Holographic Quantitative Structure-Activity Relationships of Tryptamine Derivatives at NMDA, 5HT1A and 5HT2A Receptors

Abstract
:
1. Introduction
2. Results and Discussion
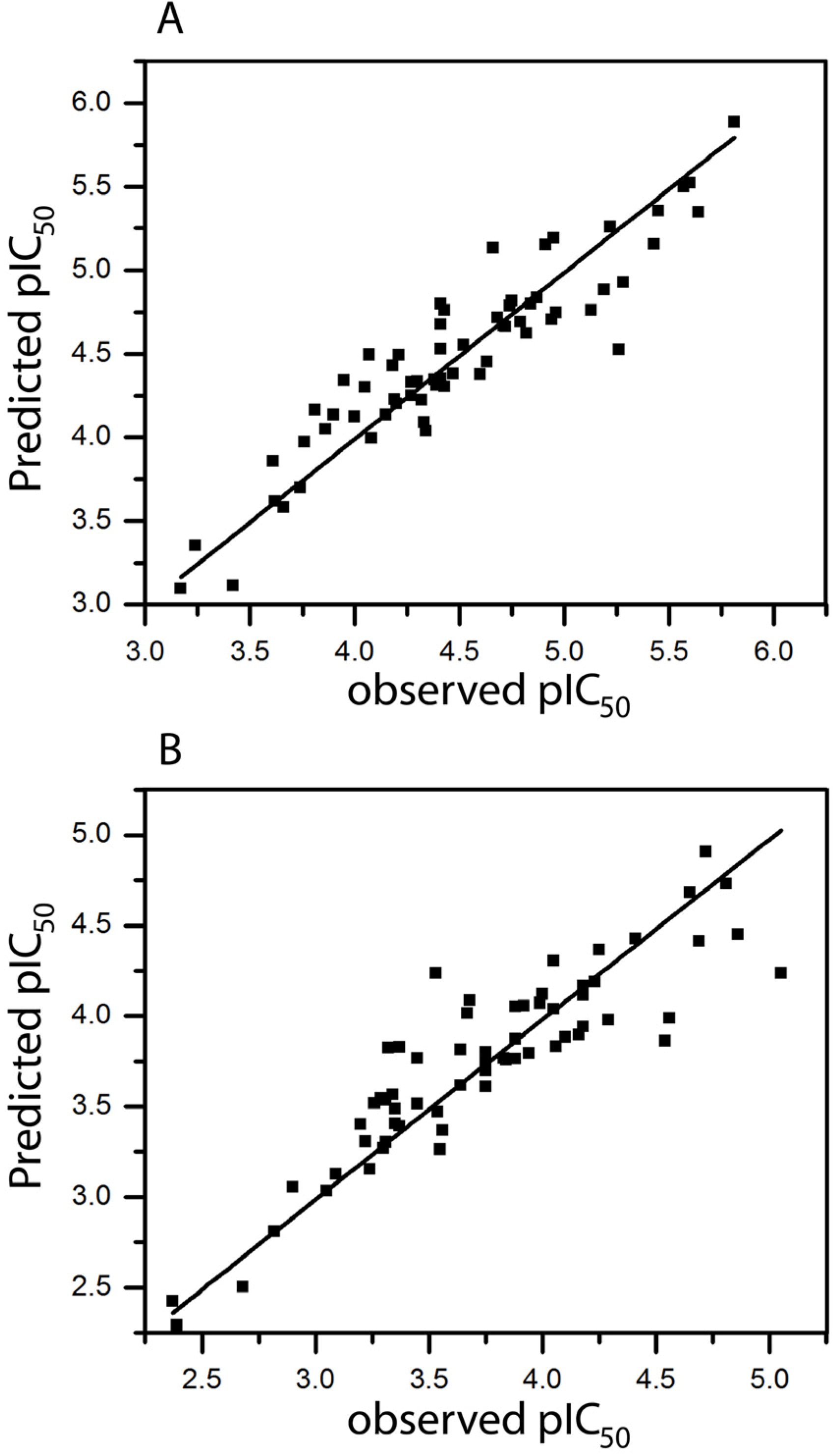
2.1. HQSAR of Ts as Inhibitors of [3H]MK-801 Binding

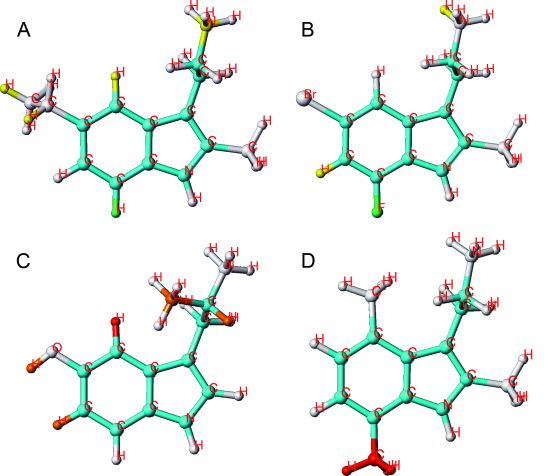{kind=link}
{kind=link}
{kind=link}
{kind=link}
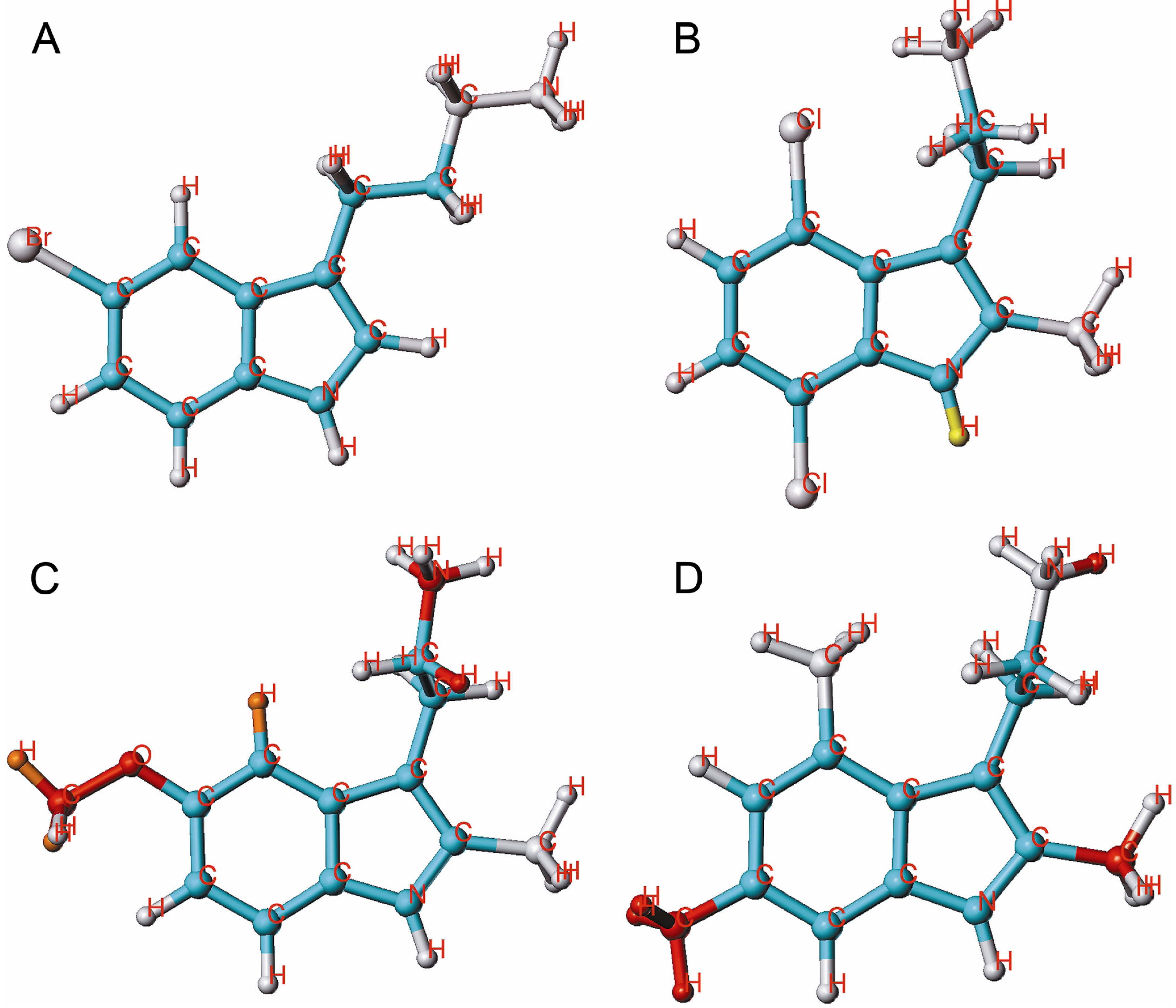{kind=link}
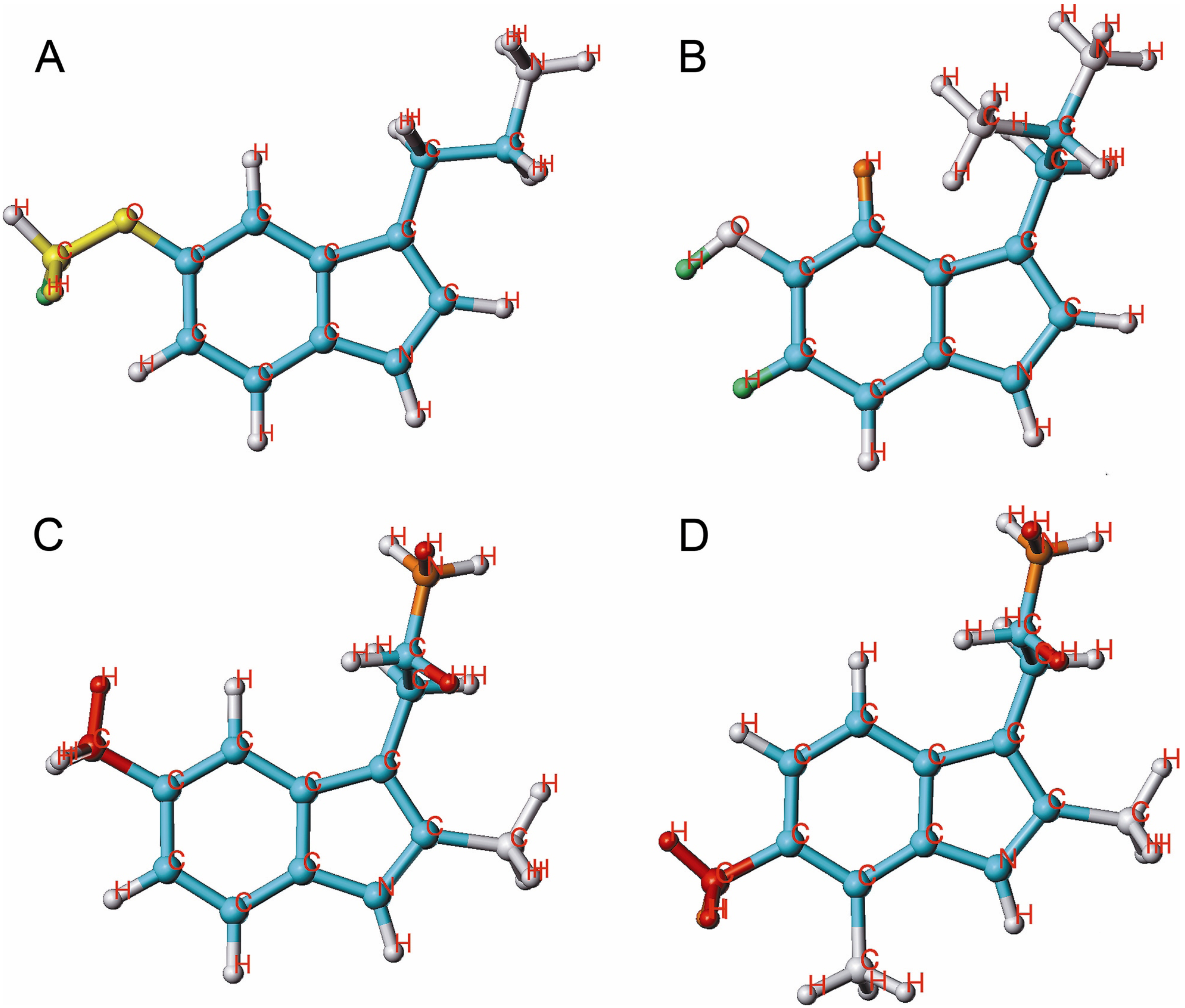{kind=link}
| Model | Field | n | q2 | r2 | SE | Ensemble | BL |
|---|---|---|---|---|---|---|---|
| 6 | DA | 3 | 0.304 | 0.487 | 0.437 | 0.257 | 307 |
| 15 | B/DA | 3 | 0.451 | 0.591 | 0.390 | 0.365 | 59 |
| 28 | A/C/DA | 6 | 0.475 | 0.856 | 0.238 | 0.375 | 353 |
| 50 | A/C/CH/DA | 5 | 0.460 | 0.820 | 0.263 | 0.339 | 353 |
| 58 | A/B/C/H/DA | 6 | 0.539 | 0.832 | 0.256 | 0.398 | 61 |
| 63 | A/B/C/H/CH/DA | 6 | 0.632 | 0.855 | 0.238 | 0.399 | 61 |
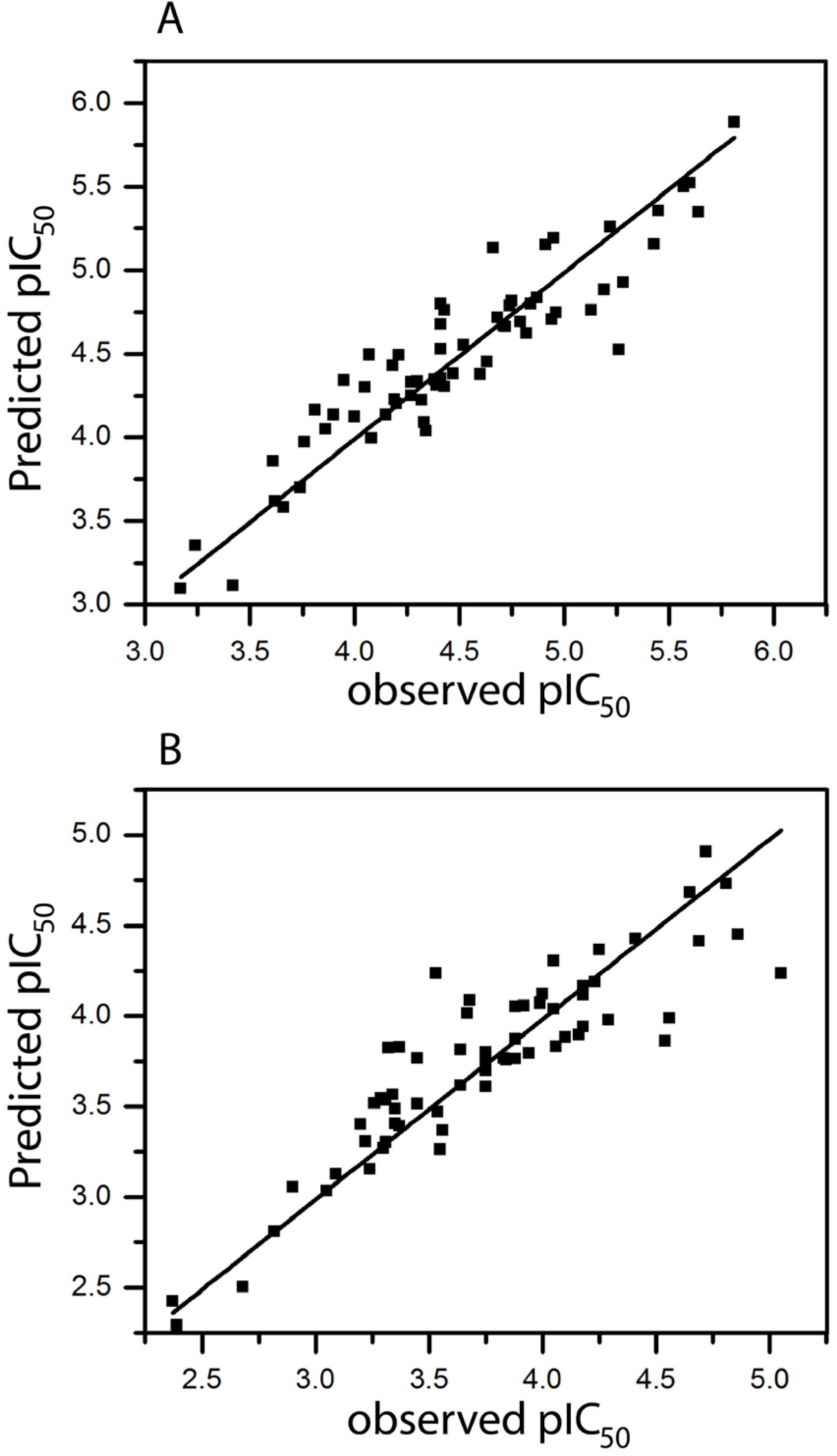
2.2. HQSAR of [3H]MK-801 Binding in the Presence of Spermine
| Model | Field | n | q2 | r2 | SE | Ensemble | BL |
|---|---|---|---|---|---|---|---|
| 3 | C | 6 | 0.292 | 0.640 | 0.366 | 0.207 | 61 |
| 9 | A/H | 3 | 0.356 | 0.535 | 0.405 | 0.292 | 353 |
| 28 | A/C/DA | 6 | 0.453 | 0.831 | 0.250 | 0.326 | 353 |
| 51 | A/H/CH/DA | 3 | 0.425 | 0.587 | 0.382 | 0.362 | 401 |
| 58 | A/B/C/H/DA | 6 | 0.525 | 0.791 | 0.278 | 0.357 | 61 |
| 63 | A/B/C/H/CH/DA | 6 | 0.600 | 0.803 | 0.270 | 0.366 | 61 |
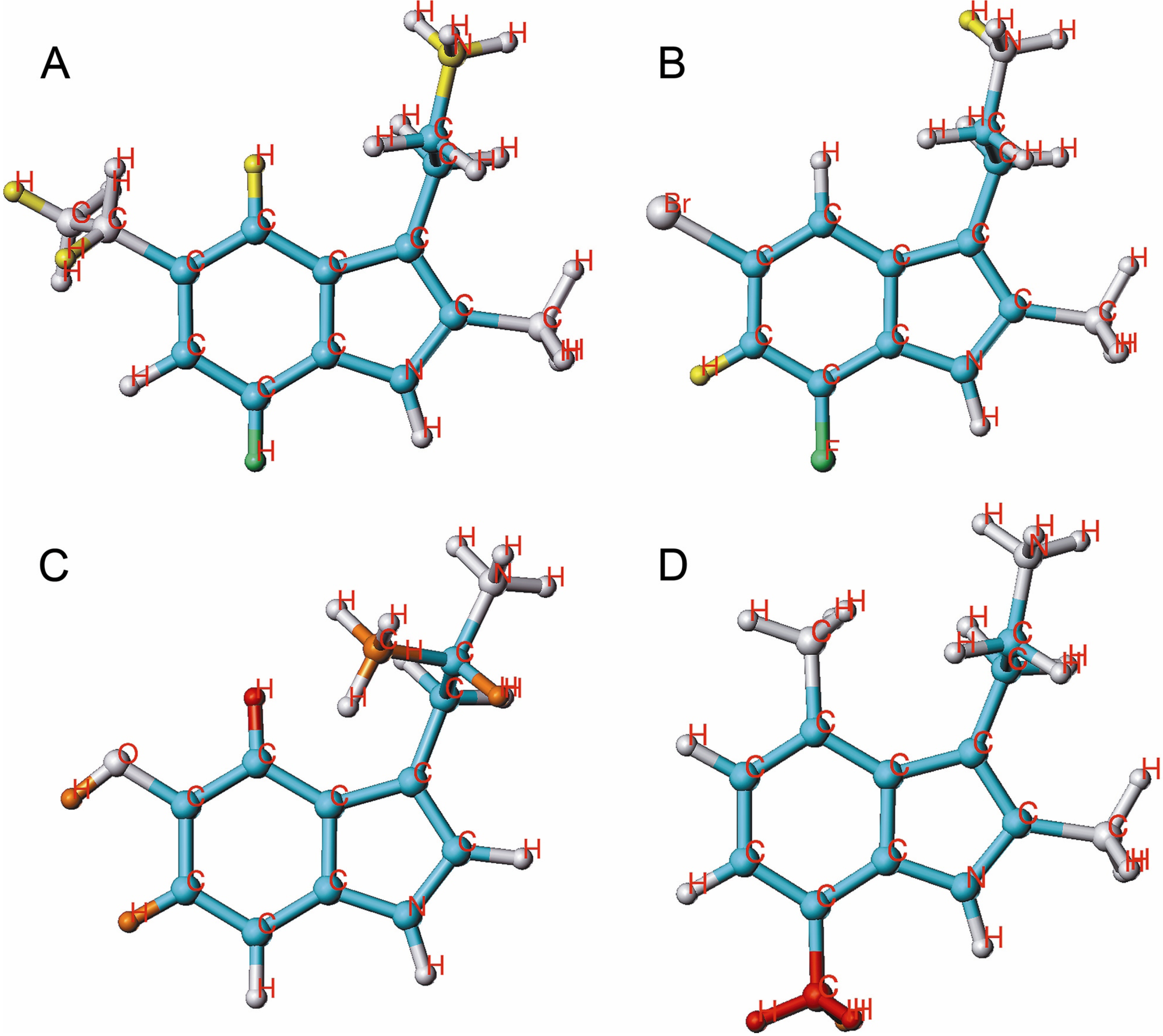
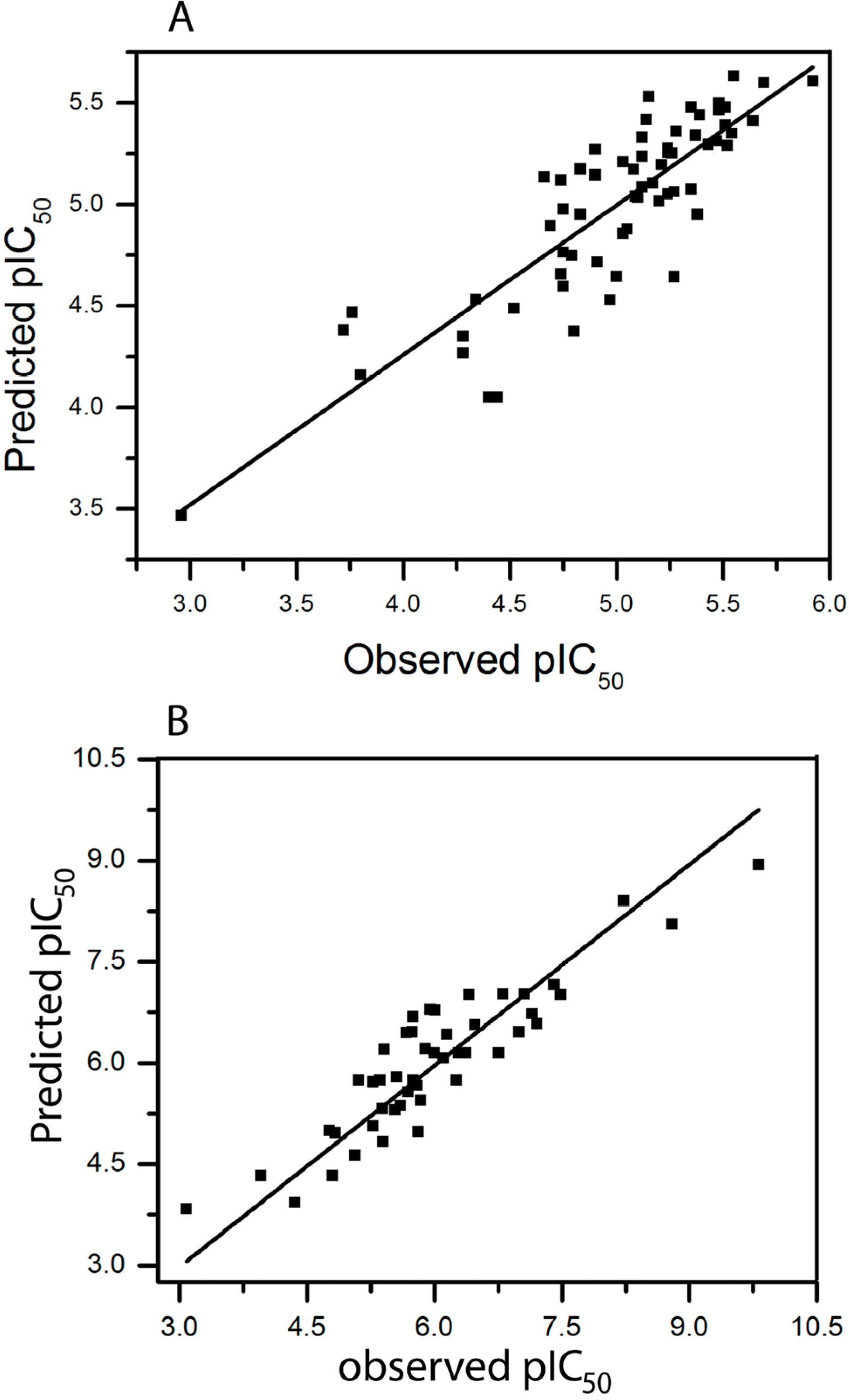
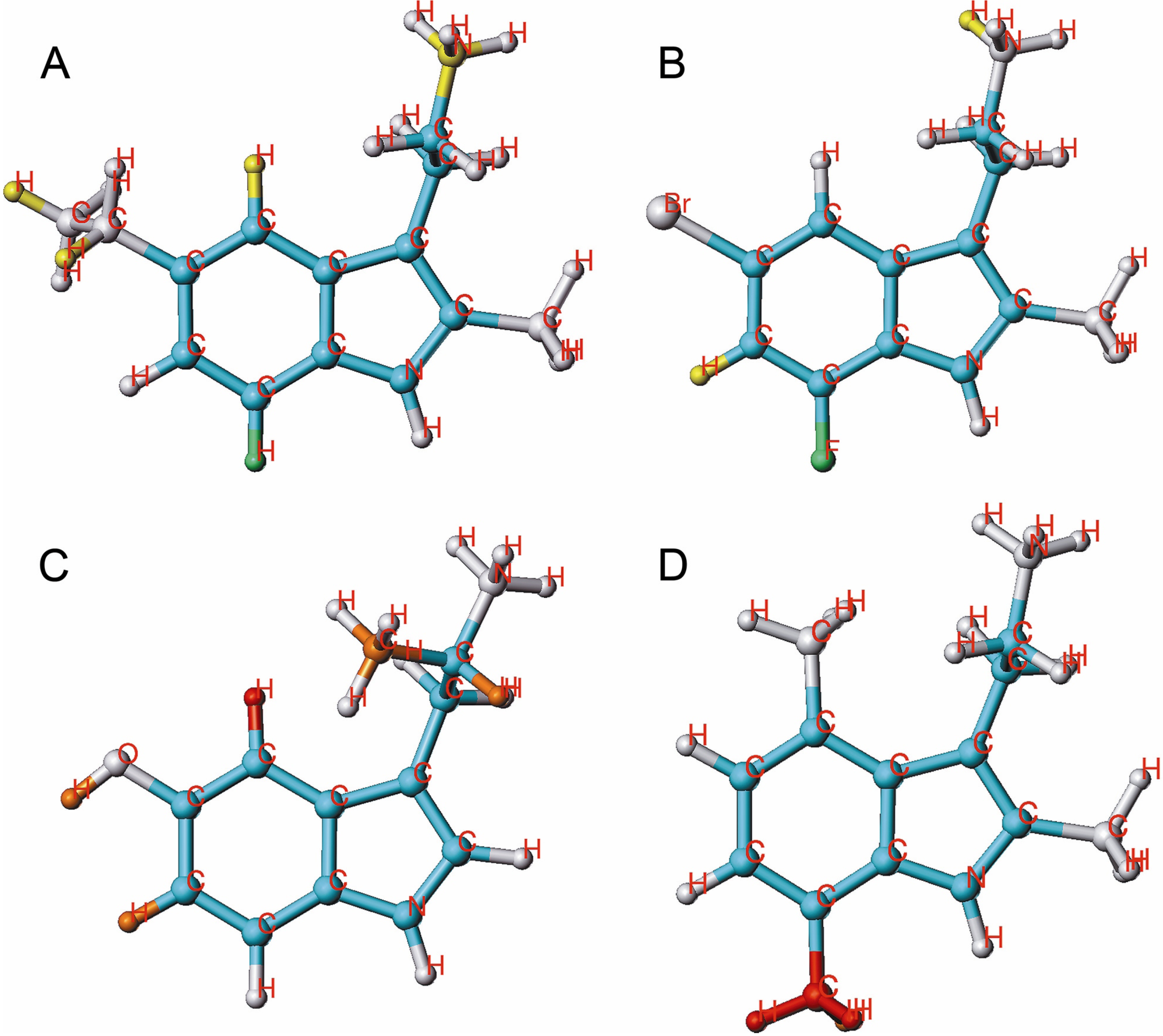
2.3. HQSAR of Ts as Inhibitors of [3H]ketanserin Binding
| Model | Field | n | q2 | r2 | SE | Ensemble | BL |
|---|---|---|---|---|---|---|---|
| 3 | C | 6 | 0.423 | 0.779 | 0.258 | 0.355 | 199 |
| 16 | C/H | 6 | 0.423 | 0.779 | 0.258 | 0.355 | 199 |
| 17 | C/CH | 6 | 0.423 | 0.779 | 0.258 | 0.355 | 199 |
| 30 | A/H/DA | 5 | 0.456 | 0.768 | 0.262 | 0.313 | 97 |
| 56 | C/H/CH/DA | 6 | 0.455 | 0.842 | 0.219 | 0.334 | 61 |
| 58 | A/B/C/H/DA | 3 | 0.489 | 0.738 | 0.274 | 0.341 | 199 |
| 63 | A/B/C/H/CH/DA | 5 | 0.465 | 0.794 | 0.247 | 0.328 | 199 |
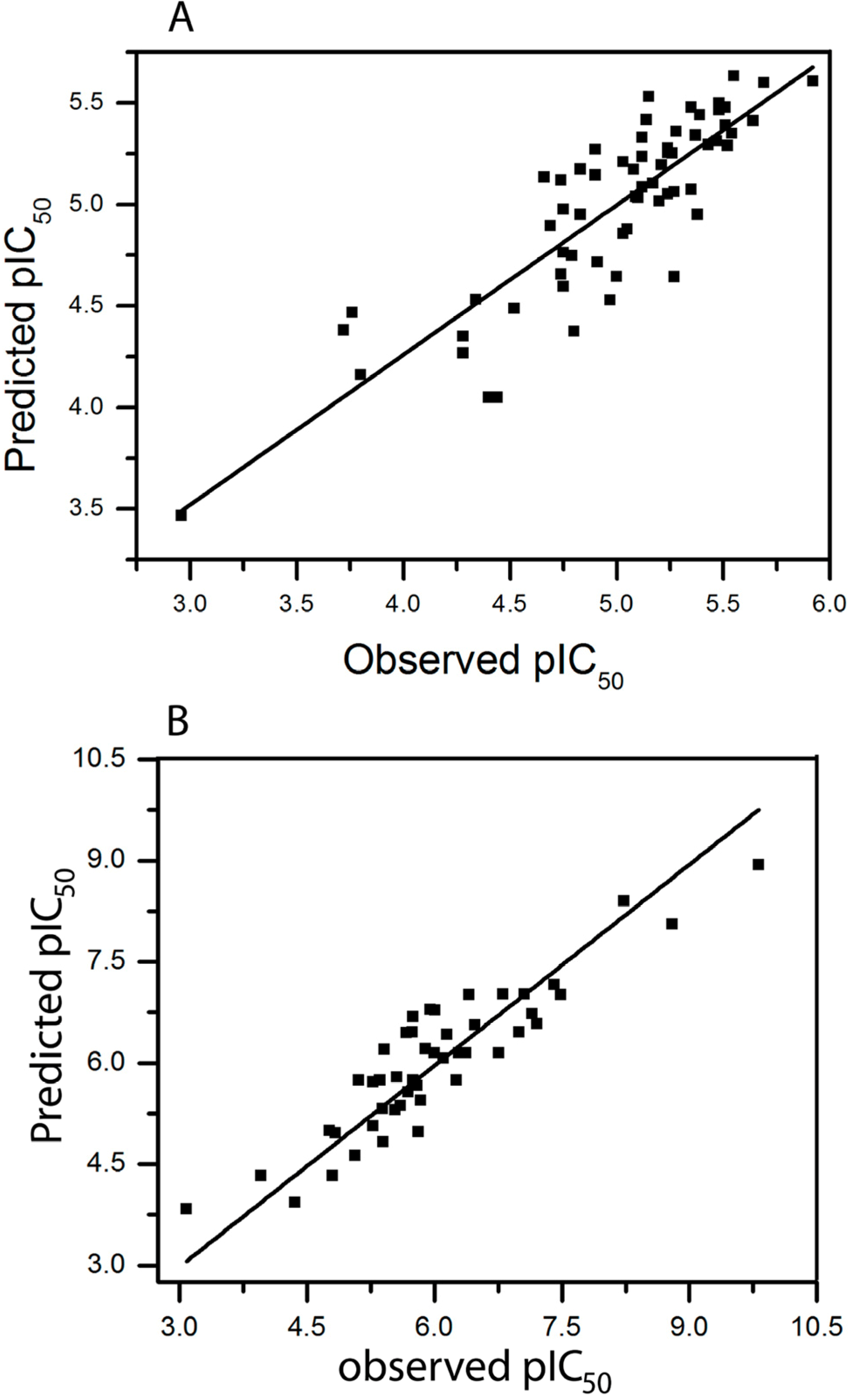

2.4. HQSAR of Ts as Inhibitors of [3H]8-OH-DPAT Binding
| Model | Field | n | q2 | r2 | SE | ensemble | BL |
|---|---|---|---|---|---|---|---|
| 3 | C | 4 | 0.575 | 0.753 | 0.610 | 0.521 | 83 |
| 15 | B/DA | 4 | 0.626 | 0.822 | 0.519 | 0.532 | 257 |
| 36 | B/H/DA | 4 | 0.626 | 0.822 | 0.519 | 0.532 | 257 |
| 55 | B/H/CH/DA | 4 | 0.626 | 0.821 | 0.519 | 0.533 | 257 |
| 61 | A/C/H/CH/DA | 5 | 0.574 | 0.830 | 0.512 | 0.436 | 71 |
| 63 | A/B/C/H/CH/DA | 4 | 0.530 | 0.792 | 0.560 | 0.445 | 97 |
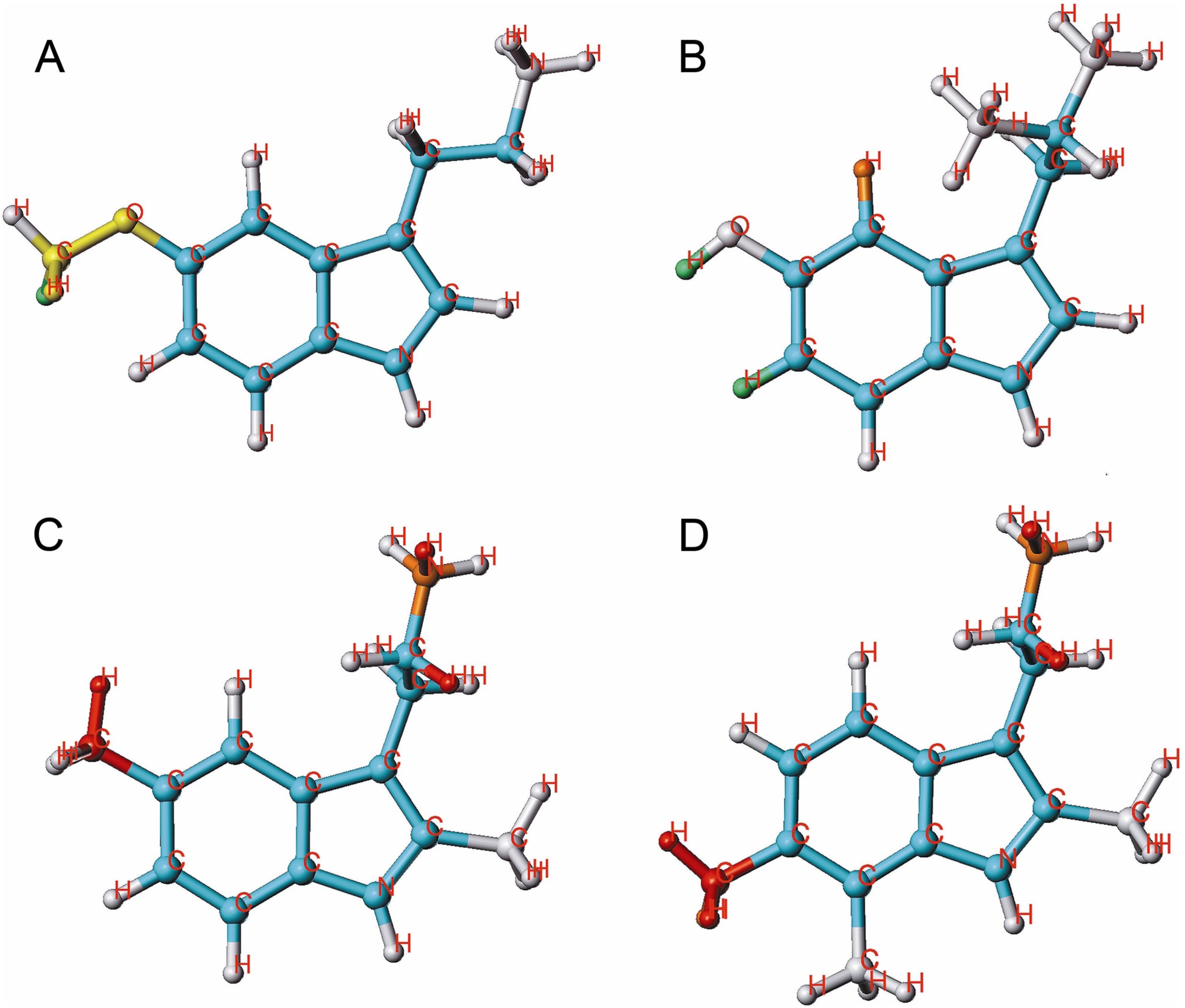
3. Experimental
3.1. Data Set
3.2. HQSAR Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
Appendix
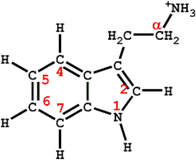
| No | Molecular name | pIC50 | |||
|---|---|---|---|---|---|
| [3H]MK-801 | [3H]MK-801 in the presence of 30 µM spermine | [3H]ketanserin | [3H]8-OH-DPAT | ||
| 1 | tryptamine | 4.07 | 3.32 | 4.79 | 6.48 |
| 2 | homo-T | 4.30 | 3.54 | 5.12 | 6.15 |
| 3 | 1-Me-T | 4.27 | 3.37 | 5.10 | 6.11 |
| 4 | 2-Me-T | 4.96 | 4.56 | 4.52 | 5.81 |
| 5 | 5-Me-T | 5.19 | 5.05 | 5.00 | 7.21 |
| 6 | 5-Bu-T | 4.82 | 4.18 | 5.12 | 7.15 |
| 7 | 6-Me-T | 3.95 | 3.37 | 4.34 | 5.41 |
| 8 | 7-Me-T | 4.34 | 3.56 | 4.75 | 5.75 |
| 9 | α-Me-T | 3.62 | 2.82 | 4.90 | 5.80 |
| 10 | N-Me-T | 3.42 | 2.68 | 4.91 | 7.00 |
| 11 | 5-F-T | 4.41 | 3.67 | 5.21 | 6.81 |
| 12 | 5-F-hT | 4.18 | 3.34 | 5.51 | 6.41 |
| 13 | 5-Cl-T | 4.75 | 3.92 | 5.17 | 7.06 |
| 14 | 5-Br-hT | 5.13 | 4.10 | 5.64 | 7.49 |
| 15 | 5-OH-T | 3.76 | 3.22 | 5.27 | 8.80 |
| 16 | 5-MeO-T | 4.72 | 3.75 | 4.97 | 8.23 |
| 17 | 5-NH2CO-T | 3.66 | 3.24 | 3.80 | 9.82 |
| 18 | 1,2-Me2-T | 4.79 | 3.84 | 4.83 | 5.84 |
| 19 | 2,5-Me2-T | 5.43 | 4.69 | 4.80 | 4.77 |
| 20 | 2-Me-5-Et-T | 5.81 | 4.72 | 5.03 | 5.28 |
| 21 | 2-Me-5-Bu-T | 4.91 | 4.05 | 5.08 | - |
| 22 | 2-Me-5-iBu-T | 5.22 | 4.18 | 5.20 | 5.60 |
| 23 | 2-Me-5CN-T | 4.66 | 3.53 | 3.76 | 5.40 |
| 24 | 2-Me-5-F-T | 4.95 | 4.25 | 4.66 | 5.36 |
| 25 | 2-Me-5-Cl-T | 5.45 | 4.41 | 5.24 | 5.11 |
| 26 | 2-Me-5-Br-T | 5.60 | 4.65 | 5.35 | 5.75 |
| 27 | 2-Me-5-I-T | 5.64 | 4.86 | 5.27 | 6.26 |
| 28 | 2-Me-5-OH-T | 4.27 | 3.35 | 3.72 | 5.67 |
| 29 | 2-Me-5-MeO-T | 5.28 | 4.29 | 4.28 | 5.95 |
| 30 | 2,7-Me2-T | 3.81 | 3.20 | 4.75 | 5.28 |
| 31 | 2-Me-7-CF3-T | 4.05 | 3.26 | 5.48 | - |
| 32 | 2-Me-7-Et-T | 4.00 | 3.35 | 4.74 | - |
| 33 | 2-Me-7-iPrp-T | 4.39 | 3.64 | 4.69 | 5.39 |
| 34 | 2-Me-7-F-T | 4.74 | 4.05 | 4.83 | - |
| 35 | 2-Me-7-Cl-T | 4.38 | 3.75 | 5.54 | 5.69 |
| 36 | 2-Me-7-Br-T | 4.21 | 3.45 | 5.51 | - |
| 37 | 2-Me-7-I-T | 4.43 | 3.75 | 5.52 | - |
| 38 | 5-Me-7-Br-T | 4.68 | 4.00 | 5.37 | 6.01 |
| 39 | α -Me-5-F-T | 4.19 | 3.31 | 5.15 | 5.90 |
| 40 | α -Me-5-OH-T | 3.17 | 2.39 | 5.09 | 7.41 |
| 41 | 4,6-Cl2-T | 4.32 | 3.83 | 5.26 | 5.74 |
| 42 | 2,4,6-Me3-T | 3.61 | 3.31 | 4.28 | - |
| 43 | 2,4,7-Me3-T | 3.74 | 3.09 | 5.38 | 5.54 |
| 44 | 2,5,7-Me3-T | 4.20 | 3.29 | 4.74 | 4.84 |
| 45 | 2,6,7-Me3-T | 4.15 | 3.30 | 4.75 | 5.07 |
| 46 | 2,5-Me2–7-Br-T | 4.41 | 3.68 | 5.47 | - |
| 47 | 2,7-Me2–4-F-T | 5.26 | 4.54 | 5.48 | 6.00 |
| 48 | 2,7-Me2–4-Cl-T | 3.90 | 3.45 | 5.69 | 6.29 |
| 49 | 2,7-Me2–5-Cl-T | 4.52 | 3.75 | 5.12 | - |
| 50 | 2,7-Me2–6-Cl-T | 4.33 | 3.55 | 4.90 | - |
| 51 | 2-Me-5-Br-7-Et-T | 4.43 | 3.88 | 5.03 | - |
| 52 | 2-Me-5,7-F2-T | 4.84 | 3.99 | 5.24 | - |
| 53 | 2-Me-4,7-Cl2-T | 4.41 | 3.88 | 5.92 | 6.37 |
| 54 | 2-Me-5,7-Cl2-T | 4.63 | 3.94 | 5.28 | - |
| 55 | 2-Me-6,7-Cl2-T | 4.60 | 3.88 | 5.43 | - |
| 56 | 2-Me-4,7-Br2-T | 4.47 | 4.06 | 5.55 | 6.76 |
| 57 | 2-Me-5-F-7-Br-T | 4.41 | 3.64 | 5.35 | - |
| 58 | 2-Me-5-Br-7-F-T | 5.57 | 4.81 | 5.24 | 5.56 |
| 59 | 2-Me-5-Cl-7-Br-T | 4.94 | 4.16 | 5.14 | - |
| 60 | 2-Me-5,7-Br2-T | 4.87 | 4.18 | 5.39 | - |
| 61 | L-Trp-Me | 3.86 | 2.90 | 4.40 | 4.80 |
| 62 | D-Trp-Me | 3.24 | 2.37 | 4.44 | 3.96 |
| 63 | L-Trp-NH2 | 4.08 | 3.05 | 2.96 | 3.08 |
| 64 | Trp-Oct | 4.71 | 4.23 | 5.05 | 4.36 |
References
- Madden, D.R. The structure and function of glutamate receptor ion channels. Nat. Rev. Neurosci. 2002, 3, 91–101. [Google Scholar] [CrossRef]
- Mayer, M.L. Glutamate receptors at atomic resolution. Nature 2006, 440, 456–462. [Google Scholar] [CrossRef]
- Trumpp-Kallmeyer, S.; Hoflack, J.; Bruinvels, A.; Hibert, M. Modeling of G-protein-coupled receptors: application to dopamine, adrenaline, serotonin, acetylcholine, and mammalian opsin receptors. J. Med. Chem. 1992, 35, 3448–3462. [Google Scholar] [CrossRef]
- Shapiro, D.A.; Kristiansen, K.; Kroeze, W.K.; Roth, B.L. Differential modes of agonist binding to 5-hydroxytryptamine2A serotonin receptors revealed by mutation and molecular modeling of conserved residues in transmembrane region 5. Mol. Pharmacol. 2000, 58, 877–886. [Google Scholar]
- Ebersole, B.J.; Visiers, I.; Weinstein, H.; Sealfon, S.C. Molecular basis of partial agonism: Orientation of indoleamine ligands in the binding pocket of the human serotonin 5-HT2A receptor determines relative efficacy. Mol. Pharmacol. 2003, 63, 36–43. [Google Scholar] [CrossRef]
- Kanagarajadurai, K.; Malini, M.; Bhattacharya, A.; Panicker, M.M.; Sowdhamini, R. Molecular modeling and docking studies of human 5-hydroxytryptamine 2A (5-HT2A) receptor for the identification of hotspots for ligand binding. Mol. BioSyst. 2009, 5, 1877–1888. [Google Scholar] [CrossRef]
- McRobb, F.M.; Capuano, B.; Crosby, I.T.; Chalmers, D.K.; Yuriev, E. Homology modeling and docking evaluation of aminergic G protein-coupled receptors. J. Chem. Inf. Model. 2010, 50, 626–637. [Google Scholar] [CrossRef]
- Dabrowska, J.; Brylinski, M. Stereoselectivity of 8-OH-DPAT toward the serotonin 5-HT1A receptor: biochemical and molecular modeling study. Biochem. Pharmacol. 2006, 72, 498–511. [Google Scholar] [CrossRef]
- Nowak, M.; Kołaczkowski, M.; Pawłowski, M.; Bojarski, A.J. Homology modeling of the serotonin 5-HT1A receptor using automated docking of bioactive compounds with defined geometry. J. Med. Chem. 2006, 49, 205–214. [Google Scholar] [CrossRef]
- Becker, O.M.; Dhanoa, D.S.; Marantz, Y.; Chen, D.; Shacham, S.; Cheruku, S.; Heifetz, A.; Mohanty, P.; Fichman, M.; Sharadendu, A.; et al. An integrated in silico 3D model-driven discovery of a novel, potent, and selective amidosulfonamide 5-HT1A agonist (PRX-00023) for the treatment of anxiety and depression. J. Med. Chem. 2006, 49, 3116–3135. [Google Scholar] [CrossRef]
- Wong, E.H.F.; Kemp, J.A.; Priestley, T.; Knight, A.R.; Woodruff, G.N.; Iversen, L.L. The antoconvulsant MK-801 is a potent N-methyl-D-aspartate antagonist. Proc. Natl. Acad. Sci. USA 1986, 83, 7104–7108. [Google Scholar] [CrossRef]
- Leysen, J.E.; Awouters, F.; Kennis, L.; Laduron, P.M.; Vandenberk, J.; Janssen, P.A. Receptor binding profile of R 41468, a novel antagonist at 5-HT2 receptors. Life Sci. 1981, 20, 1015–1018. [Google Scholar]
- Hall, M.D.; El Mestikawy, S.; Emerit, M.B.; Pichat, L.; Hamon, M.; Gozlan, H. [3H]8-hydroxy-2-(di-n-propylamino)tetralin binding to pre- and postsynaptic 5-hydroxytryptamine sites in various regions of the rat brain. J. Neurochem. 1985, 44, 1685–1696. [Google Scholar] [CrossRef]
- Tong, W.; Lowis, D.R.; Perkins, R.; Chen, Y.; Welsh, W.J.; Goddette, D.W.; Heritage, T.W.; Sheehan, D.M. Evaluation of quantitative structure-activity relationship methods for large-scale prediction of chemicals binding to the estrogen receptor. J. Chem. Inf. Comput. Sci. 1998, 38, 669–677. [Google Scholar] [CrossRef]
- HQSARTM. In Manual SYBYL-X 1.0; Tripos Inc: St. Louis, MO, USA, 2009.
- Castilho, M.S.; Guido, R.V.C.; Andricopulo, A.D. 2D Quantitative structure-activity relationship studies on a series of cholesteryl ester transfer protein inhibitors. Bioorg. Med. Chem. 2007, 15, 6242–6252. [Google Scholar] [CrossRef]
- Berger, M.L. Tryptamine derivatives as non-competitive N-methyl-D-aspartate receptor blockers: Studies using [(3)H]MK-801 binding in rat hippocampal membranes. Neurosci. Lett. 2000, 296, 29–32. [Google Scholar] [CrossRef]
- Mony, L.; Zhu, S.; Carvalho, S.; Paoletti, P. Molecular basis of positive allosteric modulation of GluN2B NMDA receptors by polyamines. EMBO J. 2011, 30, 3134–3146. [Google Scholar] [CrossRef]
- Berger, M.L.; Palangsuntikul, R.; Rebernik, P.; Wolschann, P.; Berner, H. Screening of 64 Tryptamines at NMDA, 5-HT1A and 5-HT2A receptors: A comparative binding and modeling study. Curr. Med. Chem. 2012, 19, 3044–3057. [Google Scholar] [CrossRef]
- Lowis, D.R. Tripos technical Notes. In HQSAR: a New, Highly Predictive QSAR Technique; Tripos inc: St. Louis, MO, USA, 1997; p. 1. [Google Scholar]
- Flower, D.R. On the properties of bit string-based measures of chemical similarity. J. Chem. Inf. Comput. Sci. 1998, 38, 379–386. [Google Scholar] [CrossRef]
- Salum, L.B.; Andricopulo, A.D. Fragment-based QSAR: Perspectives in drug design. Mol. Divers. 2009, 13, 277–285. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Palangsuntikul, R.; Berner, H.; Berger, M.L.; Wolschann, P. Holographic Quantitative Structure-Activity Relationships of Tryptamine Derivatives at NMDA, 5HT1A and 5HT2A Receptors. Molecules 2013, 18, 8799-8811. https://doi.org/10.3390/molecules18088799
Palangsuntikul R, Berner H, Berger ML, Wolschann P. Holographic Quantitative Structure-Activity Relationships of Tryptamine Derivatives at NMDA, 5HT1A and 5HT2A Receptors. Molecules. 2013; 18(8):8799-8811. https://doi.org/10.3390/molecules18088799
Chicago/Turabian StylePalangsuntikul, Rungtiva, Heinz Berner, Michael L. Berger, and Peter Wolschann. 2013. "Holographic Quantitative Structure-Activity Relationships of Tryptamine Derivatives at NMDA, 5HT1A and 5HT2A Receptors" Molecules 18, no. 8: 8799-8811. https://doi.org/10.3390/molecules18088799
APA StylePalangsuntikul, R., Berner, H., Berger, M. L., & Wolschann, P. (2013). Holographic Quantitative Structure-Activity Relationships of Tryptamine Derivatives at NMDA, 5HT1A and 5HT2A Receptors. Molecules, 18(8), 8799-8811. https://doi.org/10.3390/molecules18088799