Indomethacin Inhibits Cancer Cell Migration via Attenuation of Cellular Calcium Mobilization



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
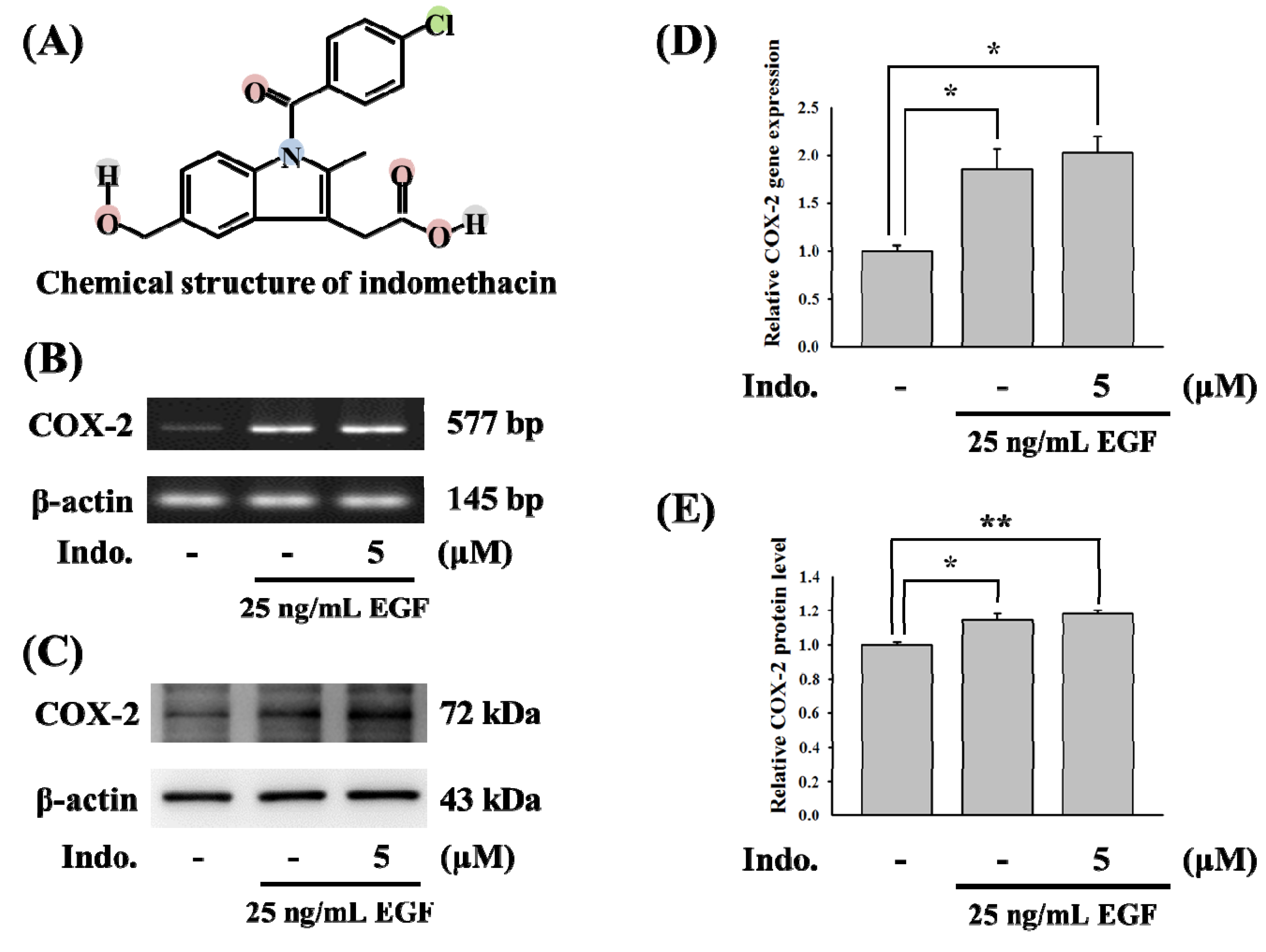
2.1. Effects of Indomethacin on COX-2 Gene Expression

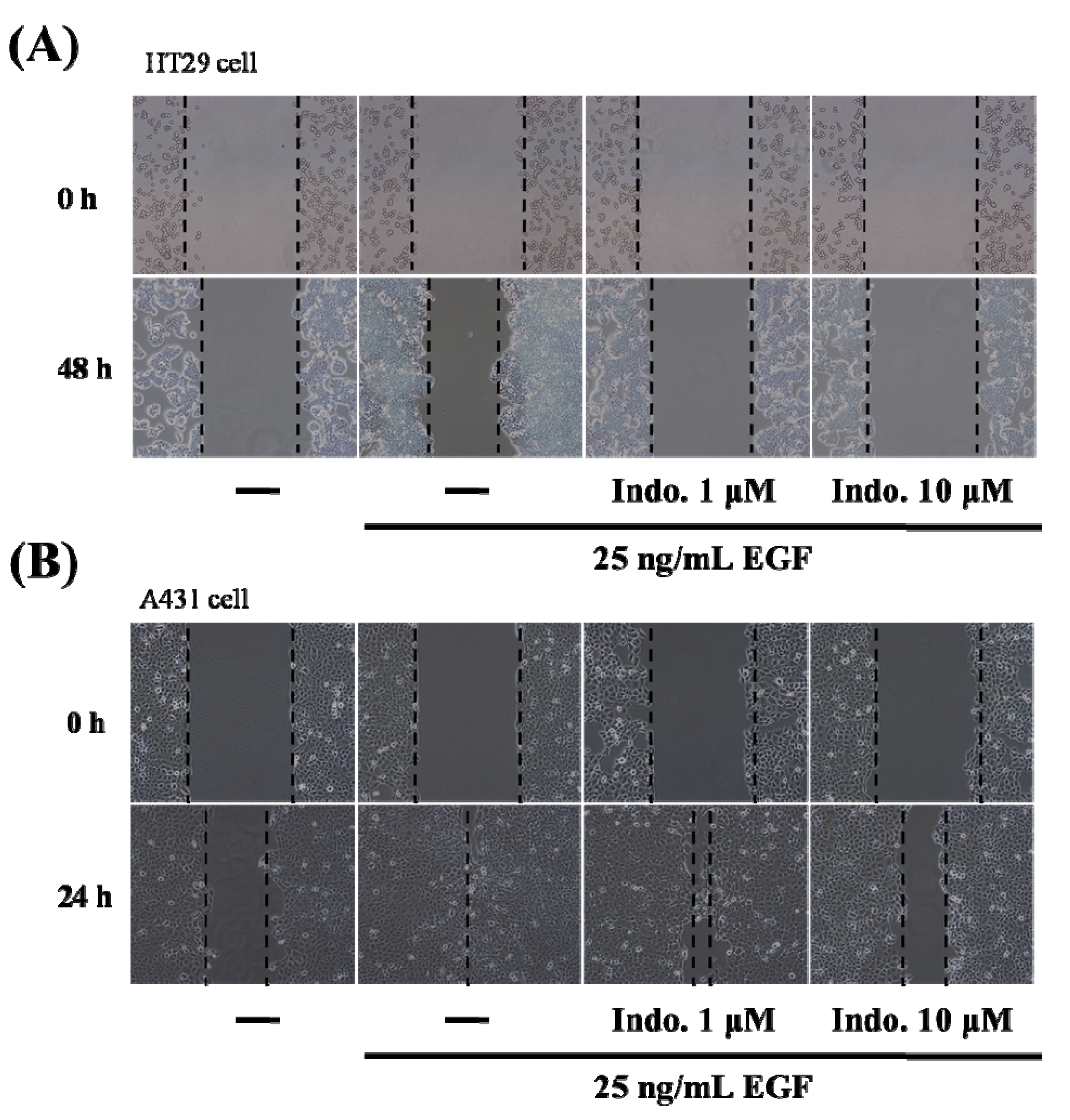
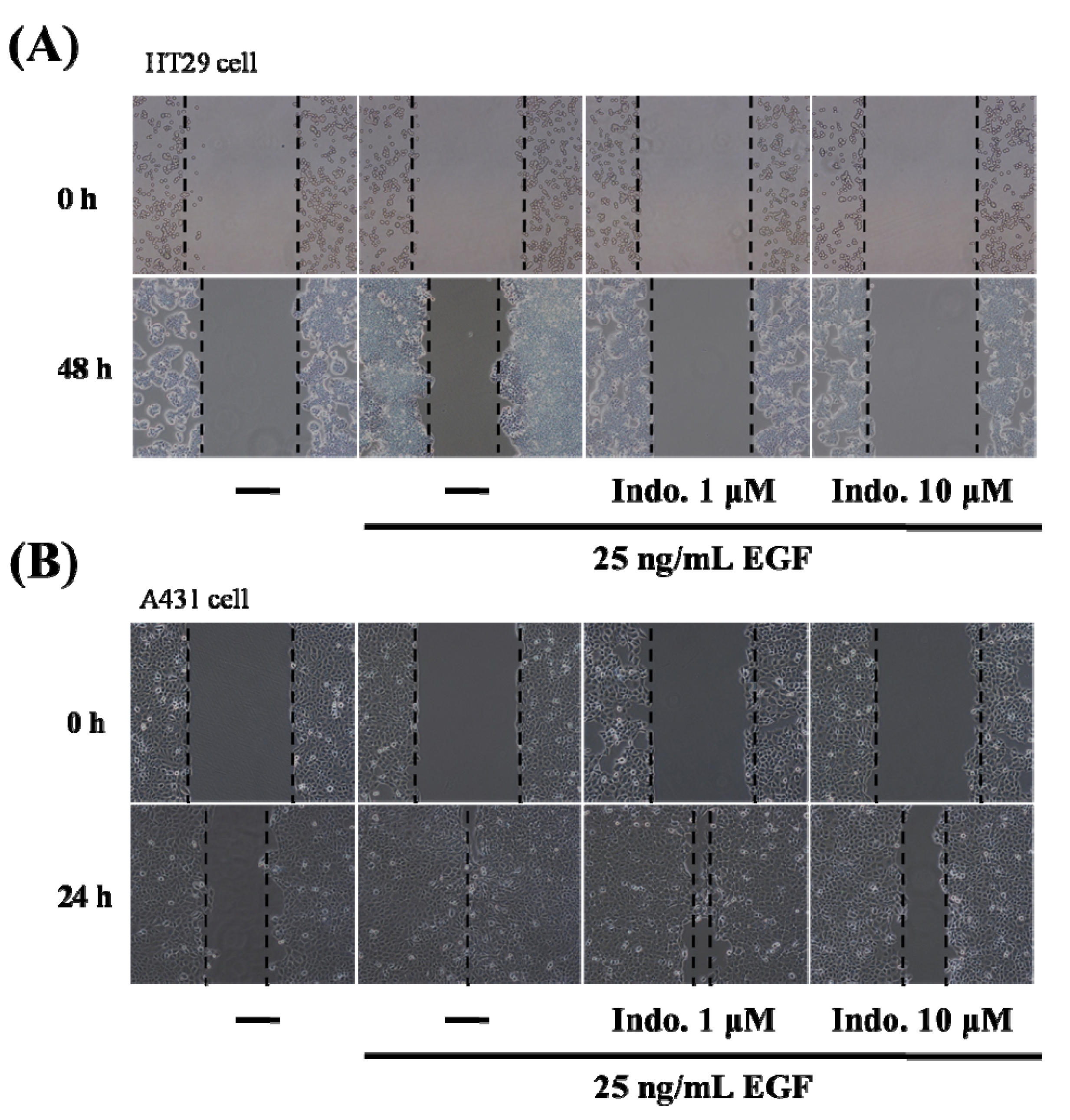
2.2. Indomethacin Inhibits Cancer Cell Migration
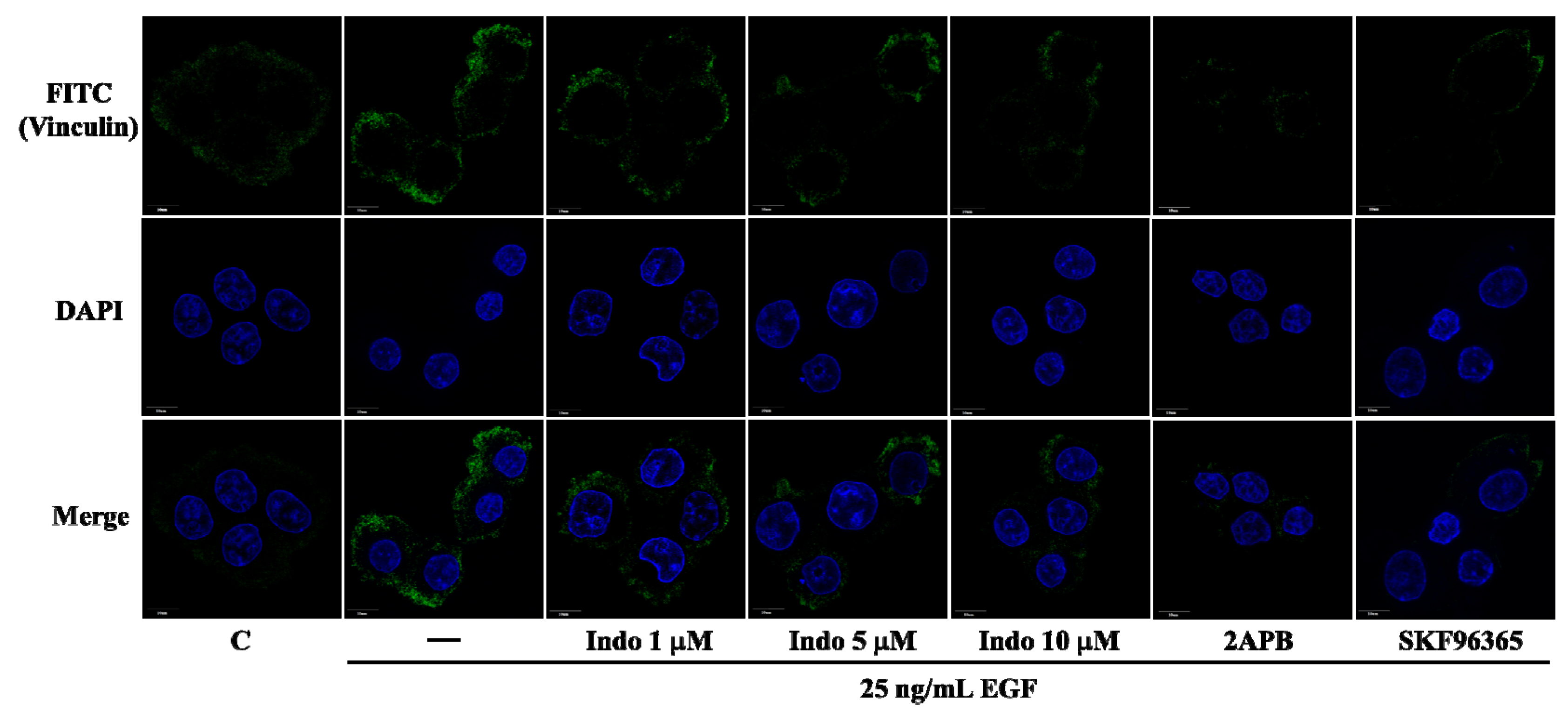
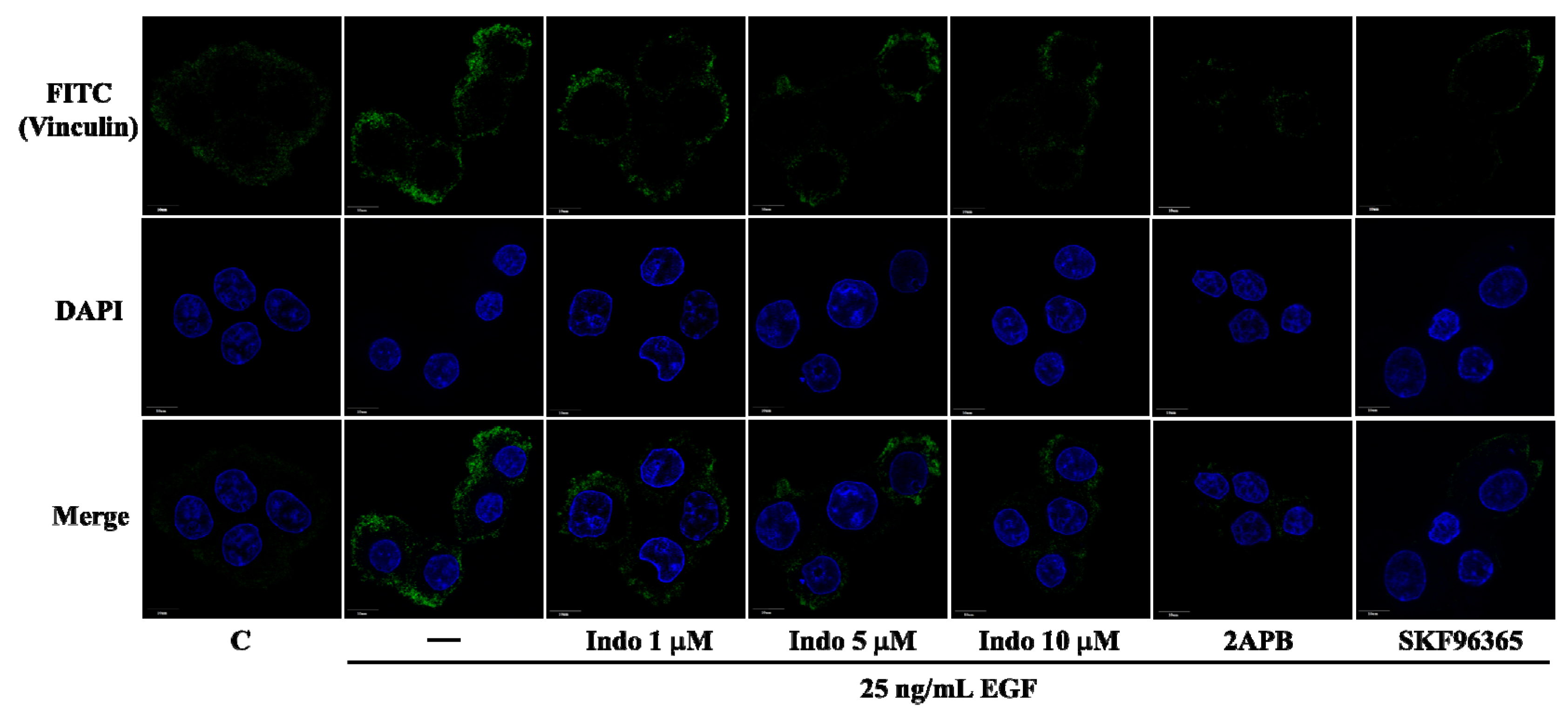
2.3. Effects of Indomethacin on Focal Complexes Formation
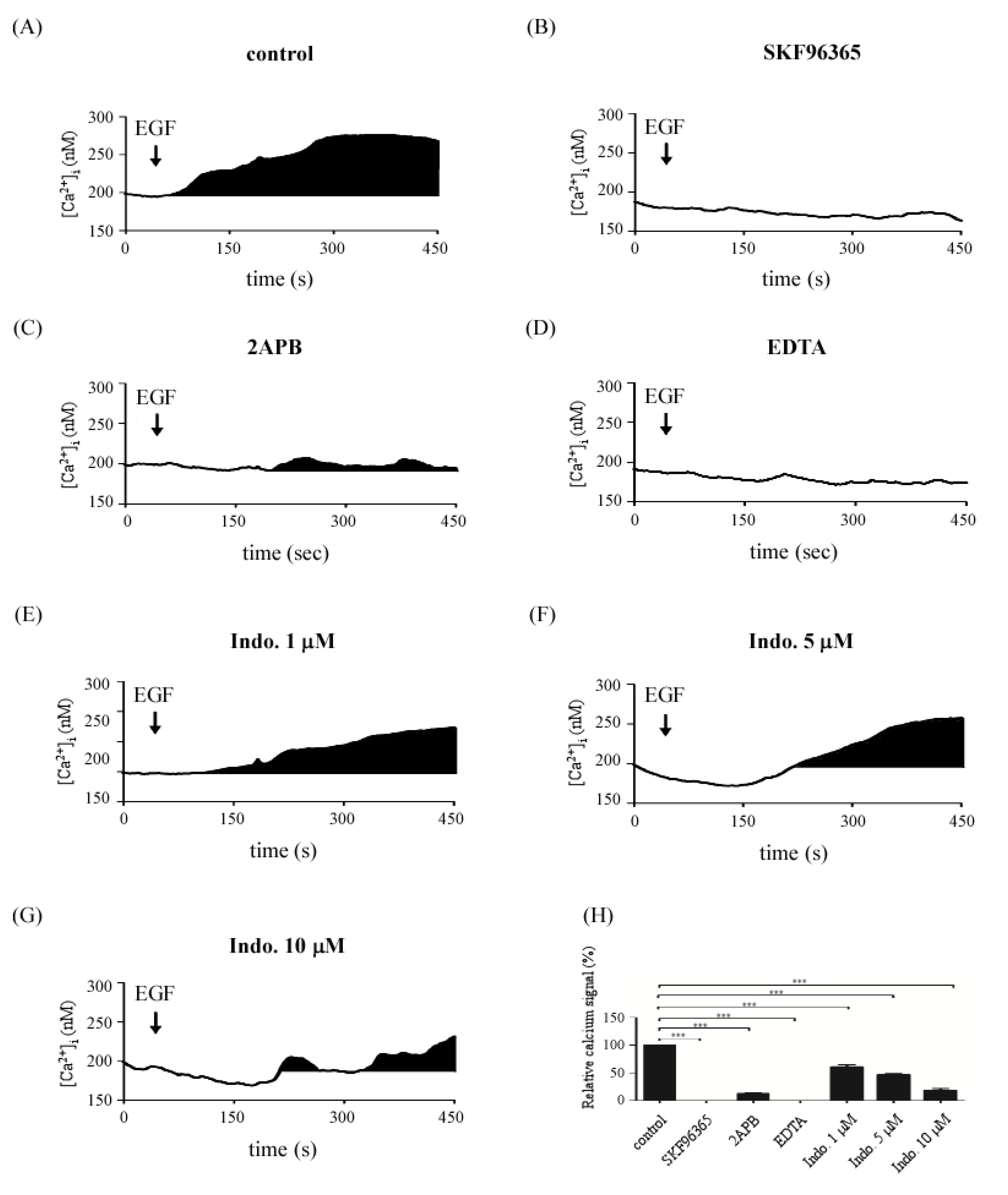
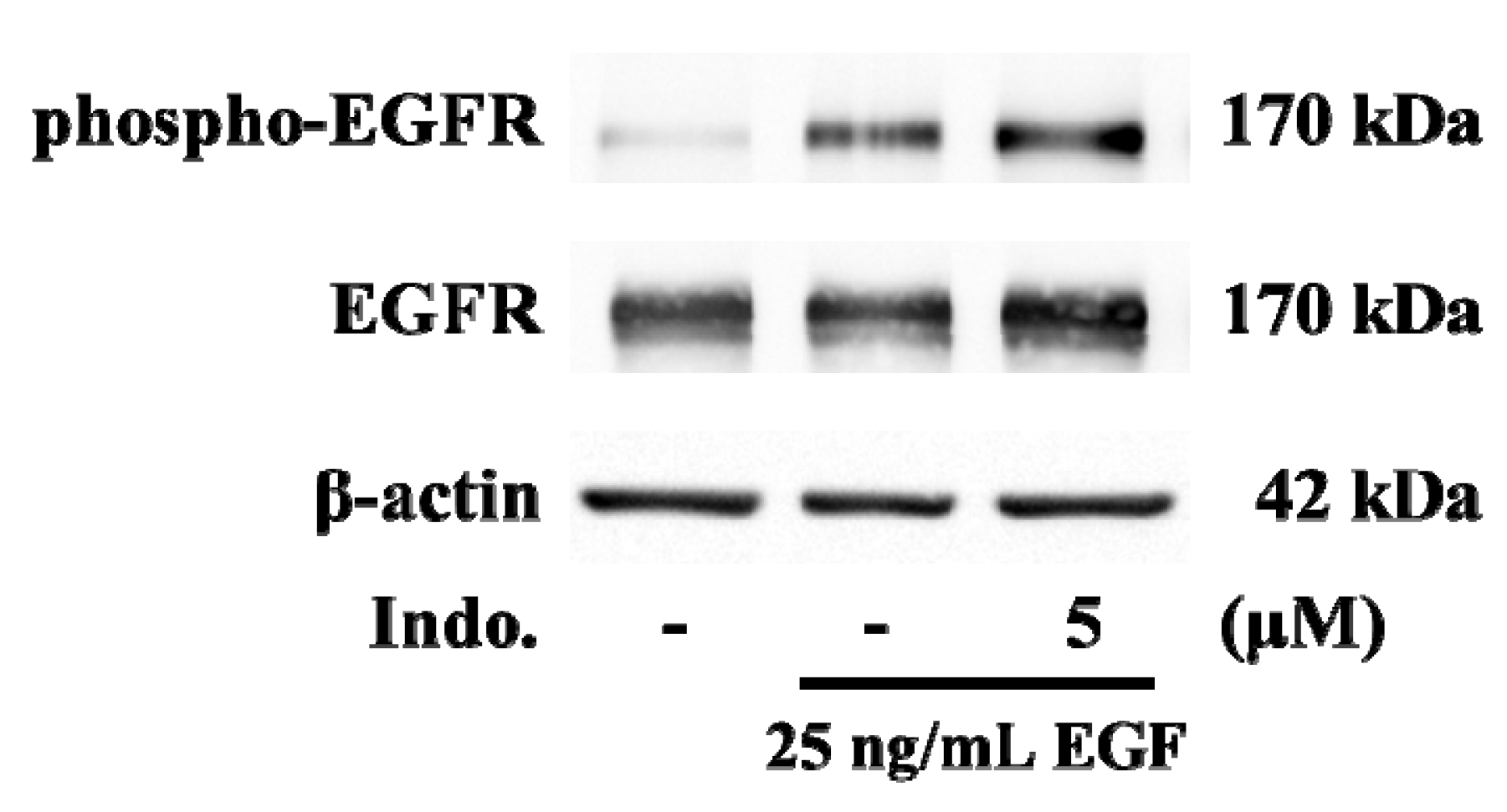
2.4. Effects of Indomethacin on EGF-Medicated Ca2+ Signaling
2.5. Effects of Indomethacin on EGF-Medicated EGFR Phosphorylation
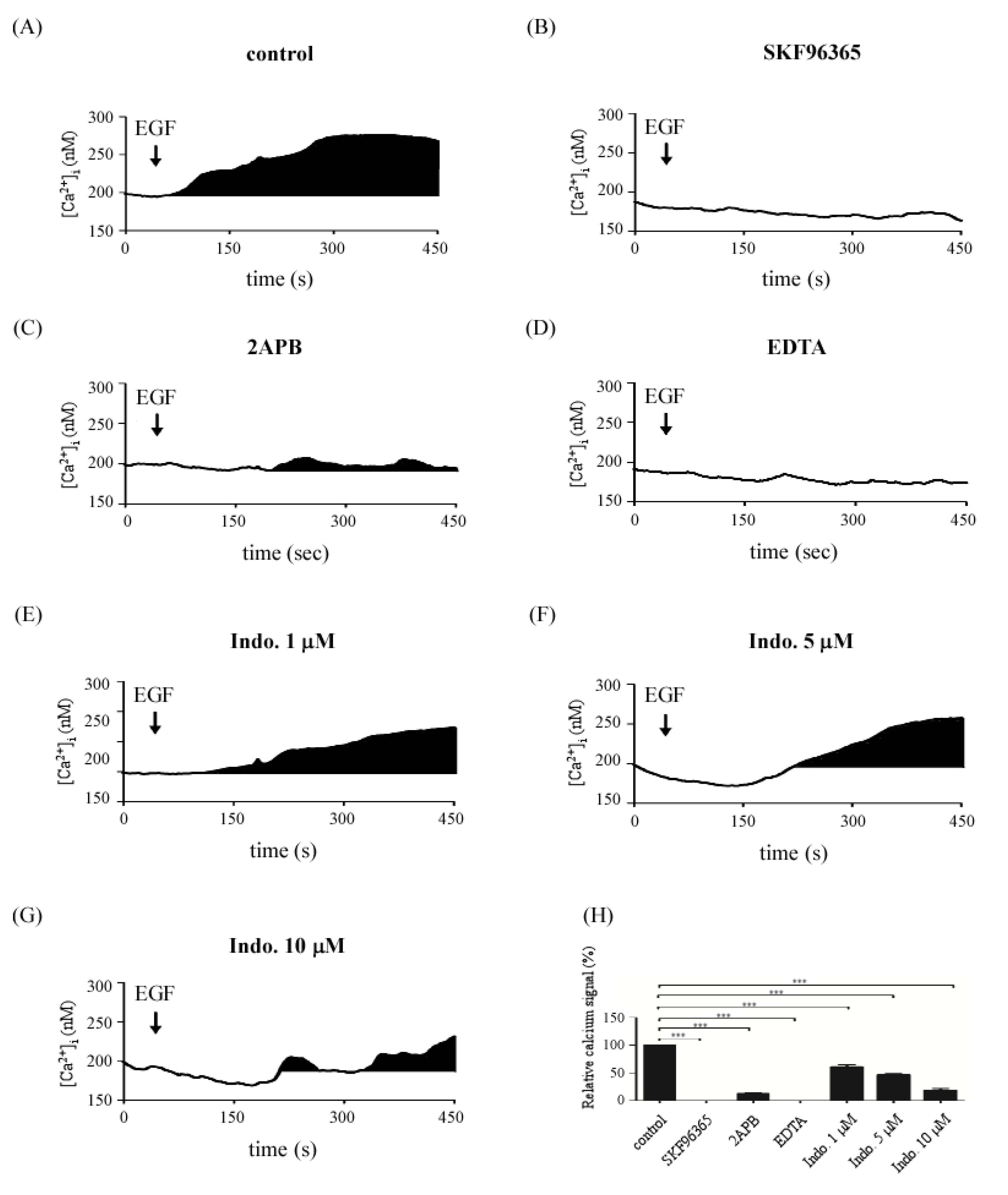
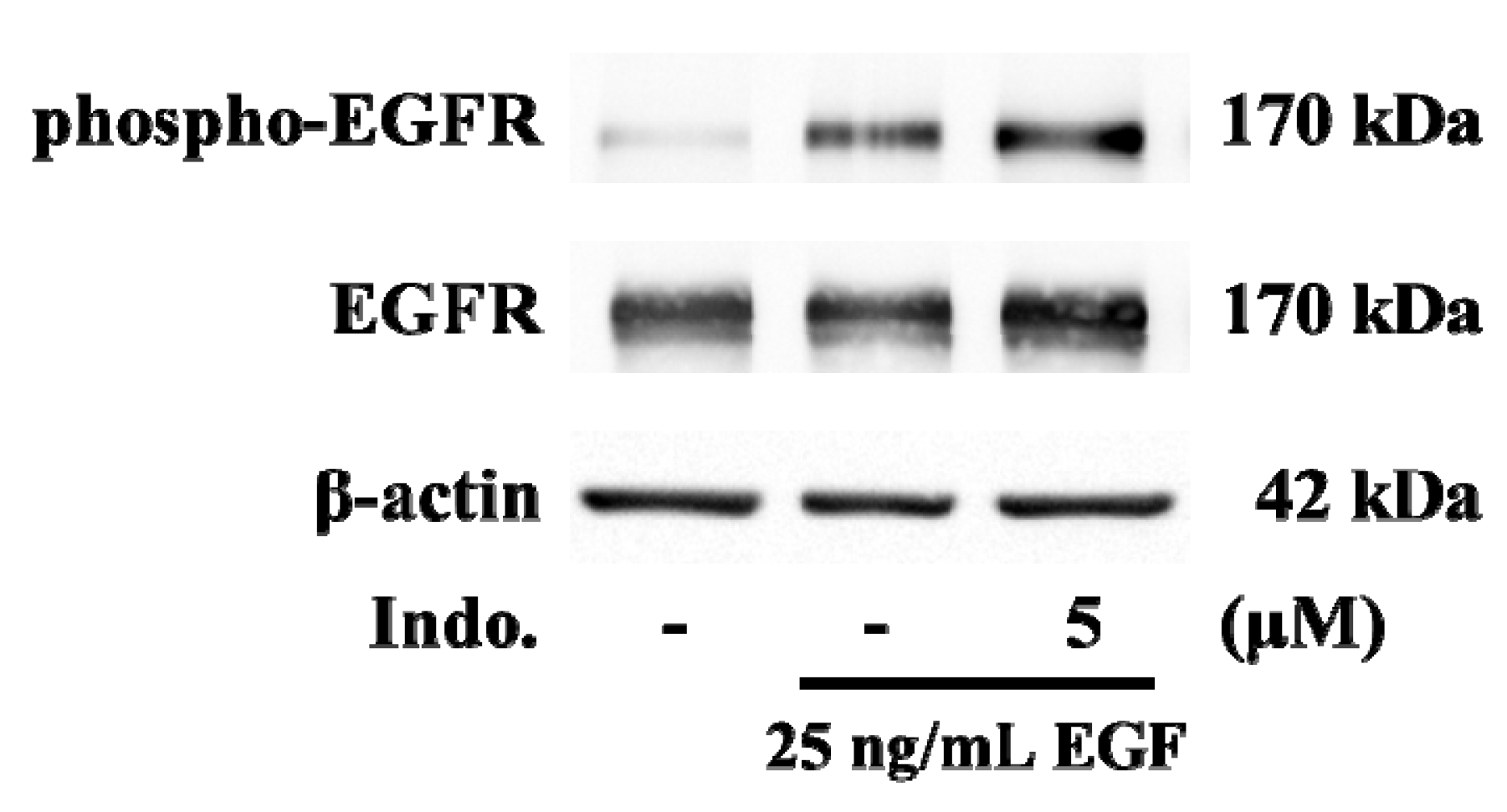
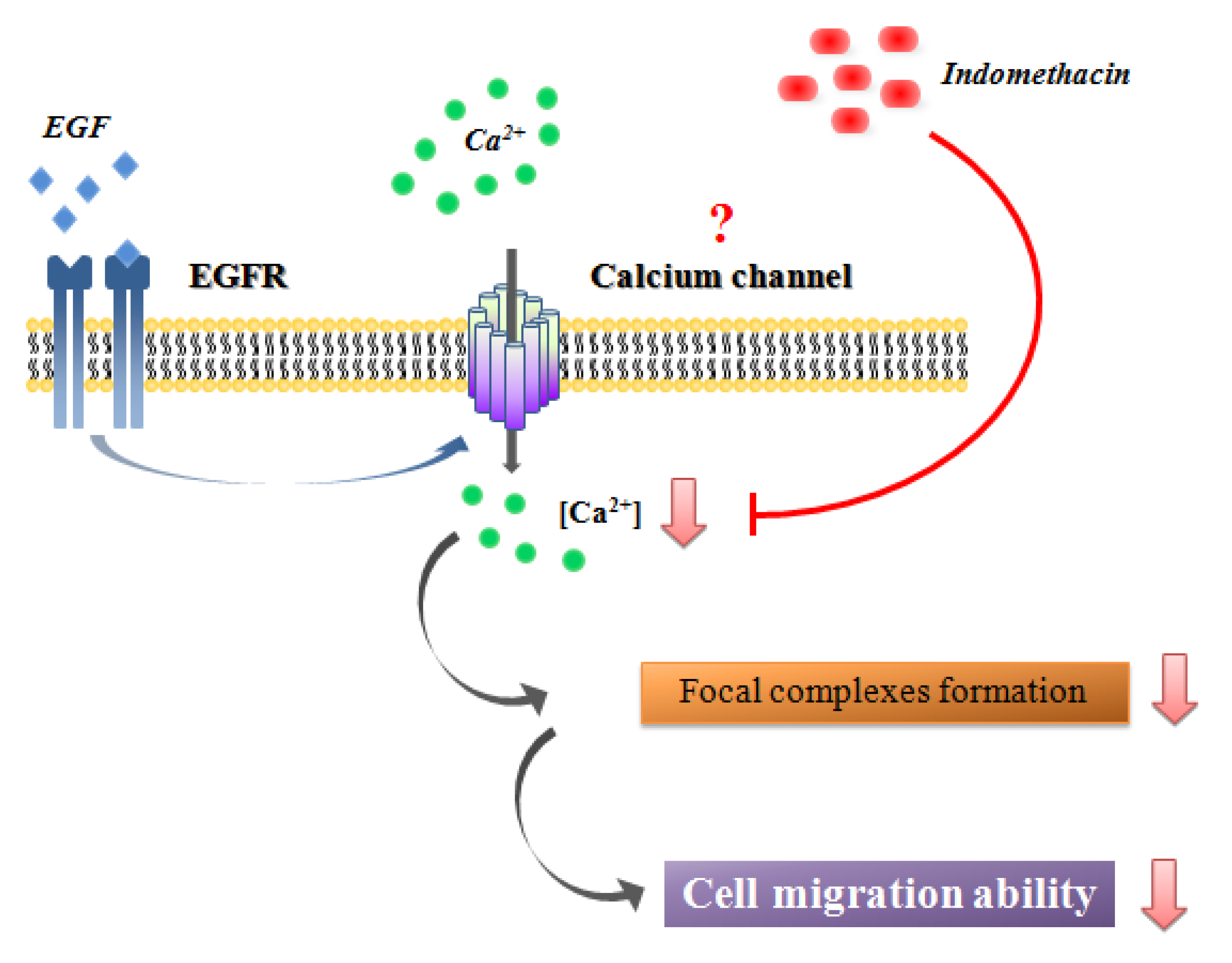
2.6. Discussion
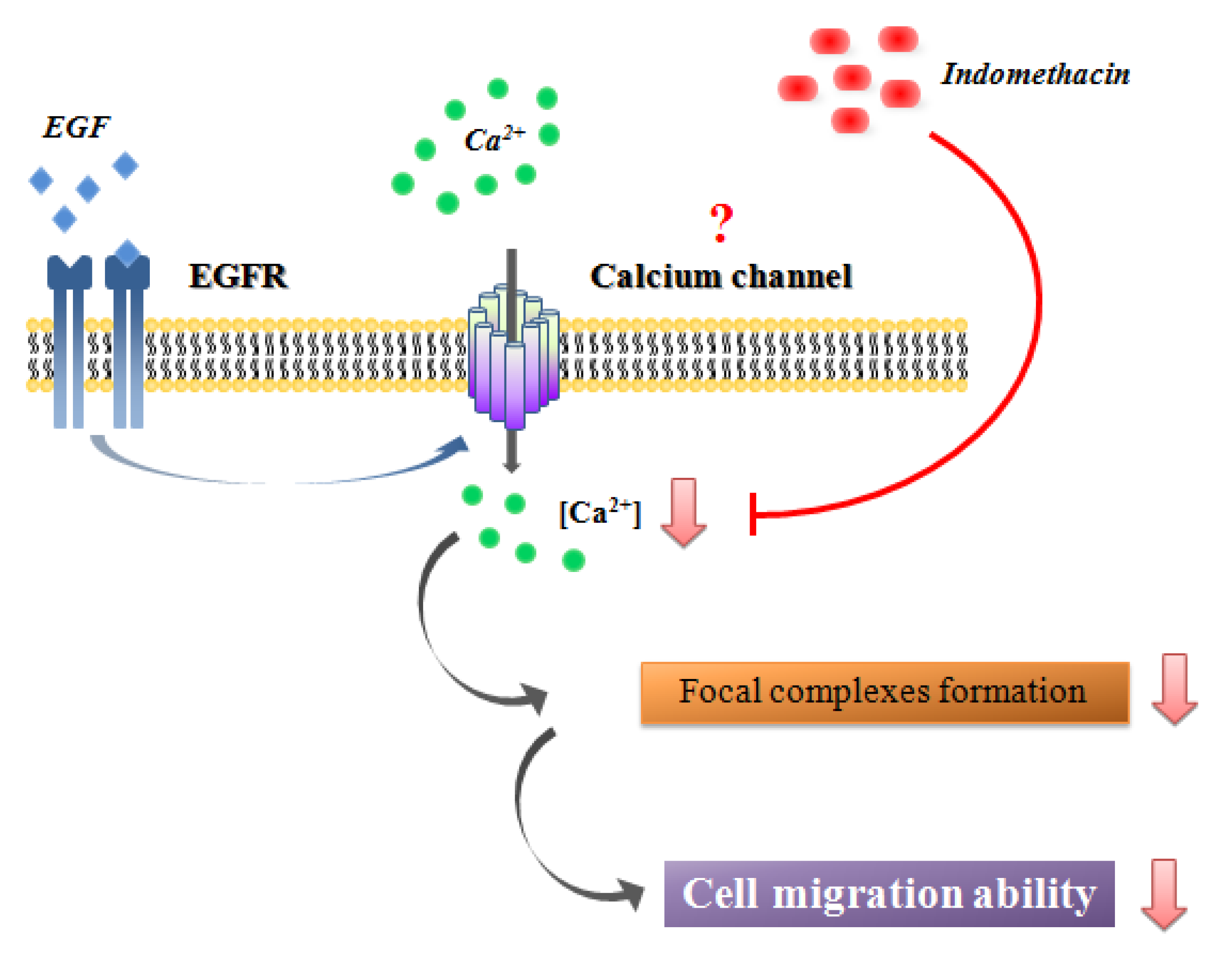
3. Experimental
3.1. Cell Culture
3.2. Calcium Imaging
3.3. Reverse Transcriptase PCR
3.4. Western Blotting
3.5. Wound Healing Assay
3.6. Immunofluorescence
3.7. Data Analysis
4. Conclusion
Acknowledgments
Conflicts of Interest
Reference
- Bayer, B.M.; Beaven, M.A. Evidence that indomethacin reversibly inhibits cell growth in the G1 phase of the cell cycle. Biochem. Pharmacol. 1979, 28, 441–443. [Google Scholar] [CrossRef]
- Hial, V.; De Mello, M.C.; Horakova, Z.; Beaven, M.A. Antiproliferative activity of anti-inflammatory drugs in two mammalian cell culture lines. J. Pharmacol. Exp. Ther. 1977, 202, 446–454. [Google Scholar]
- Takeuchi, K.; Tanaka, A.; Kato, S.; Amagase, K.; Satoh, H. Roles of COX inhibition in pathogenesis of NSAID-induced small intestinal damage. Clin. Chim. Acta 2010, 411, 459–466. [Google Scholar] [CrossRef]
- Eli, Y.; Przedecki, F.; Levin, G.; Kariv, N.; Raz, A. Comparative effects of indomethacin on cell proliferation and cell cycle progression in tumor cells grown in vitro and in vivo. Biochem. Pharmacol. 2001, 61, 565–571. [Google Scholar] [CrossRef]
- Vane, J.R.; Bakhle, Y.S.; Botting, R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 97–120. [Google Scholar] [CrossRef]
- Colville-Nash, P.R.; Gilroy, D.W. Cyclooxygenase enzymes as targets for therapeutic intervention in inflammation. Drug News Perspect. 2000, 13, 587–597. [Google Scholar] [CrossRef]
- Harris, R.E. Cyclooxygenase-2 (cox-2) and the inflammogenesis of cancer. Subcell Biochem. 2007, 42, 93–126. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, L.A.; Huerta-Alvarez, C. Reduced risk of colorectal cancer among long-term users of aspirin and nonaspirin nonsteroidal antiinflammatory drugs. Epidemiology 2001, 12, 88–93. [Google Scholar] [CrossRef]
- Harris, R.E.; Beebe-Donk, J.; Alshafie, G.A. Similar reductions in the risk of human colon cancer by selective and nonselective cyclooxygenase-2 (COX-2) inhibitors. BMC Cancer 2008, 8, 237. [Google Scholar] [CrossRef]
- Peng, X.; Nelson, E.S.; Maiers, J.L.; DeMali, K.A. New insights into vinculin function and regulation. Int. Rev. Cell. Mol. Biol. 2011, 287, 191–231. [Google Scholar] [CrossRef]
- Goldmann, W.H.; Auernheimer, V.; Thievessen, I.; Fabry, B. Vinculin, Cell mechanics and tumour cell invasion. Cell Biol. Int. 2013, 37, 397–405. [Google Scholar]
- Wang, J.Y.; Chen, B.K.; Wang, Y.S.; Tsai, Y.T.; Chen, W.C.; Chang, W.C.; Hou, M.F.; Wu, Y.C. Involvement of store-operated calcium signaling in EGF-mediated COX-2 gene activation in cancer cells. Cell Signal. 2012, 24, 162–169. [Google Scholar] [CrossRef]
- Schafer, C.; Rymarczyk, G.; Ding, L.; Kirber, M.T.; Bolotina, V.M. Role of molecular determinants of store-operated Ca(2+) entry (Orai1, phospholipase A2 group 6, and STIM1) in focal adhesion formation and cell migration. J. Biol. Chem. 2012, 287, 40745–40757. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef]
- Honegger, A.M.; Schmidt, A.; Ullrich, A.; Schlessinger, J. Evidence for epidermal growth factor (EGF)-induced intermolecular autophosphorylation of the EGF receptors in living cells. Mol. Cell. Biol. 1990, 10, 4035–4044. [Google Scholar]
- Singh, B.; Berry, J.A.; Shoher, A.; Ramakrishnan, V.; Lucci, A. COX-2 overexpression increases motility and invasion of breast cancer cells. Int. J. Oncol. 2005, 26, 1393–1399. [Google Scholar]
- Richardson, C.M.; Sharma, R.A.; Cox, G.; O'Byrne, K.J. Epidermal growth factor receptors and cyclooxygenase-2 in the pathogenesis of non-small cell lung cancer: potential targets for chemoprevention and systemic therapy. Lung Cancer 2003, 39, 1–13. [Google Scholar] [CrossRef]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef]
- Muller, M.R.; Rao, A. NFAT, immunity and cancer: a transcription factor comes of age. Nat. Rev. Immunol. 2010, 10, 645–656. [Google Scholar] [CrossRef]
- Gao, H.; Chen, X.; Du, X.; Guan, B.; Liu, Y.; Zhang, H. EGF enhances the migration of cancer cells by up-regulation of TRPM7. Cell Calcium 2011, 50, 559–568. [Google Scholar] [CrossRef]
- Oike, H.; Wakamori, M.; Mori, Y.; Nakanishi, H.; Taguchi, R.; Misaka, T.; Matsumoto, I.; Abe, K. Arachidonic acid can function as a signaling modulator by activating the TRPM5 cation channel in taste receptor cells. Biochim. Biophys. Acta 2006, 1761, 1078–1084. [Google Scholar]
- Chang, W.C.; Nelson, C.; Parekh, A.B. Ca2+ influx through CRAC channels activates cytosolic phospholipase A2, leukotriene C4 secretion, and expression of c-fos through ERK-dependent and -independent pathways in mast cells. FASEB J. 2006, 20, 2381–2383. [Google Scholar] [CrossRef]
- Huang, W.C.; Chai, C.Y.; Chen, W.C.; Hou, M.F.; Wang, Y.S.; Chiu, Y.C.; Lu, S.R.; Chang, W.C.; Juo, S.H.; Wang, J.Y.; et al. Histamine regulates cyclooxygenase 2 gene activation through Orai1-mediated NFkappaB activation in lung cancer cells. Cell Calcium 2011, 50, 27–35. [Google Scholar] [CrossRef]
- Kokoska, E.R.; Smith, G.S.; Miller, T.A. Nonsteroidal anti-inflammatory drugs attenuate proliferation of colonic carcinoma cells by blocking epidermal growth factor-induced Ca++ mobilization. J. Gastrointest. Surg. 2000, 4, 150–161. [Google Scholar] [CrossRef]
- Kokoska, E.R.; Smith, G.S.; Wolff, A.B.; Deshpande, Y.; Miller, T.A. Nonsteroidal anti-inflammatory drugs attenuate epidermal growth factor-induced proliferation independent of prostaglandin synthesis inhibition. J. Surg. Res. 1999, 84, 186–192. [Google Scholar] [CrossRef]
- Carrasco-Pozo, C.; Pastene, E.; Vergara, C.; Zapata, M.; Sandoval, C.; Gotteland, M. Stimulation of cytosolic and mitochondrial calcium mobilization by indomethacin in Caco-2 cells: modulation by the polyphenols quercetin, resveratrol and rutin. Biochim. Biophys. Acta 2012, 1820, 2052–2061. [Google Scholar]
- Batzer, A.G.; Rotin, D.; Urena, J.M.; Skolnik, E.Y.; Schlessinger, J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell. Biol. 1994, 14, 5192–5201. [Google Scholar]
- Okabayashi, Y.; Kido, Y.; Okutani, T.; Sugimoto, Y.; Sakaguchi, K.; Kasuga, M. Tyrosines 1148 and 1173 of activated human epidermal growth factor receptors are binding sites of Shc in intact cells. J. Biol. Chem. 1994, 269, 18674–18678. [Google Scholar]
- Hsu, W.L.; Tsai, M.H.; Lin, M.W.; Chiu, Y.C.; Lu, J.H.; Chang, C.H.; Yu, H.S.; Yoshioka, T. Differential effects of arsenic on calcium signaling in primary keratinocytes and malignant (HSC-1) cells. Cell Calcium 2012, 52, 161–169. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guo, Y.-C.; Chang, C.-M.; Hsu, W.-L.; Chiu, S.-J.; Tsai, Y.-T.; Chou, Y.-H.; Hou, M.-F.; Wang, J.-Y.; Lee, M.-H.; Tsai, K.-L.; et al. Indomethacin Inhibits Cancer Cell Migration via Attenuation of Cellular Calcium Mobilization. Molecules 2013, 18, 6584-6596. https://doi.org/10.3390/molecules18066584
Guo Y-C, Chang C-M, Hsu W-L, Chiu S-J, Tsai Y-T, Chou Y-H, Hou M-F, Wang J-Y, Lee M-H, Tsai K-L, et al. Indomethacin Inhibits Cancer Cell Migration via Attenuation of Cellular Calcium Mobilization. Molecules. 2013; 18(6):6584-6596. https://doi.org/10.3390/molecules18066584
Chicago/Turabian StyleGuo, Yuh-Cherng, Che-Mai Chang, Wen-Li Hsu, Siou-Jin Chiu, Yao-Ting Tsai, Yii-Her Chou, Ming-Feng Hou, Jaw-Yan Wang, Mei-Hsien Lee, Ke-Li Tsai, and et al. 2013. "Indomethacin Inhibits Cancer Cell Migration via Attenuation of Cellular Calcium Mobilization" Molecules 18, no. 6: 6584-6596. https://doi.org/10.3390/molecules18066584
APA StyleGuo, Y.-C., Chang, C.-M., Hsu, W.-L., Chiu, S.-J., Tsai, Y.-T., Chou, Y.-H., Hou, M.-F., Wang, J.-Y., Lee, M.-H., Tsai, K.-L., & Chang, W.-C. (2013). Indomethacin Inhibits Cancer Cell Migration via Attenuation of Cellular Calcium Mobilization. Molecules, 18(6), 6584-6596. https://doi.org/10.3390/molecules18066584