Biohybrid -Se-S- Coupling Reactions of an Amino Acid Derived Seleninate


Abstract
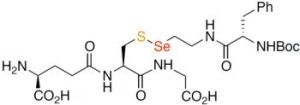
:
{kind=link}
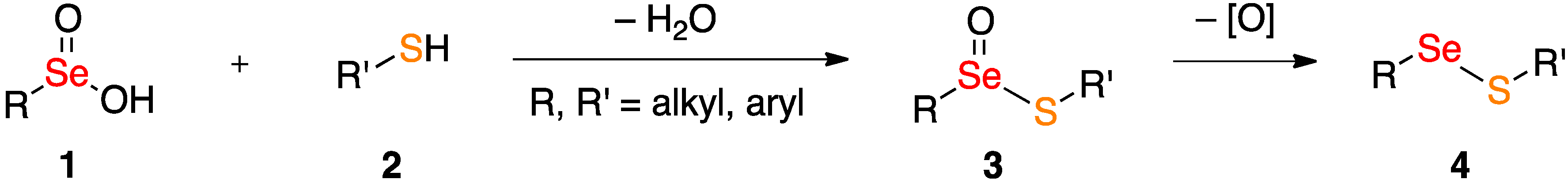{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
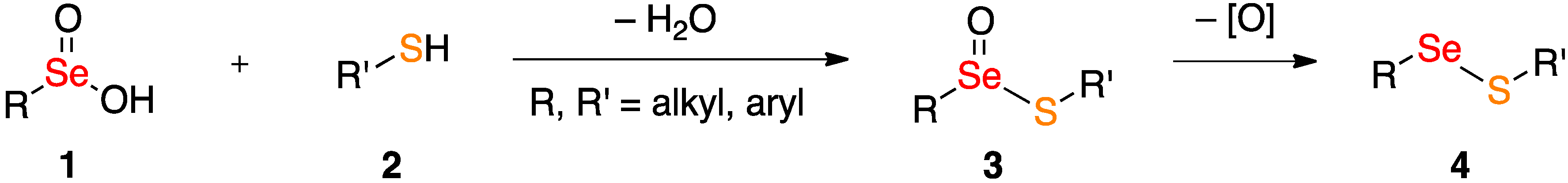
2. Results and Discussion
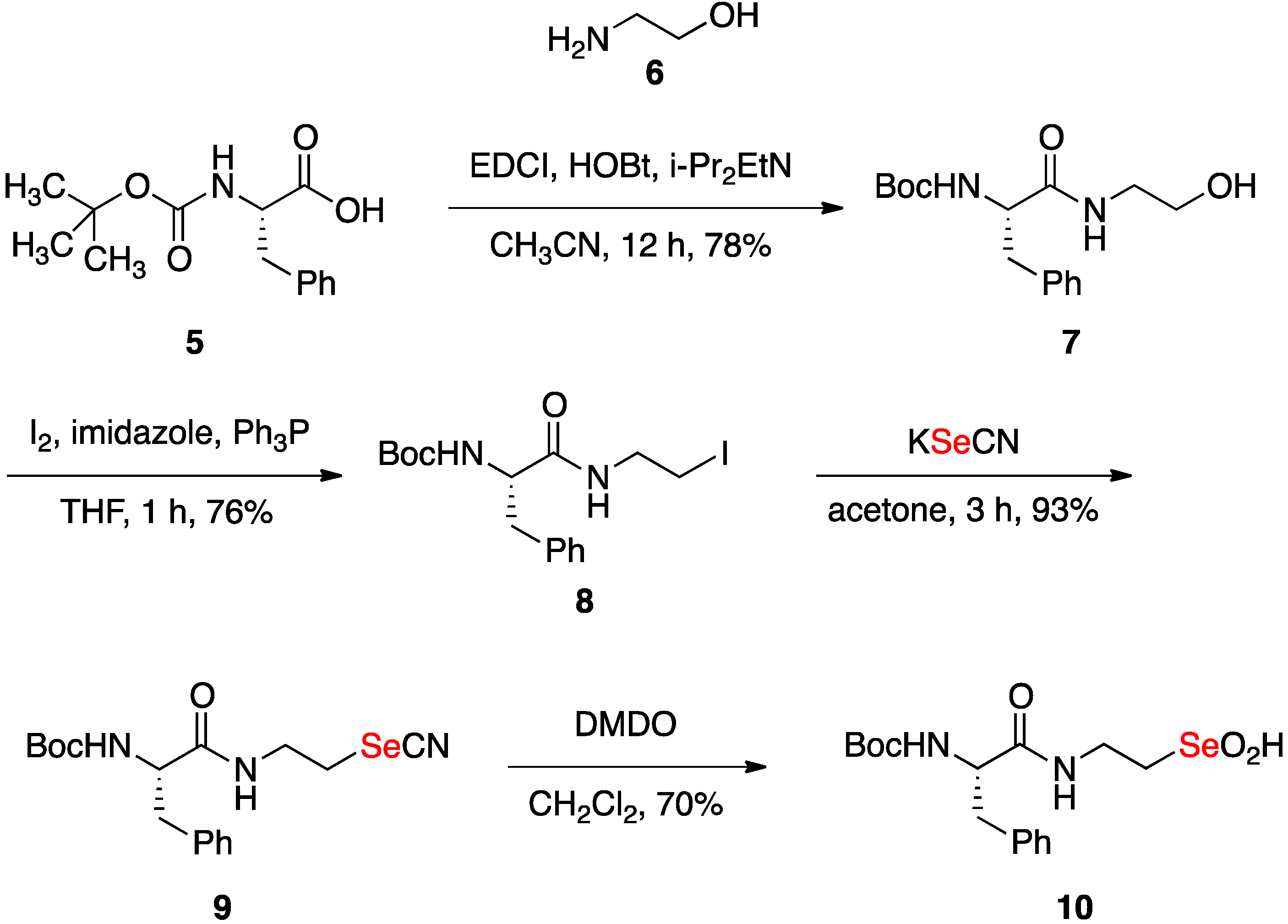
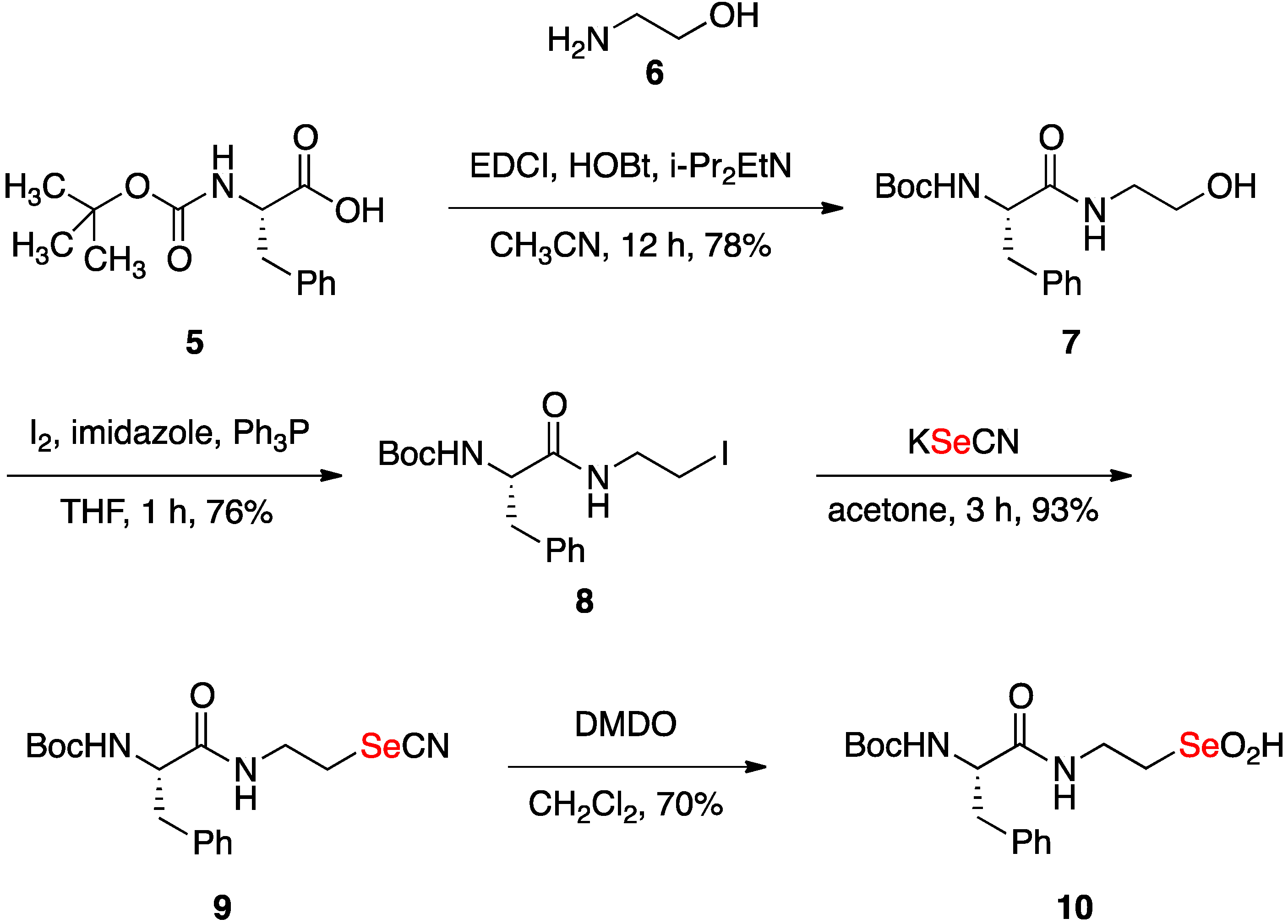
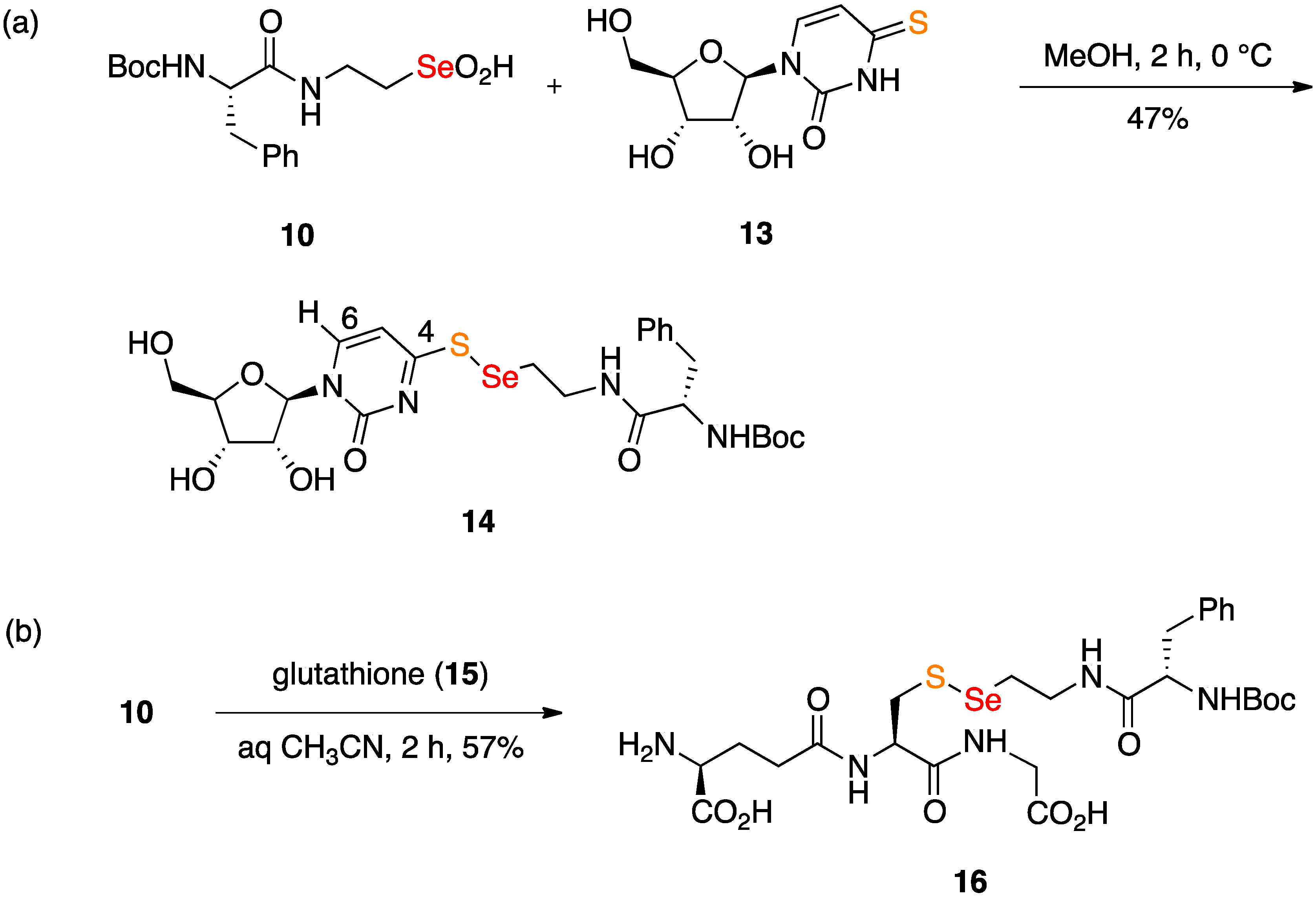
2.1. Synthesis of Seleninic Acid 10
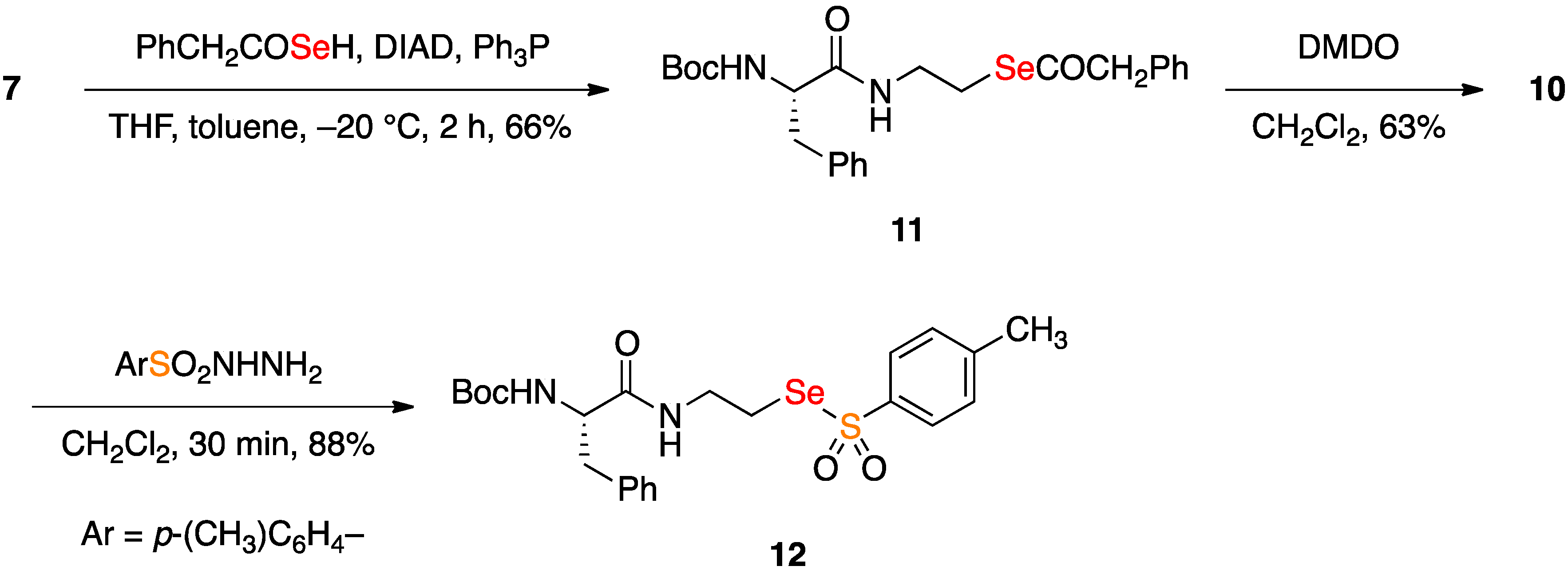
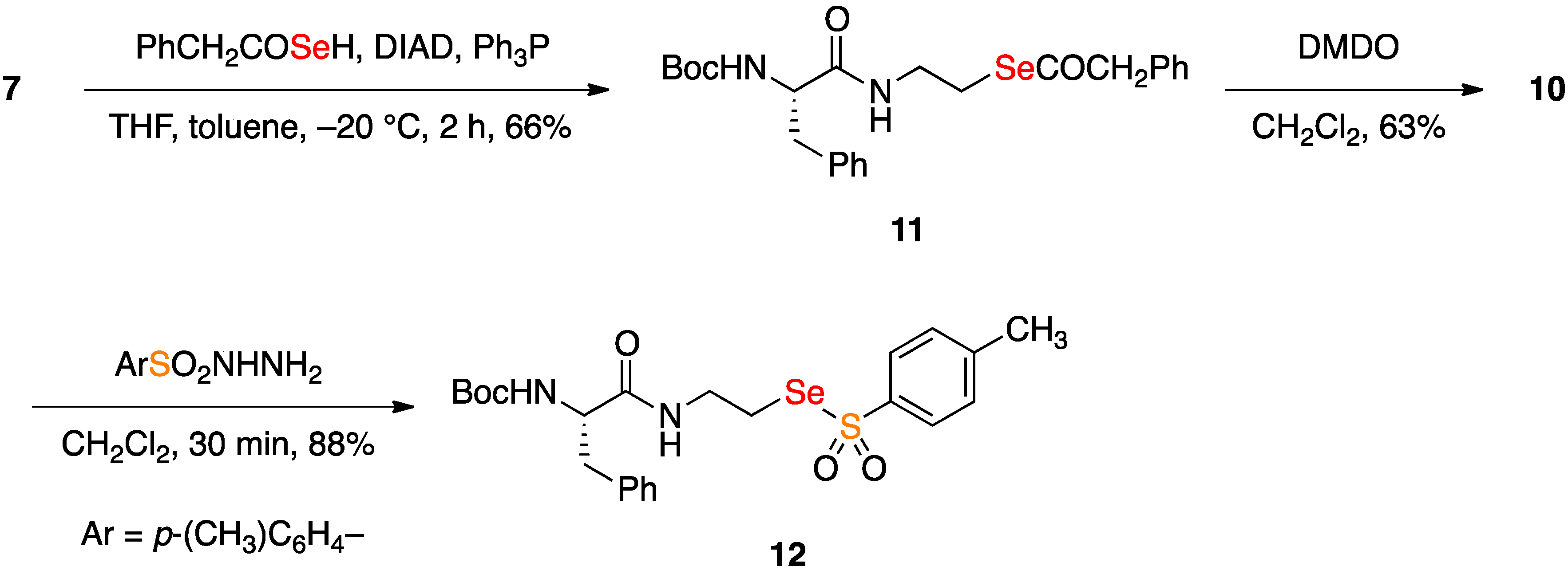
2.2. Coupling Reactions of Seleninic Acid 10
3. Experimental
3.1. General
4. Conclusions
Supplementary Materials
Acknowledgments
- Sample Availability: Samples of the compounds 7 and 10 are available from the authors.
References
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. Engl. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Finn, M.G.; Fokin, V.V. Click chemistry: Function follows form. Chem. Soc. Rev. 2010, 39, 1231–1232. [Google Scholar]
- Meldal, M.; Tornoe, C.W. Cu-Catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. Engl. 2009, 48, 6974–6998. [Google Scholar] [CrossRef]
- Jewett, J.J.; Bertozzi, C.R. Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 2010, 39, 1272–1297. [Google Scholar] [CrossRef]
- Dirks, A.J.; Cornelissen, J.J.L.M.; van Delft, F.L.; van Hest, J.C.M.; Nolte, R.J.M.; Rowan, A.E.; Turjes, F.P.J.T. From (bio)molecules to biohybrid materials with the click chemistry approach. QSAR Comb. Sci. 2007, 26, 1200–1210. [Google Scholar] [CrossRef]
- Giffin, M.J.; Heaslet, H.; Brik, A.; Lin, Y.-C.; Cauvi, G.; Wong, C.-H.; McRee, D.E.; Elder, J.H.; Stout, C.D.; Torbett, B.E. A copper(I)-catalyzed 1,2,3-triazole azide-alkyne click compound is a potent inhibitor of a multidrug-resistant HIV-1 protease variant. J. Med. Chem. 2008, 51, 6263–6270. [Google Scholar] [CrossRef]
- Manetsch, R.; Krasinski, A.; Radic, Z.; Raushel, J.; Taylor, P.; Sharpless, K.B.; Kolb, H.C. In situ click chemistry: Enzyme inhibitors made to their own specifications. J. Am. Chem. Soc. 2004, 126, 12809–12818. [Google Scholar]
- Colombano, G.; Travelli, C.; Galli, U.; Caldarelli, A.; Chini, M.G.; Canonico, P.L.; Sorba, G.; Bifulco, G.; Tron, G.C.; Genazzani, A.A. A novel potent nicotinamide phosphoribosyltransferase inhibitor synthesized via click chemistry. J. Med. Chem. 2010, 53, 616–623. [Google Scholar]
- El-Sagheer, A.H.; Brown, T. Click nucleic acid ligation: Applications in biology and nanotechnology. Acc. Chem. Res. 2012, 45, 1258–1267. [Google Scholar] [CrossRef]
- Nimmo, C.M.; Shoichet, M.S. Regenerative biomaterials that “click”: Simple, aqueous-based protocols for hydrogel synthesis, surface immobilization, and 3D patterning. Bioconjug. Chem. 2011, 22, 2199–2209. [Google Scholar] [CrossRef]
- Azagarsamy, M.A.; Anseth, K.S. Bioorthogonal click chemistry: An indispensable tool to create multifaceted cell culture scaffolds. ACS Macro Lett. 2013, 2, 5–9. [Google Scholar] [CrossRef]
- Best, M.D. Click Chemistry and Bioorthogonal Reactions: Unprecedented selectivity in the labeling of biological molecules. Biochemistry 2009, 48, 6571–6584. [Google Scholar] [CrossRef]
- Canalle, L.A.; Vong, T.; Adams, P.H.H.M.; van Delft, F.L.; Raats, J.M.H.; Chivi, R.G.S.; van Hest, J.C.M. Clickable enzyme-linked immunosorbent assay. Biomolecules 2011, 12, 3692–3697. [Google Scholar]
- Mindt, T.L.; Struthers, H.; Brnas, L.; Anguelov, T.; Schweinsberg, C.; Maes, V.; Tourwe, D.; Schibli, R. Click to chelate”: Synthesis and installation of metal chelates into biomolecules in a single step. J. Am. Chem. Soc. 2006, 128, 15096–15097. [Google Scholar]
- Cunningham, C.W.; Mukhopadhyay, A.; Lushington, G.H.; Blagg, B.S.J.; Prisinzano, T.E.; Krise, J.P. Uptake, distribution and diffusivity of reactive fluorophores in cells: Implications toward target identification. Mol. Pharm. 2010, 7, 1301–1310. [Google Scholar]
- Liu, X.-M.; Quan, L.-D.; Tian, J.; Laquer, F.C.; Cibiroski, P.; Wang, D. Syntheses of click PEG-dexamethasone conjugates for the treatment of rheumatoid arthritis. Biomacromolecules 2010, 11, 2621–2628. [Google Scholar] [CrossRef]
- Kamphuis, M.M.J.; Johnston, A.P.R.; Such, G.K.; Dam, H.H.; Evans, R.A.; Scott, A.M.; Nice, E.C.; Heath, J.K.; Caruso, F. Targeting of cancer cells using click-functionalized polymer capsules. J. Am. Chem. Soc. 2010, 132, 15881–15883. [Google Scholar]
- Ochs, C.J.; Such, G.K.; Yan, Y.; van Koerverden, M.P.; Caruso, F. Biodegradable click capsules with engineered drug-loaded multilayers. ACS Nano 2010, 4, 1653–1663. [Google Scholar]
- Kice, J.L.; Lee, T.W.S. Oxidation-reduction reactions of organoselenium compounds. 1. Mechanism of the reaction between seleninic acids and thiols. J. Am. Chem. Soc. 1978, 100, 5094–5102. [Google Scholar] [CrossRef]
- Abdo, M.; Knapp, S. Biomimetic seleninates and selenonates. J. Am. Chem. Soc. 2008, 130, 9234–9235. [Google Scholar] [CrossRef]
- Abdo, M.; Liu, S.; Zhou, B.; Walls, C.D.; Wu, L.; Knapp, S.; Zhang, Y.-Y. Seleninate in place of phosphate: Irreversible inhibition of protein tyrosine phosphatases. J. Am. Chem. Soc. 2008, 130, 13196–13197. [Google Scholar]
- Abdo, M.; Knapp, S. Mechanism of a redox coupling of seleninic acid with thiol. J. Org. Chem. 2012, 77, 3433–3438. [Google Scholar] [CrossRef]
- Kice, J.L.; Purkiss, D.W. The induced decomposition of S-tert-butyl benzenethioseleninate. J. Org. Chem. 1987, 52, 3448–3451. [Google Scholar] [CrossRef]
- Abdo, M.; Zhang, Y.; Schramm, V.L.; Knapp, S. Electrophilic aromatic selenylation: New OPRT inhibitors. Org. Lett. 2010, 12, 2982–2985. [Google Scholar] [CrossRef]
- Knapp, S.; Darout, E. New reactions of selenocarboxylates. Org. Lett. 2005, 7, 203–205. [Google Scholar] [CrossRef]
- Back, T.G.; Collins, S.; Krishna, M.V. Reactions of sulfonhydrazides with benzeneseleninic acid, selenium halides, and sulfur halides. A convenient preparation of selenosulfonates and thiosulfonates. Can. J. Chem. 1987, 65, 38–42. [Google Scholar] [CrossRef]
- Back, T.G.; Collins, S. A convenient synthesis of selenolsulfonates from the oxidation of sulfonhydrazides with benzeneseleninic acid. Tetrahedron Lett. 1980, 21, 2213–2214. [Google Scholar] [CrossRef]
- Coleman, R.S.; Siedlicki, J.M. Synthesis of a 4-thio-2'-deoxyuridine containing oligonucleotide. Development of the thiocarbonyl group as a linker element. J. Am. Chem. Soc. 1992, 114, 9229–9230. [Google Scholar] [CrossRef]
- Nogueira, C.W.; Rocha, J.B.T. Diphenyl diselenide a Janus-faced molecule. J. Braz. Chem. Soc. 2010, 21, 2055–2071. [Google Scholar] [CrossRef]
- Flemer, S., Jr. Selenol protecting groups in organic chemistry: Special emphasis on selenocysteine Se-protection in solid phase peptide synthesis. Molecules 2011, 15, 3232–3251. [Google Scholar] [CrossRef]
- Boutureira, O.; Bernardes, G.J.L.; Fernandez-Gonzalez, M.; Anthony, D.C.; Davis, B.G. Selenenylsulfide-linked homogeneous glycopeptides and glycoproteins: Synthesis of human “hepatic Se metabolite A”. Angew. Chem. Int. Ed. Engl. 2012, 51, 1432–1436. [Google Scholar]
- Sarma, B.K.; Mugesh, G. Antioxidant activity of the anti-inflammatory compound ebselen: A reversible cyclization pathway via selenenic and seleninic acid intermediates. Chem. Eur. J. 2008, 14, 10603–10614. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, X.; Yang, X.; Liang, X.; Guo, Z. Fast cleavage of a diselenide induced by a platinum(II)-methionine complex and its biological implications. J. Inorg. Biochem. 2010, 104, 1178–1184. [Google Scholar] [CrossRef]
- Crich, D.; Karatholuvhu, M.; Krishnamurthy, V.; Hutton, T.K.; Brebion, F.; Subramanian, V. Dechalcogenative methods for the preparation of allylic sulfides. WO2008134058 (A1), 6 November 2008. [Google Scholar]
- Salvadori, S.; Marastoni, M.; Balboni, G.; Sarto, G.P.; Tomatis, R. Synthesis and opioid activity of dermorphin tetrapeptides bearing D-methionine S-oxide at position 2. J. Med. Chem. 1986, 29, 889–894. [Google Scholar] [CrossRef]
- Higashiura, K.; Toyomaki, Y.; Ienaga, K. The chemical conversion of C-terminal glycines in peptides into taurine. J. Chem. Soc. Chem. Commun. 1989, 1989, 521–522. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Abdo, M.; Sun, Z.; Knapp, S. Biohybrid -Se-S- Coupling Reactions of an Amino Acid Derived Seleninate. Molecules 2013, 18, 1963-1972. https://doi.org/10.3390/molecules18021963
Abdo M, Sun Z, Knapp S. Biohybrid -Se-S- Coupling Reactions of an Amino Acid Derived Seleninate. Molecules. 2013; 18(2):1963-1972. https://doi.org/10.3390/molecules18021963
Chicago/Turabian StyleAbdo, Mohannad, Zhexun Sun, and Spencer Knapp. 2013. "Biohybrid -Se-S- Coupling Reactions of an Amino Acid Derived Seleninate" Molecules 18, no. 2: 1963-1972. https://doi.org/10.3390/molecules18021963
APA StyleAbdo, M., Sun, Z., & Knapp, S. (2013). Biohybrid -Se-S- Coupling Reactions of an Amino Acid Derived Seleninate. Molecules, 18(2), 1963-1972. https://doi.org/10.3390/molecules18021963