Abstract
In this study, a series of conformationally restricted thieno[3,2-d]pyrimidinones, thieno[3,2-d]pyrimidines and quinazolinones was designed and synthesized with the goal of improving the biological activity as 17β-hydroxysteroid dehydrogenase type 2 inhibitors of the corresponding amidothiophene derivatives. Two moderately active compounds were discovered and this allowed the identification of the biologically active open conformer as well as the extension of the enzyme binding site characterisation.
1. Introduction
Osteoporosis is a systemic skeletal disease characterised by reduction of bone mineral density and deterioration of the bone microarchitecture leading to an increase in bone fragility and thus elevated bone fracture risk [1]. The decrease in active steroid levels like estradiol (E2) and testosterone (T) is often associated with the development of osteoporosis (e.g., in women at menopause [2]). As this disease affects elderly men and especially women and the population is getting older, it has become a serious public health issue and new therapeutic alternatives have to be developed.
17β-Hydroxysteroid dehydrogenase type 2 (17β-HSD2, (EC1.1.1.51) [3]) belongs to the short-chain steroid dehydrogenase/reductase protein family (SDR). This enzyme catalyses the NAD+-dependent oxidation of the potent estradiol (E2) into estrone (E1) as well as testosterone (T) into androstenedione (Δ4-dione). 17β-HSD2 is a membrane-associated protein located in the endoplasmic reticulum and contains 387 amino acids. This enzyme is highly expressed in a wide variety of tissues such as placenta, liver, small intestine, endometrium, urinary tract and to a lesser extend also in kidney, pancreas, colon, uterus, breast, prostate and bone. The 3D-structure of this enzyme remains unknown.
Few 17β-HSD2 inhibitors have been described in the literature [4,5]. The concept of improving bone quality by inhibiting 17β-HSD2 has been validated by Bagi et al. in a monkey osteoporosis model [2] using a cis-pyrrolidinone derivative as 17β-HSD2 inhibitor [6,7,8]. However, the treatment with this compound led to few effects on bones. Therefore, it is necessary to develop new potent 17β-HSD2 inhibitors with better in vivo efficacy, which are selective toward 17β-HSD1, the isoenzyme catalysing the reverse reaction, the reduction of E1 into E2 (Figure 1).
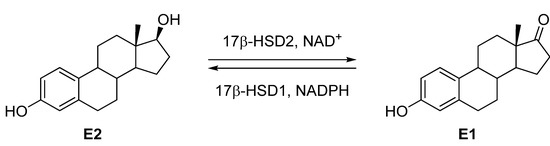
Figure 1.
Interconversion of Estradiol (E2) to Estrone (E1) by 17β-HSD2 and 17β-HSD1.
Figure 1.
Interconversion of Estradiol (E2) to Estrone (E1) by 17β-HSD2 and 17β-HSD1.
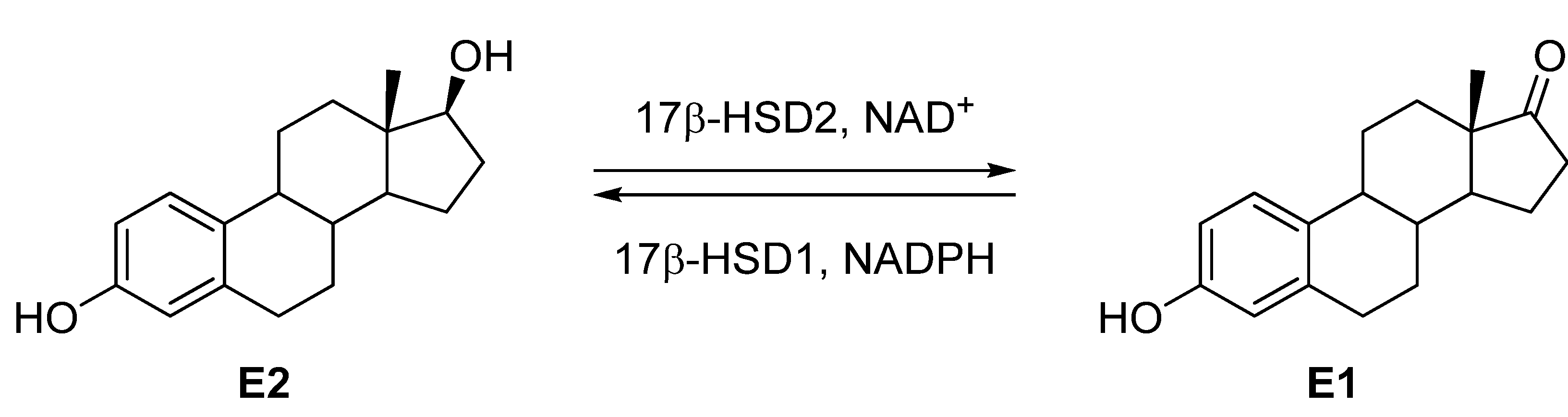
Our group has been working for 20 years on the development of inhibitors of steroidogenic enzymes (e.g., 5α-reductase [9], aromatase [10,11], CYP17 [12,13], CYP11B2 [14,15,16,17], CYP11B1 [18,19]) and in the last years a lot of experience was gained in the design of potent and selective inhibitors of 17β-HSD1 [20,21,22,23,24,25], and 17β-HSD2 [26,27,28,29,30,31,32]. Recently, we described [27] N-(3-methoxybenzyl)-5-(3-methoxyphenyl)-N-methylthiophene-2-carboxamide (compound A, Table 1) and N-(3-hydroxybenzyl)-5-(3-hydroxyphenyl)-N-methylthiophene-2-carboxamide (compound B, Table 1) as 17β-HSD2 inhibitors (63% and 70% 17β-HSD2 inhibition at a concentration of 1 µM, for A and B, respectively). Both compounds show a good selectivity toward 17β-HSD1 (0% and 21% inhibition at 1 µM, respectively). Compounds A and B consist of a central thiophene moiety connected to a substituted phenyl ring on one side and to a substituted benzyl on the other side linked to the thiophene via a methylated carboxamide bond. A and B show some flexibility at this amide bond, allowing for several conformations.
In the 1H-NMR spectra of A or B it can be seen that the peaks corresponding to N-Me group (3.15/3.16 ppm) and to the CH2 of the benzyl group (4.76/4.79 ppm) appear as very broad singlets, indicating that in solution at room temperature A and B exist as a mixture of several conformers in equilibrium. However, when the compounds bind to the enzyme binding site, only one conformer is expected to be present and therefore only one conformer is biologically active.

Table 1.
In vitro 17β-HSD2 and 17β-HSD1 inhibitory potencies of thiophene-2-carboxamide derivatives A and B.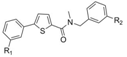
| Cmpd | R1 | R2 | Percentage of inhibition at 1µM a | |
|---|---|---|---|---|
| 17β-HSD2 b | 17β-HSD1 c | |||
| A | OMe | OMe | 63% | 0% |
| B | OH | OH | 70% | 21% |
a Mean value of three determinations, standard deviation less than 15%. b Human placental, microsomal fraction, substrate E2 [500 nM], cofactor NAD+ [1500 nM]. c Human placental, cytosolic fraction, substrate E1 [500 nM], cofactor NADH [500 nM].
Rigidification is an often applied method to increase the potency of active molecules. In addition to freezing a compound in one conformation when several exist, it is also a good strategy to identify the active conformation of a compound when it binds in the active site of a protein. Rigidification can be a useful tool for structural optimisation and also to characterize the binding site in the protein.
The goal of this study was the structural optimization of the 17β-HSD2 inhibitors A and B, using the rigidification strategy as well as the identification of the active conformation taken by A and B in the protein. In this study the design and the synthesis of new rigidified 17β-HSD2 inhibitors, mimicking the different geometries which can be adopted by the conformers derived from A and B as well as the biological evaluation of the synthesized compounds (17β-HSD2 potency as well as selectivity toward 17β-HSD1) are reported.
2. Results and Discussion
Compounds A and B show the highest flexibility at the amide bond, involving two sigma bonds (σ1 between the thiophene and the carbonyl moiety and σ2 between the carbonyl and the amino group, Figure 2). Since σ bonds are freely rotatable, rotation about the two sigma bonds (σ1 and σ2) of the amide group leads to four different conformers I-IV as depicted in Figure 2. The conformational freezing of A and B can be achieved by cyclisation leading to thiophene- or phenyl-fused derivatives: the thieno[3,2-d]pyrimidin-4-one analogues 1a–b and 3a–d, locked conformers I and III, respectively and quinazolin-4-one derivatives 2a–b and 4a–b,locked conformers II and IV, respectively (Figure 3). As restrained conformers II and IV are not possible with the thiophene core (S is bivalent), the heterocycle was replaced by the bioisosteric phenyl ring (Figure 3). In order to investigate the necessity of the benzyl group and its position, compounds 5a–d were designed (conformer I or III), and synthesized (Figure 3).
Figure 2.
Four major conformers of thiophene-2-carboxamide derivatives A and B.
Figure 2.
Four major conformers of thiophene-2-carboxamide derivatives A and B.
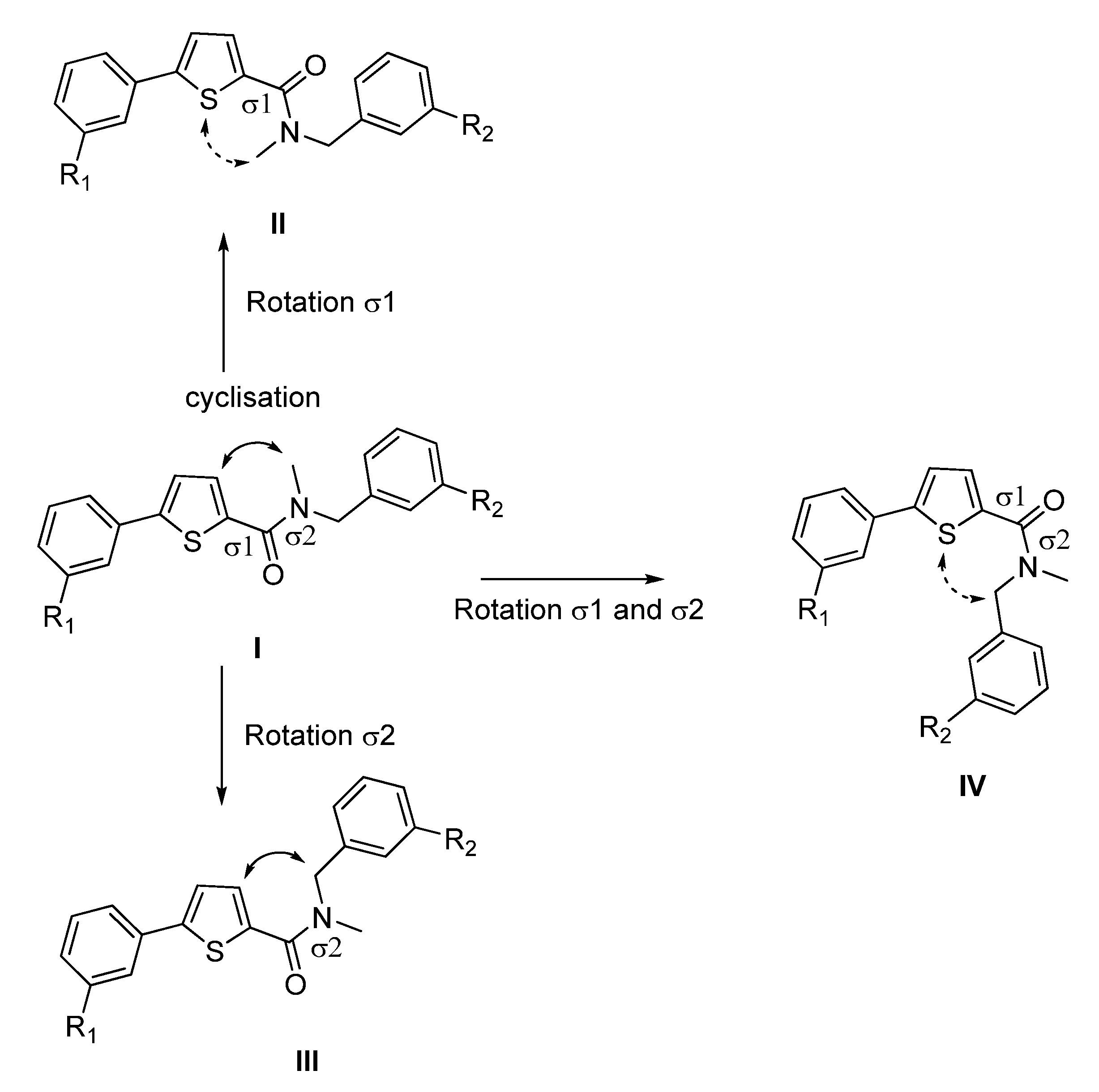
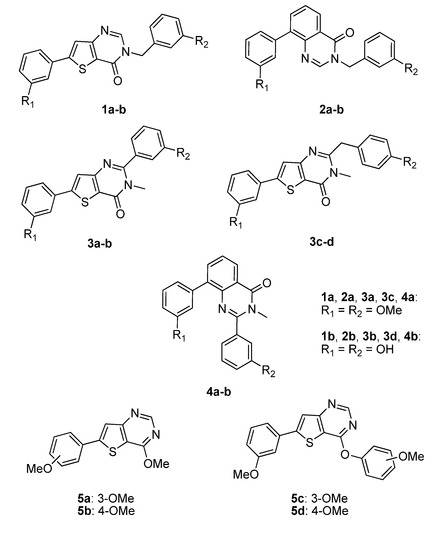
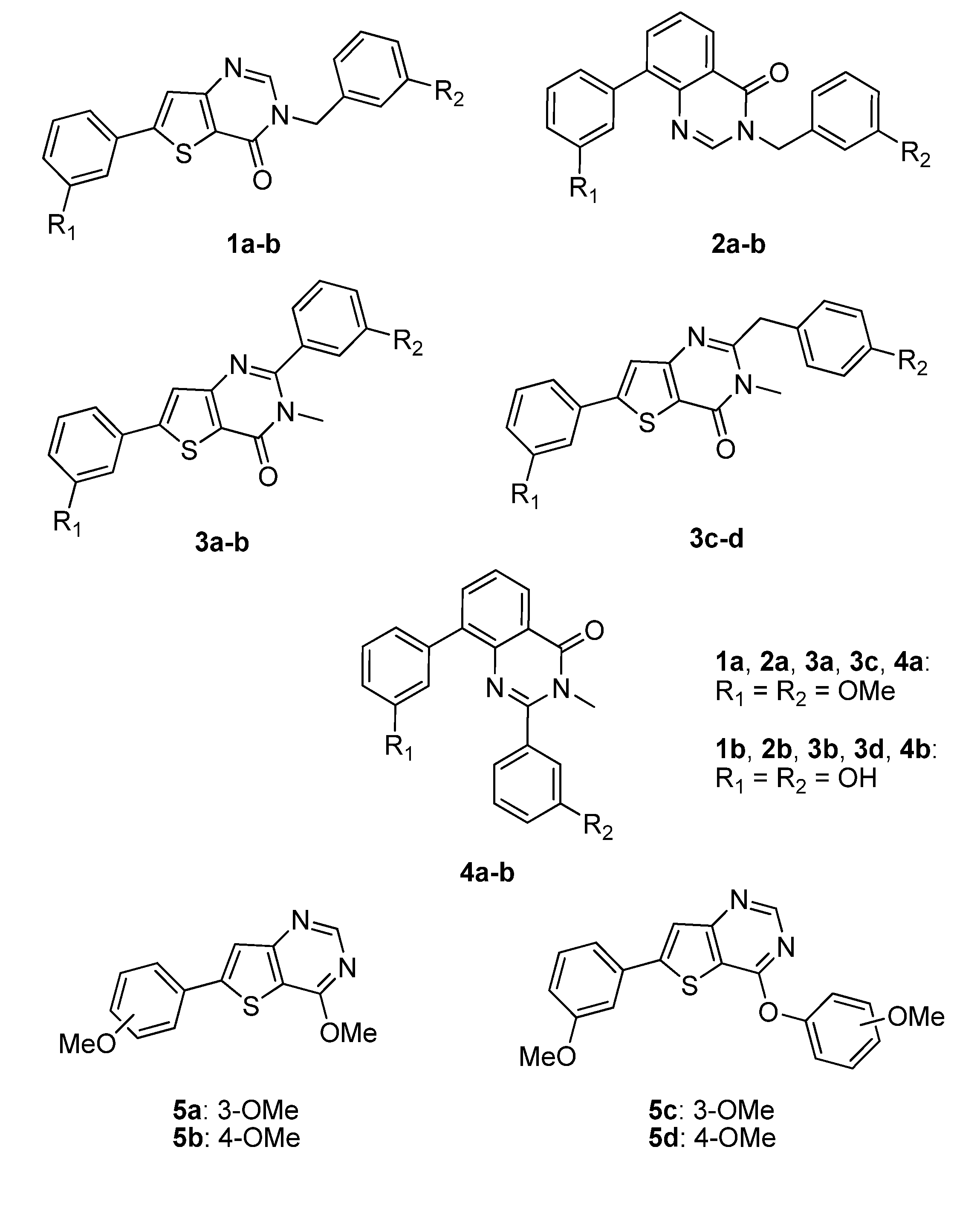
Figure 3.
Thieno[3,2-d]pyrimidin-4-ones 1a–b, 3a–d and thieno[3,2-d]pyrimidine 5a–d as conformationally restrained analogues of conformer I and III, and quinazolin-4-one 2a–b, 4a–b as conformationally restrained analogues of conformer II and IV, respectively.
Figure 3.
Thieno[3,2-d]pyrimidin-4-ones 1a–b, 3a–d and thieno[3,2-d]pyrimidine 5a–d as conformationally restrained analogues of conformer I and III, and quinazolin-4-one 2a–b, 4a–b as conformationally restrained analogues of conformer II and IV, respectively.
Compounds 1a–b were synthesized by a two- or a three-step procedure, respectively, as depicted on Scheme 1, starting from compound 6. The starting materials methyl 3-amino-5-(3-methoxyphenyl)- thiophene-2-carboxylate (6) and 3-amino-5-(3-methoxyphenyl)thiophene-2-carboxamide (11a) were synthesized using the multi-step procedure well described in the literature [33]. The condensation of molecule 6 with N,N-dimethylformamide dimethyl acetal (DMF-DMA) gave methyl 3-dimethyl-aminomethylidene-amino-5-(3-methoxyphenyl)thiophene-2-carboxylate (7) in very good yield [34] which was used directly in the next step without purification to afford compound 1a. The condensation of 6-(3-methoxyphenyl)-4H-thieno[3,2-d][1,3]oxazin-4-one (6a) with 3-methoxybenzyl amine as an alternative route to produce 1a failed. After O-demethylation using boron trifluoride methyl sulfide complex BF3.SMe2, the thieno[3,2-d]pyrimidin-4(3H)-one 1b was obtained (Scheme 1).
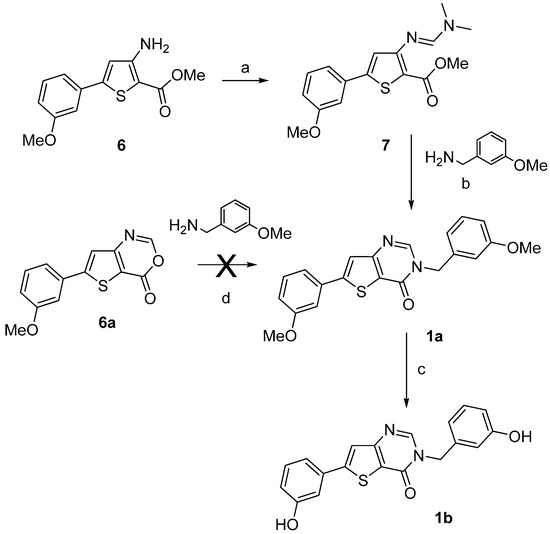
Scheme 1.
Synthesis of the thieno[3,2-d]pyrimidin-4-ones derivatives 1a–b.
Scheme 1.
Synthesis of the thieno[3,2-d]pyrimidin-4-ones derivatives 1a–b.
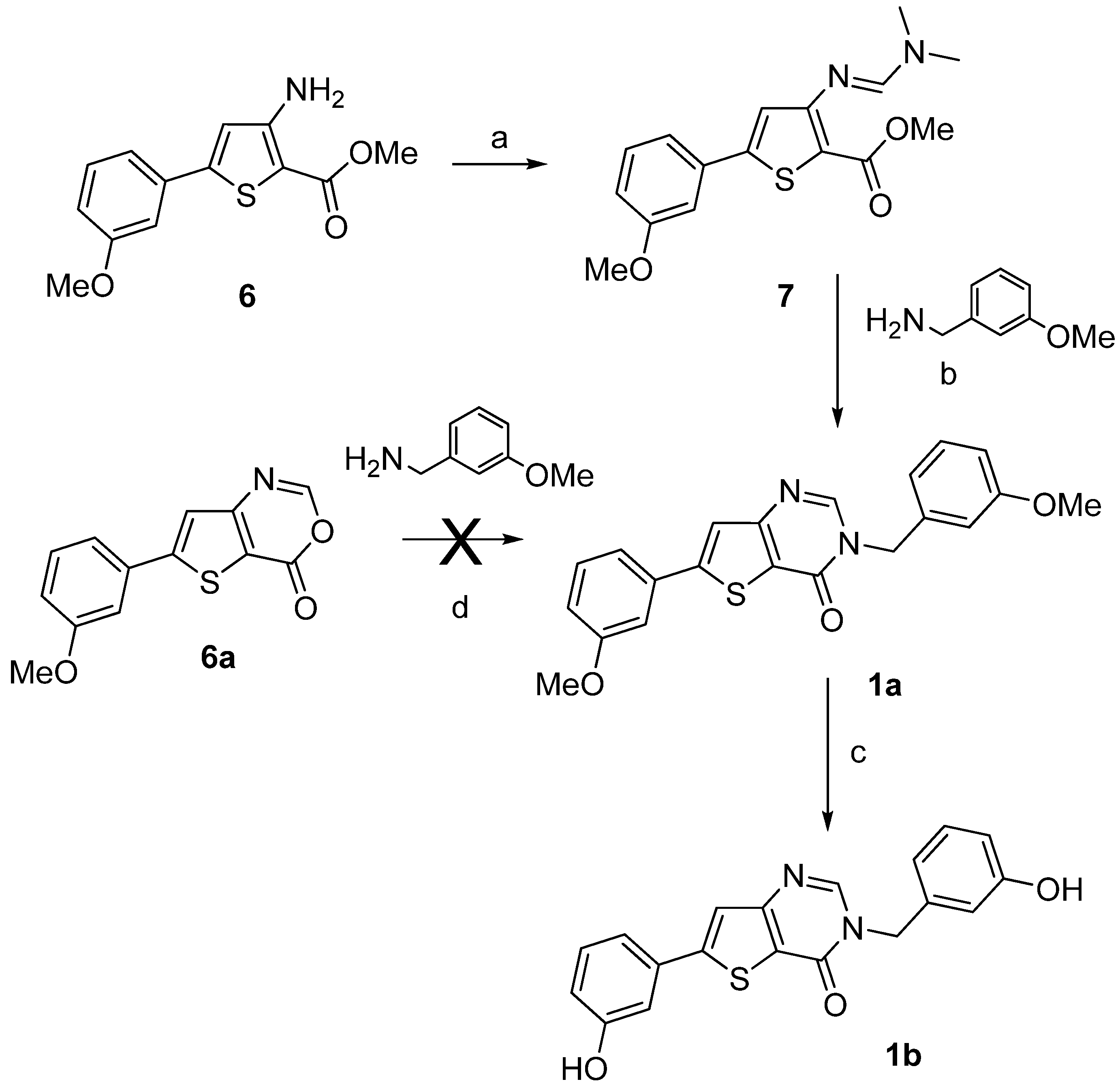
Reagents and conditions: (a) DMF-DMA, EtOH, microwave irradiation (100 °C, 80 W, 15 min), 98%; (b) DMF, microwave irradiation (100 °C, 80 W, 15 min), 31%; (c) BF3.SMe2, CH2Cl2, rt, 82%; (d) MeOH, reflux.
Synthesis of derivatives 2a–b was achieved by a three- or a four-step reaction pathway starting from the 2-amino-3-bromobenzoic acid (8) as shown on Scheme 2. The starting material 8 was prepared as described in the literature [35]. It reacted with triethyl orthoformate [CH(OEt)3] to give the cyclized benzoxazin-4-one 9, which was condensed with 3-methoxybenzylamine to afford the quinazolin-4-one 10 (Scheme 2) [36]. Suzuki-Miyaura cross coupling led to the methoxy derivative 2a in very good yield. Ether cleavage using the above described procedure afforded the hydroxylated quinazolin-4-one 2b. It is worth mentioning, that the condensation of methyl 2-bromo-6-{[(1E)-(dimethylamino)methylidene]amino}benzoate (9a) with the 3-methoxybenzyl amine, which was successfully applied in the synthesis of the thiophene fused 1a, failed to react to the quinazolin-4-one 10.
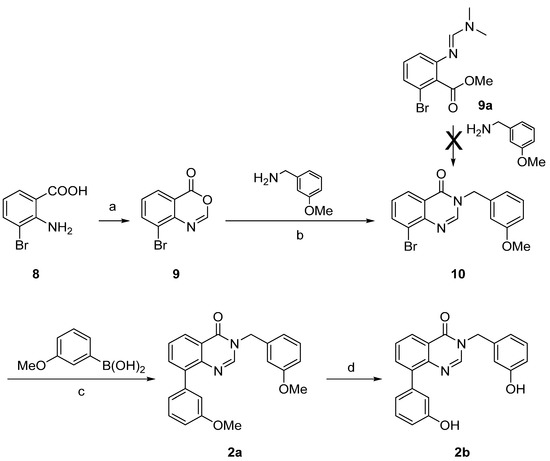
Scheme 2.
Synthesis of the quinazolin-4-ones derivatives 2a–b.
Scheme 2.
Synthesis of the quinazolin-4-ones derivatives 2a–b.
Reagents and conditions: (a) CH(OEt)3, microwave irradiation (160 °C, 80 W, 10 min), 61%; (b) dry toluene, reflux, 66%; (c) Pd(PPh3)4, Cs2CO3, DME/EtOH/H2O (1:1:1), microwave irradiation (150 °C, 150 W, 15 min), 98%; (d) BF3.SMe2, CH2Cl2, rt, 97%.
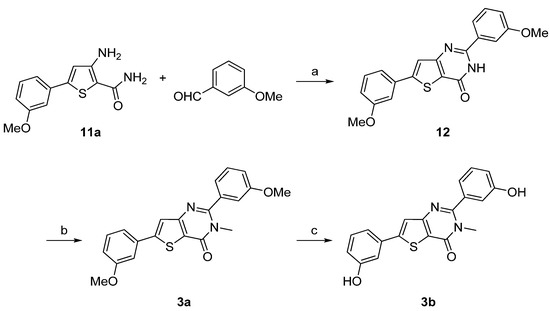
Synthesis of 3a–d was performed starting from the 3-amino-5-arylthiophene (11a) as depicted in Scheme 3 and Scheme 4 following a two- or a three-step procedure. Compound 11a was condensed with 3-methoxybenzaldehyde in presence of concentrated hydrochloric acid to give first the hydrogenated thienopyrimidinone which was then autoxidized to the corresponding thienopyrimidinone 12 on heating in acidic medium [37]. Then, N-methylation was performed as previously described to afford 3a. Ether cleavage led to the thieno[3,2-d]pyrimidin-4(3H)-one 3b (Scheme 3).
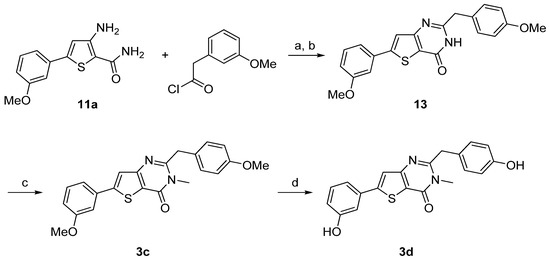
3-Amino-5-arylthiophene (11a) was also condensed with 3-methoxyphenylacetyl chloride in presence of triethylamine. The cyclization step was realized with sodium hydroxide to obtain 13. N-methylation was performed using methyliodide in presence of potassium carbonate as base to give 3c and ether cleavage was accomplished using the same procedure as described above providing 3d in very good yield (Scheme 4).
Scheme 3.
Synthesis of the thieno[3,2-d]pyrimidin-4-ones derivatives 3a–b.
Scheme 3.
Synthesis of the thieno[3,2-d]pyrimidin-4-ones derivatives 3a–b.
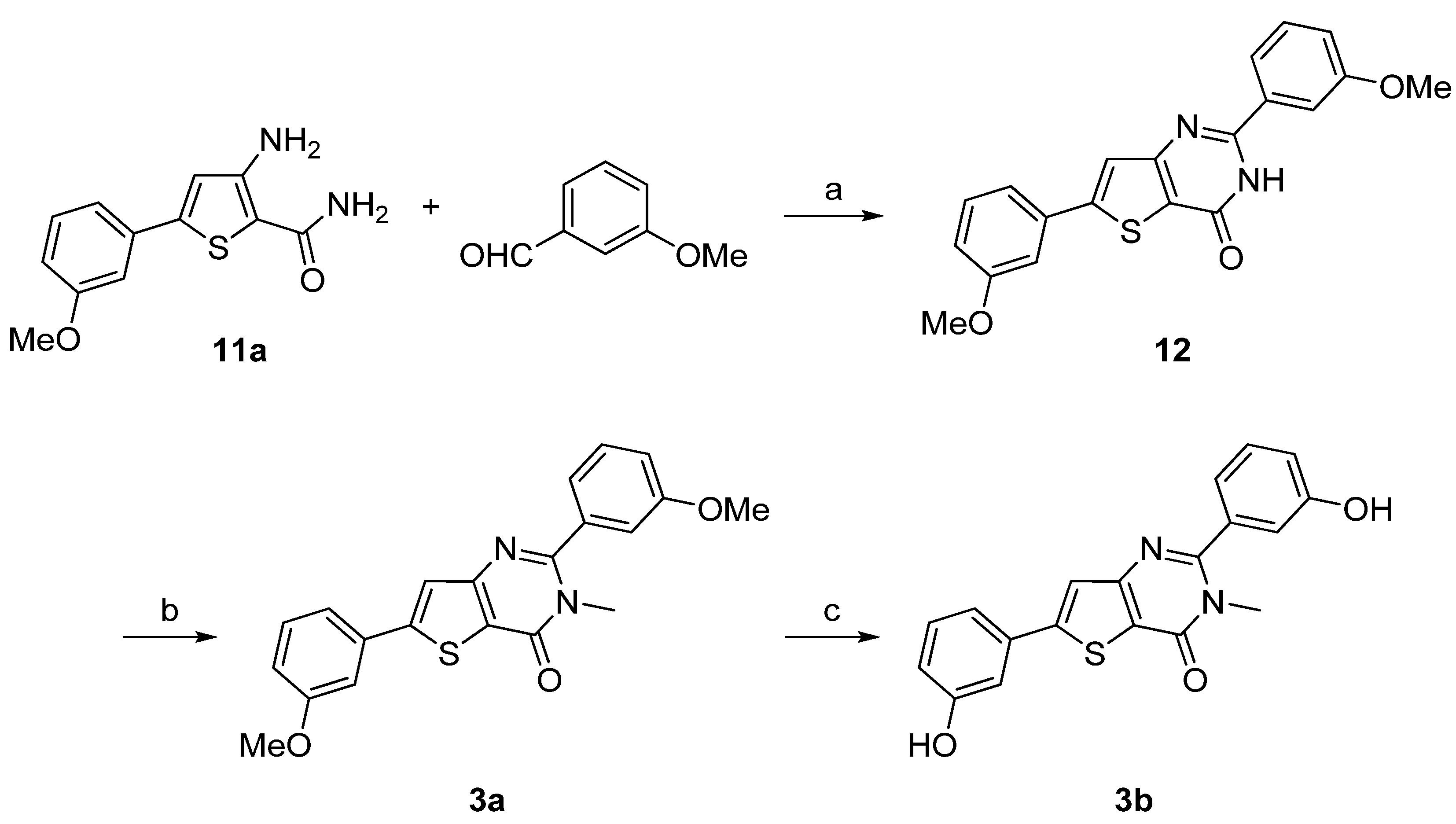
Reagents and conditions: (a) HCl 37%, MeOH, reflux, 38%; (b) MeI, K2CO3, CH3CN, reflux, 48%; (c) BF3.SMe2, CH2Cl2, rt, 57%.
Scheme 4.
Synthesis of the thieno[3,2-d]pyrimidin-4-ones derivatives 3c–d.
Scheme 4.
Synthesis of the thieno[3,2-d]pyrimidin-4-ones derivatives 3c–d.
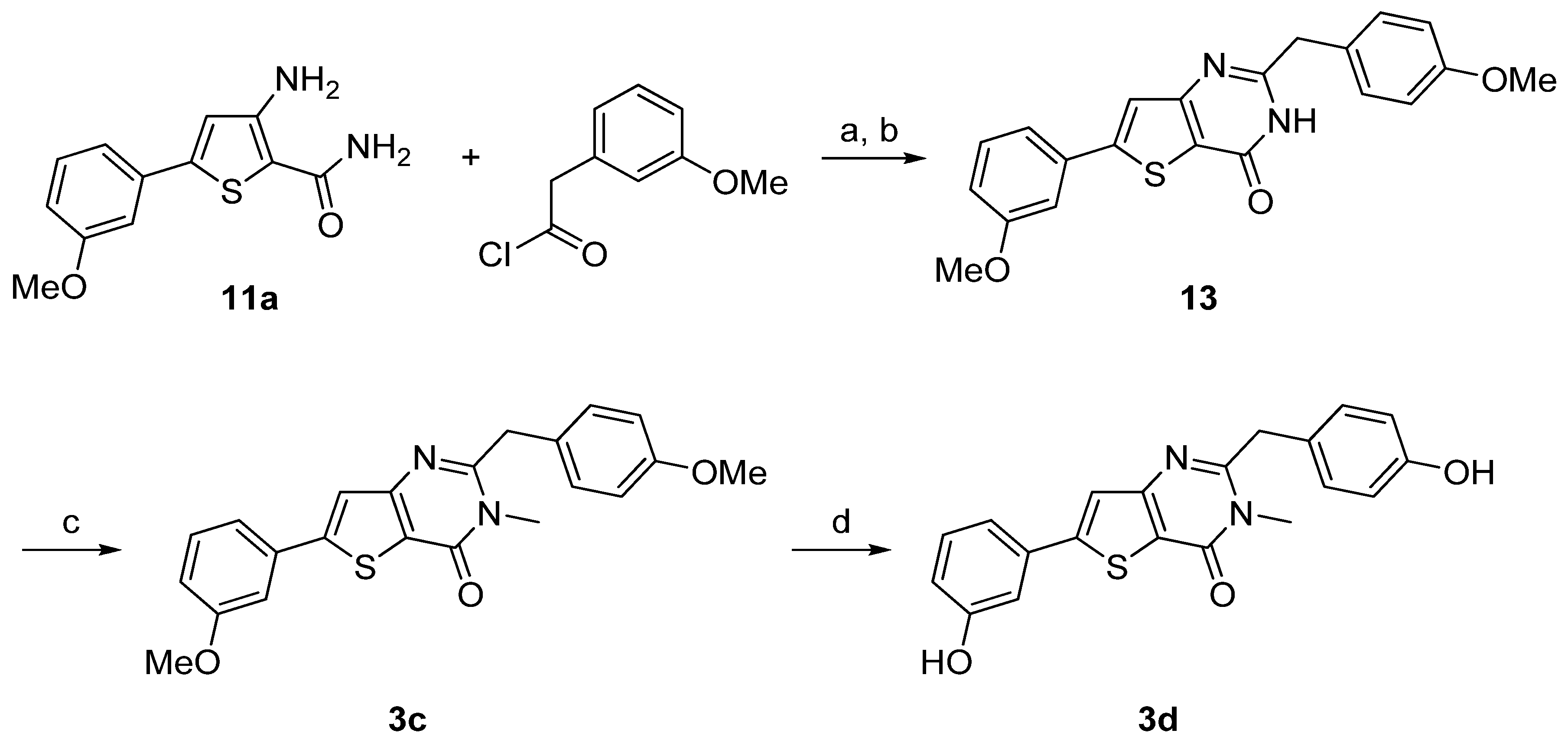
Reagents and conditions: (a) Et3N, dry THF, rt; (b) NaOH 2N, few drops of DMF, reflux, 49% (over two steps); (c) MeI, K2CO3, CH3CN, reflux, 74%; (d) BF3.SMe2, CH2Cl2, rt, 98%.
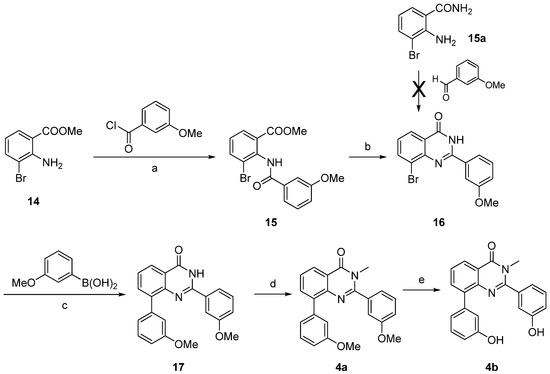
The 2-substituted quinazolin-4-one 4b was synthesized starting from 2-amino-3-bromobenzoic acid methyl ester (14) (Scheme 5), which was reacted with 3-methoxybenzoyl chloride to afford the amide 15. This intermediate was condensed with formamide to afford the 2-substituted quinazolin-4-one 16 [38]. Compound 16 could not be obtained from condensation of 2-amino-6-bromobenzamide (15a) with 3-methoxybenzaldehyde as described for the synthesis of the thiophene fused derivative 13, even using different catalysts (p-toluenesulfonic acid or sodium metabisulfite). This indicates a different reactivity of the amino group depending on its location on a thiophene or a phenyl ring. Subsequently, the Suzuki-Miyaura cross coupling led to compound 17, which was then N-methylated at the quinazolin-4-one into 4a. Treatment with BF3.SMe2 was used to cleave the methoxy groups affording compound 4b.
Scheme 5.
Synthesis of the quinazolin-4-ones derivatives 4a–b.
Scheme 5.
Synthesis of the quinazolin-4-ones derivatives 4a–b.
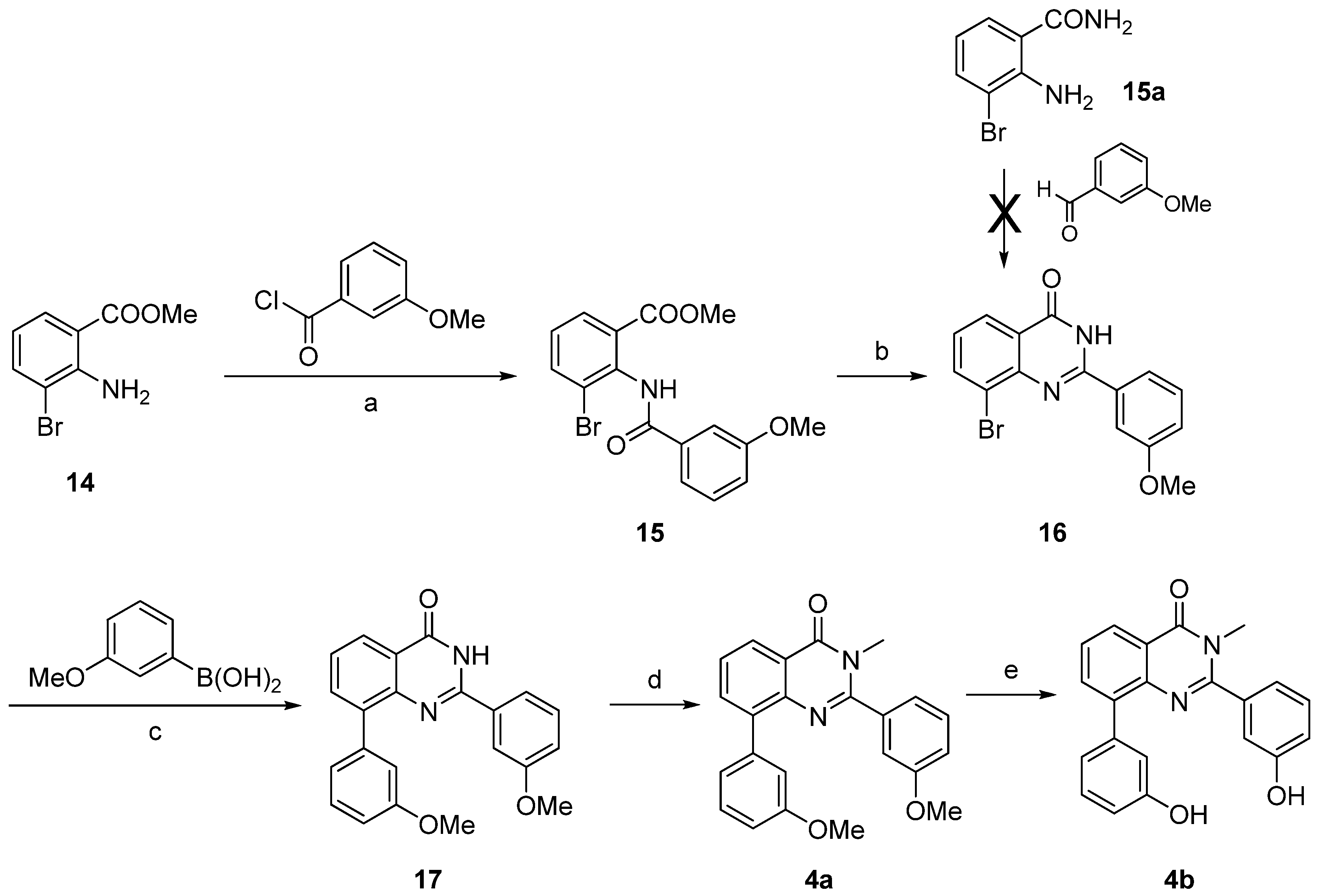
Reagents and conditions: (a) Et3N, dry CH2Cl2, rt, 15%; (b) H2NCHO, 180 °C, 81%; (c) Pd(PPh3)4, Cs2CO3, DME/EtOH/H2O (1:1:1), microwave irradiation (150 °C, 150 W, 15 min), 77%; (d) MeI, K2CO3, CH3CN, reflux, 64%; (e) BF3.SMe2, CH2Cl2, rt, 98%.
The synthesis of fused-compounds 5a–b and 5c–d was performed following two different synthetic routes depicted in Scheme 6. The 3-amino-5-arylthiophene amides 11a,b were condensed with formic acid under microwave irradiation to afford the corresponding thieno[3,2-d]pyrimidin-4-one 18a,b as described in the literature [39]. The conversion of these analogues 18a,b to thieno[3,2-d]pyrimidines 19a,b was performed using phosphorus oxychloride. The nucleophilic aromatic substitution of 19a, 19b was accomplished with sodium methylate and gave the desired 4-methoxy-thieno[3,2-d]pyrimidines 5a and 5b (Scheme 6). Compound 19a was reacted with the corresponding sodium methoxyphenolates providing the substituted 4-phenoxythieno[3,2-d]pyrimidines 5c,d, respectively (Scheme 6).
The synthesized compounds were tested for their ability to inhibit 17β-HSD2 and 17β-HSD1 using placental enzymes as previously described (compound concentration: 1 µM) [40]. Results are reported in Table 2 as percentage of inhibition. Compounds showing an inhibitory activity below 10% were considered as inactive.
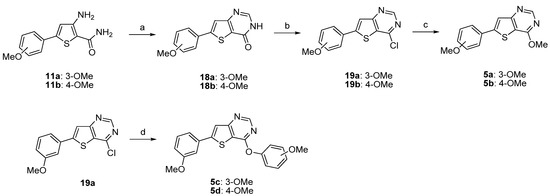
Scheme 6.
Synthesis of the thieno[3,2-d]pyrimidin-4-ones derivatives 5a–d.
Scheme 6.
Synthesis of the thieno[3,2-d]pyrimidin-4-ones derivatives 5a–d.
Reagents and conditions: (a) HCOOH, H2SO4, 50 °C overnight, 18a: 76%, 18b: 98%; (b) POCl3, microwave irradiation (95 °C, 80 W, 20 min), 19a: 98%, 19b: 99%; (c) MeONa in MeOH, DMF, rt, 2 h, 5a: 91%, 5b: 40%; (d) sodium 3-methoxyphenolate or sodium 4-methoxyphenolate, DMF dry, rt, 2 h, 5c: 88%, 5d: 87%.
Table 2.
In vitro inhibitory potencies toward 17β-HSD2 and 17β-HSD1 of compounds 1a–b, 2a–b, 3a–d, 4a–b and 5a–d, 12 and 13.
| Compound | Percentage of inhibition at 1 µM a | |
|---|---|---|
| 17β-HSD2 b | 17β-HSD1 c | |
| 1a | n.i. | n.i. |
| 1b | n.i. | n.i. |
| 2a | n.i. | n.i. |
| 2b | 11% | n.i. |
| 3a | n.i. | n.i. |
| 3b | 36% | 50% |
| 3c | n.i. | n.i. |
| 3d | 25% | 48% |
| 4a | n.i. | n.i. |
| 4b | n.i. | n.i. |
| 5a | n.i. | n.i. |
| 5b | n.i. | n.i. |
| 5c | n.i. | n.i. |
| 5d | n.i. | n.i. |
| 13 | n.i. | n.i. |
| 12 | n.i. | n.i. |
a Mean value of three determinations, standard deviation less than 15%. b Human placental, microsomal fraction, substrate E2 [500 nM], cofactor NAD+ [1500 nM]. c Human placental, cytosolic fraction, substrate E1 [500 nM], cofactor NADH [500 nM]. d n.i.: no inhibition or inhibition below 10%.
The most active compounds 3b and 3d show moderate 17β-HSD2 inhibition (36% and 25%, respectively, Table 2). These two compounds are fused thiophenes and bear two hydroxyphenyl moieties. The corresponding methoxy derivatives are inactive. Compounds 3b/3d both result from the freezing of conformer III and differ only in the presence of a phenyl or a benzyl group in position 2 of the pyrimidinone. As they are the only active compounds identified in this study, this indicates that conformation III is likely to reflect the favourite geometry adapted by compounds A and B in the active site of the enzyme, where it is expected that the compounds are present as only one conformer. However, it is striking that compounds 3b/3d and B present such a difference in activity (3b: 36% @ 1 μM, 3d: 25% @ 1 μM vs B: 70% @ 1 μM). It should be noticed that the corresponding methoxy derivatives 3a and 3c are inactive, while methoxy compound A has a similar biological activity as the hydroxy compound B (A: 63% @ 1 μM). One explanation for this finding it that A and B might have a different binding mode in the enzyme binding site.
In order to better understand the biological activities, manual superimposition of 3b and 3d with B were made using MOE 2010.10 (considering the lowest energy for each compound, Figure 4). The thiophene moiety of 3b/3d was superimposed on the one of B as well as the corresponding carbonyl and N-amide moieties. In the superimposition pictures it can be seen that the compounds do not overlap exactly with each other (however, they show a better hit than the other conformers). The hydroxyphenyl in the 5 position of the fused thiophenes 3b/3d superimposes well on B. The main differences concern the second hydroxyphenyl/benzyl which is in a completely different position and the presence of the nitrogen N1 of the thienopyrimidinone (absent in A/B). It might be that this polar N with hydrogen bond acceptor activity is located in a lipophilic pocket or that there is no space in this area to accept the closed thienopyrimidinone ring. These differences might explain the contrasting biological activities observed for 3b/3d and B.
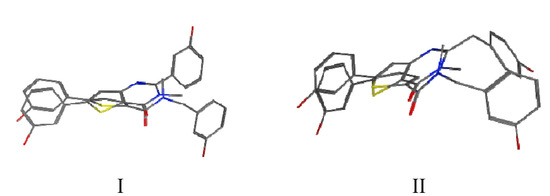
Figure 4.
Superimposition between compounds 3b and B (Figure 4-I) and between compounds 3d and B (Figure 4-II).
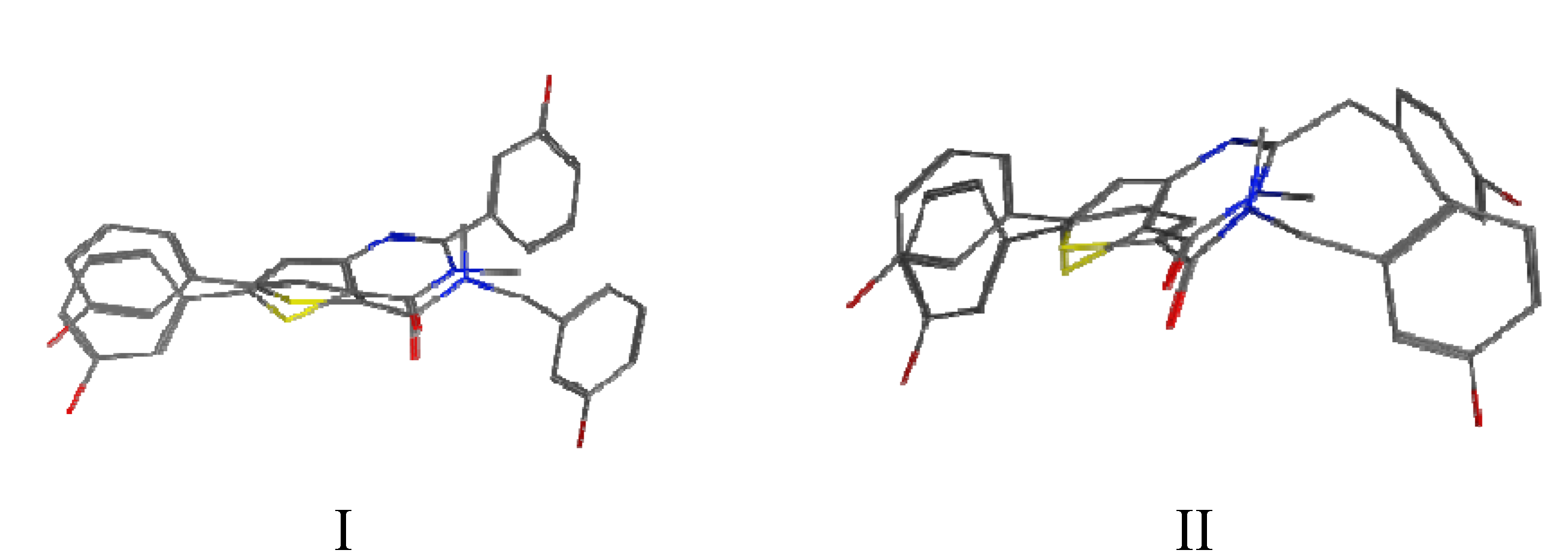
In Figure 4, it can also be seen that the planarity induced by the rigidified compounds is comparable to the planarity of the amide and therefore does not seem to have an impact on the potency of the compounds. In this series of compounds the planar triazole derivative C (Figure 5), a cyclised bioisostere of the amide function, was synthesized and was also a moderate 17β-HSD2 inhibitor with inhibitory activity in the same range as 3b/3d (C: 42% @ 1 μM, [31]).
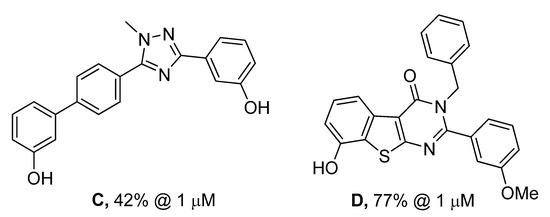
Figure 5.
Described analogues of 3b/3d with rigidified amide bond moiety.
Figure 5.
Described analogues of 3b/3d with rigidified amide bond moiety.
In order to investigate the role of the anisole moiety linked to the thienopyrimidinone, compounds 5a,c, without OMe-Ph and with OMe-Ph on the O next to the thiophene, respectively, both derived from rigidification of conformer I or III, were synthesized. Derivative 5a, without OMe-Ph was inactive, indicating the importance of this group, which certainly in case of A is able to interact with the active site. Compound 5c, with the OMe-Ph at the O next to the thiophene, was also inactive. This anisole moiety, which covers a different area in the active site, might induce a steric clash, preventing the binding of the compound in the enzyme binding site. It might indicate that the binding cavity is elongated and cannot accommodate globular compounds. This could also explain the inactivity of compounds 4a/4b, which have a different geometry compared to 3b/3d.
The thienopyrimidinones 3b/3d show a structural similarity to compound D [41], previously described as 17β-HSD1 inhibitor (17β-HSD1: 95% inh @ 1 μM, 17β-HSD2: 77% inh @ 1 μM). D differs from 3b/3d in the geometry of the pyrimidinone and the OH-Ph fused to the thiophene. As observed for D, 3b/3d also shows a slightly better 17β-HSD1 inhibition profile.
The synthesis of the different rigidified conformers highlights a difference in the reactivity of the amino group depending on its position on the phenyl ring or on the thiophene moiety. All experiments show that the amino group on the thiophene is more reactive than the amino group at the phenyl in spite of the fact that phenyl and thiophene are considered to have similar chemical properties.
3. Experimental
Chemical names follow IUPAC nomenclature. Starting materials were purchased from Aldrich, Acros, Combi-Blocks or Fluka and were used without purification. Flash column chromatography (FC) was performed on silica gel (70–200 μm), and reaction progress was monitored by TLC on Alugram SIL G/UV254 (Macherey-Nagel). Visualization was accomplished with UV light.
1H-NMR and 13C-NMR spectra were measured on a Bruker AM500 spectrometer (at 500 MHz and 125 MHz, respectively) at 300 K in acetone-d6 or DMSO-d6. Chemical shifts are reported in δ (parts per million: ppm), by reference to the hydrogenated residues of deuteriated solvent as internal standard: 2.05 ppm (1H-NMR) and 29.8 and 206.3 ppm (13C-NMR) for acetone-d6, 2.50 ppm (1H-NMR) and 39.5 ppm (13C-NMR) for DMSO-d6 and 7.26 ppm (1H-NMR) and 77.2 ppm (13C-NMR) for CDCl3. Signals are described as br (broad), s (singlet), d (doublet), t (triplet), dd (doublet of doublets), ddd (doublet of doublet of doublets), dt (doublet of triplets) and m (multiplet). All coupling constants (J) are given in Hertz (Hz). Melting points (mp) were measured in open capillaries on a Stuart Scientific SMP3 apparatus and are uncorrected.
Mass spectrometry was performed on a TSQ® Quantum (ThermoFisher, Dreieich, Germany). The triple quadrupole mass spectrometer was equipped with an electrospray interface (ESI). The purity of the compounds was assessed by LC/MS. The Surveyor®-LC-system consisted of a pump, an auto sampler, and a PDA detector. The system was operated by the standard software Xcalibur®. A RP C18 NUCLEODUR® 100-5 (3 mm) column (Macherey-Nagel GmbH, Dühren, Germany) was used as stationary phase. All solvents were HPLC grade. In a gradient run the percentage of acetonitrile (containing 0.1% trifluoroacetic acid) was increased from an initial concentration of 0% at 0 min to 100% at 15 min and kept at 100% for 5 min. The injection volume was 15 µL and flow rate was set to 800 µL/min. MS analysis was carried out at a needle voltage of 3,000 V and a capillary temperature of 350 °C. Mass spectra were acquired in positive mode from 100 to 1,000 m/z and UV spectra were recorded at the wave length of 254 nm and in some cases at 360 nm. All tested compounds exhibited ≥95% chemical purity. Compounds 6 [20], 8 [22], 11a–b [20] were prepared according to previously described procedures.
3.1. General Procedure for Suzuki — Miyaura Coupling (2a, 17) — Method A
A solution of 2a or 17 (1 eq.), 3-methoxyphenyl boronic acid (1.5 eq.), cesium carbonate (3 eq.) and tetrakis(triphenylphosphine)palladium(0) (0.02 eq.) in oxygen free DME/EtOH/H2O solution (3 mL, 1:1:1) was heated under microwave irradiation at 150 °C/150 W for 15 min. After cooling to room temperature, the mixture water/EtOAc (2 mL, 1:1) was added to quench the reaction. The solution was extracted three times by EtOAc (3 × 10 mL). The organic layer was dried over MgSO4, filtered and the solution was concentrated under reduced pressure. The residue was purified by silica gel column chromatography using n-hexane and EtOAc as eluent.
3.2. General Procedure for Cleavage of Ethers 1b–4b — Method B
To a solution of 1a–4a (1 eq.) in CH2Cl2 (10 mL) was added dropwise boron fluoride-dimethyl sulfide complex BF3.SMe2 (3 eq./each methoxy group) at 0 °C. After 1 h at 0 °C, the ice bath was removed and the reaction mixture was stirred overnight at room temperature. The solution was quenched by addition of 10 mL MeOH. After 30 min, the solvent were concentrated under reduced pressure at 35 °C and the residue was triturated with cold water. The aqueous layer was extracted three times with EtOAc (3 × 15 mL), the organic layer was dried over MgSO4, filtered and the solution concentrated under reduced pressure. The residue was purified by silica gel column chromatography using n-hexane and EtOAc as eluent or triturated in a mixture of diethylether/petroleum ether and filtered off.
Methyl 3-Amino-5-(3-methoxyphenyl)thiophene-2-Carboxylate (6). To a solution of 3-chloro-3-(3-methoxyphenyl)prop-2-enenitrile (3.56 g, 18.40 mmol) and dry potassium carbonate (3.05 g, 22.08 mmol) in dry NMP (20 mL) was added dropwise under N2 atmosphere methyl thioglycolate (2.01 mL, 22.08 mmol). The reaction mixture was heated at 60 °C overnight. After cooling, the reaction mixture was poured into ice water solution and stirred. The aqueous layer was extracted three times with EtOAc (3 × 30 mL). The organic layer was washed once with water, dried over MgSO4, filtered and the solution was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (n-hexane/EtOAc 70:30) to afford 6 as a yellow solid. Yield: 50%. Mp: 72–74 °C. 1H-NMR (CDCl3) δ 3.84 (s, 3H), 3.85 (s, 3H), 5.47 (br, 2H), 6.76 (s, 1H), 6.90 (dd, J = 1.9, 8.2 Hz, 1H), 7.10 (t, J = 1.9 Hz, 1H), 7.16–7.19 (m, 1H), 7.30 (t, J = 8.0 Hz, 1H); 13C-NMR (CDCl3) δ 51.5, 55.6, 111.8, 114.8, 115.9, 118.8, 130.2, 134.9, 149.2, 154.4, 160.2, 165.2. LC-MS (ESI): [M+H]+ = 264.29.
Methyl 3-{[(1E)-(dimethylamino)methylidene]amino}-5-(3-methoxyphenyl)thiophene-2-carboxylate (7). To a solution of methyl 3-amino-5-(3-methoxyphenyl)thiophene-2-carboxylate 6 (250 mg, 0.95 mmol) in EtOH (10 mL) was added N-dimethoxymethyl-N,N-dimethylamine (189 µL, 1.42 mmol). The reaction mixture was heated under microwave irradiation at 100 °C/80 W for 30 min. The solution was concentrated under reduced pressure to give 7 as an orange oil. Yield: 99%. The title compound was used for the next step without further purification.
3-(3-Methoxybenzyl)-6-(3-methoxyphenyl)thieno[3,2-d]pyrimidin-4(3H)-one (1a). A solution of 7 (302 mg, 0.95 mmol) and 3-methoxybenzylamine (146 µL, 1.14 mmol) in DMF (4 mL) was heated under microwave irradiation at 100 °C/80 W for 30 min. Water was added to the solution to quench the reaction. The aqueous layer was extracted three times with EtOAc (3 × 15 mL). The organic layer was washed once with brine and once with water, dried over MgSO4, filtered and the solution was concentrated under reduced pressure. The residue was purified by silica gel column chromatography using n-hexane and EtOAc as eluent (n-hexane/EtOAc 60:40) to afford 1a as a pale yellow solid. Yield: 31%. Mp: 144–146 °C. 1H-NMR (CDCl3) δ 3.78 (s, 3H), 3.90 (s, 3H), 5.29 (s, 2H), 6.88 (dd, J = 2.3, 8.2 Hz, 1H), 6.99–7.05 (m, 3H), 7.28 (t, J = 8.0 Hz, 1H), 7.36–7.44 (m, 3H), 7.68 (s, 1H), 8.48 (s, 1H); 13C-NMR (CDCl3) δ 49.6, 55.4, 55.8, 112.5, 114.1, 114.7, 116.2, 119.5, 120.9, 122.1, 123.2, 130.7, 131.3, 135.3, 139.4, 150.1, 152.6, 157.4, 158.9, 161.0, 161.4. LC-MS (ESI): [M+H]+ = 379.31.
3-(3-Hydroxybenzyl)-6-(3-hydroxyphenyl)thieno[3,2-d]pyrimidin-4(3H)-one (1b). Synthesized according to Method B using 1a (52 mg, 0.14 mmol) and BF3.SMe2 (88 µL, 0.84 mmol). The residue was triturated in a mixture of diethyl ether/petroleum ether to afford 1b as a pale brown solid. Yield: 82%. Mp: 270–272 °C. 1H-NMR (DMSO-d6) δ 5.15 (s, 2H), 6.67 (ddd, J = 0.7, 2.4, 8.0 Hz, 1H), 6.71 (t, J = 1.7 Hz, 1H), 6.77 (d, J = 7.7 Hz, 1H), 6.87 (dt, J = 2.0, 7.2 Hz, 1H), 7.14 (t, J = 7.8 Hz, 1H), 7.17–7.19 (m, 1H), 7.27–7.32 (m, 2H), 7.76 (s, 1H), 8.60 (s, 1H), 9.44 (s, 1H), 9.78 (s, 1H); 13C-NMR (DMSO-d6) δ 48.4, 112.8, 114.2, 114.7, 116.8, 117.0, 118.0, 120.9, 121.4, 129.7, 130.5, 133.6, 138.1, 149.8, 151.5, 156.2, 157.6, 157.7, 158.0. LC-MS (ESI): [M]+ = 350.84.
8-Bromo-4H-3,1-benzoxazin-4-one (9). A solution of 2-amino-3-bromobenzoic acid (250 mg, 1.16 mmol) and triethyl orthoformate (2 mL) was heated under microwave irradiation at 160 °C/80 W for 15 min. After cooling, the precipitate formed was collected by filtration and washed with petroleum ether to afford 9 as colorless needles. Yield: 61%. Mp: 314–316 °C. 1H-NMR (acetone-d6) δ 7.57 (t, J = 7.9 Hz, 1H), 8.14 (s, 1H), 8.17 (dd, J = 1.5, 7.9 Hz, 1H), 8.20 (dd, J = 1.5, 8.0 Hz, 1H); 13C-NMR (acetone-d6) δ 121.7, 122.2, 128.6, 130.8, 140.9, 144.6, 152.1, 158.4.
8-Bromo-3-(3-methoxybenzyl)quinazolin-4(3H)-one (10). A solution of 9 (100 mg, 0.44 mmol) and 3-methoxybenzylamine (73 mg, 0.53 mmol) in 2 mL dry toluene was heated at reflux overnight. After cooling, the solvent was removed under reduced pressure. The residue was triturated with diethyl ether and filtered off to give 10 as a colorless solid. Yield: 66%. Mp: 139–140 °C. 1H-NMR (acetone-d6) δ 3.78 (s, 3H), 5.27 (s, 2H), 6.88 (ddd, J = 0.6, 2.6, 8.4 Hz, 1H), 6.99–7.02 (m, 1H), 7.04 (t, J = 2.1 Hz, 1H), 7.28 (t, J = 8.0 Hz, 1H), 7.45 (t, J = 7.9 Hz, 1H), 8.11 (dd, J = 1.5 Hz, 7.8 Hz, 1H), 8.23 (dd, J = 1.5 Hz, 8.0 Hz, 1H), 8.53 (s, 1H); 13C-NMR (acetone-d6) δ 50.1, 55.5, 114.2, 114.7, 120.9, 123.1, 124.8, 127.1, 128.7, 130.7, 138.7, 139.1, 146.9, 149.1, 160.8, 161.0. LC-MS (ESI): [M]+ = 345.46, 347.40.
3-(3-Methoxybenzyl)-8-(3-methoxyphenyl)quinazolin-4(3H)-one (2a). Synthesized according to Method A using 10 (100 mg, 0.29 mmol) and 3-methoxyphenylboronic acid (66 mg, 0.43 mmol). The residue was purified by silica gel column chromatography (n-hexane/EtOAc 70:30) to afford 2a as colorless oil. Yield: 98%. 1H-NMR (acetone-d6) δ 3.75 (s, 3H), 3.81 (s, 3H), 5.23 (s, 2H), 6.85 (ddd, J = 0.8, 2.6, 8.3 Hz, 1H), 6.94 (ddd, J = 1.0, 2.7, 8.3 Hz, 1H), 6.98–7.01 (m, 1H), 7.04 (t, J = 2.1 Hz, 1H), 7.16 (ddd, J = 1.0, 1.5, 7.6 Hz, 1H), 7.21 (dd, J = 1.6, 2.5 Hz, 1H), 7.25 (t, J = 7.9 Hz, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.81 (dd, J = 1.6, 7.4 Hz, 1H), 8.27 (dd, J = 1.6, 8.0 Hz, 1H), 8.36 (s, 1H); 13C-NMR (acetone-d6) δ 49.9, 55.56, 55.58, 113.5, 114.1, 114.8, 117.4, 120.9, 123.7, 123.9, 126.8, 127.7, 129.6, 130.7, 135.8, 139.4, 140.3, 140.9, 146.4, 147.5, 160.1, 161.0, 161.5. LC-MS (ESI): [M]+ = 372.77.
3-(3-Hydroxybenzyl)-8-(3-hydroxyphenyl)quinazolin-4(3H)-one (2b). Synthesized according to Method B using 2a (125 mg, 0.31 mmol) and BF3.SMe2 (196 µL, 1.86 mmol). The residue was triturated in diethyl ether to afford 2b as a colorless solid. Yield: 97%. Mp: 155–158 °C. 1H-NMR (acetone-d6) δ 5.43 (s, 2H), 6.84 (ddd, J = 0.8, 2.4, 8.1 Hz, 1H), 6.99–7.04 (m, 5H), 7.20–7.23 (m, 1H), 7.36–7.40 (m, 1H), 7.90 (t, J = 7.7 Hz, 1H), 8.01 (dd, J = 1.5, 7.5 Hz, 1H), 8.40 (dd, J = 1.5, 8.1 Hz, 1H), 8.43 (br, 1H), 8.67 (br, 1H), 9.48 (s, 1H); 13C-NMR (acetone-d6) δ 52.6, 116.5, 117.2, 117.4, 120.7, 121.6, 121.8, 127.9, 130.5, 130.9, 131.6, 135.2, 136.3, 136.5, 136.7, 137.9, 152.1, 158.7, 158.9, 159.3. LC-MS (ESI): [M]+ = 344.81.
3-Amino-5-(3-methoxyphenyl)thiophene-2-carboxamide (11a). A solution of sodium sulfide nonahydrate Na2S.9H2O (24 g, 100 mmol) in DMF (100 mL) was heated at 40 °C for 30 min. To this solution 3-chloro-3-(3-methoxyphenyl)prop-2-enenitrile (9.68 g, 50 mmol) in DMF (20 mL) was added. After 2 hours at 50 °C, 2-chloroacetamide (8.42 g, 100 mmol) in DMF (20 mL) was added to the reaction mixture. The solution was stirred overnight at 50 °C. Freshly prepared EtONa in EtOH (2.30 g of sodium in 30 mL of absolute EtOH) was added to the reaction mixture, which was stirred and kept at 50 °C for 2 h. After cooling to room temperature, the mixture was poured into ice water solution under stirring. The aqueous layer was extracted three times with EtOAc (3 × 50 mL). The organic layer was washed once with water, dried over MgSO4, filtered and the solution was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc 100%) to afford 11a as a pale brown solid. Yield: 21%. Mp: 104–106 °C. 1H-NMR (acetone-d6) δ 3.85 (s, 3H), 6.28 (br, 2H), 6.67 (br, 2H), 6.93 (s, 1H), 6.95 (ddd, J = 0.9, 2.6, 8.3 Hz, 1H), 7.16 (t, J = 2.1 Hz, 1H), 7.22 (ddd, J = 0.9, 1.6, 7.7 Hz, 1H), 7.34 (t, J = 8.1 Hz, 1H); 13C-NMR (acetone-d6) δ 60.0, 111.1, 116.3, 119.37, 119.43, 123.2, 135.3, 140.4, 154.2, 157.0, 165.4, 166.2. LC-MS (ESI): [M]+ = 248.94.
2,6-Bis(3-methoxyphenyl)thieno[3,2-d]pyrimidin-4(3H)-one (12). A solution of 3-amino-5-(3-methoxyphenyl)thiophene-2-carboxamide 11a (200 mg, 0.80 mmol), 4-methoxybenzaldehyde (117 µL, 0.96 mmol) and concentrated hydrochloric acid 37% (250 µL) was refluxed for 2 hours. After cooling, the precipitate was collected by filtration, rinsed with cold MeOH and washed twice with diethyl ether to give 12 as a pale brown solid. Yield: 61%. Mp: 241–243 °C. 1H-NMR (DMSO-d6) δ 3.86 (s, 3H), 3.88 (s, 3H), 7.03 (ddd, J = 1.3, 2.5, 7.9 Hz, 1H), 7.14 (ddd, J = 0.9, 2.7, 8.3 Hz, 1H), 7.36 (t, J = 1.9 Hz, 1H), 7.37–7.47 (m, 3H), 7.71 (t, J = 1.8 Hz, 1H), 7.74 (ddd, J = 0.9, 1.5, 7.7 Hz, 1H), 7.81 (s, 1H); 13C-NMR (DMSO-d6) δ 56.0, 56.1, 112.3, 113.5, 116.0, 118.2, 119.2, 120.7, 121.1, 122.0, 130.2, 131.0, 134.4, 134.5, 151.5, 155.1, 158.5, 158.7, 160.1, 160.6. LC-MS (ESI): [M+H]+ = 365.26.
2,6-Bis(3-methoxyphenyl)-3-methylthieno[3,2-d]pyrimidin-4(3H)-one (3a). Methyl iodide (20 µL, 0.32 mmol) was added to a solution of 12 (75 mg, 0.21 mmol) and potassium carbonate (58 mg, 0.42 mmol) in dry CH3CN (2 mL). The reaction mixture was refluxed for 3 h. After cooling to room temperature, the reaction was stopped by adding water. The aqueous layer was extracted three times with EtOAc (3 × 5 mL). The organic layer was dried over MgSO4, filtered and the solution was concentrated under reduced pressure. The residue was purified by silica TLC using n-hexane and EtOAc as eluent (n-hexane/EtOAc 60:40) to afford 3a as a colorless solid. Yield: 48%. Mp: 148–150 °C. 1H-NMR (CDCl3) δ 3.54 (s, 3H), 3.871 (s, 3H), 3.873 (s, 3H), 6.96 (ddd, J = 0.8, 2.5, 8.2 Hz, 1H), 7.05–7.08 (m, 2H), 7.11 (dt, J = 1.2, 7.8 Hz, 1H), 7.23 (t, J = 2.0 Hz, 1H), 7.30 (ddd, J = 1.0, 1.4, 7.7 Hz, 1H), 7.37 (t, J = 8.0 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 7.51 (s, 1H). 13C-NMR (CDCl3) δ 34.2, 55.4, 55.5, 112.1, 113.5, 114.9, 115.9, 119.0, 120.2, 120.8, 121.2, 130.1, 130.3, 134.5, 136.3, 152.5, 156.5, 157.8, 158.5, 159.9, 160.1. LC-MS (ESI): [M+H]+ = 379.29.
2,6-Bis(3-hydroxyphenyl)-3-methylthieno[3,2-d]pyrimidin-4(3H)-one (3b). Synthesized according to Method B using 3a (27 mg, 0.08 mmol) and BF3.SMe2 (51 µL, 0.48 mmol). The residue was triturated in a mixture of diethyl ether/petroleum ether to afford 3b as a pale pink solid. Yield: 57%. Mp: 303–304 °C. 1H-NMR (DMSO-d6) δ 3.38 (s, 3H), 6.87 (dt, J = 2.0, 7.2 Hz, 1H), 6.94 (ddd, J = 0.8, 2.5, 8.1 Hz, 1H), 7.00 (t, J = 1.7 Hz, 1H), 7.04–7.06 (m, 1H), 7.19–7.20 (m, 1H), 7.27–7.36 (m, 3H), 7.74 (s, 1H), 9.78 (br, 2H); 13C-NMR (DMSO-d6) δ 33.8, 112.7, 115.1, 116.79, 116.81, 117.0, 118.7, 119.8, 120.8, 129.6, 130.5, 133.7, 136.2, 151.2, 156.4, 157.3, 157.6, 157.99, 158.02. LC-MS (ESI): [M]+ = 350.85.
2-(4-Methoxybenzyl)-6-(3-methoxyphenyl)thieno[3,2-d]pyrimidin-4(3H)-one (13). To a solution of 3-amino-5-(3-methoxyphenyl)thiophene-2-carboxamide 11a (100 mg, 0.40 mmol) in dry THF (3 mL) was added Et3N (67 µL, 0.48 mmol) followed by 4-methoxyphenylacetyl chloride (73 µL, 0.48 mmol). The reaction mixture was stirred at room temperature for 1 h. The solvent was removed under reduced pressure before NaOH 2N (1.5 mL) and DMF (1.5 mL) were added to the residue. The solution was refluxed for 30 min, cooled to room temperature and poured into cold water. The precipitate formed was filtered off, washed with water and diethyl ether to give 13 as a colorless solid. Yield: 49%. Mp: 258–261 °C. 1H-NMR (DMSO-d6) δ 3.71 (s, 3H), 3.83 (s, 3H), 3.89 (s, 2H), 6.87 (d, J = 8.6 Hz, 2H), 7.00–7.03 (m, 1H), 7.28 (d, J = 8.6 Hz, 2H), 7.33–7.42 (m, 2H), 7.79 (s, 1H), 12.59 (br, 1H); 13C-NMR (DMSO-d6) δ 50.0, 60.3, 60.6, 114.6, 116.5, 118.3, 118.6, 119.2, 120.6, 123.7, 125.0, 133.6, 135.2, 135.8, 139.1, 155.8, 163.4, 163.9, 165.0. LC-MS (ESI): [M+H]+ = 379.22.
2-(4-Methoxybenzyl)-6-(3-methoxyphenyl)-3-methylthieno[3,2-d]pyrimidin-4(3H)-one (3c). Methyl iodide (11 µL, 0.18 mmol) was added to a solution of 13 (46 mg, 0.12 mmol) and potassium carbonate (66 mg, 0.24 mmol) in dry CH3CN (2 mL). The reaction mixture was refluxed for 3 hours. After cooling to room temperature, the reaction was stopped by adding water. The aqueous layer was extracted three times with EtOAc (3 × 5 mL). The organic layer was dried over MgSO4, filtered and the solution was concentrated under reduced pressure. The residue was purified by preparative TLC using n-hexane and EtOAc as eluent (n-hexane/EtOAc 60:40) to afford 3c as a pale yellow solid. Yield: 74%. Mp: 133–134 °C. 1H-NMR (CDCl3) δ 3.61 (s, 3H), 3.87 (s, 3H), 3.96 (s, 3H), 4.27 (s, 2H), 6.95 (d, J = 8.7 Hz, 2H), 7.02–7.05 (m, 1H), 7.25 (d, J = 8.7 Hz, 2H), 7.30–7.32 (m, 1H), 7.34–7.40 (m, 1H), 7.45 (t, J = 7.7 Hz, 1H), 7.57 (s, 1H); 13C-NMR (CDCl3) δ 30.9, 42.0, 55.5, 55.6, 112.3, 114.7, 115.2, 119.2, 120.8, 121.1, 126.8, 129.6, 130.5, 134.8, 152.5, 156.7, 157.9, 158.7, 159.1, 160.3. LC-MS (ESI): [M+H]+ = 393.28.
2-(4-Hydroxybenzyl)-6-(3-hydroxyphenyl)-3-methylthieno[3,2-d]pyrimidin-4(3H)-one (3d). Synthesized according to Method B using 3c (35 mg, 0.01 mmol) and BF3.SMe2 (56 µL, 0.54 mmol). The residue was triturated in a mixture diethyl ether/petroleum ether to afford 3d as a pale brown solid. Yield: 98%. Mp: 308–310 °C. 1H-NMR (DMSO-d6) δ 3.46 (s, 3H), 4.16 (s, 2H), 6.72 (d, J = 7.1 Hz, 2H), 6.83–6.88 (m, 1H), 7.07 (d, J = 7.1 Hz, 2H), 7.17 (br, 1H), 7.26–7.33 (m, 2H), 7.72 (s, 1H), 9.35 (s, 1H), 9.79 (s, 1H); 13C-NMR (DMSO-d6) δ 30.7, 75.4, 111.8, 113.2, 116.0, 117.2, 117.5, 119.7, 121.3, 125.8, 130.1, 130.9, 134.2, 151.5, 156.7, 157.0, 158.1, 158.5, 159.3. LC-MS (ESI): [M+H]+ = 364.89.
Methyl 2-amino-3-bromobenzoate (14). A solution of 2-amino-3-bromobenzoic acid 8 (1.033 g, 4.78 mmol) and thionyl chloride (30 mL) was heated at reflux for 4 hours. After cooling, the solvent was reduced under reduced pressure. The residue was poured into MeOH at 0 °C. Excess solvent was removed under reduced pressure. The precipitate was filtered and purified by recrystallization in a MeOH/water mixture to afford 14 as a yellow solid. Yield: 89%. Mp: 39–40 °C. 1H-NMR (Acetone-d6) δ 3.87 (s, 3H), 6.57 (t, J = 7.8 Hz, 1H), 6.63 (br, 2H), 7.64 (dd, J = 1.6, 7.9 Hz, 1H), 7.85 (dd, J = 1.5, 8.1 Hz, 1H); 13C-NMR (Acetone-d6) δ 52.3, 110.8, 112.4, 117.0, 131.6, 138.2, 148.8, 168.5.
Methyl 3-bromo-2-{[(3-methoxyphenyl)carbonyl]amino}benzoate (15). To a solution of 14 (983 mg, 4.27 mmol) in dry CH2Cl2 (5 mL) was added Et3N (653 µL, 4.70 mmol) followed by 3-methoxyphenylacetyl chloride (600 µL, 4.27 mmol). The reaction mixture was stirred at room temperature overnight. Water was added to the solution to stop the reaction. The aqueous layer was extracted three times with CH2Cl2 (3 × 20 mL). The organic layer was dried over MgSO4, filtered and the solution was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (n-hexane/EtOAc 95: 5 to 70:30) to afford 15 as a white viscous oil. Yield: 15%. 1H-NMR (acetone-d6) δ 3.78 (s, 3H), 3.89 (s, 3H), 7.18 (ddd, J = 1.0, 2.6, 8.3 Hz, 1H), 7.35 (t, J = 7.9 Hz, 1H), 7.46 (t, J = 7.7 Hz, 1H), 7.59 (dd, J = 1.6, 2.6 Hz, 1H), 7.64 (ddd, J = 1.0, 1.5, 7.6 Hz, 1H), 7.90 (dd, J = 1.5, 7.8 Hz, 1H), 7.93 (dd, J = 1.5, 8.0 Hz, 1H), 9.48 (br, 1H); 13C-NMR (acetone-d6) δ 52.7, 55.8, 113.7, 118.6, 120.6, 123.8, 128.4, 130.57, 130.59, 131.0, 136.9, 137.2, 137.4, 160.9, 166.1, 166.8.
8-Bromo-2-(3-methoxyphenyl)quinazolin-4(3H)-one (16). A solution of 15 in formamide (5 mL) was heated at 180 °C for 6 hours. After cooling, the precipitate formed was dispersed with cold water and filtered off. The solid was washed twice with water to give 16 as a pale brown solid. The title compound was used for the next step without further purification. Yield: 81%. Mp: 236–239 °C. 1H-NMR (DMSO-d6) δ 3.87 (s, 3H), 7.18 (ddd, J = 1.0, 2.5, 8.2 Hz, 1H), 7.42 (t, J = 7.8 Hz, 1H), 7.49 (t, J = 8.0 Hz, 1H), 7.83 (t, J = 1.8 Hz, 1H), 7.88 (ddd, J = 0.9, 1.4, 7.9 Hz, 1H), 8.14 (dd, J = 1.3, 5.4 Hz, 1H), 8.16 (dd, J = 1.4, 5.3 Hz, 1H), 12.75 (br, 1H); 13C-NMR (DMSO-d6) δ 55.4, 112.9, 117.8, 120.3, 122.2, 122.6, 125.7, 127.4, 129.8, 133.6, 137.9, 146.0, 152.6, 159.3, 161.9. LC-MS (ESI): [M]+ = 331.54, 333.35.
2,8-Bis(3-methoxyphenyl)quinazolin-4(3H)-one (17). Synthesized according to Method A using 16 (144 mg, 0.43 mmol) and 3-methoxyphenylboronic acid (98 mg, 0.65 mmol). The residue was purified by silica gel column chromatography (n-hexane/EtOAc 70:30) to afford 17 as a colorless solid. Yield: 77%. Mp: 198–201 °C. 1H-NMR (DMSO-d6) δ 3.81 (s, 3H), 3.82 (s, 3H), 6.99 (dd, J = 2.0, 8.2 Hz, 1H), 7.09–7.13 (m, 1H), 7.28 (d, J = 7.7 Hz, 1H), 7.33 (t, J = 2.0 Hz, 1H), 7.39–7.44 (m, 2H), 7.59 (t, J = 7.7 Hz, 1H), 7.74–7.77 (m, 2H), 7.90 (dd, J = 1.4, 7.4 Hz, 1H), 8.19 (dd, J = 1.4, 7.9 Hz, 1H), 12.61 (br, 1H); 13C-NMR (DMSO-d6) δ 55.0, 55.1, 112.3, 112.9, 116.2, 117.8, 119.8, 121.7, 122.9, 125.4, 126.5, 128.7, 129.7, 134.1, 135.1, 138.4, 139.6, 145.5, 150.8, 158.7, 159.3, 162.4. LC-MS (ESI): [M]+ = 358.76.
2,8-Bis(3-methoxyphenyl)-3-methylquinazolin-4(3H)-one (4a). Methyl iodide (29 µL, 0.47 mmol) was added to a solution of 17 (112 mg, 0.31 mmol) and potassium carbonate (86 mg, 0.62 mmol) in dry CH3CN (3 mL). The reaction mixture was refluxed for 3 h. After cooling to room temperature, the solvent was removed under reduced pressure and water was added to the residue to dissolve the inorganic salt. The aqueous layer was extracted three times with EtOAc (3 × 5 mL). The organic layer was dried over MgSO4, filtered and the solution was concentrated under reduced pressure. The residue was purified by silica gel column chromatography using n-hexane and EtOAc as eluent (n-hexane/EtOAc 70:30) to afford 4a as a colorless solid. Yield: 64%. Mp: 241–244 °C. 1H-NMR (acetone-d6) δ 3.49 (s, 3H), 3.81 (s, 3H), 3.86 (s, 3H), 6.88 (ddd, J = 1.0, 2.6, 8.2 Hz, 1H), 7.07 (ddd, J = 0.9, 2.6, 8.3 Hz, 1H), 7.23 (dt, J = 1.2, 7.7 Hz, 1H), 7.25 (dt, J = 1.1, 7.6 Hz, 1H), 7.28–7.29 (m, 1H), 7.30 (t, J = 7.8 Hz, 1H), 7.35 (dd, J = 1.7, 2.5 Hz, 1H), 7.43 (t, J = 8.0 Hz, 1H), 7.59 (t, J = 7.7 Hz, 1H), 7.87 (dd, J = 1.6, 7.5 Hz, 1H), 8.26 (dd, J = 1.6, 7.9 Hz, 1H); 13C-NMR (acetone-d6) δ 34.4, 55.5, 55.8, 113.8, 114.8, 116.3, 117.3, 121.3, 122.4, 123.5, 126.7, 127.3, 129.5, 130.4, 135.6, 138.3, 139.9, 140.9, 145.5, 156.2, 160.0, 160.6, 163.0. LC-MS (ESI): [M]+ = 372.71.
2,8-Bis(3-hydroxyphenyl)-3-methylquinazolin-4(3H)-one (4b). Synthesized according to Method B using 4a (65 mg, 0.17 mmol) and BF3.SMe2 (107 µL, 1.02 mmol). The residue was triturated in diethyl ether to afford 4b as a colorless solid. Yield: 98%. Mp: 241–244 °C. 1H-NMR (acetone-d6) δ 3.48 (s, 3H), 6.80 (ddd, J = 1.0, 2.6, 8.2 Hz, 1H), 6.98 (ddd, J = 1.0, 2.4, 8.2 Hz, 1H), 7.11–7.15 (m, 3H), 7.18–7.19 (m, 1H), 7.21 (t, J = 7.8 Hz, 1H), 7.33 (t, J = 8.1 Hz, 1H), 7.56–7.59 (m, 1H), 7.81 (dd, J = 1.7, 7.5 Hz, 1H), 8.25 (dd, J = 1.6, 8.0 Hz, 1H), 8.35 (s, 1H), 8.75 (s, 1H); 13C-NMR (acetone-d6) δ 34.4, 115.0, 116.3, 117.6, 118.5, 120.4, 122.3, 122.7, 126.6, 127.3, 129.5, 130.4, 135.6, 138.2, 140.2, 141.0, 145.6, 156.3, 157.7, 158.3, 163.1. LC-MS (ESI): [M]+ = 344.77.
3.3. General Procedure for the Preparation of Compounds 18a–b — Method C
A solution of 3-amino-5-(3-methoxyphenyl)thiophene-2-carboxamide 11a (4 mmol) or 3-amino-5-(4-methoxyphenyl)thiophene-2-carboxamide 11b (4 mmol) in formic acid (10 mL) and with catalytic amounts of concentrated sulfuric acid (30 drops) was heated at 50 °C overnight. After cooling, the reaction was quenched with cold water and the solid formed was collected by filtration. The residue was triturated in diethyl ether and filtered off.
6-(3-Methoxyphenyl)thieno[3,2-d]pyrimidin-4(3H)-one (18a). Synthesized according to Method C using 11a (993 mg, 4 mmol) and HCOOH (10 mL). The residue was triturated in diethyl ether to afford 18a as a pale green solid. Yield: 76%. Mp: 237–245 °C. 1H-NMR (DMSO-d6) δ 3.85 (s, 3H), 7.03 (dt, J = 2.4, 6.9, 1H), 7.38–7.43 (m, 3H), 7.87 (s, 1H), 8.17 (s, 1H), 12.55 (br, 1H); 13C-NMR (DMSO-d6) δ 55.4, 111.5, 115.4, 118.5, 121.5, 122.2, 130.5, 133.8, 147.1, 150.4, 157.0, 158.4, 159.8. LC-MS (ESI): [M+H]+ = 259.18.
6-(4-Methoxyphenyl)thieno[3,2-d]pyrimidin-4(3H)-one (18b). Synthesized according to Method C using 11b (993 mg, 4 mmol) and HCOOH (10 mL). The residue was triturated in diethyl ether to afford 18b as a pale green solid. Yield: 98%. Mp: 296–299 °C. 1H-NMR (DMSO-d6) δ 3.82 (s, 3H), 7.04 (d, J = 8.9, 2H), 7.69 (s, 1H), 7.78 (d, J = 8.9, 2H), 8.14 (s, 1H), 12.45 (br, 1H); 13C-NMR (DMSO-d6) δ 55.4, 114.7, 119.7, 121.2, 125.1, 127.6, 146.9, 150.8, 156.9, 158.6, 160.4. LC-MS (ESI): [M+H]+ = 259.19.
3.4. General Procedure for the Preparation of Compounds 19a–b — Method D
A solution of 6-(3-methoxyphenyl)thieno[3,2-d]pyrimidin-4(3H)-one (18a, 0.58 mmol) or 6-(4-methoxyphenyl)thieno[3,2-d]pyrimidin-4(3H)-one (18b, 0.58 mmol) in phosphorus oxychloride (5 mL) was heated under microwave irradiation at 95 °C/80 W for 20 min. Excess of phosphorus oxychloride was removed under reduced pressure. Ice was added to the residue and the precipitate formed was collected by filtration. The compounds were pure enough and could be used in the next step without further purification.
4-Chloro-6-(3-methoxyphenyl)thieno[3,2-d]pyrimidine (19a). Synthesized according to Method D using 18a (150 mg, 0.58 mmol) and POCl3 (5 mL) afforded 19a as a brown solid. Yield: 98%. Mp: 153–156 °C. 1H-NMR (DMSO-d6) δ 3.87 (s, 3H), 7.10–7.14 (m, 1H), 7.47 (t, J = 8.2, 1H), 7.51–7.54 (m, 2H), 8.28 (s, 1H), 9.03 (s, 1H); 13C-NMR (DMSO-d6) δ 55.5, 111.9, 116.8, 119.2, 120.8, 129.4, 130.7, 132.9, 153.0, 154.3, 154.8, 159.9, 162.5. LC-MS (ESI): [M+H]+ = 277.17.
4-Chloro-6-(4-methoxyphenyl)thieno[3,2-d]pyrimidine (19b). Synthesized according to Method D using 18b (150 mg, 0.58 mmol) and POCl3 (5 mL) afforded 19b as a brown solid. Yield: 99%. Mp: 153–156 °C. 1H-NMR (DMSO-d6) δ 3.84 (s, 3H), 7.09 (d, J = 8.7, 2H), 7.92 (d, J = 8.9, 2H), 8.07 (s, 1H), 8.97 (s, 1H); 13C-NMR (DMSO-d6) δ 55.5, 114.9, 118.7, 124.1, 128.5, 128.8, 152.6, 154.6, 154.7, 161.4, 162.9. LC-MS (ESI): [M+H]+ = 277.16.
3.5. General Procedure for the Preparation of Compounds 5a–b – Method E
To a solution of 4-chloro-6-(3-methoxyphenyl)thieno[3,2-d]pyrimidine 19a (0.51 mmol) or 4-chloro-6-(4-methoxyphenyl)thieno[3,2-d]pyrimidine 19b (0.51 mmol) in dry DMF (2 mL) was added sodium methanolate in methanol (0.51 mmol) and the reaction mixture was stirred at room temperature for 2 hours. Water was added to quench the reaction. The precipitate formed was collected by filtration and washed once with water and once with petroleum ether.
4-Methoxy-6-(3-methoxyphenyl)thieno[3,2-d]pyrimidine (5a). Synthesized according to Method E using 19a (140 mg, 0.51 mmol) and NaOMe (0.51 mmol in 2 mL MeOH). The residue was triturated in petroleum ether to afford 5a as a brown solid. Yield: 91%. Mp: 159–162 °C. 1H-NMR (DMSO-d6) δ 3.86 (s, 3H), 4.13 (s, 3H), 7.07 (dt, J = 2.3, 7.4 Hz, 1H), 7.42–7.46 (m, 3H), 8.08 (s, 1H), 8.77 (s, 1H); 13C-NMR (DMSO-d6) δ 54.2, 55.4, 111.8, 115.9, 116.1, 118.8, 120.5, 130.6, 133.6, 151.3, 154.7, 159.9, 162.4, 163.5. LC-MS (ESI): [M+H]+ = 273.22.
4-Methoxy-6-(4-methoxyphenyl)thieno[3,2-d]pyrimidine (5b). Synthesized according to Method E using 19b (140 mg, 0.51 mmol) and NaOMe (0.51 mmol in 2 mL MeOH). The residue was triturated in petroleum ether to afford 5b as a brown solid. Yield: 40%. Mp: 184–186 °C. 1H-NMR (DMSO-d6) δ 3.83 (s, 3H), 4.12 (s, 3H), 7.08 (d, J = 8.8 Hz, 2H), 7.86 (d, J = 8.8 Hz, 2H), 7.91 (s, 1H), 8.74 (s, 1H); 13C-NMR (DMSO-d6) δ 54.1, 55.4, 114.8, 115.4, 118.6, 124.8, 128.0, 151.6, 154.7, 160.8, 162.8, 163.3. LC-MS (ESI): [M+H]+ = 273.21.
3.6. General Procedure for the Preparation of Compounds 5c–d – Method F
To a solution of 4-chloro-6-(3-methoxyphenyl)thieno[3,2-d]pyrimidine (19a, 1 eq.) in dry DMF (2 mL) was added freshly prepared dry sodium 3-methoxyphenolate or sodium 4-methoxyphenolate (1.2 eq.) The reaction mixture was stirred at room temperature for 2 h. Water was added to stop the reaction. Organic layer was washed once with NaOH 2N and once with brine, dried over MgSO4, filtered and the solution was concentrated under reduced pressure. The precipitate formed was triturated with a mixture of petroleum ether / diethyl ether and filtered off.
4-(3-Methoxyphenoxy)-6-(3-methoxyphenyl)thieno[3,2-d]pyrimidine (5c). Synthesized according to Method F using 19a (100 mg, 0.36 mmol) and sodium 3-methoxyphenolate (56 mg, 0.43 mmol). The residue was triturated in petroleum ether to afford 5c as a pale brown solid. Yield: 88%. Mp: 129–132 °C. 1H-NMR (DMSO-d6) δ 3.78 (s, 3H), 3.87 (s, 3H), 6.92 (dd, J = 2.4, 8.2 Hz, 2H), 6.97 (t, J = 2.3 Hz, 1H), 7.09 (ddd, J = 1.1, 1.3, 8.0 Hz, 1H), 7.39 (t, J = 8.2 Hz, 1H), 7.46 (t, J = 8.2 Hz, 1H), 7.49–7.51 (m, 2H), 8.16 (s, 1H), 8.70 (s, 1H); 13C-NMR (DMSO-d6) δ 54.4, 55.4, 108.0, 111.86, 111.87, 114.0, 116.1, 116.3, 118.9, 120.5, 130.2, 130.6, 133.4, 152.3, 152.7, 154.6, 159.9, 160.4, 163.1, 163.6. LC-MS (ESI): [M+H]+ = 365.26.
4-(4-Methoxyphenoxy)-6-(3-methoxyphenyl)thieno[3,2-d]pyrimidine (5d). Synthesized according to Method F using 19a (30 mg, 0.11 mmol) and sodium 4-methoxyphenolate (17 mg, 0.13 mmol). The residue was triturated in petroleum ether to afford 5d as a pale brown solid. Yield: 87%. Mp: 139–146 °C. 1H-NMR (DMSO-d6) δ 3.85 (s, 3H), 3.90 (s, 3H), 6.98–7.01 (m, 3H), 7.15–7.17 (m, 1H), 7.20 (t, J = 7.9 Hz, 1H), 7.29 (t, J = 2.0 Hz, 1H), 7.36–7.42 (m, 2H), 7.70 (s, 1H), 8.71 (s, 1H); 13C-NMR (DMSO-d6) δ 55.4, 55.6, 112.5, 114.8, 115.4, 117.4, 119.3, 120.0, 122.7, 130.4, 134.3, 145.4, 153.2, 154.8, 157.5, 160.2, 163.6, 164.0. LC-MS (ESI): [M+H]+ = 365.25.
4. Conclusions
In this study, a series of six thieno[3,2-d]pyrimidin-4-ones, four thieno[3,2-d]pyrimidines and four quinazolin-4-ones was synthesized and tested for 17β-HSD2 and 17β-HSD1 inhibition. Two compounds 3b and 3d were identified as moderate 17β-HSD2 inhibitors, 3b being the best one with an inhibition of 36% at 1 µM. These compounds resulted from the rigidification of conformer III, which can be considered as the active conformer in the enzyme binding site. The inactive globular compounds like 4a/4b or 5c/5d are indicative of the enzyme binding site being rather elongated and are tools to further characterize the 17β-HSD2 active site. Compounds 3b/3d need further optimization focussing on conformer III and exchanging the geometry of the pyrimidinone or replacing the polar nitrogen of the pyrimidinone moiety by a carbon or reducing the size of the ring fused to the thiophene.
Acknowledgments
We thank Josef Zapp for the NMR measurements, and Isabella Mang, Laura Schrobildgen, and Nina Hanke for performing the biological tests. We acknowledge the Deutsche Forschungsgemeinschaft (DFG) for financial support (Grants HA1315/12-1).
References
- Cummings, S.R.; Melton, L.J. Epidemiology and outcomes of osteoporotic fractures. Lancet 2002, 359, 1761–1767. [Google Scholar] [CrossRef]
- Bagi, C.M.; Wood, J.; Wilkie, D.; Dixon, B. Effect of 17beta-hydroxysteroid dehydrogenase type 2 inhibitor on bone strength in ovariectomized cynomolgus monkeys. J. Musculoskelet. Neuronal Interact. 2008, 8, 267–280. [Google Scholar]
- Wu, L.; Einstein, M.; Geissler, W.M.; Chan, H.K.; Elliston, K.O.; Andersson, S. Expression cloning and characterization of human 17 beta-hydroxysteroid dehydrogenase type 2, a microsomal enzyme possessing 20 alpha-hydroxysteroid dehydrogenase activity. J. Biol. Chem. 1993, 268, 12964–12969. [Google Scholar]
- Poirier, D.; Bydal, P.; Tremblay, M.R.; Sam, K.M.; Luu-The, V. Inhibitors of type ii 17beta-hydroxysteroid dehydrogenase. Mol. Cell. Endocrinol. 2001, 171, 119–128. [Google Scholar] [CrossRef]
- Bydal, P.; Auger, S.; Poirier, D. Inhibition of type 2 17beta-hydroxysteroid dehydrogenase by estradiol derivatives bearing a lactone on the d-ring: Structure-activity relationships. Steroids 2004, 69, 325–342. [Google Scholar] [CrossRef]
- Cook, J.H.; Baryza, J.; Brennan, C.; Lowe, D.; Wang, M.; Redman, A.; Scott, W.J.; Wood, J.E. Disubstituted cis-pyrrolidinones as inhibitors of 17β-hydroxysteroid dehydrogenase ii. Part 1: Synthetic approach. Tetrahedron Lett. 2005, 46, 1525–1528. [Google Scholar] [CrossRef]
- Gunn, D.; Akuche, C.; Baryza, J.; Blue, M.L.; Brennan, C.; Campbell, A.M.; Choi, S.; Cook, J.; Conrad, P.; Dixon, B.; et al. 4,5-disubstituted cis-pyrrolidinones as inhibitors of type ii 17beta-hydroxysteroid dehydrogenase. Part 2. Sar. Bioorg. Med. Chem. Lett. 2005, 15, 3053–3057. [Google Scholar] [CrossRef]
- Wood, J.; Bagi, C.M.; Akuche, C.; Bacchiocchi, A.; Baryza, J.; Blue, M.L.; Brennan, C.; Campbell, A.M.; Choi, S.; Cook, J.H.; et al. 4,5-disubstituted cis-pyrrolidinones as inhibitors of type ii 17beta-hydroxysteroid dehydrogenase. Part 3. Identification of lead candidate. Bioorg. Med. Chem. Lett. 2006, 16, 4965–4968. [Google Scholar] [CrossRef]
- Picard, F.; Baston, E.; Reichert, W.; Hartmann, R.W. Synthesis of n-substituted piperidine-4-(benzylidene-4-carboxylic acids) and evaluation as inhibitors of steroid-5alpha-reductase type 1 and 2. Bioorg. Med. Chem. 2000, 8, 1479–1487. [Google Scholar] [CrossRef]
- Leze, M.P.; Le Borgne, M.; Pinson, P.; Palusczak, A.; Duflos, M.; Le Baut, G.; Hartmann, R.W. Synthesis and biological evaluation of 5-[(aryl)(1h-imidazol-1-yl)methyl]-1h-indoles: Potent and selective aromatase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 1134–1137. [Google Scholar]
- Gobbi, S.; Cavalli, A.; Negri, M.; Schewe, K.E.; Belluti, F.; Piazzi, L.; Hartmann, R.W.; Recanatini, M.; Bisi, A. Imidazolylmethylbenzophenones as highly potent aromatase inhibitors. J. Med. Chem. 2007, 50, 3420–3422. [Google Scholar] [CrossRef]
- Hu, Q.; Negri, M.; Jahn-Hoffmann, K.; Zhuang, Y.; Olgen, S.; Bartels, M.; Muller-Vieira, U.; Lauterbach, T.; Hartmann, R.W. Synthesis, Biological evaluation, and molecular modeling studies of methylene imidazole substituted biaryls as inhibitors of human 17alpha-hydroxylase-17,20-lyase (cyp17)—part ii: Core rigidification and influence of substituents at the methylene bridge. Bioorg. Med. Chem. 2008, 16, 7715–7727. [Google Scholar] [CrossRef]
- Hille, U.E.; Hu, Q.; Vock, C.; Negri, M.; Bartels, M.; Muller-Vieira, U.; Lauterbach, T.; Hartmann, R.W. Novel cyp17 inhibitors: Synthesis, biological evaluation, structure-activity relationships and modelling of methoxy- and hydroxy-substituted methyleneimidazolyl biphenyls. Eur. J. Med. Chem. 2009, 44, 2765–2775. [Google Scholar] [CrossRef]
- Voets, M.; Antes, I.; Scherer, C.; Muller-Vieira, U.; Biemel, K.; Marchais-Oberwinkler, S.; Hartmann, R.W. Synthesis and evaluation of heteroaryl-substituted dihydronaphthalenes and indenes: Potent and selective inhibitors of aldosterone synthase (cyp11b2) for the treatment of congestive heart failure and myocardial fibrosis. J. Med. Chem. 2006, 49, 2222–2231. [Google Scholar] [CrossRef]
- Lucas, S.; Heim, R.; Negri, M.; Antes, I.; Ries, C.; Schewe, K.E.; Bisi, A.; Gobbi, S.; Hartmann, R.W. Novel aldosterone synthase inhibitors with extended carbocyclic skeleton by a combined ligand-based and structure-based drug design approach. J. Med. Chem. 2008, 51, 6138–6149. [Google Scholar] [CrossRef]
- Lucas, S.; Heim, R.; Ries, C.; Schewe, K.E.; Birk, B.; Hartmann, R.W. In vivo active aldosterone synthase inhibitors with improved selectivity: Lead optimization providing a series of pyridine substituted 3,4-dihydro-1h-quinolin-2-one derivatives. J. Med. Chem. 2008, 51, 8077–8087. [Google Scholar] [CrossRef]
- Heim, R.; Lucas, S.; Grombein, C.M.; Ries, C.; Schewe, K.E.; Negri, M.; Muller-Vieira, U.; Birk, B.; Hartmann, R.W. Overcoming undesirable cyp1a2 inhibition of pyridylnaphthalene-type aldosterone synthase inhibitors: Influence of heteroaryl derivatization on potency and selectivity. J. Med. Chem. 2008, 51, 5064–5074. [Google Scholar] [CrossRef]
- Hille, U.E.; Zimmer, C.; Haupenthal, J.; Hartmann, R.W. Optimization of the first selective steroid-11β-hydroxylase (cyp11b1) inhibitors for the treatment of cortisol dependent diseases. Med. Chem. Lett. 2011, 2, 559–564. [Google Scholar] [CrossRef]
- Hille, U.E.; Zimmer, C.; Vock, C.; Hartmann, R.W. First selective cyp11b inhibitors for the treatment of cortisol dependent diseases. ACS Med. Chem. Lett. 2011, 2, 2–6. [Google Scholar] [CrossRef]
- Marchais-Oberwinkler, S.; Wetzel, M.; Ziegler, E.; Kruchten, P.; Werth, R.; Henn, C.; Hartmann, R.W.; Frotscher, M. New drug-like hydroxyphenylnaphthol steroidomimetics as potent and selective 17beta-hydroxysteroid dehydrogenase type 1 inhibitors for the treatment of estrogen-dependent diseases. J. Med. Chem. 2011, 54, 534–547. [Google Scholar] [CrossRef]
- Oster, A.; Klein, T.; Henn, C.; Werth, R.; Marchais-Oberwinkler, S.; Frotscher, M.; Hartmann, R.W. Bicyclic substituted hydroxyphenylmethanone type inhibitors of 17 beta-hydroxysteroid dehydrogenase type 1 (17 beta-hsd1): The role of the bicyclic moiety. ChemMedChem 2011, 6, 476–487. [Google Scholar] [CrossRef]
- Spadaro, A.; Negri, M.; Marchais-Oberwinkler, S.; Bey, E.; Frotscher, M. Hydroxybenzothiazoles as new nonsteroidal inhibitors of 17beta-hydroxysteroid dehydrogenase type 1 (17beta-hsd1). PLoS One 2012, 7, e29252. [Google Scholar]
- Henn, C.; Einspanier, A.; Marchais-Oberwinkler, S.; Frotscher, M.; Hartmann, R.W. Lead optimization of 17beta-hsd1 inhibitors of the (hydroxyphenyl)naphthol sulfonamide type for the treatment of endometriosis. J. Med. Chem. 2012, 55, 3307–3318. [Google Scholar] [CrossRef]
- Bey, E.; Marchais-Oberwinkler, S.; Kruchten, P.; Frotscher, M.; Werth, R.; Oster, A.; Algul, O.; Neugebauer, A.; Hartmann, R.W. Design, synthesis and biological evaluation of bis(hydroxyphenyl) azoles as potent and selective non-steroidal inhibitors of 17beta-hydroxysteroid dehydrogenase type 1 (17beta-hsd1) for the treatment of estrogen-dependent diseases. Bioorg. Med. Chem. 2008, 16, 6423–6435. [Google Scholar] [CrossRef]
- Bey, E.; Marchais-Oberwinkler, S.; Negri, M.; Kruchten, P.; Oster, A.; Klein, T.; Spadaro, A.; Werth, R.; Frotscher, M.; Birk, B.; et al. New insights into the sar and binding modes of bis(hydroxyphenyl)thiophenes and -benzenes: Influence of additional substituents on 17beta-hydroxysteroid dehydrogenase type 1 (17beta-hsd1) inhibitory activity and selectivity. J. Med. Chem. 2009, 52, 6724–6743. [Google Scholar] [CrossRef]
- Wetzel, M.; Marchais-Oberwinkler, S.; Hartmann, R.W. 17beta-hsd2 inhibitors for the treatment of osteoporosis: Identification of a promising scaffold. Bioorg. Med. Chem. 2011, 19, 807–815. [Google Scholar] [CrossRef]
- Xu, K.; Al-Soud, Y.A.; Wetzel, M.; Hartmann, R.W.; Marchais-Oberwinkler, S. Triazole ring-opening leads to the discovery of potent nonsteroidal 17beta-hydroxysteroid dehydrogenase type 2 inhibitors. Eur. J. Med. Chem. 2011, 46, 5978–5990. [Google Scholar] [CrossRef]
- Wetzel, M.; Marchais-Oberwinkler, S.; Perspicace, E.; Moller, G.; Adamski, J.; Hartmann, R.W. Introduction of an electron withdrawing group on the hydroxyphenylnaphthol scaffold improves the potency of 17beta-hydroxysteroid dehydrogenase type 2 (17beta-hsd2) inhibitors. J. Med. Chem. 2011, 54, 7547–7557. [Google Scholar] [CrossRef]
- Xu, K.; Wetzel, M.; Hartmann, R.W.; Marchais-Oberwinkler, S. Synthesis and biological evaluation of spiro-δ-lactones as inhibitors of 17β-hydroxysteroid dehydrogenase type 2 (17β-hsd2). Lett. Drug Des. Discov. 2011, 8, 406–421. [Google Scholar] [CrossRef]
- Wetzel, M.; Gargano, E.M.; Hinsberger, S.; Marchais-Oberwinkler, S.; Hartmann, R.W. Discovery of a new class of bicyclic substituted hydroxyphenylmethanones as 17beta-hydroxysteroid dehydrogenase type 2 (17beta-hsd2) inhibitors for the treatment of osteoporosis. Eur. J. Med. Chem. 2012, 47, 1–17. [Google Scholar] [CrossRef]
- Al-Soud, Y.A.; Marchais-Oberwinkler, S.; Frotscher, M.; Hartmann, R.W. Synthesis and biological evaluation of phenyl substituted 1h-1,2,4-triazoles as non-steroidal inhibitors of 17beta-hydroxysteroid dehydrogenase type 2. Arch. Pharm. (Weinheim) 2012, 345, 610–621. [Google Scholar] [CrossRef]
- Marchais-Oberwinkler, S.; Xu, K.; Wetzel, M.; Perspicace, E.; Negri, M.; Meyer, A.; Odermatt, A.; Moller, G.; Adamski, J.; Hartmann, R.W. Structural optimization of 2,5-thiophene amides as highly potent and selective 17beta-hydroxysteroid dehydrogenase type 2 inhibitors for the treatment of osteoporosis. J. Med. Chem. 2013, 56, 167–181. [Google Scholar] [CrossRef]
- Migianu, E.; Kirsch, G. Synthesis of new thieno[b]azepinediones from α-methylene ketones. Synthesis 2002, volume, 1096–1100. [Google Scholar] [CrossRef]
- Hertzog, D.L.; Al-Barazanji, K.A.; Bigham, E.C.; Bishop, M.J.; Britt, C.S.; Carlton, D.L.; Cooper, J.P.; Daniels, A.J.; Garrido, D.M.; Goetz, A.S.; et al. The discovery and optimization of pyrimidinone-containing mch r1 antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 4723–4727. [Google Scholar] [CrossRef]
- Perrissin, M.; Favre, M.; Cuong, L.D.; Huguet, F.; Gaultier, C.; Narcisse, G. Synthesis and pharmacological activities of some substituted thienopyrimidine-4-ones. Eur. J. Med. Chem. 1988, 23, 453–456. [Google Scholar] [CrossRef]
- Boehm, N.; Krasselt, U.; Leistner, S.; Wagner, G. Reaction of 4-oxo-4h-pyrido[3'2':4,5]thieno[3,2-d]-1,3-oxazines with amines. Pharmazie 1992, 47, 897–901. [Google Scholar]
- Peinador, C.; Ojea, V.; Quintela, J.M. A convenient synthesis of some new pyrido[3',2':4,5]thieno[3,2-d]pyrimidine derivatives with potential biological activity. J. Heterocycl. Chem. 1992, 29, 1693–1702. [Google Scholar] [CrossRef]
- Lessene, G.; Baell, J. Preparation of amino acid derivatives as alpha-helical mimetics. PCT Int. Appl. WO 2006002474 A1 20060112, 2006. [Google Scholar]
- Hesse, S.; Perspicace, E. Microwave-assisted synthesis of 2-aminothiophene-3-carboxylic acid derivatives, 3h-thieno[2,3-d]pyrimidin-4-one and 4-chlorothieno[2,3-d]pyrimidine. Tetrahedron Lett. 2007, 48, 5261–5264. [Google Scholar] [CrossRef]
- Kruchten, P.; Werth, R.; Marchais-Oberwinkler, S.; Frotscher, M.; Hartmann, R.W. Development of a biological screening system for the evaluation of highly active and selective 17beta-hsd1-inhibitors as potential therapeutic agents. Mol. Cell. Endocrinol. 2009, 301, 154–157. [Google Scholar] [CrossRef]
- Messinger, J.; Hirvela, L.; Husen, B.; Kangas, L.; Koskimies, P.; Pentikainen, O.; Saarenketo, P.; Thole, H. New inhibitors of 17beta-hydroxysteroid dehydrogenase type 1. Mol. Cell. Endocrinol. 2006, 248, 192–198. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1a–b, 2a–b, 3a–d, 4a–b, 5a–d are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).