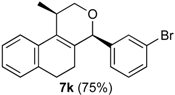
Prins cyclization of 1a and 2k. The reaction was performed following the general procedure, but using 1a (0.39 g, 2.1 mmol), 2k (0.24 mL, 2.1 mmol), CH2Cl2 (10 mL), I2 (0.026 g, 0.10 mmol). Compound cis-7k (0.55 g, 1.5 mmol, 75%) was obtained as a colorless solid.
(±)-cis-4-(3-Bromophenyl)-2,4,5,6-tetrahydro-1-methyl-1H-benzo[f]isochromene (cis-7k). m.p. 154 °C. IR (film): 3060, 2959, 2917, 1487, 1460, 788, 732 cm−1. 1H-NMR (500 MHz, CDCl3) δ: 1.45 (d, J = 6.5 Hz, 3H), 1.75–1.81 (m, 1H), 1.94–2.00 (m, 1H), 2.61–2.72 (m, 2H), 2.76–2.77 (m, 1H), 3.88 (dd, J = 11.0, 2.0 Hz, 1H), 3.95 (dd, J = 11.0, 3.0 Hz, 1H), 5.08 (s, 1H), 7.10 (d, J = 7.0 Hz, 1H), 7.16 (td, J = 7.5, 1.0 Hz, 1H), 7.21–7.26 (m, 2H), 7.33 (d, J = 6.5 Hz, 1H), 7.34 (d, J = 7.5 Hz, 1H), 7.44–7.46 (m, 1H), 7.55 (t, J = 1.5 Hz, 1H). 13C-NMR (75 MHz, CDCl3) δ: 18.8, 24.3, 27.9, 28.9, 70.4, 80.2, 122.2, 122.6, 126.5, 126.8, 127.3, 127.7, 130.1, 131.4, 131.7, 132.0, 132.4, 133.5, 135.6, 143.0. LRMS m/z (rel. int.): 356 (M+∙+2, 25), 354 (M+∙, 25), 314 (45), 312 (46), 233 (33), 215 (25), 185 (36), 183 (34), 157 (22), 155 (22), 129 (100). HRMS [ESI(+)] calcd. for [C20H19BrO+Na] 377.0518, 379.0509, found 377.0516, 379.0504.
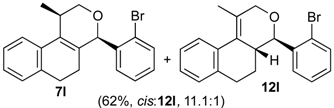
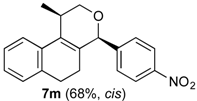
Prins cyclization of 1a and 2l. The reaction was performed following the general procedure, but using 1a (0.094 g, 0.50 mmol), 2l (0.092 g, 0.50 mmol), CH2Cl2 (5 mL), I2 (0.0063, 0.025 mmol). A mixture of 7l and 12l (cis:12l = 11.1:1, 0.11 g, 0.31 mmol, 62%) was obtained as a colorless viscous oil.
(±)-cis-4-(2-Bromophenyl)-2,4,5,6-tetrahydro-1-methyl-1H-benzo[f]isochromene (cis-7l) and (±)-(4R,4aR)-4-(2-bromophenyl)-4,4a,5,6-tetrahydro-1-methyl-2H-benzo[f]isochromene (12l). IR (film): 3062, 2962, 2928, 1470, 1438, 759, 732 cm−1. 1H-NMR (500 MHz, CDCl3) δ: cis-7l: 1.43 (d, J = 7.0 Hz, 3H), 1.75–1.82 (m, 1H), 2.00–2.08 (m, 1H), 2.60–2.73 (m, 2H), 2.77–2.81 (m, 1H), 3.88 (dd, J = 10.7, 2.5 Hz, 1H), 4.00 (dd, J = 10.7, 3.5 Hz, 1H), 5.69 (s, 1H), 7.01–7.18 (m, 3H), 7.28–7.46 (m, 3H), 7.45 (dd, J = 7.7, 1.5 Hz, 1H), 7.59 (dd, J = 7.7, 1.5 Hz, 1H). 12l: 1.93 (d, J = 1.0 Hz, 3H), 4.33 (s, 2H), 4.78 (d, J = 9.5 Hz, 2H). Other signals overlap with the major diastereomer. 13C-NMR (75 MHz, CDCl3) δ: cis-7l: 18.8, 24.3, 27.9, 28.8, 70.4, 78.7, 122.2, 125.5, 126.5, 126.7, 127.7, 127.8, 129.7, 129.9, 132.4, 132.7, 132.9, 133.6, 135.7, 139.7. 12l: 16.5, 25.7, 28.4, 41.2, 71.6, 80.4, 125.1, 125.46, 125.53, 127.9, 128.3, 128.8, 129.0, 129.4, 132.6. LRMS m/z (rel. int.): 356 (M+∙+2, 21), 354 (M+∙, 24), 314 (38), 312 (38), 232 (55), 215 (34), 185 (34), 183 (38), 157 (19), 155 (20), 129 (83), 128 (100). HRMS [ESI(+)] calcd. for [C20H19BrO+Na] 377.0517, 379.0497, found 377.0516, 379.0504.
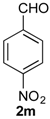
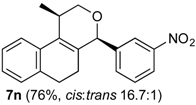
Prins cyclization of 1a and 2n. The reaction was performed following the general procedure, but using 1a (0.094 g, 0.50 mmol), 2n (0.076 g, 0.50 mmol), CH2Cl2 (5 mL), I2 (0.0063, 0.025 mmol). Compound 7n (cis:trans = 16.7:1, 0.12 g, 0.38 mmol, 76%) was obtained as a white solid.
(±)-cis-2,4,5,6-Tetrahydro-1-methyl-4-(3-nitrophenyl)-1H-benzo[f]isochromene (cis-7n). m.p. 104–105 °C. IR (film): 3061, 3020, 2963, 2927, 2249, 1727, 1486, 1451, 768, 735 cm−1. 1H-NMR (500 MHz, CDCl3) δ: 1.43 (d, J = 7.0 Hz, 3H), 1.74–1.80 (m, 1H), 2.08–2.16 (m, 1H), 2.59–2.73 (m, 2H), 2.78–2.82 (m, 1H), 3.86 (dd, J = 11.0, 2.0 Hz, 1H), 3.98 (dd, J = 11.0, 3.0 Hz, 1H), 5.24 (s, 1H), 7.11 (dd, J = 7.5, 1.0 Hz, 1H), 7.16 (td, J = 7.5, 1.5 Hz, 1H), 7.23–7.26 (m, 2H), 7.33 (d, J = 6.5 Hz, 1H), 7.44–7.48 (m, 1H), 7.58 (td, J = 7.0, 1.5 Hz 1H), 7.66 (dd, J = 8.0, 1.0 Hz, 1H), 7.84 (dd, J = 8.0, 1.0 Hz, 1H). 13C-NMR (75 MHz, CDCl3) δ: 19.0, 24.4, 27.8, 28.8, 70.5, 79.9, 122.3, 123.3, 123.7, 126.6, 127.1, 127.8, 129.5 131.1, 133.1, 133.3, 134.7, 135.5, 143.0, 148.4. LRMS m/z (rel. int.): 321 (M+∙, 11), 279 (33), 150 (28), 129 (100). HRMS [ESI(+)] calcd. for [C20H19NO3+Na] 344.1263, found 344.1253.
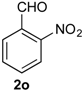
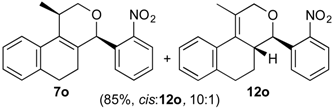
Prins cyclization of 1a and 2o. The reaction was performed following the general procedure, but using 1a (0.094 g, 0.50 mmol), 2o (0.076 g, 0.50 mmol), CH2Cl2 (5 mL), I2 (0.0063, 0.025 mmol). A mixture of 7o and 12o (cis:12o = 10:1, 0.14 g, 0.42 mmol, 85%) was obtained as a colorless viscous oil.
(±)-cis-2,4,5,6-Tetrahydro-1-methyl-4-(2-nitrophenyl)-1H-benzo[f]isochromene (cis-7o) and (±)-(4R,4aR)-4,4a,5,6-tetrahydro-1-methyl-4-(2-nitrophenyl)-2H-benzo[f]isochromene (12o). IR (film): 3065, 2962, 2928, 2253, 1527, 1488, 1450, 768, 735 cm−1. 1H-NMR (500 MHz, CDCl3) δ: cis-7o: 1.43 (d, J = 7.0 Hz, 3H), 1.74–1.80 (m, 1H), 2.08–2.16 (m, 1H), 2.59–2.73 (m, 2H), 2.78–2.82 (m, 1H), 3.86 (dd, J = 11.0, 2.0 Hz, 1H), 3.98 (dd, J = 11.0, 3.0 Hz, 1H), 5.79 (s, 1H), 7.11 (dd, J = 7.5, 1.0 Hz, 1H), 7.16 (td, J = 7.5, 1.5 Hz, 1H), 7.23–7.26 (m, 1H), 7.33 (d, J = 6.5 Hz, 1H), 7.44–7.48 (m, 1H), 7.58 (td, J = 7.0, 1.5 Hz, 1H), 7.66 (dd, J = 8.0, 1.0 Hz, 1H), 7.84 (dd, J = 8.0, 1.0 Hz, 1H). 12o: 1.68–1.73 (m, 1H), 1.58–1.64 (m 1H), 1.91 (t, J = 1.0 Hz, 3H), 4.27 (dd, J = 16.5, 1.5 Hz, 1H), 4.32 (dd, J = 16.5, 1.0 Hz, 1H), 4.92 (d, J = 9.5 Hz, 1H), 7.41 (dd, J = 7.2, 1.5 Hz, 1H), 7.63 (dd, J = 7.2, 1.0 Hz, 1H), 7.72 (dd, J = 8.0, 1.5 Hz, 1H), 7.82 (dd, J = 8.2, 1.5 Hz, 1H). Other signals overlap with the major diastereomer. 13C-NMR (75 MHz, CDCl3) δ: cis-7o: 18.9, 24.4, 27.9, 28.6, 70.7, 74.3, 122.3, 123.9, 126.6, 127.0, 127.7, 129.0 130.4, 131.8, 132.8, 133.1, 133.3, 135.1, 135.7, 150.6. 12o: 16.5, 25.7, 28.2, 40.8, 71.5, 76.2, 123.8, 125.1, 126.8, 127.1, 128.7, 128.8, 129.2, 129.3, 132.8, 134.6, 135.2, 138.0. LRMS m/z (rel. int.): 321 (M+∙, 1.7), 303 (22), 312 (46), 233 (33), 215 (25), 185 (36), 183 (34), 157 (22), 155 (22), 129 (100). HRMS [ESI(+)] calcd. for [C20H19NO3+Na] 344.1263, found 344.1263.
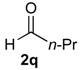
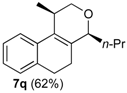
Prins cyclization of 1a and 2q. The reaction was performed following the general procedure, but using 1a (0.084 g, 0.44 mmol), 2q (0.040 mL, 0.44 mmol), CH2Cl2 (5 mL), I2 (0.0056, 0.022 mmol). Compound cis-7q (0.067 g, 0.28 mmol, 62%) was obtained as colorless viscous oil.
(±)-cis-2,4,5,6-Tetrahydro-1-methyl-4-propyl-1H-benzo[f]isochromene (cis-7q). IR (film): 3065, 2965, 2936, 1726, 1457, 760 cm−1. 1H-NMR (200 MHz, CDCl3) δ: 0.93 (t, J = 7.2 Hz, 3H), 1.33 (d, J = 6.8 Hz, 3H), 1.25–1.85 (m, 4H), 1.94–2.30 (m, 2H), 2.50–2.62 (m, 1H), 2.73–2.81 (m, 2H), 3.75 (dd, J = 10.6, 2.5 Hz, 1H), 3.81 (dd, J = 10.6, 1.8 Hz, 1H), 4.22–4.24 (m, 1H), 7.11 (dd, J = 4.7, 1.4 Hz, 2H), 7.16–7.23 (m, 1H), 7.24–7.29 (m, 1H). 13C-NMR (75 MHz, CDCl3) δ: 14.3, 18.0, 18.6, 23.9, 28.2, 29.2, 35.2, 69.8, 76.6, 121.9, 126.3, 126.4, 127.5, 131.3, 133.8, 134.3, 135.3. LRMS m/z (rel. int.): 242 (M+∙, 31), 227 (20), 199 (29), 184 (36), 170 (26), 158 (32), 157 (24), 155 (100). HRMS [ESI(+)] calcd. for [C17H22O+Na] 265.1568, found 265.1556.
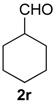
Prins cyclization of 1a and 2r. The reaction was performed following the general procedure, but using 1a (0.0932 g, 0.496 mmol), 2r (0.0600 mL, 0.496 mmol), CH2Cl2 (5 mL), I2 (0.0063, 0.024 mmol). Compound 7r (cis:trans = 7.1:1, 0.10 g, 0.36 mmol, 72%) was obtained as colorless viscous oil.
(±)-4-Cyclohexyl-2,4,5,6-tetrahydro-1-methyl-1H-benzo[f]isochromene (7r). IR (film): 3091, 3063, 2930, 2853, 1711, 1451, 1451, 763 cm−1. 1H-NMR (300 MHz, CDCl3) δ: cis-7r: 1.09–1.28 (m, 4H), 1.34 (d, J = 6.9 Hz, 3H), 1.39–1.51 (m, 1H), 1.53–1.73 (m, 6H), 1.77–1.85 (m, 2H), 1.95–2.07 (m, 1H), 2.15–2.26 (m, 1H), 2.48–2.55 (m, 1H), 2.71–2.82 (m, 2H), 3.72 (dd, J = 10.6, 2.5 Hz, 1H), 3.81 (dd, J = 10.5, 1.2 Hz, 1H), 4.10 (s, 1H), 7.10–7.13 (m, 2H), 7.14–7.25 (m, 1H), 7.27 (d, J = 7.5 Hz, 1H). trans-7r: 3.02 (d, J = 9.6 Hz, 1H), 3.99 (dd, J = 16.2, 1.5 Hz, 1H), 4. 10 (s, 1H), 4.19 (dd, J = 16.2, 0.9 Hz, 1H), other signals overlap with major diastereomer. 13C-NMR (75 MHz, CDCl3) δ: cis-7r: 18.7, 23.6, 26.0, 26.4, 26.7, 27.2, 28.2, 29.4, 30.4, 39.7, 70.0, 81.2, 121.8, 126.2, 126.4, 127.4, 131.8, 133.0, 134.5, 135.3. trans-7r: 16.5, 26.1, 26.6, 26.9, 27.6, 28.4, 31.3, 35.6, 70.7, 84.3, 124.8, 126.5, 127.2, 128.3, 128.9, 129.8, 135.5, 137.6. LRMS m/z (rel. int.): 282 (M+∙, 20), 267 (9), 224 (6), 199 (30), 197 (6), 187 (12), 186 (89), 181 (16), 171 (32), 170 (68), 169 (20), 158 (44), 157 (24), 156 (16), 155 (100). HRMS [ESI(+)] calcd. for [C20H26O+H] 283.2062, found 283.2613.
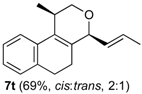
Prins cyclization of 1a and 2t. The reaction was performed following the general procedure, but using 1a (0.092 g, 0.49 mmol), 2t (0.070 mL, 0.49 mmol), CH2Cl2 (5 mL), I2 (0.0062 g, 0.024 mmol). Compound 7t (cis:trans= 2:1, 0.081 g, 0.34 mmol, 69%) was obtained as a colorless viscous oil. The relative configuration was assigned based on NOESY experiments of an enriched sample of cis-7t (cis:trans = 12:1) and enriched sample of trans-7t (cis:trans = 1:10) that were obtained after successive purifications of the product by flash column chromatography (1% AcOEt in hexanes).
(±)-cis-2,4,5,6-tetrahydro-1-methyl-4-((E)-prop-1-enyl)-1H-benzo[f]isochromene (cis-7t). IR (film): 2964, 2931, 2878, 2833, 1714, 1489, 1451, 1127, 768, 738 cm−1. 1H-NMR (300 MHz, CDCl3) δ: 1.30 (d, J = 6.9 Hz, 3H), 1.76 (dd, J = 6.4, 1.8 Hz, 3H), 1.94–2.03 (m, 1H), 2.05–2.20 (m, 1H), 2.63–2.67 (m, 1H), 3.73 (t, J = 7.2 Hz, 2H), 3.81 (s, 1H), 3.82 (s, 1H), 4.51 (d, J = 8.1 Hz, 1H), 5.46 (ddq, J = 15.1, 8.2, 1.8 Hz, 1H), 5.85 (dqd, J = 15.1, 6.4, 0.6 Hz, 1H), 7.11–7.13 (m, 2H), 7.15–7.27 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ: 17.8, 18.4, 24.2, 28.1, 28.8, 69.6, 78.7, 122.0, 126.39, 126.42, 127.6, 129.1, 130.5, 131.0, 131.2, 132.9, 135.6. LRMS m/z (rel. int.): 240 (M+∙, 85), 225 (26), 198 (82), 197 (80), 183 (99), 181 (17), 179 (18), 177 (18), 165 (55), 155 (61), 153 (28), 152 (39), 141 (57), 129 (100). HRMS [ESI(+)] calcd. for [C17H21O+H]+ 241.1592, found 241.1589.
(±)-trans-2,4,5,6-Tetrahydro-1-methyl-4-((E)-prop-1-enyl)-1H-benzo[f]isochromene (trans-7t). IR (film): 3067, 3022, 2964, 2931, 2878, 1708, 1451, 1380, 1127, 768, 738 cm−1. 1H-NMR (300 MHz, CDCl3) δ: 1.16 (d, J = 6.9 Hz, 3H), 1.72 (ddd, J = 9.0, 1.6, 0.6 Hz, 3H), 1.91–2.00 (m, 1H), 2.06–2.18 (m, 1H), 2.69–2.80 (m, 3H), 3.55 (dd, J = 11.2, 3.6 Hz, 1H), 4.03 (dd, J = 11.1, 3.9 Hz, 1H), 4.57 (d, J = 7.5 Hz, 1H), 5.51 (ddq, J = 15.4, 7.3, 1.5 Hz, 1H), 5.77 (dqd, J = 15.2, 6.4, 0.6 Hz, 1H), 7.12–7.14 (m, 2H), 7.15–7.19 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ: 17.7, 17.9, 25.5, 28.19, 28.21, 67.2, 77.4, 122.4, 126.26, 126.29, 127.5, 128.2, 130.5, 131.3, 133.1, 133.8, 136.1. LRMS m/z (rel. int.): 240 (M+∙, 97), 225 (21), 199 (16), 198 (76), 197 (70), 183 (82), 181 (16), 179 (18), 166 (17), 165 (43), 155 (43), 153 (27), 152 (24), 141 (52), 129 (100). HRMS [ESI(+)] calcd. for [C17H21O+H]+ 241.1592, found 241.1581.
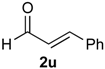
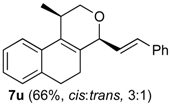
Prins cyclization of 1a and 2u. The reaction was performed following the general procedure, but using 1a (0.104 g, 0.556 mmol), 2u (0.070 mL, 0.556 mmol), CH2Cl2 (5 mL), I2 (0.0071, 0.028 mmol). Compound 7u (cis:trans = 3:1, 0.111 g, 0.368 mmol, 66%) was obtained as colorless viscous oil. The relative configuration was assigned by NMR analysis, including NOESY experiments, of enriched samples of cis-7u and trans-7u that were obtained after successive purifications of the product by flash column chromatography (1% AcOEt in hexanes).
(±)-cis-2,4,5,6-Tetrahydro-1-methyl-4-styryl-1H-benzo[f]isochromene (cis-7u). IR (film): 3070, 3020, 2954, 2917, 2849, 1461, 1375, 1117, 756 cm−1. 1H-NMR (300 MHz, CDCl3) δ: 1.34 (d, J = 6.9 Hz, 3H), 2.02–2.26 (m, 2H), 2.68–2.77 (m, 3H), 3.87 (s, 1H), 3.88 (s, 1H), 4.74 (d, J = 8.1 Hz, 1H), 6.17 (J = 13.9, 8.1 Hz,1H), 6.73 (d, J = 15.9 Hz, 1H), 7.10–7.15 (m, 2H), 7.17–7.25 (m, 2H), 7.27–7.30 (m, 2H), 7.31–7.35 (m, 1H), 7.36–7.43 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ: 18.4, 24.3, 28.1, 28.8, 69.6, 78.6, 122.1, 126.4, 126.57, 126.64, 127.6, 127.8, 128.5, 128.6, 131.7, 132.3, 133.8, 134.1, 135.6, 136.5. LRMS m/z (rel. int.): 302 (M+∙, 47), 301 (25), 287 (4), 260 (16), 259 (13), 241 (8), 210 (15), 198 (16), 197 (89), 181 (17), 165 (27), 156 (19), 155 (100). HRMS [ESI(+)] calcd. for [C22H22O+Na]+ 325.1568, found 325.1330.
(±)-trans-2,4,5,6-Tetrahydro-1-methyl-4-styryl-1H-benzo[f]isochromene (trans-7u). IR (film): 3058, 3025, 2959, 2926, 2871, 2834, 1490, 1450, 1121, 967, 765, 694 cm−1. 1H-NMR (500 MHz, CDCl3) δ: 1.21 (d, J = 7.0 Hz, 3H), 1.98–2.08 (m, 1H), 2.18–2.26 (m, 1H), 2.70–2.84 (m, 3H), 3.63 (dd, J = 11.0, 3.5 Hz, 1H), 4.10 (dd, J = 11.0, 4.0 Hz, 1H), 4.81 (d, J = 7.0 Hz, 1H), 6.25 (dd, J = 15.7, 7.0 Hz, 1H), 7.12–7.44 (m, 9H). 13C-NMR (50 MHz, CDCl3) δ: 17.8, 25.6, 28.1, 28.2, 28.2, 67.1, 77.1, 122.4, 125.2, 126.3, 126.4, 126.5, 127.6, 131.9, 132.3, 133.5, 133.6, 136.1. LRMS m/z (rel. int.): 302 (M+∙, 44), 301 (23), 260 (17), 259 (11), 210 (14), 207 (15), 197 (67), 179 (15), 165 (29), 155 (100). HRMS [ESI(+)] calcd. for [C22H22O+Na]+ 325.1568, found 325.1355.
Prins cyclization of 1b and 2b. The reaction was performed following the general procedure, but using 1b (0.104 g, 0.600 mmol), 2b (0.073 mL, 0.60 mmol), CH2Cl2 (5 mL), I2 (0.030, 0.076 mmol). Compound 13b was obtained as a pale brown oil (3:1 cis:trans, 0.142 g, 0.486 mmol, 81%).
(±)-1-(4-Methoxyphenyl)-4-methyl-1,3,4,9-tetrahydroindeno[2,1-c]pyran (13b). IR (film): 3020, 2961, 2906, 1512, 1246, 832 cm−1. 1H-NMR (200 MHz, CDCl3) δ: cis-13b: 1.48 (d, J = 7.0 Hz, 3H), 2.74–2.82 (m, 1H), 2.99 (br, 1H), 3.06–3.95 (br, 1H), 3.80 (s, 3H), 3.95–3.97 (m, 2H); trans-13b: 1.34 (d, J = 7.0 Hz, 3H), 2.88 (br, 1H), 3.20 (br, 1H), 3.44–3.53 (m, 1H), 3.84–3.88 (m, 2H). Other signals overlap with the major diastereomer. 13C-NMR (75 MHz, CDCl3) δ: cis-13b: 18.2, 28.7, 37.8, 55.3, 70.4, 78.6, 113.9, 118.5, 123.8, 124.4, 126.3, 129.1, 129.6, 133.2, 140.1, 143.1, 144.1, 159.5.
trans-13b: 16.4, 28.8, 38.3, 55.2, 67.7, 77.1, 113.8, 119.4, 123.8, 124.4, 126.2, 129.6, 132.8, 140.3, 140.4, 141.0, 143.2, 144.0, 159.5. LRMS m/z (rel. int.): 292 (M+∙, 3.9), 250 (7.1), 215 (6.0), 202 (14), 141 (19.7), 135 (100). HRMS [ESI(+)] calcd. for [C20H20O2+ Na]+ 315.1356, found 315.1355, [C20H20O2+ H]+ 293.1536, found 293.1537.
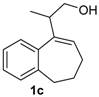
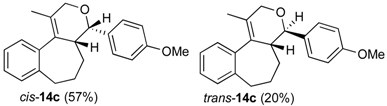
Prins cyclization of 1c and 2b. The reaction was performed following the general procedure, but using 1c (0.121 g, 0.600 mmol), 2b (0.073 mL, 0.60 mmol), CH2Cl2 (5 mL), I2 (0.030, 0.076 mmol). Compounds cis-14c (0.110 g, 0.344 mmol, 57%) and trans-14c (0.038 g, 0.118 mmol, 20%) were obtained as colorless oil.
(±)-(4S,4aR)-4-(4-Methoxyphenyl)-1-methyl-2,4,4a,5,6,7-hexahydrobenzo[3,4]cyclohepta-[1,2-c]pyran (cis-14c). IR (film): 3061, 3035, 3012, 2927, 1513, 1249, 830, 760, 751 cm−1. 1H-NMR (200 MHz, CDCl3) δ: 1.45 (s, 3H), 1.56–1.75 (m, 1H), 1.82–1.90 (m, 2H), 2.00–2.09 (m, 1H), 2.53–2.58 (m, 1H), 2.69–2.90 (m, 2H), 3.87 (s, 3H), 3.98 (d, J = 16.6 Hz, 1H), 4.17 (d, J = 16.2 Hz, 1H), 4.67 (d, J = 4.0 Hz, 1H), 6.95 (d, J = 8.6 Hz, 2H), 7.12–7.15 (m, 1H), 7.22–7.30 (m, 3H), 7.39 (d, J = 8.8 Hz, 2H). 13C-NMR (50 MHz, CDCl3) δ: 15.36, 25.68, 33.59, 34.50, 40.00, 55.18, 66.13, 78.86, 113.49, 125.80, 126.79, 127.41, 128.70, 128.72, 128.82, 132.21, 133.58, 140.85, 141.19, 158.86. LRMS m/z (rel. int.): 320 (M+∙, 0.08), 263 (1.9), 203 (1.8), 202 9 (3.2), 186 (1.1), 185 (17.3), 184 (100.0). HRMS [ESI(+)] calcd. for [C22H24O2+H]+ 321.1849, found 321.1860.
(±)-(4R,4aR)-4-(4-Methoxyphenyl)-1-methyl-2,4,4a,5,6,7-hexahydrobenzo[3,4]cyclohepta-[1,2-c]-pyran (trans-14c). IR (film): 3062, 3012, 2926, 1514, 1246, 832, 759 cm−1. 1H-NMR (200 MHz, CDCl3) δ: 1.23–1.30 (m, 1H), 1.44–1.69 (m, 2H), 1.57 (s, 3H), 1.86–1.93 (m, 1H), 2.08 (br, 1H), 2.55–2.79 (m, 2H), 3.80 (s, 3H), 4.40 (s, 2H), 4.827 (d, J = 2.6 Hz, 1H), 6.84–6.91 (m, 2H), 7.14–7.24 (m, 6H). 13C-NMR (50 MHz, CDCl3) δ: 15.1, 27.3, 30.5, 36.0, 43.3, 55.2, 70.7, 78.4, 113.4, 125.6, 126.60, 126.62, 126.66, 128.93, 133.2, 136.3, 141.7, 142.1, 158.2. LRMS m/z (rel. int.): 320 (M+∙, 0.05), 186 (0.9), 185 (10.3), 184 (100.0). HRMS [ESI(+)] calcd. for [C22H24O2+H]+ 321.1849, found 321.1864.
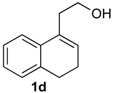
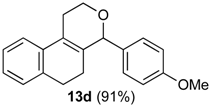
Prins cyclization of 1d and 2b. The reaction was performed following the general procedure, but using 1d (0.104 g, 0.600 mmol), 2b (0.073 mL, 0.60 mmol), CH2Cl2 (5 mL), I2 (0.0076 0.030 mmol). Compound 13d (0.159 g, 0.544 mmol, 91%) was obtained as colorless viscous oil.
(±)-4-(4-Methoxyphenyl)-1,4,5,6-tetrahydro-2H-benzo[f]isochromene (13d). IR (film): 3061, 3003, 2836, 1713, 1606, 1511, 761, 735 cm−1. 1H-NMR (200 MHz,CDCl3) δ: 1.83–1.91 (m, 2H), 2.41–2.50 (m, 1H), 2.68–2.77 (m, 3H), 3.78 (s, 3H), 3.83–3.92 (m, 1H), 4.04–4.15 (m, 1H), 5.13 (s, 1H), 6.86 (d, J = 8.4 Hz, 2H), 7.08–7.19 (m, 2H), 7.23–7.31 (m, 4H). 13C-NMR (50 MHz, CDCl3) δ: 25.11, 25.15, 27.87, 55.21, 62.24, 78.96, 113.71, 121.74, 126.45, 126.66, 126.84, 127.30, 130.00, 132.01, 133.59, 135.10, 135.14, 159.46. LRMS- m/z (rel. int.): 292 (M+∙, 92), 264 (16), 263 (31), 233 (16), 135 (100). HRMS [ESI(+)] calcd. for [C20H20O2+H]+ 293.1536, found 293.1543.
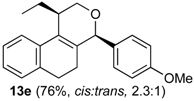
Prins cyclization of 1e and 2b. The reaction was performed following the general procedure, but using 1e (0.10 g, 0.49 mmol), 2b (0.060 mL, 0.49 mmol), CH2Cl2 (5 mL), I2 (0.0063, 0.025 mmol). Compound 13e (cis:trans = 2.3:1, 0.12 g, 0.37 mmol, 76%) was obtained as a colorless viscous oil. This mixture was subjected to another flash column chromatography (1% AcOEt in hexanes). Pure samples were obtained for characterization.
(±)-cis-1-Ethyl-2,4,5,6-tetrahydro-4-(4-methoxyphenyl)-1H-benzo[f]isochromene (cis-13e). IR (film): 3061, 3015, 2959, 2930, 2873, 1606, 1510, 1460, 1249, 1034, 766, 736 cm−1. 1H-NMR (200 MHz, CDCl3) δ: 0.99 (t, J = 7.4 Hz, 3H), 1.79–2.04 (m, 2H), 1.57–1.68 (m, 2H), 2.54–2.56 (m, 1H), 2.69–2.81 (m, 2H), 3.70 (dd, J = 11.7, 2.8 Hz, 1H), 3.88 (dd, J = 11.6, 3.6 Hz, 1H), 5.15 (s, 1H), 6.84–6.90 (m, 2H), 7.13–7.17 (m, 2H), 7.25–7.29 (4H). 13C-NMR (50 MHz, CDCl3) δ: 11.9, 24.3, 26.2, 28.2, 34.9, 55.2, 62.2, 78.1, 113.7, 122.3, 126.3, 126.5, 127.7, 130.4, 131.4, 131.7, 132.7, 136.3, 159.5. LRMS m/z (rel. int.): 320 (M+∙, 20), 264 (42), 263 (35), 233 (18), 139 (11), 135 (100). HRMS [ESI(+)] calcd. for [C22H24O2+Na]+ 343.1674, found 343.1652.
(±)-trans-1-Ethyl-2,4,5,6-tetrahydro-4-(4-methoxyphenyl)-1H-benzo[f]isochromene (trans-13e). IR (film): 2962, 2934, 2876, 1603, 1511, 1452, 1441, 1255, 1171, 1110, 831, 767 cm−1. 1H-NMR (200 MHz, CDCl3) δ: 1.08 (t, J = 7.6 Hz, 3H), 1.76–2.00 (m, 4H), 2.39–2.43 (m, 1H), 2.58–2.74 m, 2H), 3.80 (s, 3H), 3.80–3.88 (m, 1H), 4.10 (dd, J = 11.0, 1.6 Hz, 1H), 5.07 (s, 1H), 6.84–6.92 (m, 2H), 7.11–7.15 (m, 2H), 7.22–7.57 (m, 4H). 13C-NMR (50 MHz, CDCl3) δ: 12.5, 24.5, 24.7, 28.0, 36.0, 55.3, 66.3, 80.3, 113.9, 113.7, 121.9, 126.5, 126.6, 127.7, 129.8, 130.4, 131.3, 131.5, 133.1, 133.4, 133.9, 133.9, 135.8, 159.6. LRMS m/z (rel. int.): 320 (M+∙, 21), 264 (55), 263 (50), 233 (21), 135 (100). HRMS [ESI(+)] calcd. for [C22H24O2+Na]+ 343.1674, found 343.1553.
Prins cyclization of 1e and 2k. The reaction was performed following the general procedure, but using 1e (0.10 g, 0.51 mmol), 2k (0.060 mL, 0.51 mmol), CH2Cl2 (5 mL), I2 (0.0065 g, 0.026 mmol). Compound 15e (0.15 g, 0.41 mmol, 80%) was obtained as a colorless solid.
(±)-4-(3-Bromophenyl)-1-ethyl-2,4,5,6-tetrahydro-1H-benzo[f]isochromene (15e). m.p. 125.2 °C. IR (film): 3102, 3053, 2956, 2926, 1488, 1461, 884, 793, 766, 727, 696 cm−1. 1H-NMR (200 MHz, CDCl3) δ: 1.10 (d, J = 7.4 Hz, 3H), 1.70–2.07 (m, 4H), 2.40–2.44 (m, 1H), 2.61–2.71 (m, 2H), 3.84 (dd, J = 11.1, 2.4 Hz, 1H), 4.12 (dd, J = 11.1, 1.6 Hz, 1H), 5.07 (s, 1H), 7.08–7.22 (m, 3H), 7.24–7.27 (m, 2H), 7.33 (dt, J = 7.8, 1.4 Hz, 1H), 7.45 (ddd, J = 7.6, 2.0, 1.4 Hz, 1H), 7.53 (t, J = 1.8 Hz, 1H). 13C-NMR (50 MHz, CDCl3) δ: 12.5, 24.3, 24.7, 27.9, 36.0, 66.4, 80.2, 122.0, 122.6, 126.5, 126.8, 127.3, 127.7, 130.1, 131.4, 131.7, 131.8, 132.2, 133.5, 135.6, 143.2. LRMS m/z (rel. int.): 370 (M+∙, 18), 368 (M+∙, 18), 314 (46), 312 (49), 233 (37), 215 (24), 185 (48), 183 (44), 157 (25), 155 (28), 129 (100). HRMS [ESI(+)] calcd. for [C21H21BrO+Na] 391.0673, 393.0653, found 391.0664, 393.0646.
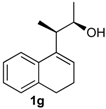
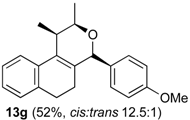
Prins cyclization of 1g and 2b. The reaction was performed following the general procedure, but using 1g (0.10 g, 0.49 mmol), 2b (0.060 mL, 0.49 mmol), CH2Cl2 (5 mL), I2 (0.0063, 0.025 mmol). Compound 13g (cis:trans = 12.5:1, 0.83 g, 0.35 mmol, 52%) was obtained as a colorless viscous oil.
(±)-2,4,5,6-Tetrahydro-4-(4-methoxyphenyl)-1,2-dimethyl-1H-benzo[f]isochromene (13g). IR (film): 3062, 3031, 2971, 2934, 2887, 2834, 1609, 1511, 1488, 1452, 1245, 1033, 832, 767, 732 cm−1. 1H-NMR (500 MHz, CDCl3) δ: cis-13g: 1.28 (d, J = 6.5 Hz, 3H), 1.30 (d, J = 6.5 Hz, 3H), 1.75–1.81 (m, 1H), 1.89–1.96 (m, 1H), 2.59–2.70 (m, 3H), 3.79 (s, 3H), 4.01 (qd, J = 6.5, 2.5 Hz, 1H), 5.13 (s, 1H), 6.85–6.88 (m, 2H), 7.08 (dd, J = 7.2, 1.0 Hz, 2H), 7.13 (td, J = 7.2, 1.0 Hz, 2H), 7.22 (dd, J = 8.0, 1.0 Hz, 1H), 7.24–7.33 (m, 3H).
trans-13g: 1.11 (d, J = 4.5 Hz, 3H), 1.12 (d, J = 3.5 Hz, 3H), 3.76 (s, 3H), 3.96 (q, J = 3.0 Hz, 1H), 5.20 (s, 1H), other signals overlap with the major diastereomer. 13C-NMR (50 MHz, CDCl3) δ: cis-13g: 13.6, 18.5, 24.3, 28.1, 32.8, 55.3, 73.0, 81.4, 113.9, 121.8, 126.4, 126.5, 127.7, 129.8, 133.06, 133.12, 133.3, 133.9, 135.7, 159.5. LRMS m/z (rel. int.): 320 (M+∙, 15), 264 (44), 263 (40), 233 (19), 135 (100). HRMS [ESI(+)] calcd . for [C22H24O2+Na]+ 343.1674, found 343.1667.
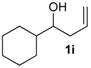
Prins cyclization of 1h and 2b. The reaction was performed following the general procedure, but using 1h (0.10 g, 0.70 mmol), 2b (0.085 mL, 0.70 mmol), CH2Cl2 (5 mL), I2 (0.089, 0.35 mmol). Compound 5h (0.092 g, 0.23 mmol, 33%) was obtained as a white solid.
(±)-(2R,4R,6R)-Tetrahydro-4-iodo-2-(4-methoxyphenyl)-6-phenyl-2H-pyran (5h). m.p. 115–117 °C. IR (film): 3065, 3033, 3004, 2921, 2249, 1514, 1249, 909, 733 cm−1. 1H-NMR (500 MHz, CDCl3) δ: 2.22–2.36 (m, 2H), 2.57–2.66 (m, 2H), 3.79 (s, 3H), 4.49–4.59 (m, 3H), 6.86.12 (t, J = 2.1 Hz, 1H), 6.89 (t, J = 3.0 Hz, 1H), 7.24–7.32 (m, 1H), 7.33–7.41 (m, 6H). 13C-NMR (75 MHz, CDCl3) δ: 21.7, 29.7, 47.0, 47.1, 55.3, 80.6, 80.9, 113.8, 125.7, 127.1, 127.7, 128.4, 133.4, 141.2, 159.1. LRMS m/z (rel. int.): 394 (M+∙, 0.1), 267 (5), 161 (15), 160 (19), 159 (19), 144 (15), 137 (43), 136 (44), 135 (81), 131 (56), 130 (51), 129 (72), 127 (86), 115 (60), 107 (16), 106 (22), 105 (35), 92 (18), 91 (50), 78 (19), 77 (100). HRMS [ESI(+)] calcd. for [C18H19IO2+H]+ 395.0508, found 395.0500.
Prins cyclization of 1i and 2b. The reaction was performed following the general procedure, but using 1i (0.0762 g, 0.495 mmol), 2b (0.0600 mL, 0.495 mmol), CH2Cl2 (5 mL), I2 (0.00626, 0.247 mmol). Compound 5i (0.0822 g, 0.205 mmol, 41%) was obtained as colorless viscous oil.
(±)-(2S,4R,6R)-2-Cyclohexyltetrahydro-4-iodo-6-(4-methoxyphenyl)-2H-pyran (5i). IR (film): 2924, 2851, 1612, 1513, 1248, 1066, 1035, 826, 551 cm−1. 1H-NMR (300 MHz, CDCl3) δ: 1.00–1.15 (m, 3H), 1.15–1.25 (m, 3H), 1.42–1.53 (m, 1H), 1.62–1.80 (m, 5H), 1.84–1.91 (m, 1H), 1.94–2.20 (m, 1H), 2.34–2.57 (m, 1H), 3.22 (ddd, J = 11.1, 6.0, 1.8 Hz, 1H), 3.79 (s, 3H), 4.28 (dd, J = 11.1, 1.8 Hz, 1H), 4.40 (tt, J = 12.3, 4.2 Hz, 1H), 6.84–6.88 (m, 2H), 7.23–7.28 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ: 23.8, 26.1, 26.2, 26.5, 28.6, 28.9, 42.0, 42.8, 47.4, 55.3, 80.0, 83.3, 113.7, 126.9, 133.9, 159.0. LRMS m/z (rel. int.): 400 (M+∙, 0.2), 273 (10), 161 (8), 138 (9), 137 (100). HRMS [ESI(+)] calcd. for [C18H25IO2+H]+ 401.0977, found 401.1062.
Prins cyclization of 1j and 2b. The reaction was performed following the general procedure, but using 1j (0.0426 g, 0.495 mmol), 2b (0.0600 mL, 0.495 mmol), CH2Cl2 (5 mL), I2 (0.00626, 0.247 mmol). Compound 5j (0.0471 g, 0142 mmol, 29%) was obtained as colorless viscous oil.
(±)-(2R,4R,6R)-Tetrahydro-4-iodo-2-(4-methoxyphenyl)-6-methyl-2H-pyran (5j). IR (film): 3067, 3036, 2970, 2954, 2835, 1613, 1514, 1250, 1178, 1055, 1036, 827, 774, 548 cm−1. 1H-NMR (300 MHz, CDCl3) δ: 1.25 (d, J = 6.0 Hz, 3H), 2.00 (td, J = 11.5, 11.0 Hz, 1H), 2.18 (td, J = 11.5, 11.0 Hz, 1H), 2.38 (dqt, J = 12.5, 2.0 Hz, 1H), 2.49 (td, J = 12.5, 2.0 Hz, 1H), 3.59–3.66 (m, 1H), 4.31 (dd, J = 11.0, 2.0 Hz, 1H), 4.40 (tt, J = 12.5, 4.5 Hz, 1H), 6.85–6.86 (m, 1H), 6.87–6.88 (m, 1H), 7.24–7.25 (m, 1H), 7.26–7.27 (m, 1H). 13C-NMR (50 MHz, CDCl3) δ: 21.5, 22.3, 46.6, 46.8, 55.3, 75.2, 80.4, 113.8, 125.3, 127.2, 133.5. LRMS m/z (rel. int.): 332 (M+∙, 0.4), 206 (6), 205 (46), 161 (8), 146 (3), 137 (100). HRMS [ESI(+)] calcd. for [C13H17IO2+Na]+ 355.0171, found 355.0164.
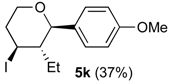
Prins cyclization of 1k and 2b. The reaction was performed following the general procedure, but using 1k (0.0496 g, 0.495 mmol), 2h (0.0600 mL, 0.495 mmol), CH2Cl2 (5 mL), I2 (0.00626, 0.247 mmol). Compound 5k (0.0642 g, 0185 mmol, 37%) was obtained as colorless viscous oil.
(±)-(2R,3S,4S)-3-Ethyltetrahydro-4-iodo-2-(4-methoxyphenyl)-2H-pyran (5k). IR (film): 2958, 2933, 2872, 2848, 1516, 1444, 1257, 1088, 1029, 826, 814, 545 cm−1. 1H-NMR (300 MHz, CDCl3) δ: 0.66 (t, J = 7.8 Hz, 3H), 1.36–1.30 (m, 1H), 1.48–1.62 (m, 2H), 2.02–2.12 (m, 1H), 2.45–2.53 (m, 1H), 2.57–2.71 (m, 1H), 3.51 (td, J = 11.8, 2.1 Hz, 1H), 3.80 (s, 3H), 3.81–3.85 (m, 1H), 4.12 (d, J = 9.9 Hz, 1H), 4.35 (td, J = 11.7, 4.5), 6.85–6.90 (m, 2H), 7.23–7.28 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ: 8.3, 25.0, 33.2, 41.5, 51.5, 55.3, 69.7, 83.2, 113.8, 128.3, 132.7, 159.5. LRMS m/z (rel. int.): 346 (M+, 0.05), 220 (3), 219 (20), 137 (52), 135 (15), 83 (38), 77 (11), 67 (85), 55 (100). HRMS [ESI(+)] calcd. for [C14H19IO2+Na]+ 369.0327, found 369. 0333.
Prins cyclization of 1l and 2b with 0.5 equiv of Iodine. The reaction was performed following the general procedure, but using
1l (0.0496 g, 0.495 mmol),
2b (0.0600 mL, 0.495 mmol), CH
2Cl
2 (5 mL), I
2 (0.0626, 0.247 mmol). Compound
5l [
30] (0.0689 g, 0.199 mmol, 40%) was obtained as colorless viscous oil.
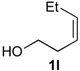
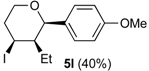
Prins cyclization of 1l and 2b with 1 equiv of Iodine. The reaction was performed following the general procedure, but using
1l (0.0496 g, 0.495 mmol),
2b (0.0600 mL, 0.496 mmol), CH
2Cl
2 (5 mL), I
2 (0.1252, 0.495 mmol). Compound
5l [
30] (0.139 g, 0.402 mmol, 81%) was obtained as colorless viscous oil.
Prins cyclization of 1l and 2b with 1 equiv of Iodine and 2 equiv of 2b. The reaction was performed following the general procedure, but using
1l (0.0992 g, 0.990 mmol),
2b (0.0600 mL, 0.495 mmol), CH
2Cl
2 (5 mL), I
2 (0.125, 0.495 mmol). Compound
5l[
30] (0.144 g, 0.417 mmol, 84%) was obtained as colorless viscous oil.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}