Synthesis and Antiviral Activity of N-Phenylbenzamide Derivatives, a Novel Class of Enterovirus 71 Inhibitors

Abstract
:
1. Introduction
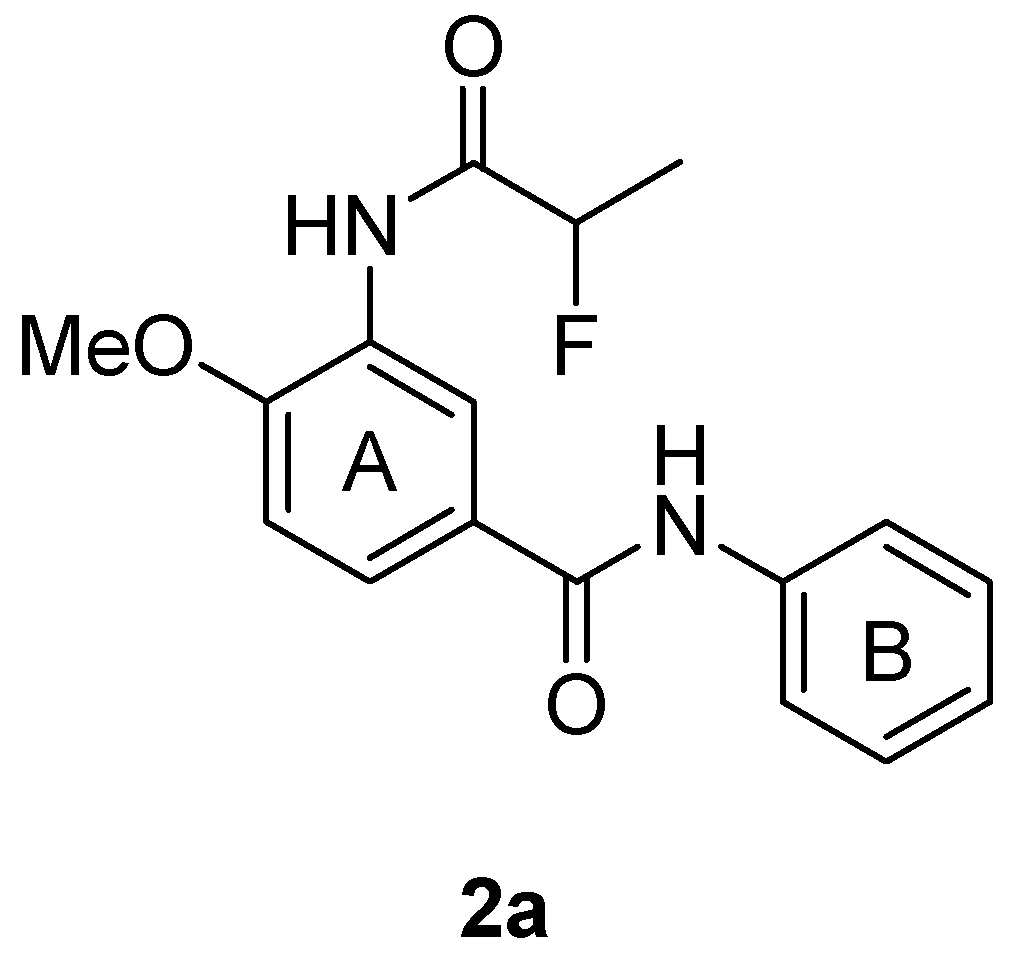
2. Results and Discussion
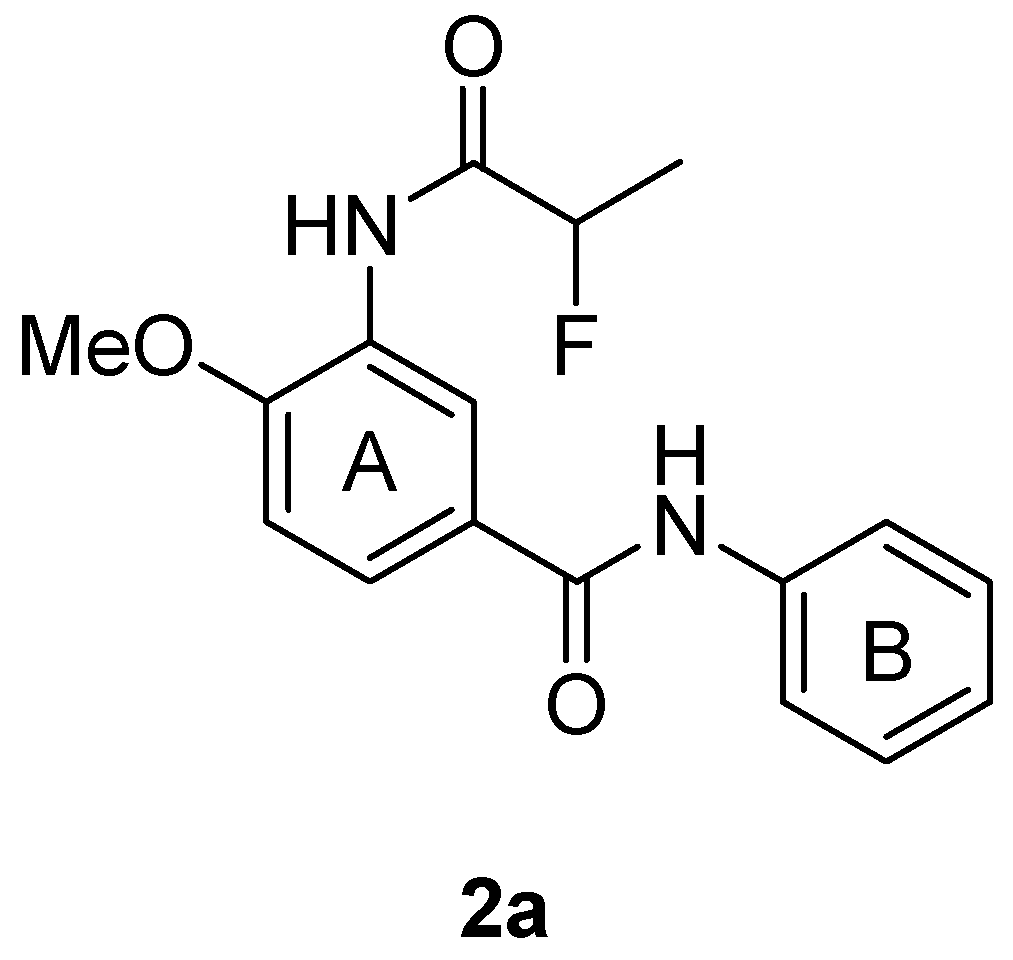
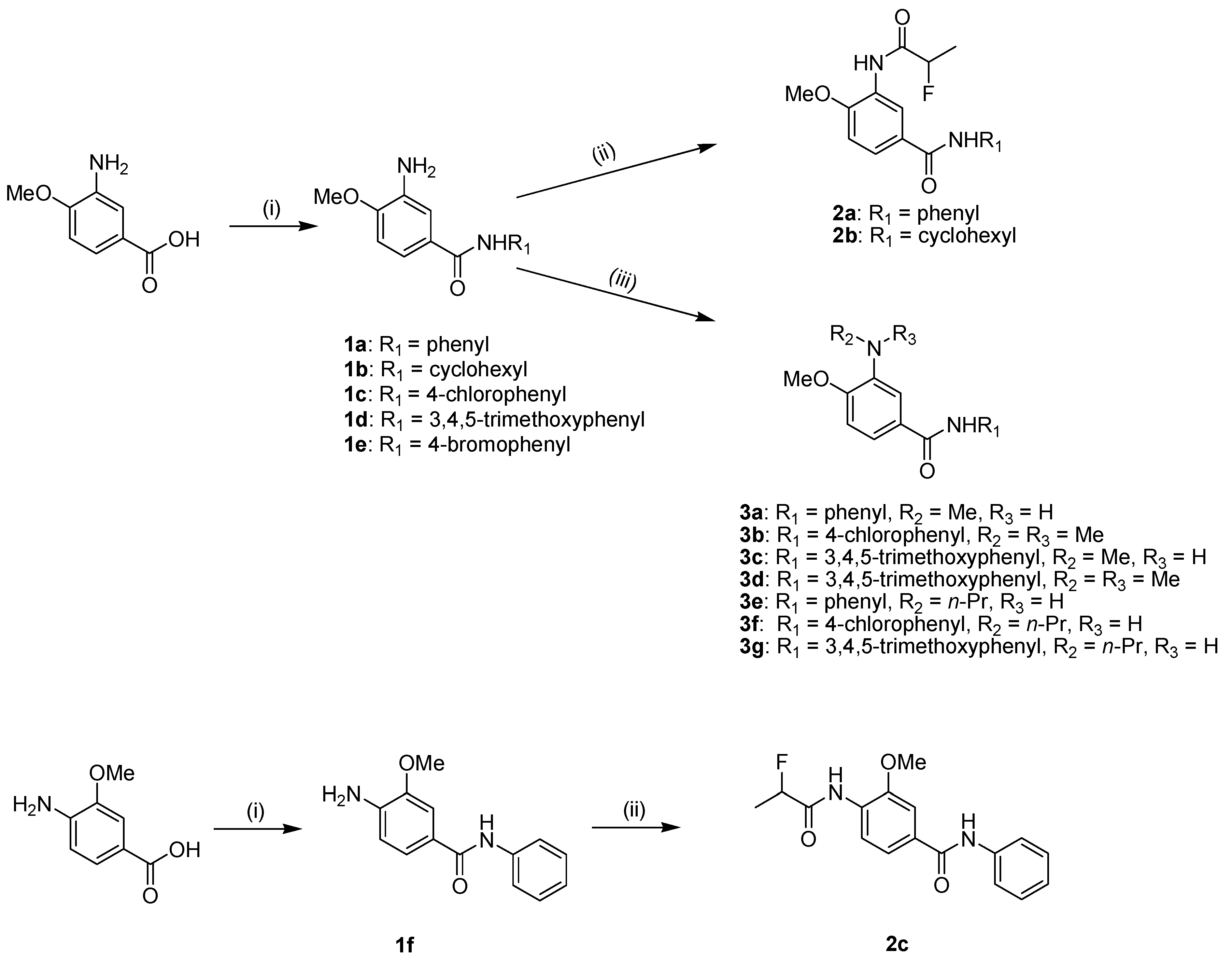
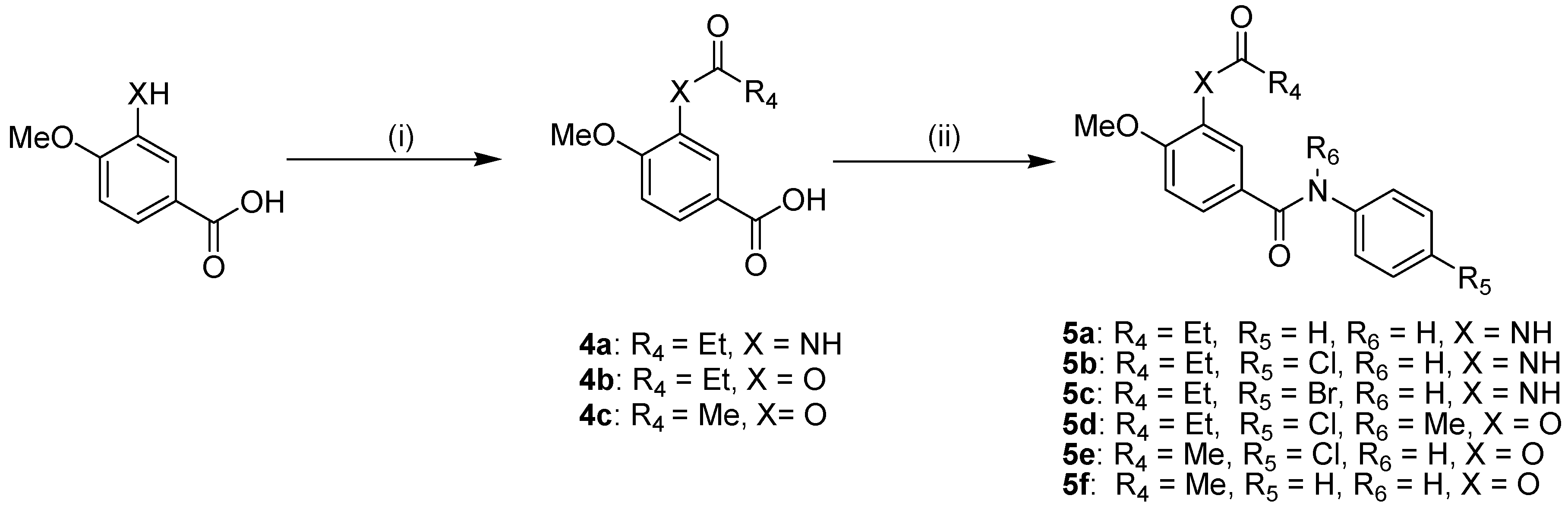
2.1. Chemistry
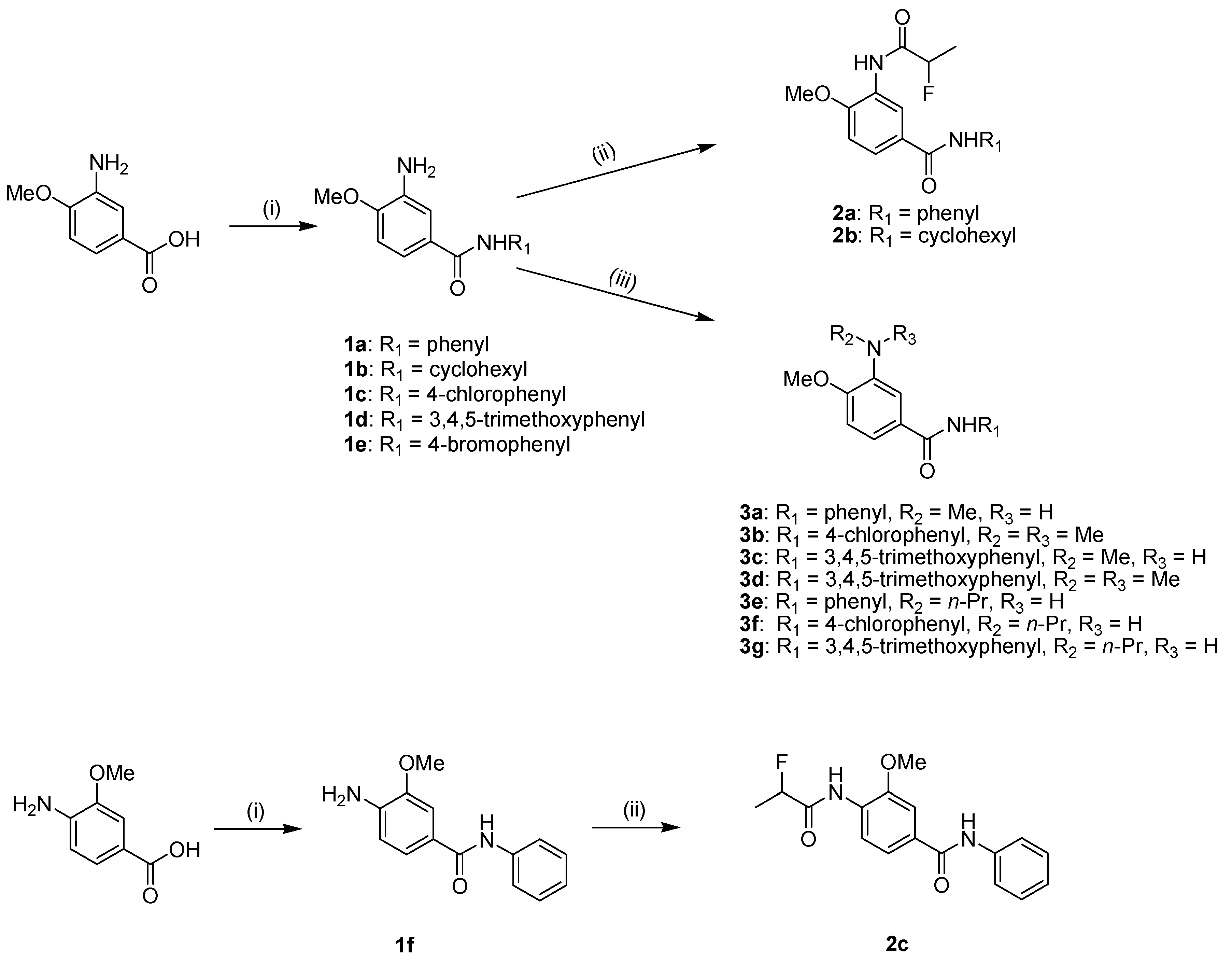
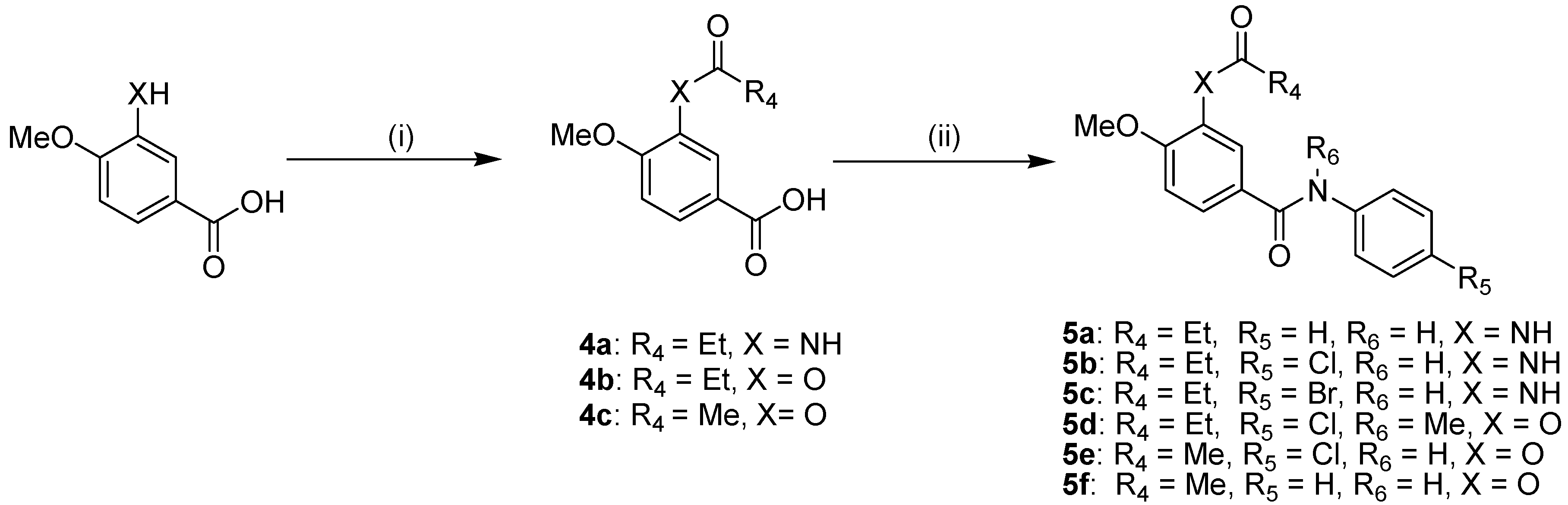
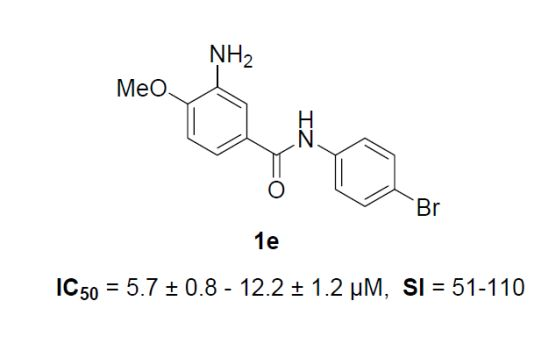
2.2. Anti-EV 71 Activity
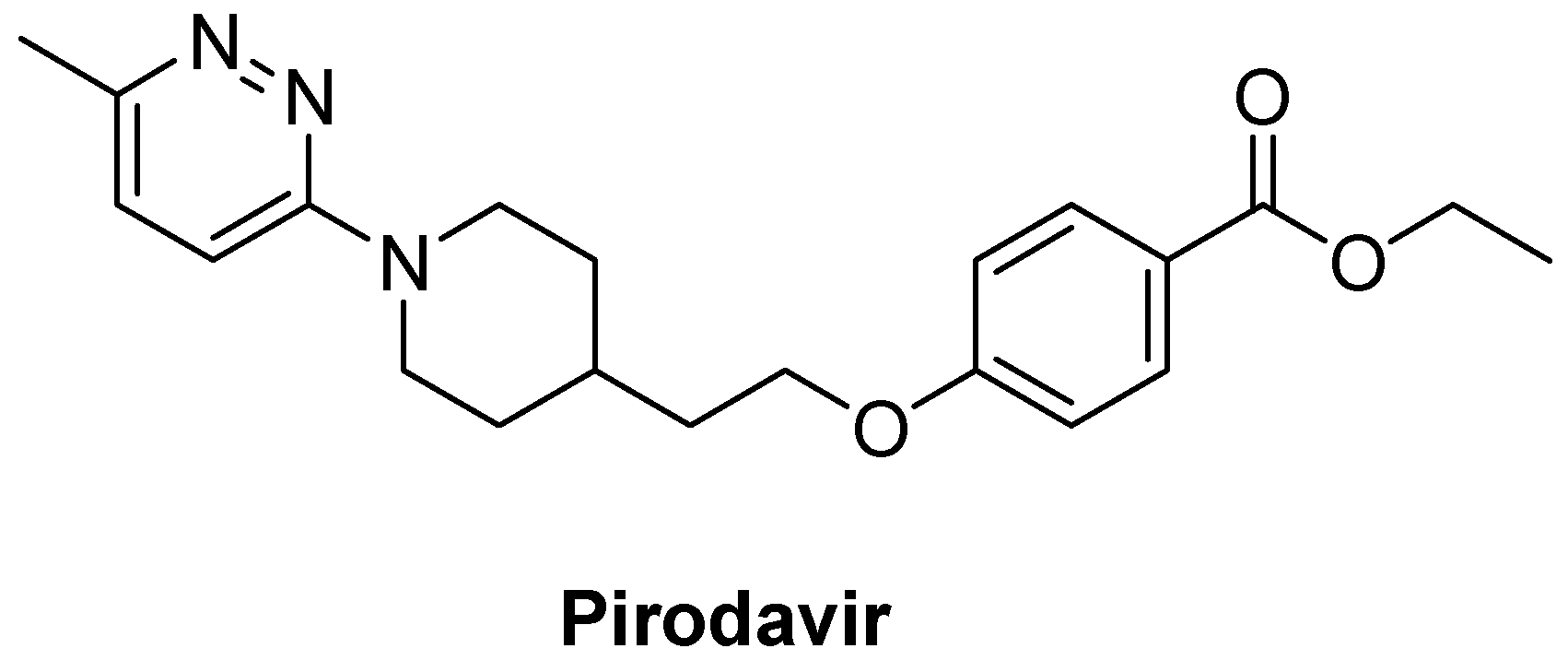

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpds | TC50 (μM) | SZ-98 | JS-52-3 | H | BrCr | ||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | SI | IC50 (μM) | SI | IC50 (μM) | SI | IC50 (μM) | SI | ||
| 1a | >820 | 350 ± 24 | 2.3 | 160 ± 7.9 | 5.2 | 190 ± 14 | 4.3 | 160 ± 12 | 5.2 |
| 1c | 520 ± 29 | 15 ± 0.6 | 36 | 46 ± 5.7 | 12 | 34 ± 3.6 | 16 | 56 ± 4.2 | 10 |
| 1e | 620 ± 0.0 | 12 ± 1.1 | 51 | 9.8 ± 0.4 | 64 | 5.7 ± 0.8 | 110 | 9.1 ± 1.4 | 68 |
| 2a | 630 ± 0.0 | >630 | - | 160 ± 5.7 | >3.9 | 18 ± 1.2 | >35 | >630 | - |
| 2b | >620 | >620 | - | 210 ± 0.0 | >3.0 | 430 ± 36 | >1.4 | 360 ± 26 | >1.7 |
| 2c | >630 | 110 ± 12 | >5.8 | 34 ± 2.8 | 41 ± 2.9 | >16 | 90 ± 9.5 | >7.0 | |
| 3a | 780 ± 0.0 | >260 | - | 260 ± 0.0 | 3.0 | 260 ± 0.0 | 3.0 | 180 ± 14 | 4.3 |
| 3b | 510 ± 38 | 73 ± 8.6 | 7.0 | 42 ± 3.1 | 12 | 31 ± 4.3 | 16 | 50 ± 3.8 | 10 |
| 3c | 280 ± 5.9 | 64 ± 0.0 | 4.3 | 37 ± 1.9 | 7.5 | 37 ± 1.2 | 7.5 | 44 ± 1.0 | 6.2 |
| 3d | >560 | 320 ± 32 | >1.7 | >560 | - | 190 ± 0.0 | >3.0 | 190 ± 0.0 | >3.0 |
| 3e | 410 ± 22 | >78 | - | 34 ± 4.6 | 12 | 20 ± 1.2 | 20 | 45 ± 0.0 | 9.0 |
| 3f | 120 | >70 | - | >70 | - | >70 | - | >70 | - |
| 3g | 530 | >59 | - | 34 ± 2.5 | 16 | 15 ± 0.0 | 35 | 20 ± 3.1 | 27 |
| 5a | >670 | >75 | - | 58 ± 4.9 | >12 | 96 ± 10 | >7.0 | 45 ± 10 | 9.0 |
| 5b | >600 | 38 ± 2.9 | > 16 | 39 ± 1.9 | >16 | 22 ± 2.9 | >27 | >67 | - |
| 5c | 530 ± 0.0 | 11 ± 0.8 | 47 | 20 ± 3.1 | 27 | 8.4 ± 1.2 | 63 | 34 ± 0.0 | 16 |
| 5d | 280 ± 21 | 190 ± 12 | 1.4 | 44 ± 3.7 | 6.2 | 65 ± 0.0 | 4.3 | 53 ± 2.4 | 5.2 |
| 5e | 470 ± 22 | 13 ± 2.4 | 37 | 32 ± 3.6 | 14 | 22 ± 4.7 | 21 | 32 ± 3.9 | 15 |
| 5f | 360 ± 39 | 90 ± 11 | 4.0 | 98 ± 4.7 | 3.7 | 47 ± 6.1 | 7.6 | 95 ± 9.0 | 3.8 |
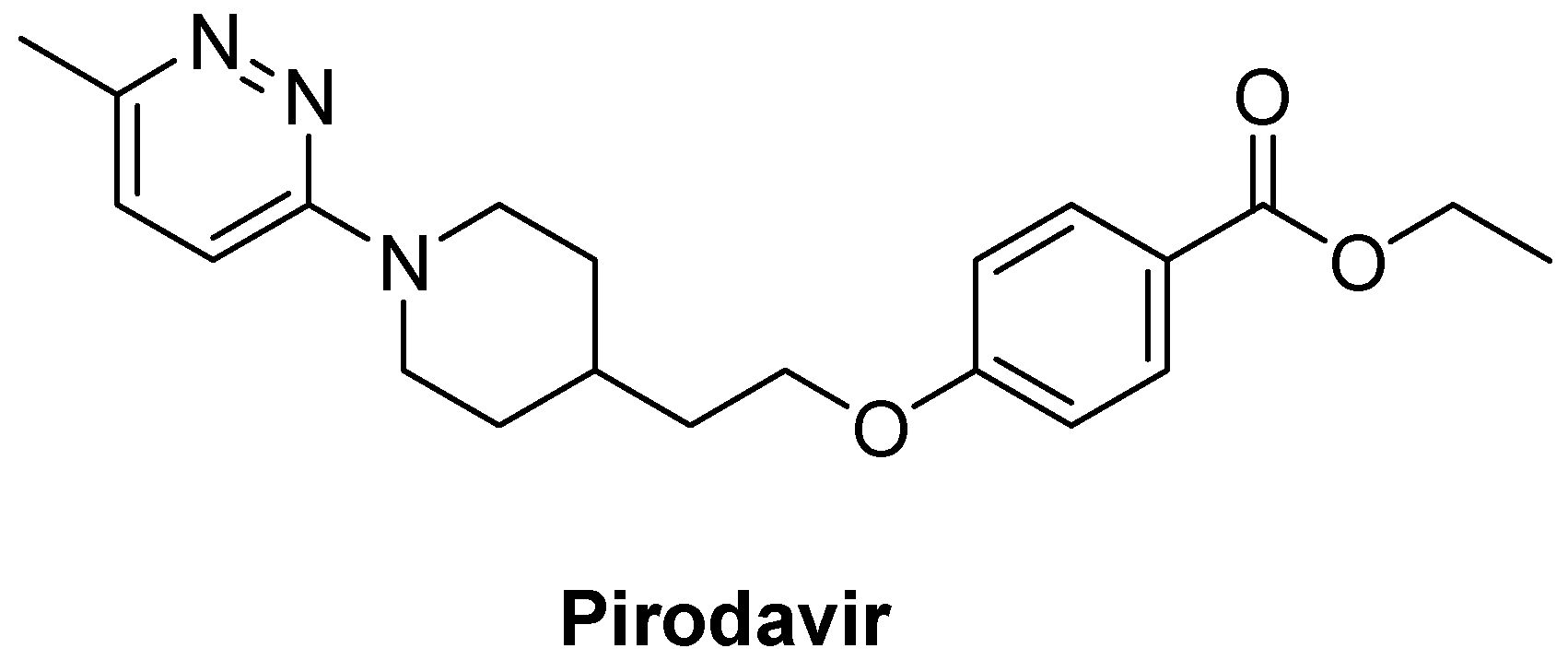
| Pirodavir | 31 ± 2.2 | 1.2 ± 0.2 | 25 | 1.0 ± 0.2 | 30 | 0.6 ± 0.1 | 52 | 1.0 ± 0.2 | 30 |
3. Experimental
3.1. General
3.1.1. General Procedure for the Synthesis of Compound 1b–f, 5b, 5e, 5f
3.1.2. General Procedure for the Synthesis of Compounds 2b, c
3.1.3. General Procedure for the Synthesis of Compound 3a–g
3.2. Biological Activity Test Procedures
3.2.1. Cytotoxicity Determination
3.2.2. Anti EV 71 Activity Assay
4. Conclusions
Acknowledgments
References
- Schmidt, N.J.; Lennette, E.H.; Ho, H.H. An apparently new enterovirus isolated from patients with disease of the central nervous system. J. Infect. Dis. 1974, 129, 304–309. [Google Scholar] [CrossRef]
- Samuda, G.M.; Chang, W.K.; Yeung, C.Y.; Tang, P.S. Monoplegia caused by Enterovirus 71: An outbreak in Hong Kong. Pediatr. Infect. Dis. J. 1987, 6, 206–208. [Google Scholar] [CrossRef]
- Singh, S.; Chow, V.T.; Phoon, M.C.; Chan, K.P.; Poh, C.L. Direct detection of enterovirus 71 (EV71) in clinical specimens from a hand, foot, and mouth disease outbreak in Singapore by reverse transcription-PCR with universal enterovirus and EV71-specific primers. J. Clin. Microbiol. 2002, 40, 2823–2827. [Google Scholar] [CrossRef]
- Yang, F.; Ren, L.; Xiong, Z.; Li, J.; Xiao, Y.; Zhao, R.; He, Y.; Bu, G.; Zhou, S.; Wang, J.; et al. Enterovirus 71 outbreak in the People's Republic of China in 2008. J. Clin. Microbiol. 2009, 47, 2351–2352. [Google Scholar] [CrossRef]
- Lee, T.C.; Guo, H.R.; Jenny Su, H.J.; Yang, Y.C.; Chang, S.L.; Chen, K.T. Diseases caused by enterovirus 71 infection. Pediatr. Infect. Dis. J. 2009, 28, 904–910. [Google Scholar] [CrossRef]
- Brown, B.A.; Oberste, M.S.; Alexander, J.P, Jr.; Kennett, M.L.; Pallansch, M.A. Molecular epidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998. J. Virol. 1999, 73, 9969–9975. [Google Scholar]
- Shin, S.R.; Ho, M.S.; Lin, K.H.; Wu, S.L.; Chen, Y.T.; Wu, C.N.; Lin, T.Y.; Chang, L.Y.; Tsao, K.C.; Ning, H.C.; et al. Genetic analysis of enterovirus 71 isolated from fatal and non-fatal cases of hand, Foot and mouth disease during an epidemic in Taiwan, 1998. Virus Res. 2000, 68, 127–136. [Google Scholar] [CrossRef]
- Hao, L.H.; Li, Y.P.; He, W.Y.; Wang, H.Q.; Shang, G.Z.; Jiang, J.D.; Li, Y.H.; Li, Z.R. Synthesis and antiviral activity of substituted bisaryl amide compounds as novel influenza virus inhibitors. Eur. J. Med. Chem. 2012, 55, 117–124. [Google Scholar] [CrossRef]
- Humphrey, J.M.; Chamberlin, A.R. Chemical synthesis of natural product peptides: Coupling methods for the incorporation of noncoded amino acids into peptides. Chem. Rev. 1997, 97, 2243–2266. [Google Scholar] [CrossRef]
- Huang, Y.P.; Lin, T.L.; Kuo, C.Y.; Lin, M.W.; Yao, C.Y.; Liao, H.W.; Hsu, L.C.; Yang, C.F.; Yang, J.Y.; Chen, P.J.; et al. The circulation of subgenogroups B5 and C5 of enterovirus 71 in Taiwan from 2006 to 2007. Virus. Res. 2008, 137, 206–212. [Google Scholar] [CrossRef]
- Tan, X.J.; Huang, X.J.; Zhu, S.L.; Chen, H.; Yu, Q.L.; Wang, H.Y.; Huo, X.X.; Zhou, J.H.; Wu, Y.; Yan, D.M.; et al. The persistent circulation of enterovirus 71 in People's Republic of China: causing emerging nationwide epidemics since 2008. PLoS One 2008, 6, e25662. [Google Scholar]
- Andries, K.; Dewindt, B.; Snoeks, J.; Willebrords, R.; van Eemeren, K.; Stokbroekx, R.; Janssen, P.A. In vitro activity of pirodavir (R 77975), a substituted phenoxy-pyridazinamine with broad-spectrum antipicornaviral activity. Antimicrob. Agents Chemother. 1992, 36, 100–107. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1a, 1c, 1e, 2a–c, 3a–g, and 5a–f are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ji, X.-Y.; Wang, H.-Q.; Hao, L.-H.; He, W.-Y.; Gao, R.-M.; Li, Y.-P.; Li, Y.-H.; Jiang, J.-D.; Li, Z.-R. Synthesis and Antiviral Activity of N-Phenylbenzamide Derivatives, a Novel Class of Enterovirus 71 Inhibitors. Molecules 2013, 18, 3630-3640. https://doi.org/10.3390/molecules18033630
Ji X-Y, Wang H-Q, Hao L-H, He W-Y, Gao R-M, Li Y-P, Li Y-H, Jiang J-D, Li Z-R. Synthesis and Antiviral Activity of N-Phenylbenzamide Derivatives, a Novel Class of Enterovirus 71 Inhibitors. Molecules. 2013; 18(3):3630-3640. https://doi.org/10.3390/molecules18033630
Chicago/Turabian StyleJi, Xing-Yue, Hui-Qiang Wang, Lan-Hu Hao, Wei-Ying He, Rong-Mei Gao, Yan-Ping Li, Yu-Huan Li, Jian-Dong Jiang, and Zhuo-Rong Li. 2013. "Synthesis and Antiviral Activity of N-Phenylbenzamide Derivatives, a Novel Class of Enterovirus 71 Inhibitors" Molecules 18, no. 3: 3630-3640. https://doi.org/10.3390/molecules18033630
APA StyleJi, X.-Y., Wang, H.-Q., Hao, L.-H., He, W.-Y., Gao, R.-M., Li, Y.-P., Li, Y.-H., Jiang, J.-D., & Li, Z.-R. (2013). Synthesis and Antiviral Activity of N-Phenylbenzamide Derivatives, a Novel Class of Enterovirus 71 Inhibitors. Molecules, 18(3), 3630-3640. https://doi.org/10.3390/molecules18033630