Solution NMR Studies on the Orientation of Membrane-Bound Peptides and Proteins by Paramagnetic Probes

Abstract
:
{kind=link}
{kind=link}
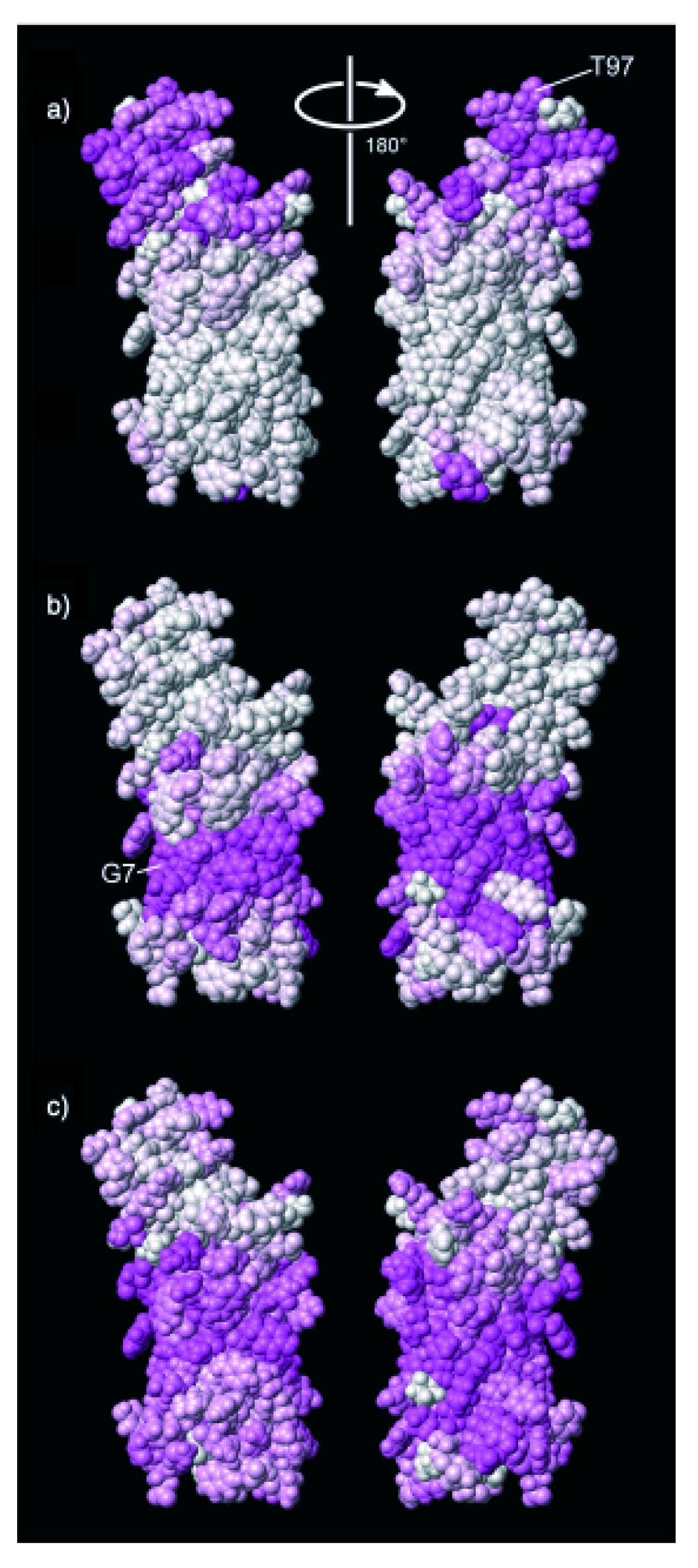
{kind=link}
{kind=link}
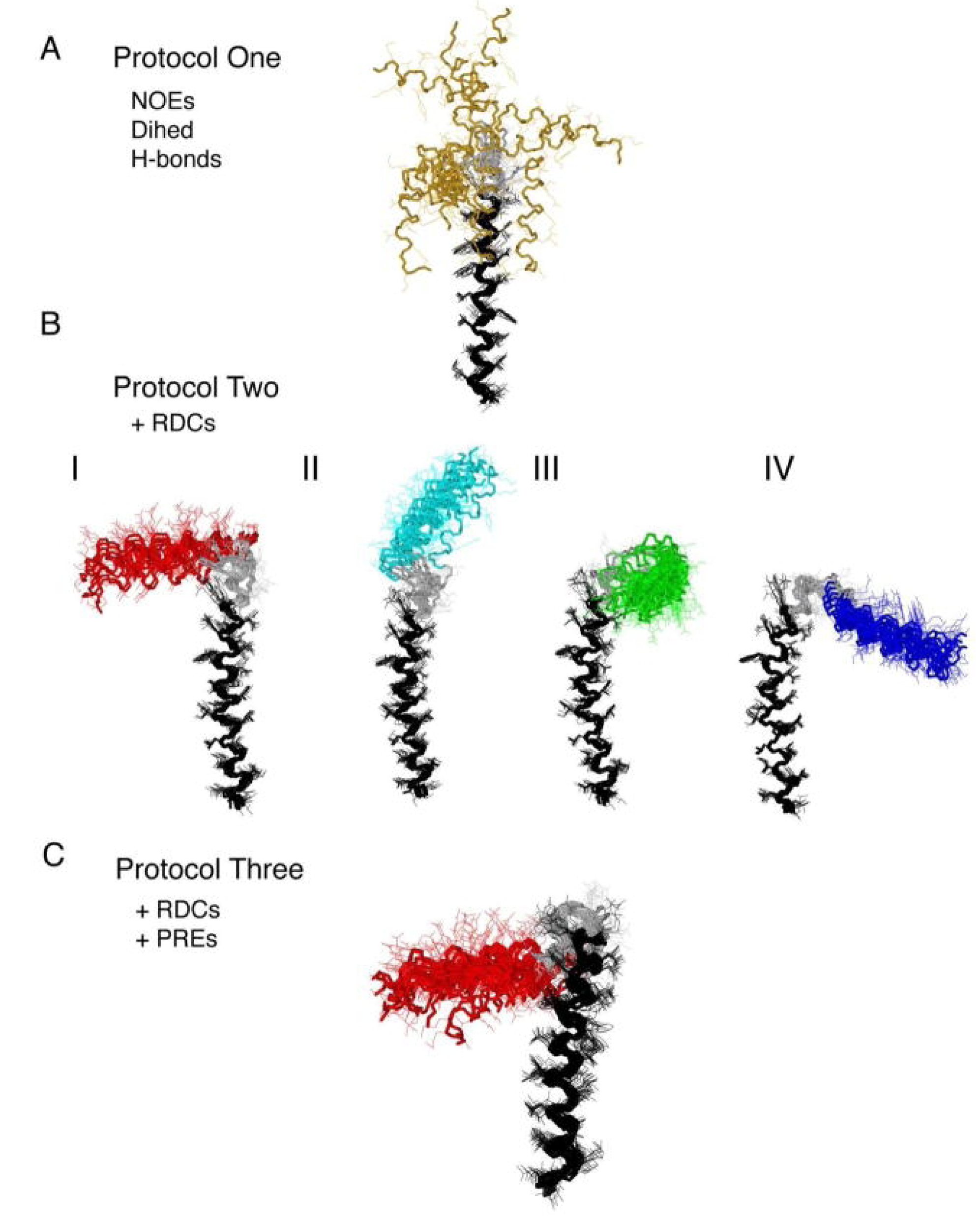
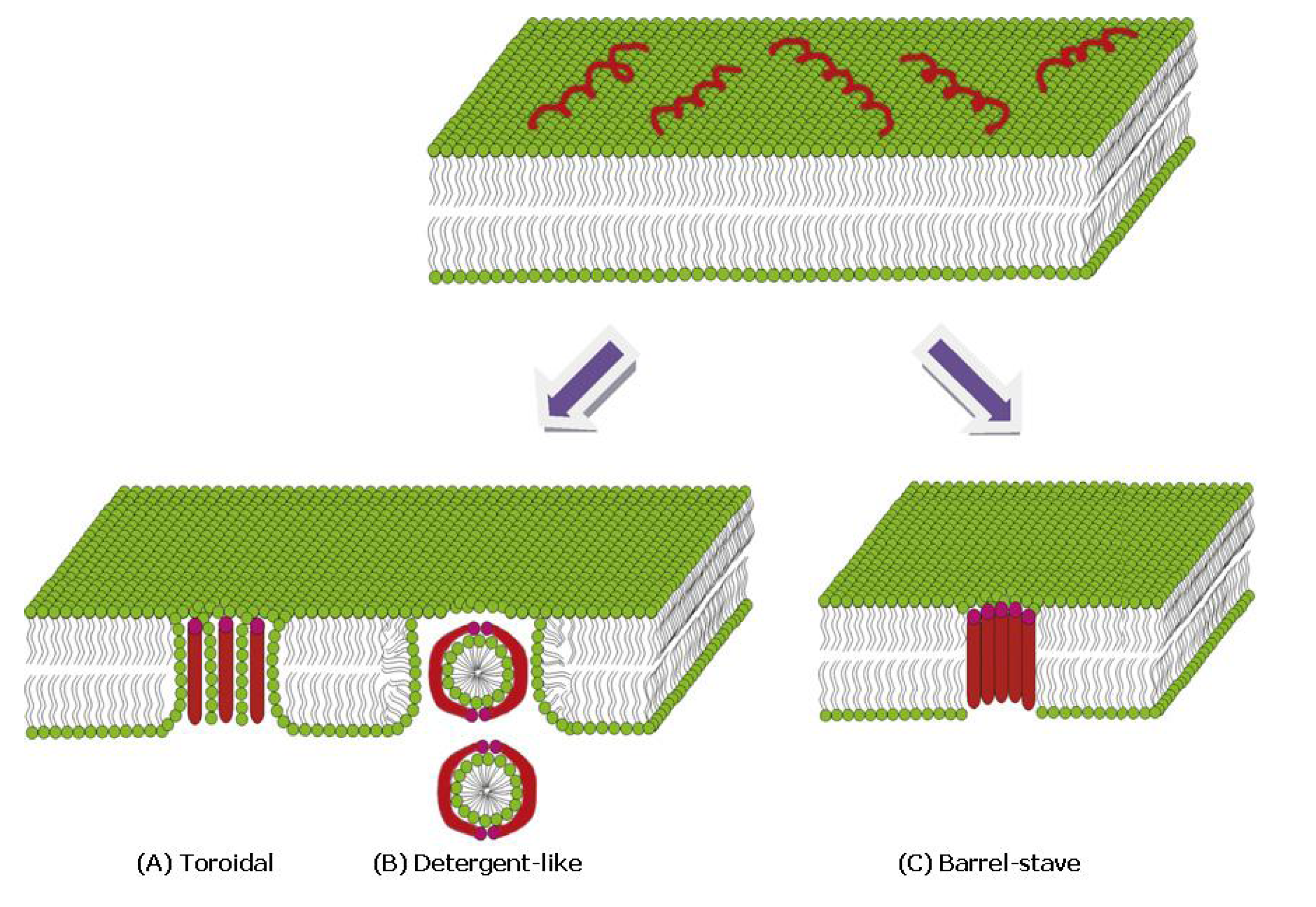{kind=link}
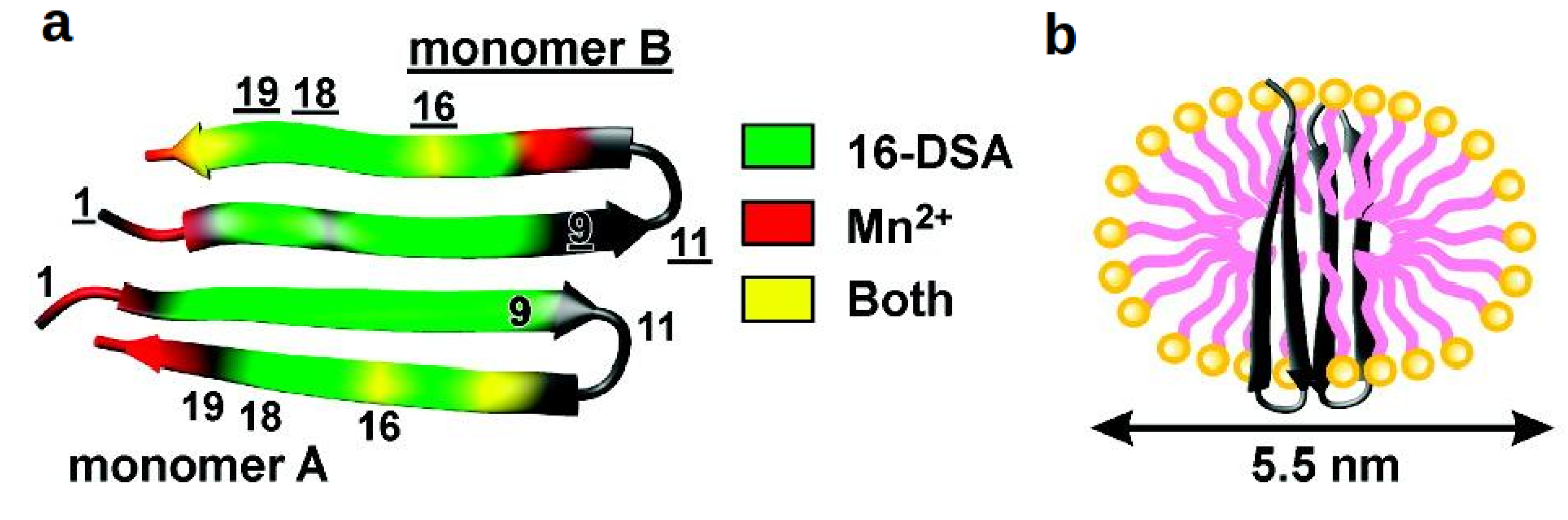{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Paramagnetic Agents
2.1. Paramagnetically Tagged Lipids
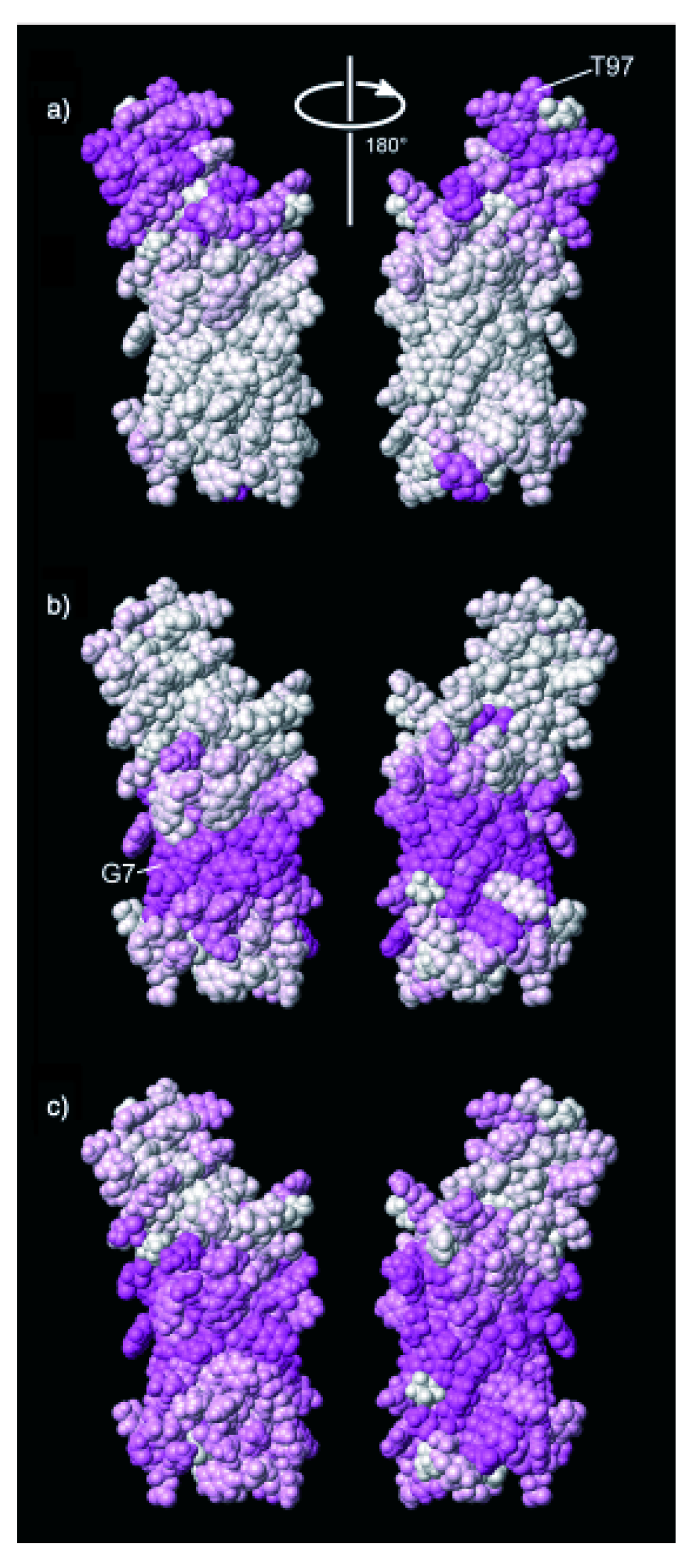
2.2. A Paramagnetic Insertion Gradient by Dissolved Oxygen
2.3. Inert Gadolinium-Based Solvent PREs and Paramagnetic Relaxation Waves
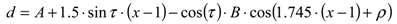
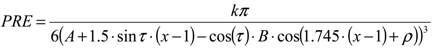
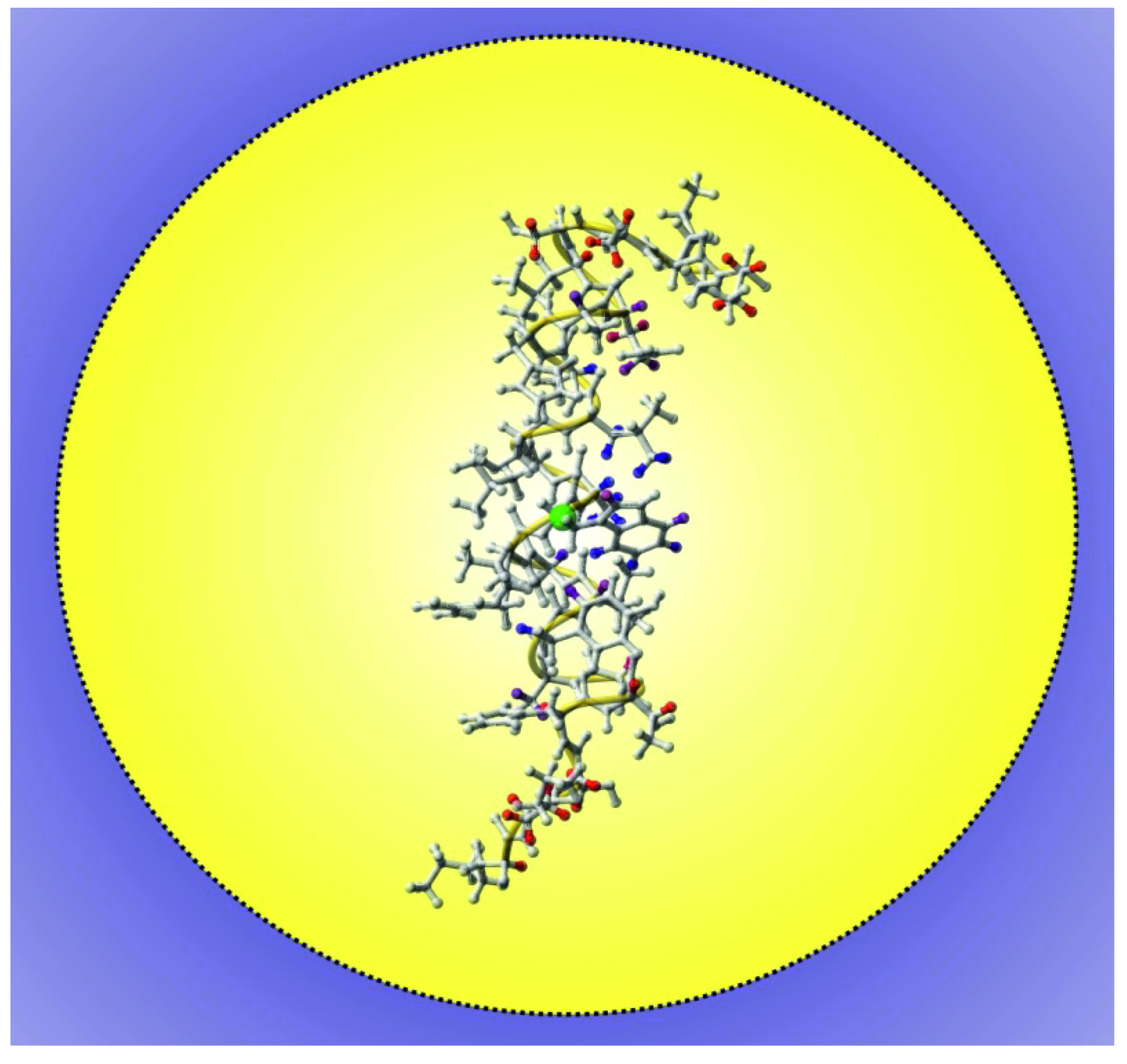
3. Using PREs for the Structure Calculation of Membrane-Bound Peptides and Proteins
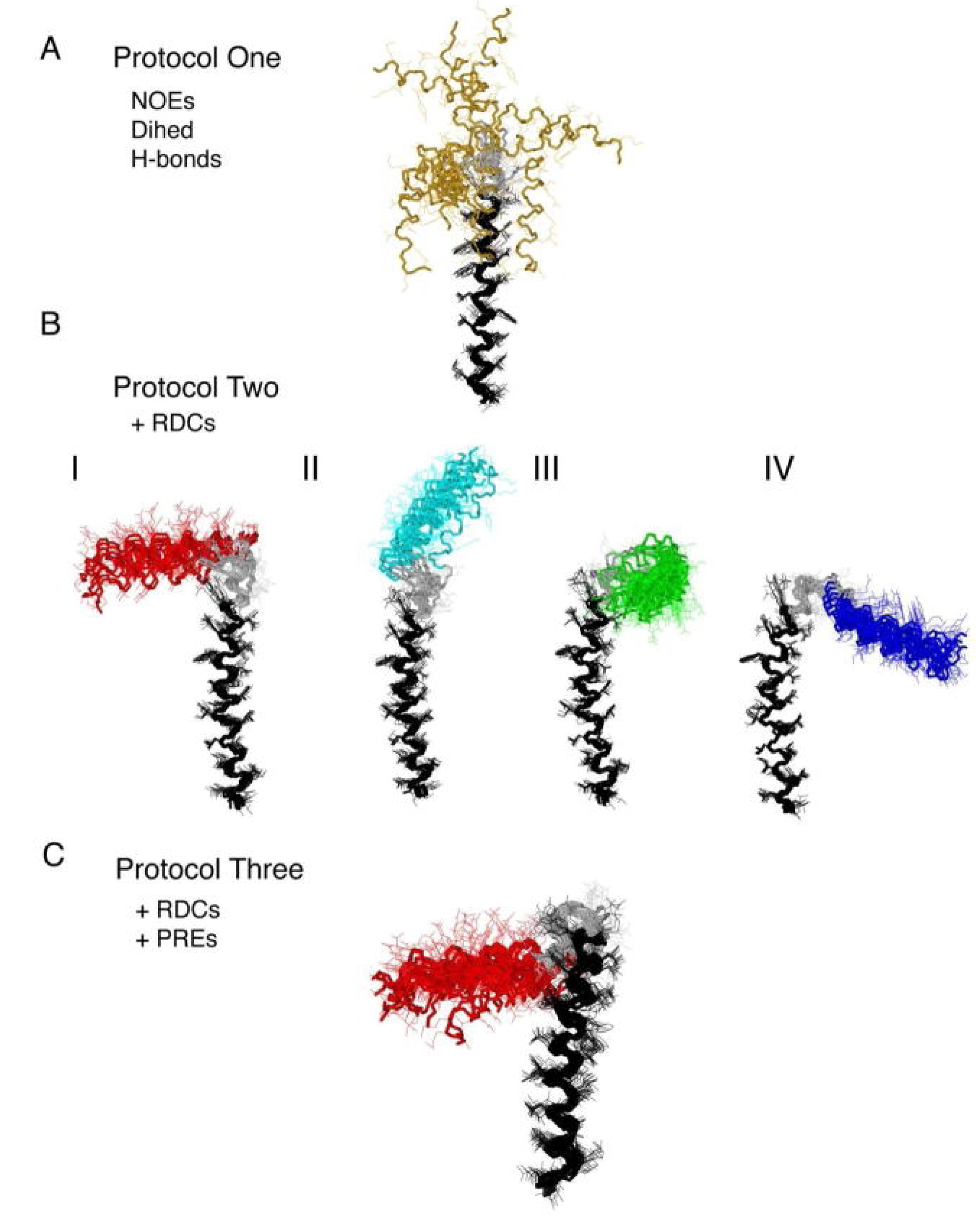
4. Applications
4.1. Phospholamban
4.2. The Heat Shock Protein Hsp12
4.3. Antimicrobial Peptides
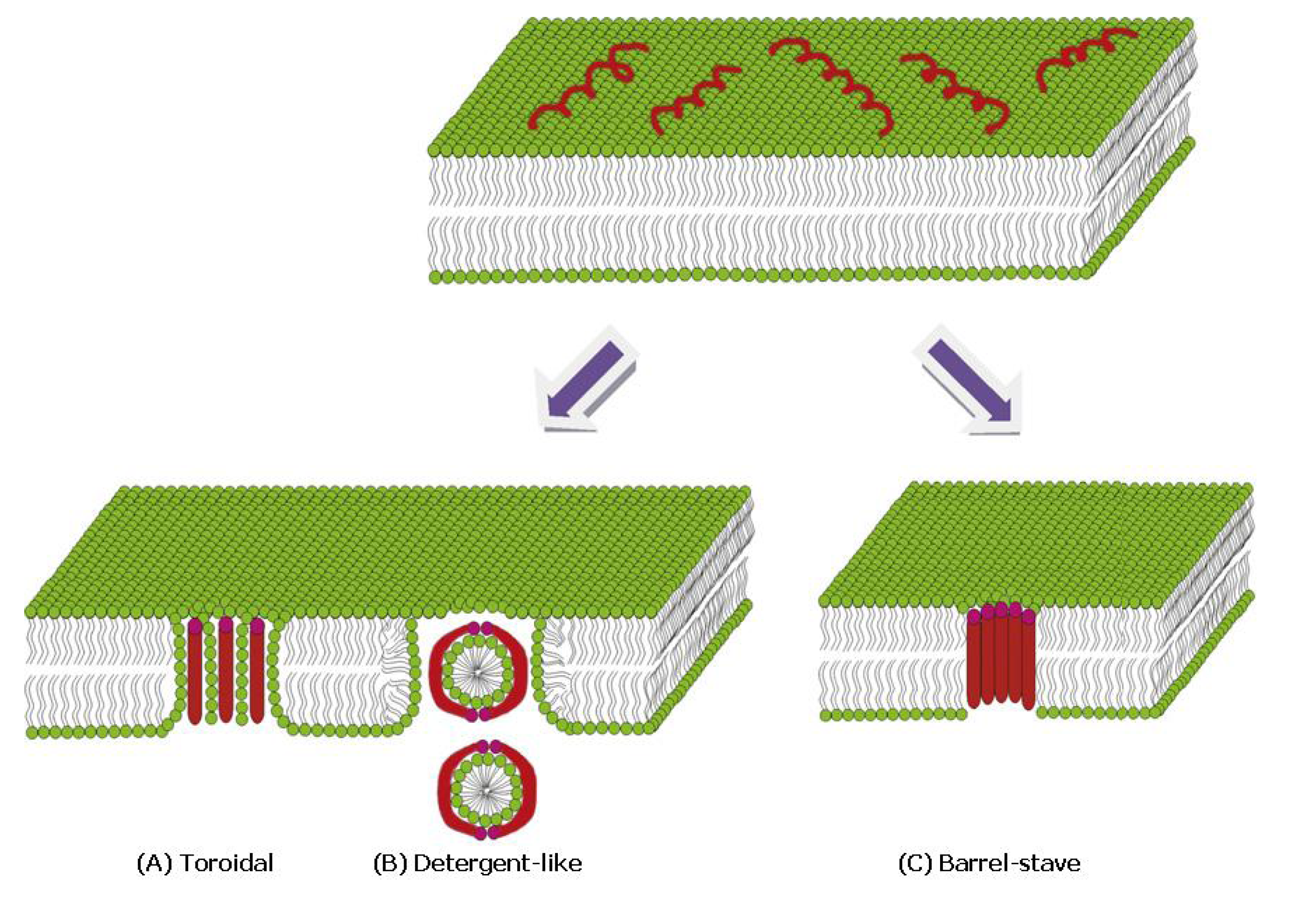
4.3.1. α-Helical Peptides
4.3.1.1. Temporins
4.3.1.2. Melittin
4.3.1.3. Magainin
4.3.2. β-Structures: Arenicin-2

4.3.3. Cyclotides
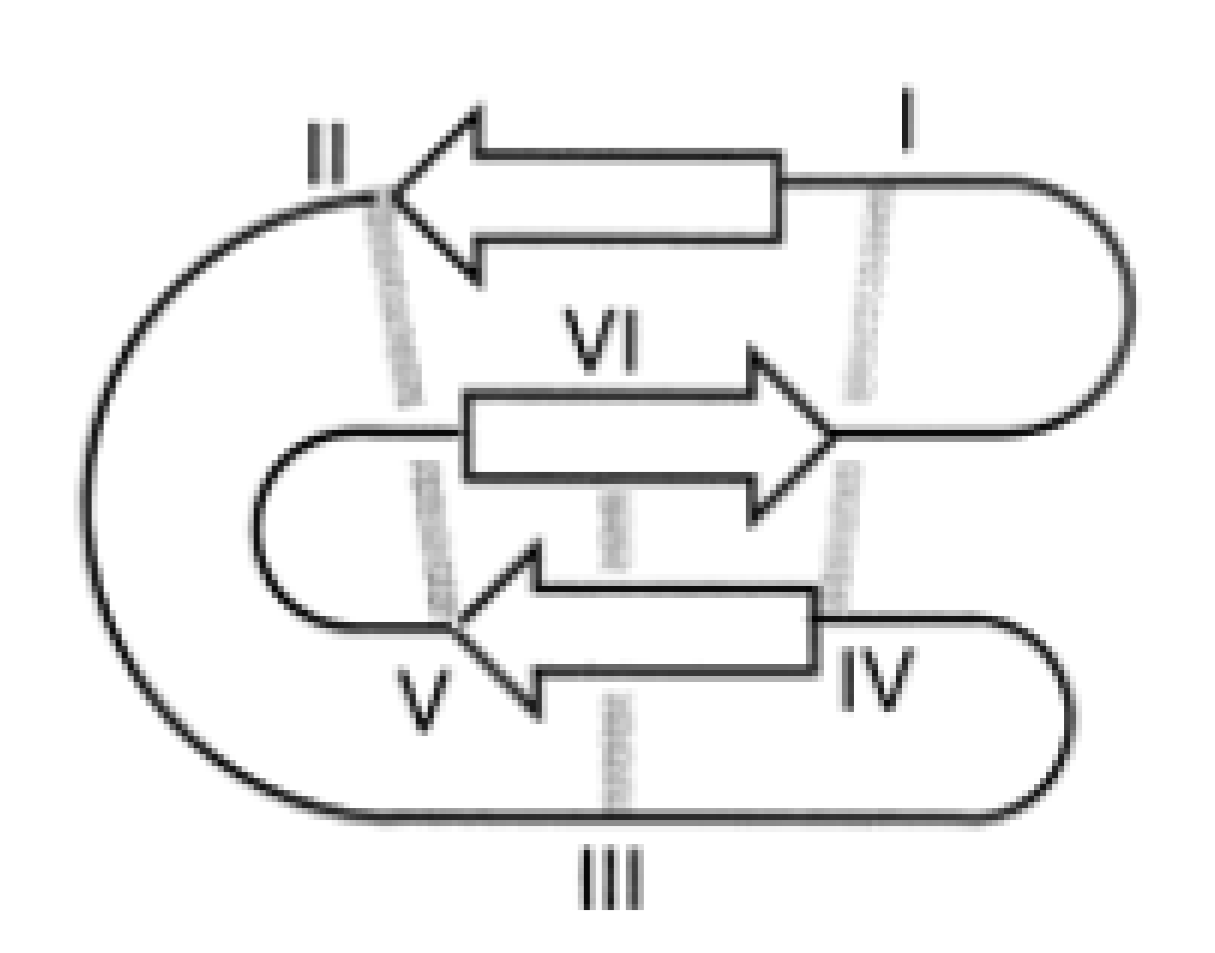
4.3.4. AMPs and MD Simulations
4.4. Amyloid Peptides
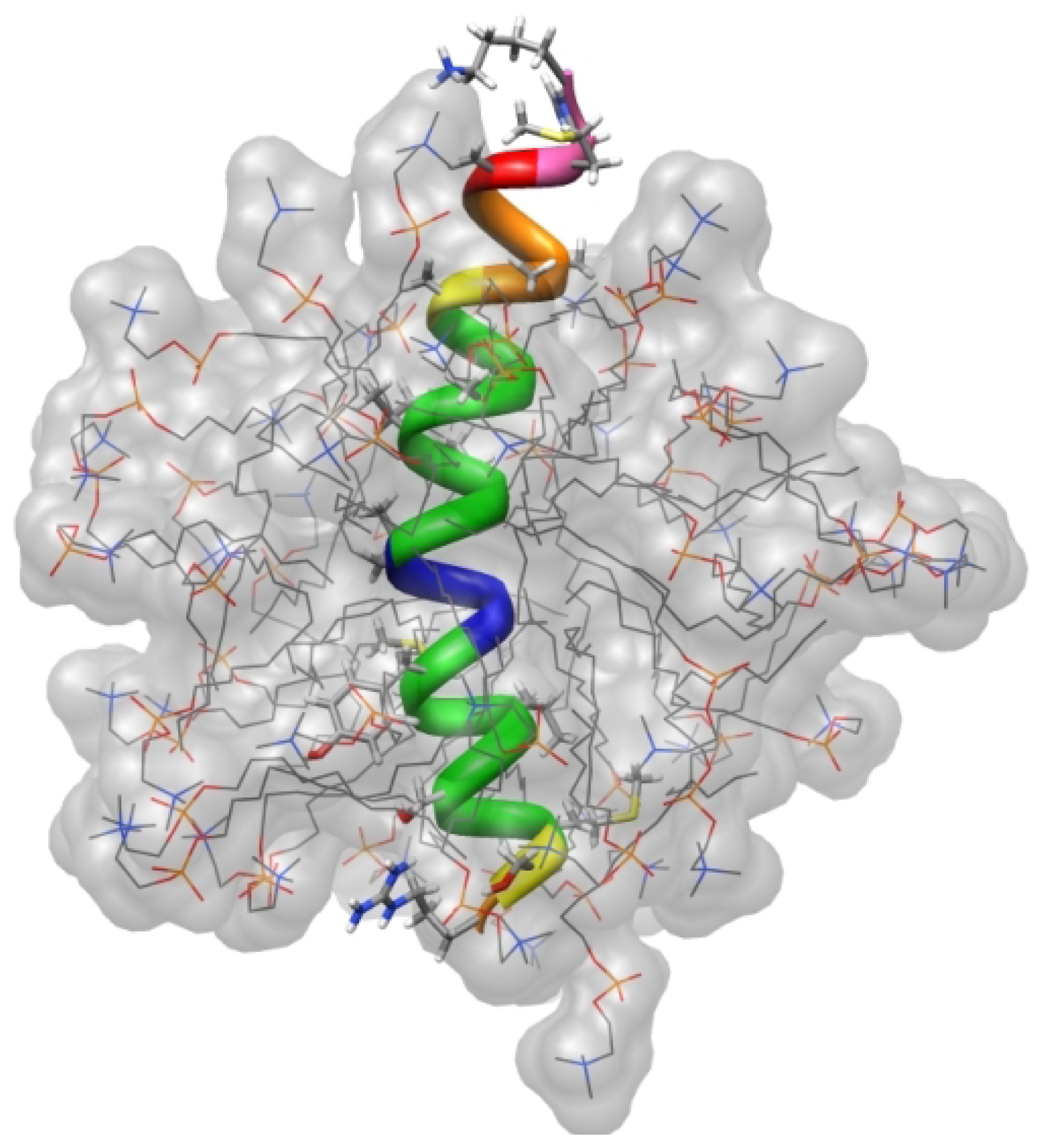
4.5. Cell Penetrating Peptides
5. Larger Membrane-Mimetic Systems
6. Conclusion and Outlook
Acknowledgments
Conflicts of Interest
References
- Lehnert, U.; Xia, Y.; Royce, T.E.; Goh, C.S.; Liu, Y.; Senes, A.; Yu, H.; Zhang, Z.L.; Engelman, D.M.; Gerstein, M. Computational analysis of membrane proteins: Genomic occurrence, Structure prediction and helix interactions. Q. Rev. Biophys. 2004, 37, 121–146. [Google Scholar] [CrossRef]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef]
- Raman, P.; Cherezov, V.; Caffrey, M. The membrane proztein data bank. Cell. Mol. Life Sci. 2006, 63, 36–51. [Google Scholar] [CrossRef]
- Klammt, C.; Maslennikov, I.; Bayrhuber, M.; Eichmann, C.; Vajpai, N.; Chiu, E.J.; Blain, K.Y.; Esquivies, L.; Kwon, J.H.; Balana, B.; et al. Facile backbone structure determination of human membrane proteins by NMR spectroscopy. Nat. Methods 2012, 9, 834–839. [Google Scholar] [CrossRef]
- Hohlweg, W.; Kosol, S.; Zangger, K. Determining the orientation and localization of membrane-bound peptides. Curr. Protein Pept. Sci. 2012, 13, 267–279. [Google Scholar]
- Pervushin, K.; Riek, R.; Wider, G.; Wüthrich, K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA 1997, 94, 12366–12371. [Google Scholar] [CrossRef]
- Kainosho, M.; Torizawa, T.; Iwashita, Y.; Terauchi, T.; Mei Ono, A.; Güntert, P. Optimal isotope labelling for NMR protein structure determinations. Nature 2006, 440, 52–57. [Google Scholar] [CrossRef]
- Göbl, C.; Dulle, M.; Hohlweg, W.; Grossauer, J.; Falsone, S.F.; Glatter, O.; Zangger, K. Influence of phosphocholine alkyl chain length on peptide-micelle interactions and micellar size and shape. J. Phys. Chem. B 2010, 114, 4717–4724. [Google Scholar] [CrossRef]
- Kallick, D.A.; Tessmer, M.R.; Watts, C.R.; Li, C.Y. The use of dodecylphosphocholine micelles in solution NMR. J. Magn. Reson. B 1995, 109, 60–65. [Google Scholar] [CrossRef]
- Keifer, P.A.; Peterkofsky, A.; Wang, G. Effects of detergent alkyl chain length and chemical structure on the properties of a micelle-bound bacterial membrane targeting peptide. Anal. Biochem. 2004, 331, 33–39. [Google Scholar]
- Dürr, U.H.; Gildenberg, M.; Ramamoorthy, A. The magic of bicelles lights up membrane protein structure. Chem. Rev. 2012, 112, 6054–6074. [Google Scholar] [CrossRef]
- Warschawski, D.E.; Arnold, A.A.; Beaugrand, M.; Gravel, A.; Chartrand, E.; Marcotte, I. Choosing membrane mimetics for NMR structural studies of transmembrane proteins. Biochim. Biophys. Acta 2011, 1808, 1957–1974. [Google Scholar] [CrossRef]
- Hagn, F.; Etzkorn, M.; Raschle, T.; Wagner, G. Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J. Am. Chem. Soc. 2013, 135, 1919–1925. [Google Scholar] [CrossRef]
- Puthenveetil, R.; Vinogradova, O. Optimization of the design and preparation of nanoscale phospholipid bilayers for its application to solution NMR. Proteins 2013, 81, 1222–1231. [Google Scholar] [CrossRef]
- Bertini, I.; Luchinat, C.; Parigi, G. Solution NMR of Paramagnetic Molecules; Elsevier: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Brown, L.R.; Bosch, C.; Wüthrich, K. Location and orientation relative to the micelle surface for glucagon in mixed micelles with dodecylphosphocholine: EPR and NMR studies. Biochim. Biophys. Acta 1981, 642, 296–312. [Google Scholar] [CrossRef]
- Respondek, M.; Madl, T.; Göbl, C.; Golser, R.; Zangger, K. Mapping the orientation of helices in micelle-bound peptides by paramagnetic relaxation waves. J. Am. Chem. Soc. 2007, 129, 5228–5234. [Google Scholar] [CrossRef]
- Fernandez, C.; Hilty, C.; Wider, G.; Wüthrich, K. Lipid-protein interactions in DHPC micelles containing the integral membrane protein OmpX investigated by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2002, 99, 13533–13537. [Google Scholar] [CrossRef]
- Hilty, C.; Wider, G.; Fernandez, C.; Wüthrich, K. Membrane protein - lipid interactions in mixed micelles studied by NMR spectroscopy with the use of paramagnetic reagents. ChemBioChem 2004, 5, 467–473. [Google Scholar] [CrossRef]
- Yamamoto, K.; Vivekanandan, S.; Ramamoorthy, A. Fast NMR data acquisition from bicelles containing a membrane-associated peptide at natural-abundance. J. Phys. Chem. B 2011, 115, 12448–12455. [Google Scholar] [CrossRef]
- Yamamoto, K.; Xu, J.; Kawulka, K.E.; Vederas, J.C.; Ramamoorthy, A. Use of a copper-chelated lipid speeds up NMR measurements from membrane proteins. J. Am. Chem. Soc. 2010, 132, 6929–6931. [Google Scholar] [CrossRef]
- Nielsen, R.D.; Che, K.; Gelb, M.H.; Robinson, B.H. A ruler for determining the position of proteins in membranes. J. Am. Chem. Soc. 2005, 127, 6430–6442. [Google Scholar]
- Prosser, R.S.; Luchette, P.A. An NMR study of the origin of dioxygen-induced spin-lattice relaxation enhancement and chemical shift perturbation. J. Magn. Reson. 2004, 171, 225–232. [Google Scholar] [CrossRef]
- Prosser, R.S.; Luchette, P.A.; Westerman, P.W. Using O2 to probe membrane immersion depth by 19F NMR. Proc. Natl. Acad. Sci. USA 2000, 97, 9967–9971. [Google Scholar] [CrossRef]
- Luchette, P.A.; Prosser, R.S.; Sanders, C.R. Oxygen as a paramagnetic probe of membrane protein structure by cysteine mutagenesis and 19F NMR spectroscopy. J. Am. Chem. Soc. 2002, 124, 1778–1781. [Google Scholar] [CrossRef]
- Evanics, F.; Hwang, P.M.; Cheng, Y.; Kay, L.E.; Prosser, R.S. Topology of an outer-membrane enzyme: Measuring oxygen and water contacts in solution NMR studies of PagP. J. Am. Chem. Soc. 2006, 128, 8256–8264. [Google Scholar]
- Fazal, M.A.; Roy, B.C.; Sun, S.; Mallik, S.; Rodgers, K.R. Surface recognition of a protein using designed transition metal complexes. J. Am. Chem. Soc. 2001, 123, 6283–6290. [Google Scholar] [CrossRef]
- Niccolai, N.; Spiga, O.; Bernini, A.; Scarselli, M.; Ciutti, A.; Fiaschi, I.; Chiellini, S.; Molinari, H.; Temussi, P.A. NMR studies of protein hydration and TEMPOL accessibility. J. Mol. Biol. 2003, 332, 437–447. [Google Scholar] [CrossRef]
- Pintacuda, G.; Otting, G. Identification of protein surfaces by NMR measurements with a pramagnetic Gd(III) chelate. J. Am. Chem. Soc. 2002, 124, 372–373. [Google Scholar] [CrossRef]
- Kosol, S.; Schrank, E.; Bukvic-Krajacic, M.; Wagner, G.E.; Meyer, H.; Göbl, C.; Zangger, K.; Novak, P. Probing the interactions of macrolide antibiotics with membrane-mimetics by NMR spectroscopy. J. Med. Chem. 2012, 55, 5632–5636. [Google Scholar] [CrossRef]
- Zangger, K.; Respondek, M.; Göbl, C.; Hohlweg, W.; Rasmussen, K.; Grampp, G.; Madl, T. Positioning of micelle-bound peptides by paramagnetic relaxation enhancements. J. Phys. Chem. B 2009, 113, 4400–4406. [Google Scholar] [CrossRef]
- Madl, T.; Bermel, W.; Zangger, K. Use of relaxation enhancements in a paramagnetic environment for the structure determination of proteins using NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2009, 48, 8259–8262. [Google Scholar] [CrossRef]
- Fröhlich, R.F.; Schrank, E.; Zangger, K. 2,2,2-Trifluoroethyl 6-thio-beta-D-glucopyranoside as a selective tag for cysteines in proteins. Carbohydr. Res. 2012, 361, 100–104. [Google Scholar] [CrossRef]
- Pintacuda, G.; Otting, G. Identification of Protein Surfaces by NMR Measurements with a Paramagnetic Gd(III) Chelate. J. Am. Chem. Soc. 2001, 124, 372–373. [Google Scholar] [CrossRef]
- Zangger, K.; Gossler, R.; Khatai, L.; Lohner, K.; Jilek, A. Structures of the glycine-rich diastereomeric peptides bombinin H2 and H4. Toxicon 2008, 52, 246–254. [Google Scholar] [CrossRef]
- Meyer, N.H.; Zangger, K. Simplifying Proton NMR Spectra by Instant Homonuclear Broadband Decoupling. Angew. Chem. Int. Ed. Engl. 2013. [Google Scholar] [CrossRef]
- Kosol, S.; Zangger, K. Dynamics and orientation of a cationic antimicrobial peptide in two membrane-mimetic systems. J. Struct. Biol. 2010, 170, 172–179. [Google Scholar] [CrossRef]
- Grossauer, J.; Kosol, S.; Schrank, E.; Zangger, K. The peptide hormone ghrelin binds to membrane-mimetics via its octanoyl chain and an adjacent phenylalanine. Bioorg. Med. Chem. 2010, 18, 5483–5488. [Google Scholar] [CrossRef]
- Göbl, C.; Kosol, S.; Stockner, T.; Rückert, H.M.; Zangger, K. Solution structure and membrane binding of the toxin fst of the par addiction module. Biochemistry 2010, 49, 6567–6575. [Google Scholar] [CrossRef]
- Franzmann, M.; Otzen, D.; Wimmer, R. Quantitative use of paramagnetic relaxation enhancements for determining orientations and insertion depths of peptides in micelles. Chembiochem 2009, 10, 2339–2347. [Google Scholar] [CrossRef]
- Cohen, L.S.; Arshava, B.; Neumoin, A.; Becker, J.M.; Guntert, P.; Zerbe, O.; Naider, F. Comparative NMR analysis of an 80-residue G protein-coupled receptor fragment in two membrane mimetic environments. Biochim. Biophys. Acta 2011, 1808, 2674–2684. [Google Scholar] [CrossRef]
- Page, R.C.; Lee, S.; Moore, J.D.; Opella, S.J.; Cross, T.A. Backbone structure of a small helical integral membrane protein: A unique structural characterization. Protein Sci. 2009, 18, 134–146. [Google Scholar]
- Shi, L.; Traaseth, N.J.; Verardi, R.; Gustavsson, M.; Gao, J.; Veglia, G. Paramagnetic-based NMR restraints lift residual dipolar coupling degeneracy in multidomain detergent-solubilized membrane proteins. J. Am. Chem. Soc. 2011, 133, 2232–2241. [Google Scholar]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell. Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef]
- Simmerman, H.K.; Kobayashi, Y.M.; Autry, J.M.; Jones, L.R. A leucine zipper stabilizes the pentameric membrane domain of phospholamban and forms a coiled-coil pore structure. J. Biol. Chem. 1996, 271, 5941–5946. [Google Scholar]
- Karim, C.B.; Marquardt, C.G.; Stamm, J.D.; Barany, G.; Thomas, D.D. Synthetic null-cysteine phospholamban analogue and the corresponding transmembrane domain inhibit the Ca-ATPase. Biochemistry 2000, 39, 10892–10897. [Google Scholar] [CrossRef]
- Traaseth, N.J.; Shi, L.; Verardi, R.; Mullen, D.G.; Barany, G.; Veglia, G. Structure and topology of monomeric phospholamban in lipid membranes determined by a hybrid solution and solid-state NMR approach. Proc. Natl. Acad. Sci. USA 2009, 106, 10165–10170. [Google Scholar]
- Traaseth, N.J.; Verardi, R.; Torgersen, K.D.; Karim, C.B.; Thomas, D.D.; Veglia, G. Spectroscopic validation of the pentameric structure of phospholamban. Proc. Natl. Acad. Sci. USA 2007, 104, 14676–14681. [Google Scholar]
- Zamoon, J.; Mascioni, A.; Thomas, D.D.; Veglia, G. NMR solution structure and topological orientation of monomeric phospholamban in dodecylphosphocholine micelles. Biophys. J. 2003, 85, 2589–2598. [Google Scholar] [CrossRef]
- Al-Abdul-Wahid, M.S.; Verardi, R.; Veglia, G.; Prosser, R.S. Topology and immersion depth of an integral membrane protein by paramagnetic rates from dissolved oxygen. J. Biomol. NMR 2011, 51, 173–183. [Google Scholar] [CrossRef]
- Praekelt, U.M.; Meacock, P.A. HSP12, a new small heat shock gene of Saccharomyces cerevisiae: analysis of structure, regulation and function. Mol. Gen. Genet. 1990, 223, 97–106. [Google Scholar] [CrossRef]
- Singarapu, K.K.; Tonelli, M.; Chow, D.C.; Frederick, R.O.; Westler, W.M.; Markley, J.L. Structural characterization of Hsp12, the heat shock protein from Saccharomyces cerevisiae, in aqueous solution where it is intrinsically disordered and in detergent micelles where it is locally alpha-helical. J. Biol. Chem. 2011, 286, 43447–43453. [Google Scholar]
- Welker, S.; Rudolph, B.; Frenzel, E.; Hagn, F.; Liebisch, G.; Schmitz, G.; Scheuring, J.; Kerth, A.; Blume, A.; Weinkauf, S.; Haslbeck, M.; Kessler, H.; Buchner, J. Hsp12 is an intrinsically unstructured stress protein that folds upon membrane association and modulates membrane function. Mol. Cell. 2010, 39, 507–520. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Lehrer, R. Cationic peptides: A new source of antibiotics. Trends Biotech. 1998, 16, 82–88. [Google Scholar] [CrossRef]
- Abbassi, F.; Lequin, O.; Piesse, C.; Goasdoue, N.; Foulon, T.; Nicolas, P.; Ladram, A. Temporin-SHf, a new type of phe-rich and hydrophobic ultrashort antimicrobial peptide. J. Biol. Chem. 2010, 285, 16880–16892. [Google Scholar]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipathic, Alpha-helical antimicrobial peptides. Biopolymers 2000, 55, 4–30. [Google Scholar] [CrossRef]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Nicolas, P. Multifunctional host defense peptides: Intracellular-targeting antimicrobial peptides. FEBS J. 2009, 276, 6483–6496. [Google Scholar] [CrossRef]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef]
- Seo, M.D.; Won, H.S.; Kim, J.H.; Mishig-Ochir, T.; Lee, B.J. Antimicrobial peptides for therapeutic applications: A review. Molecules 2012, 17, 12276–12286. [Google Scholar] [CrossRef]
- Li, Y.; Xiang, Q.; Zhang, Q.; Huang, Y.; Su, Z. Overview on the recent study of antimicrobial peptides: Origins, Functions, Relative mechanisms and application. Peptides 2012, 37, 207–215. [Google Scholar] [CrossRef]
- Matsuzaki, K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetype. Biochim. Biophys. Acta 1999, 1462, 1–10. [Google Scholar] [CrossRef]
- Dempsey, C.E. The actions of melittin on membranes. Biochim. Biophys. Acta 1990, 1031, 143–161. [Google Scholar] [CrossRef]
- Pistolesi, S.; Pogni, R.; Feix, J.B. Membrane insertion and bilayer perturbation by antimicrobial peptide CM15. Biophys. J. 2007, 93, 1651–1660. [Google Scholar] [CrossRef]
- Bhargava, K.; Feix, J.B. Membrane binding, Structure, and localization of cecropin-mellitin hybrid peptides: A site-directed spin-labeling study. Biophys. J. 2004, 86, 329–336. [Google Scholar] [CrossRef]
- Ramamoorthy, A.; Marassi, F.M.; Zasloff, M.; Opella, S.J. Three-dimensional solid-state NMR spectroscopy of a peptide oriented in membrane bilayers. J. Biomol. NMR 1995, 6, 329–334. [Google Scholar]
- Porcelli, F.; Buck-Koehntop, B.A.; Thennarasu, S.; Ramamoorthy, A.; Veglia, G. Structures of the dimeric and monomeric variants of magainin antimicrobial peptides (MSI-78 and MSI-594) in micelles and bilayers, determined by NMR spectroscopy. Biochemistry 2006, 45, 5793–5799. [Google Scholar]
- Lequin, O.; Bruston, F.; Convert, O.; Chassaing, G.; Nicolas, P. Helical structure of dermaseptin B2 in a membrane-mimetic environment. Biochemistry 2003, 42, 10311–10323. [Google Scholar] [CrossRef]
- Saravanan, R.; Bhattacharjya, S. Oligomeric structure of a cathelicidin antimicrobial peptide in dodecylphosphocholine micelle determined by NMR spectroscopy. Biochim. Biophys. Acta 2011, 1808, 369–381. [Google Scholar] [CrossRef]
- Van Den Hooven, H.W.; Spronk, C.A.; van de Kamp, M.; Konings, R.N.; Hilbers, C.W.; van de van, F.J. Surface location and orientation of the lantibiotic nisin bound to membrane-mimicking micelles of dodecylphosphocholine and of sodium dodecylsulphate. Eur. J. Biochem. 1996, 235, 394–403. [Google Scholar]
- Simmaco, M.; Mignogna, G.; Canofeni, S.; Miele, R.; Mangoni, M.L.; Barra, D. Temporins, antimicrobial peptides from the European red frog Rana temporaria. Eur. J. Biochem. 1996, 242, 788–792. [Google Scholar]
- Abbassi, F.; Galanth, C.; Amiche, M.; Saito, K.; Piesse, C.; Zargarian, L.; Hani, K.; Nicolas, P.; Lequin, O.; Ladram, A. Solution structure and model membrane interactions of temporins-SH, antimicrobial peptides from amphibian skin. A NMR spectroscopy and differential scanning calorimetry study. Biochemistry 2008, 47, 10513–10525. [Google Scholar] [CrossRef]
- Saravanan, R.; Bhunia, A.; Bhattacharjya, S. Micelle-bound structures and dynamics of the hinge deleted analog of melittin and its diastereomer: Implications in cell selective lysis by D-amino acid containing antimicrobial peptides. Biochim. Biophys. Acta 2010, 1798, 128–139. [Google Scholar] [CrossRef]
- Lee, D.K.; Brender, J.R.; Sciacca, M.F.; Krishnamoorthy, J.; Yu, C.; Ramamoorthy, A. Lipid composition-dependent membrane fragmentation and pore-forming mechanisms of membrane disruption by pexiganan (MSI-78). Biochemistry 2013. [Google Scholar] [CrossRef]
- Ovchinnikova, T.V.; Shenkarev, Z.O.; Balandin, S.V.; Nadezhdin, K.D.; Paramonov, A.S.; Kokryakov, V.N.; Arseniev, A.S. Molecular insight into mechanism of antimicrobial action of the β-hairpin peptide arenicin: Specific oligomerization in detergent micelles. Biopolymers 2008, 89, 455–464. [Google Scholar] [CrossRef]
- Shenkarev, Z.O.; Balandin, S.V.; Trunov, K.I.; Paramonov, A.S.; Sukhanov, S.V.; Barsukov, L.I.; Arseniev, A.S.; Ovchinnikova, T.V. Molecular mechanism of action of beta-hairpin antimicrobial peptide arenicin: Oligomeric structure in dodecylphosphocholine micelles and pore formation in planar lipid bilayers. Biochemistry 2011, 50, 6255–6265. [Google Scholar] [CrossRef]
- Garcia-Olmedo, F.; Molina, A.; Alamillo, J.M.; Rodriguez-Palenzuela, P. Plant defense peptides. Biopolymers 1998, 47, 479–491. [Google Scholar] [CrossRef]
- McDonald, N.Q.; Hendrickson, W.A. A structural superfamily of growth factors containing a cystine knot motif. Cell 1993, 73, 421–424. [Google Scholar] [CrossRef]
- Craik, D.J.; Cemazar, M.; Wang, C.K.; Daly, N.L. The cyclotide family of circular miniproteins: Nature’s combinatorial peptide template. Biopolymers 2006, 84, 250–266. [Google Scholar] [CrossRef]
- Craik, D.J.; Daly, N.L.; Waine, C. The cystine knot motif in toxins and implications for drug design. Toxicon. 2001, 39, 43–60. [Google Scholar] [CrossRef]
- Craik, D.J.; Daly, N.L. NMR as a tool for elucidating the structures of circular and knotted proteins. Mol. Biosyst. 2007, 3, 257–265. [Google Scholar] [CrossRef]
- Rosengren, K.J.; Daly, N.L.; Plan, M.R.; Waine, C.; Craik, D.J. Twists, Knots, and rings in proteins. Structural definition of the cyclotide framework. J. Biol. Chem. 2003, 278, 8606–8616. [Google Scholar]
- Felizmenio-Quimio, M.E.; Daly, N.L.; Craik, D.J. Circular proteins in plants: solution structure of a novel macrocyclic trypsin inhibitor from Momordica cochinchinensis. J. Biol. Chem. 2001, 276, 22875–22882. [Google Scholar] [CrossRef]
- Craik, D.J.; Daly, N.L.; Bond, T.; Waine, C. Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J. Mol. Biol. 1999, 294, 1327–1336. [Google Scholar] [CrossRef]
- Shenkarev, Z.O.; Nadezhdin, K.D.; Lyukmanova, E.N.; Sobol, V.A.; Skjeldal, L.; Arseniev, A.S. Divalent cation coordination and mode of membrane interaction in cyclotides: NMR spatial structure of ternary complex Kalata B7/Mn2+/DPC micelle. J. Inorg. Biochem. 2008, 102, 1246–1256. [Google Scholar] [CrossRef]
- Goransson, U.; Sjogren, M.; Svangard, E.; Claeson, P.; Bohlin, L. Reversible antifouling effect of the cyclotide cycloviolacin O2 against barnacles. J. Nat. Prod. 2004, 67, 1287–1290. [Google Scholar] [CrossRef]
- Jennings, C.; West, J.; Waine, C.; Craik, D.; Anderson, M. Biosynthesis and insecticidal properties of plant cyclotides: The cyclic knotted proteins from Oldenlandia affinis. Proc. Natl. Acad. Sci. USA 2001, 98, 10614–10619. [Google Scholar] [CrossRef]
- Gran, L. On the effect of a polypeptide isolated from “Kalata-Kalata” (Oldenlandia affinis DC) on the oestrogen dominated uterus. Acta Pharmacol. Toxicol. (Copenh). 1973, 33, 400–408. [Google Scholar] [CrossRef]
- Witherup, K.M.; Bogusky, M.J.; Anderson, P.S.; Ramjit, H.; Ransom, R.W.; Wood, T.; Sardana, M. Cyclopsychotride A, a biologically active, 31-residue cyclic peptide isolated from Psychotria longipes. J. Nat. Prod. 1994, 57, 1619–1625. [Google Scholar] [CrossRef]
- Miljanich, G.P.; Ramachandran, J. Antagonists of neuronal calcium channels: Structure, Function, and therapeutic implications. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 707–734. [Google Scholar] [CrossRef]
- Tam, J.P.; Lu, Y.A.; Yang, J.L.; Chiu, K.W. An unusual structural motif of antimicrobial peptides containing end-to-end macrocycle and cystine-knot disulfides. Proc. Natl. Acad. Sci. USA 1999, 96, 8913–8918. [Google Scholar] [CrossRef]
- Gustafson, K.R.; Sowder, R.C.; Henderson, L.E.; Parsons, I.C.; Kashman, Y.; Cardellina, J.H.; McMahon, J.B.; Buckheit, R.W.; Pannell, L.K.; Boyd, M.R. Circulins A and B. Novel human immunodeficiency virus (HIV)-inhibitory macrocyclic peptides from the tropical tree Chassalia parvifolia. J. Am. Chem. Soc. 1994, 116, 9337–9338. [Google Scholar] [CrossRef]
- Hallock, Y.F.; Sowder, R.C., 2nd; Pannell, L.K.; Hughes, C.B.; Johnson, D.G.; Gulakowski, R.; Cardellina, J.H., 2nd; Boyd, M.R. Cycloviolins A-D, anti-HIV macrocyclic peptides from Leonia cymosa. J. Org. Chem. 2000, 65, 124–128. [Google Scholar] [CrossRef]
- Daly, N.L.; Clark, R.J.; Plan, M.R.; Craik, D.J. Kalata B8, a novel antiviral circular protein, exhibits conformational flexibility in the cystine knot motif. Biochem. J. 2006, 393, 619–626. [Google Scholar] [CrossRef]
- Shenkarev, Z.O.; Nadezhdin, K.D.; Sobol, V.A.; Sobol, A.G.; Skjeldal, L.; Arseniev, A.S. Conformation and mode of membrane interaction in cyclotides. Spatial structure of kalata B1 bound to a dodecylphosphocholine micelle. FEBS J. 2006, 273, 2658–2672. [Google Scholar] [CrossRef]
- Daly, N.L.; Rosengren, K.J.; Craik, D.J. Discovery, structure and biological activities of cyclotides. Adv. Drug Deliv. Rev. 2009, 61, 918–930. [Google Scholar] [CrossRef]
- Simonsen, S.M.; Sando, L.; Rosengren, K.J.; Wang, C.K.; Colgrave, M.L.; Daly, N.L.; Craik, D.J. Alanine scanning mutagenesis of the prototypic cyclotide reveals a cluster of residues essential for bioactivity. J. Biol. Chem. 2008, 283, 9805–9813. [Google Scholar]
- Lindholm, P.; Goransson, U.; Johansson, S.; Claeson, P.; Gullbo, J.; Larsson, R.; Bohlin, L.; Backlund, A. Cyclotides: A novel type of cytotoxic agents. Mol. Cancer Ther. 2002, 1, 365–369. [Google Scholar] [CrossRef]
- Wang, C.K.; Colgrave, M.L.; Ireland, D.C.; Kaas, Q.; Craik, D.J. Despite a conserved cystine knot motif, different cyclotides have different membrane binding modes. Biophys. J. 2009, 97, 1471–1481. [Google Scholar] [CrossRef]
- Tang, J.; Yin, H.; Qiu, J.; Tucker, M.J.; DeGrado, W.F.; Gai, F. Using two fluorescent probes to dissect the binding, insertion, and dimerization kinetics of a model membrane peptide. J. Am. Chem. Soc. 2009, 131, 3816–3817. [Google Scholar] [CrossRef]
- Bourbigot, S.; Dodd, E.; Horwood, C.; Cumby, N.; Fardy, L.; Welch, W.H.; Ramjan, Z.; Sharma, S.; Waring, A.J.; Yeaman, M.R.; Booth, V. Antimicrobial peptide RP-1 structure and interactions with anionic versus zwitterionic micelles. Biopolymers 2009, 91, 1–13. [Google Scholar] [CrossRef]
- Wang, Y.; Schlamadinger, D.E.; Kim, J.E.; McCammon, J.A. Comparative molecular dynamics simulations of the antimicrobial peptide CM15 in model lipid bilayers. Biochim. Biophys. Acta 2012, 1818, 1402–1409. [Google Scholar] [CrossRef]
- Dittmer, J.; Thogersen, L.; Underhaug, J.; Bertelsen, K.; Vosegaard, T.; Pedersen, J.M.; Schiott, B.; Tajkhorshid, E.; Skrydstrup, T.; Nielsen, N.C. Incorporation of antimicrobial peptides into membranes: A combined liquid-state NMR and molecular dynamics study of alamethicin in DMPC/DHPC bicelles. J. Phys. Chem. B 2009, 113, 6928–6937. [Google Scholar] [CrossRef]
- Sauve, S.; Buijs, D.; Gingras, G.; Aubin, Y. Interactions between the conserved hydrophobic region of the prion protein and dodecylphosphocholine micelles. J. Biol. Chem. 2012, 287, 1915–1922. [Google Scholar] [CrossRef]
- Liu, G.; Prabhakar, A.; Aucoin, D.; Simon, M.; Sparks, S.; Robbins, K.J.; Sheen, A.; Petty, S.A.; Lazo, N.D. Mechanistic studies of peptide self-assembly: Transient alpha-helices to stable beta-sheets. J. Am. Chem. Soc. 2010, 132, 18223–18232. [Google Scholar] [CrossRef]
- Jarvet, J.; Danielsson, J.; Damberg, P.; Oleszczuk, M.; Gräslund, A. Positioning of the Alzheimer Abeta(1–40) peptide in SDS micelles using NMR and paramagnetic probes. J. Biomol. NMR 2007, 39, 63–72. [Google Scholar] [CrossRef]
- Wahlstrom, A.; Hugonin, L.; Peralvarez-Marin, A.; Jarvet, J.; Graslund, A. Secondary structure conversions of Alzheimer’s Abeta(1–40) peptide induced by membrane-mimicking detergents. FEBS J. 2008, 275, 5117–5128. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Matsuzaki, K.; Hoshino, M. Interaction between soluble Abeta-(1–40) monomer and Abeta-(1–42) fibrils probed by paramagnetic relaxation enhancement. FEBS Lett. 2013, 587, 620–624. [Google Scholar] [CrossRef]
- Nanga, R.P.; Brender, J.R.; Xu, J.; Hartman, K.; Subramanian, V.; Ramamoorthy, A. Three-dimensional structure and orientation of rat islet amyloid polypeptide protein in a membrane environment by solution NMR spectroscopy. J. Am. Chem. Soc. 2009, 131, 8252–8261. [Google Scholar] [CrossRef]
- Patil, S.M.; Xu, S.; Sheftic, S.R.; Alexandrescu, A.T. Dynamic alpha-helix structure of micelle-bound human amylin. J. Biol. Chem. 2009, 284, 11982–11991. [Google Scholar] [CrossRef]
- Nanga, R.P.; Brender, J.R.; Vivekanandan, S.; Popovych, N.; Ramamoorthy, A. NMR structure in a membrane environment reveals putative amyloidogenic regions of the SEVI precursor peptide PAP(248–286). J. Am. Chem. Soc. 2009, 131, 17972–17979. [Google Scholar] [CrossRef]
- Mukherjee, S.; Ghosh, R.N.; Maxfield, F.R. Endocytosis. Physiol. Rev. 1997, 77, 759–803. [Google Scholar]
- Lindgren, M.; Hallbrink, M.; Prochiantz, A.; Langel, U. Cell-penetrating peptides. Trends Pharmacol. Sci. 2000, 21, 99–103. [Google Scholar] [CrossRef]
- Lindberg, M.; Jarvet, J.; Langel, U.; Graslund, A. Secondary structure and position of the cell-penetrating peptide transportan in SDS micelles as determined by NMR. Biochemistry 2001, 40, 3141–3149. [Google Scholar] [CrossRef]
- Berlose, J.P.; Convert, O.; Derossi, D.; Brunissen, A.; Chassaing, G. Conformational and associative behaviours of the third helix of antennapedia homeodomain in membrane-mimetic environments. Eur. J. Biochem. 1996, 242, 372–386. [Google Scholar]
- Drin, G.; Demene, H.; Temsamani, J.; Brasseur, R. Translocation of the pAntp peptide and its amphipathic analogue AP-2AL. Biochemistry 2001, 40, 1824–1834. [Google Scholar] [CrossRef]
- Lindberg, M.; Graslund, A. The position of the cell penetrating peptide penetratin in SDS micelles determined by NMR. FEBS Lett. 2001, 497, 39–44. [Google Scholar] [CrossRef]
- Lindberg, M.; Biverstahl, H.; Graslund, A.; Maler, L. Structure and positioning comparison of two variants of penetratin in two different membrane mimicking systems by NMR. Eur. J. Biochem. 2003, 270, 3055–3063. [Google Scholar] [CrossRef]
- Derossi, D.; Chassaing, G.; Prochiantz, A. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell. Biol. 1998, 8, 84–87. [Google Scholar]
- Ohman, A.; Lycksell, P.O.; Jureus, A.; Langel, U.; Bartfai, T.; Graslund, A. NMR study of the conformation and localization of porcine galanin in SDS micelles. Comparison with an inactive analog and a galanin receptor antagonist. Biochemistry 1998, 37, 9169–9178. [Google Scholar] [CrossRef]
- Barany-Wallje, E.; Andersson, A.; Graslund, A.; Maler, L. NMR solution structure and position of transportan in neutral phospholipid bicelles. FEBS Lett. 2004, 567, 265–269. [Google Scholar] [CrossRef]
- Lau, A.L.; Chan, S.I. Alamethicin-mediated fusion of lecithin vesicles. Proc. Natl. Acad. Sci. USA 1975, 72, 2170–2174. [Google Scholar] [CrossRef]
- Feigenson, G.W.; Meers, P.R.; Kingsley, P.B. NMR observation of gramicidin A' in phosphatidylcholine vesicles. Biochim. Biophys. Acta 1977, 471, 487–491. [Google Scholar] [CrossRef]
- Weinstein, S.; Wallace, B.A.; Blout, E.R.; Morrow, J.S.; Veatch, W. Conformation of gramicidin A channel in phospholipid vesicles: A 13C and 19F nuclear magnetic resonance study. Proc. Natl. Acad. Sci. USA 1979, 76, 4230–4234. [Google Scholar]
- Glover, K.J.; Whiles, J.A.; Wu, G.; Yu, N.; Deems, R.; Struppe, J.O.; Stark, R.E.; Komives, E.A.; Vold, R.R. Structural evaluation of phospholipid bicelles for solution-state studies of membrane-associated biomolecules. Biophys. J. 2001, 81, 2163–2171. [Google Scholar] [CrossRef]
- Tjandra, N.; Bax, A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 1997, 278, 1111–1114. [Google Scholar] [CrossRef]
- Matsumori, N.; Morooka, A.; Murata, M. Conformation and location of membrane-bound salinomycin-sodium complex deduced from NMR in isotropic bicelles. J. Am. Chem. Soc. 2007, 129, 14989–14995. [Google Scholar] [CrossRef]
- Morrison, E.A.; DeKoster, G.T.; Dutta, S.; Vafabakhsh, R.; Clarkson, M.W.; Bahl, A.; Kern, D.; Ha, T.; Henzler-Wildman, K.A. Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature 2012, 481, 45–50. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schrank, E.; Wagner, G.E.; Zangger, K. Solution NMR Studies on the Orientation of Membrane-Bound Peptides and Proteins by Paramagnetic Probes. Molecules 2013, 18, 7407-7435. https://doi.org/10.3390/molecules18077407
Schrank E, Wagner GE, Zangger K. Solution NMR Studies on the Orientation of Membrane-Bound Peptides and Proteins by Paramagnetic Probes. Molecules. 2013; 18(7):7407-7435. https://doi.org/10.3390/molecules18077407
Chicago/Turabian StyleSchrank, Evelyne, Gabriel E. Wagner, and Klaus Zangger. 2013. "Solution NMR Studies on the Orientation of Membrane-Bound Peptides and Proteins by Paramagnetic Probes" Molecules 18, no. 7: 7407-7435. https://doi.org/10.3390/molecules18077407
APA StyleSchrank, E., Wagner, G. E., & Zangger, K. (2013). Solution NMR Studies on the Orientation of Membrane-Bound Peptides and Proteins by Paramagnetic Probes. Molecules, 18(7), 7407-7435. https://doi.org/10.3390/molecules18077407