Case Studies of the Synthesis of Bioactive Cyclodepsipeptide Natural Products

Abstract
:1. Introduction
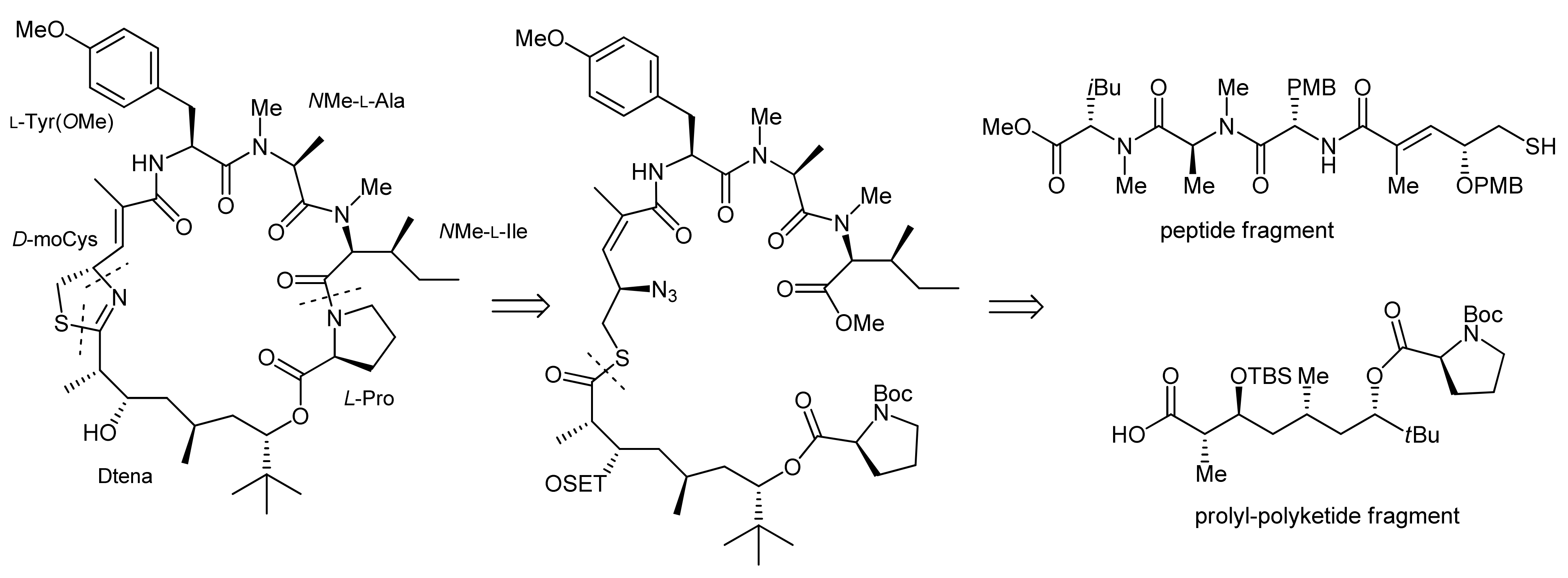
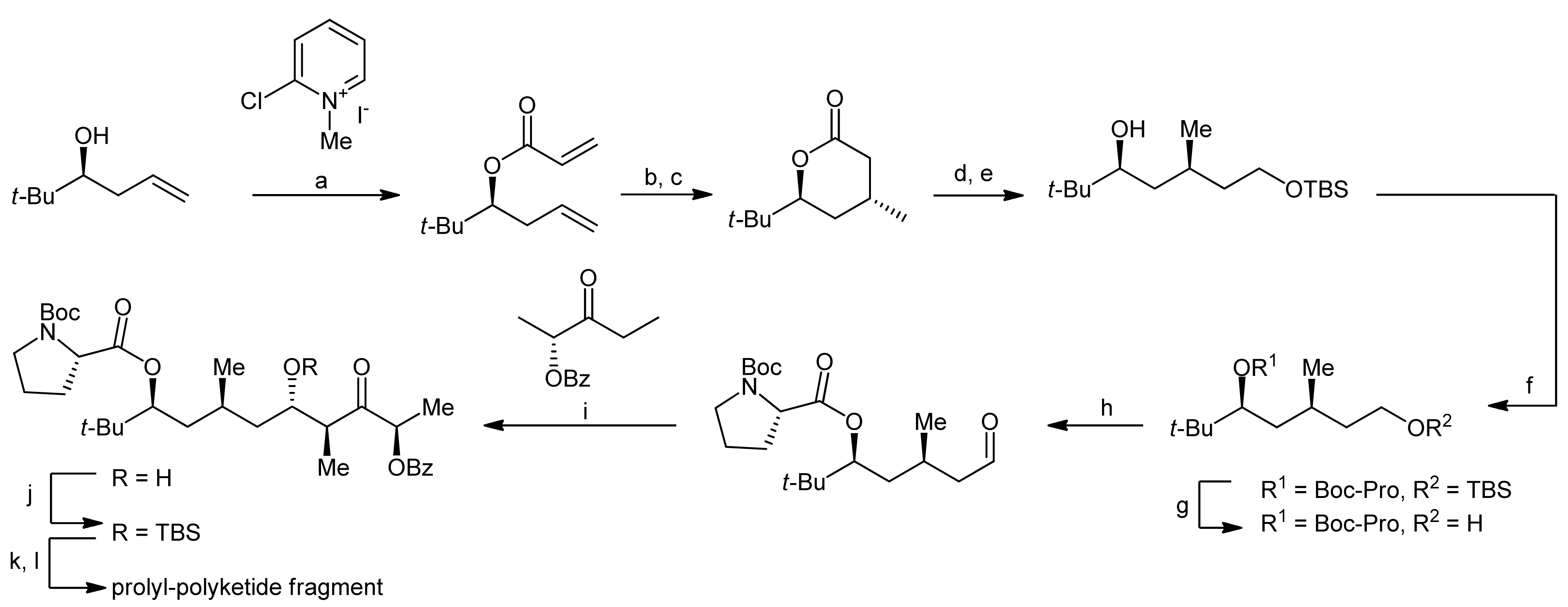
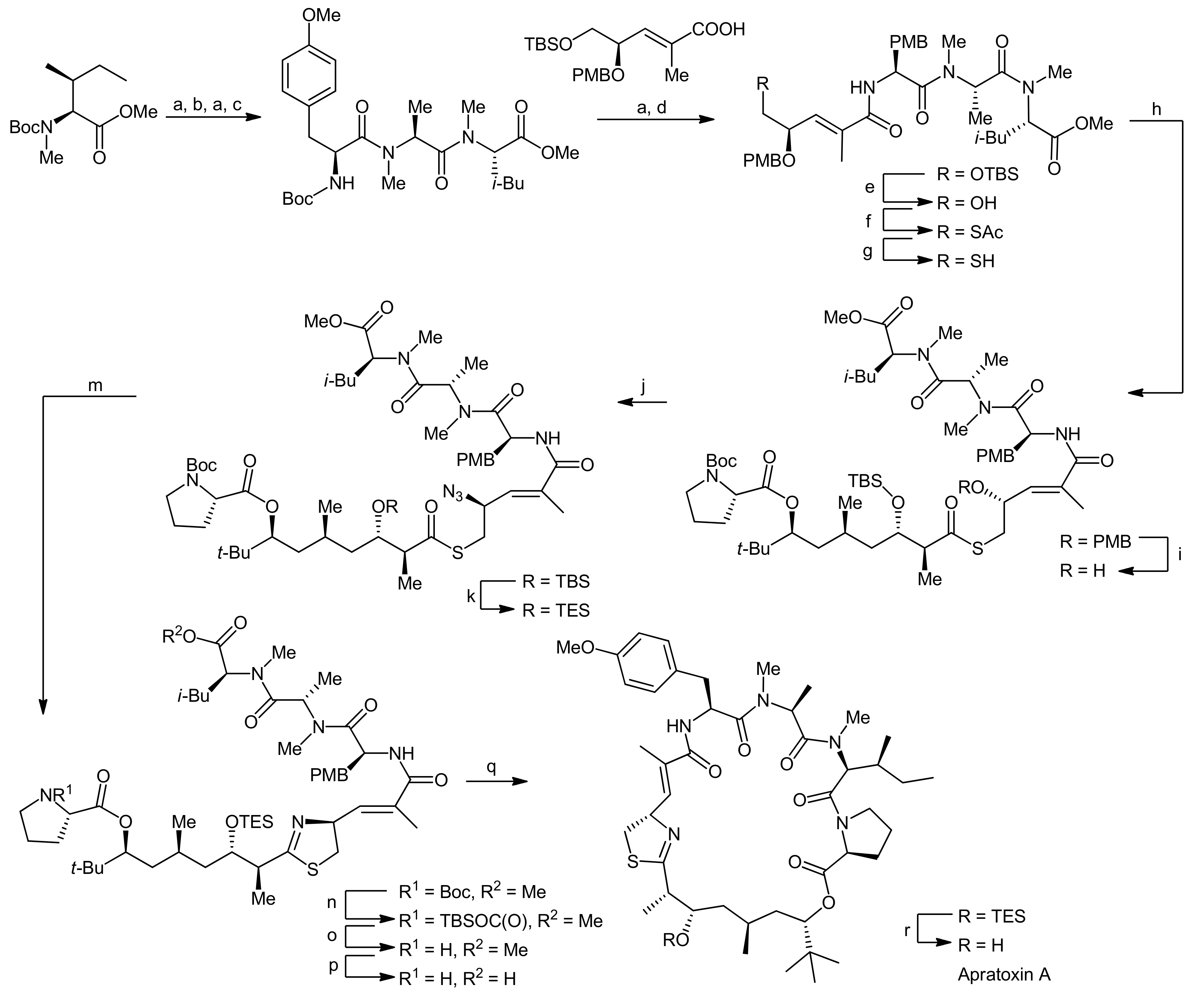
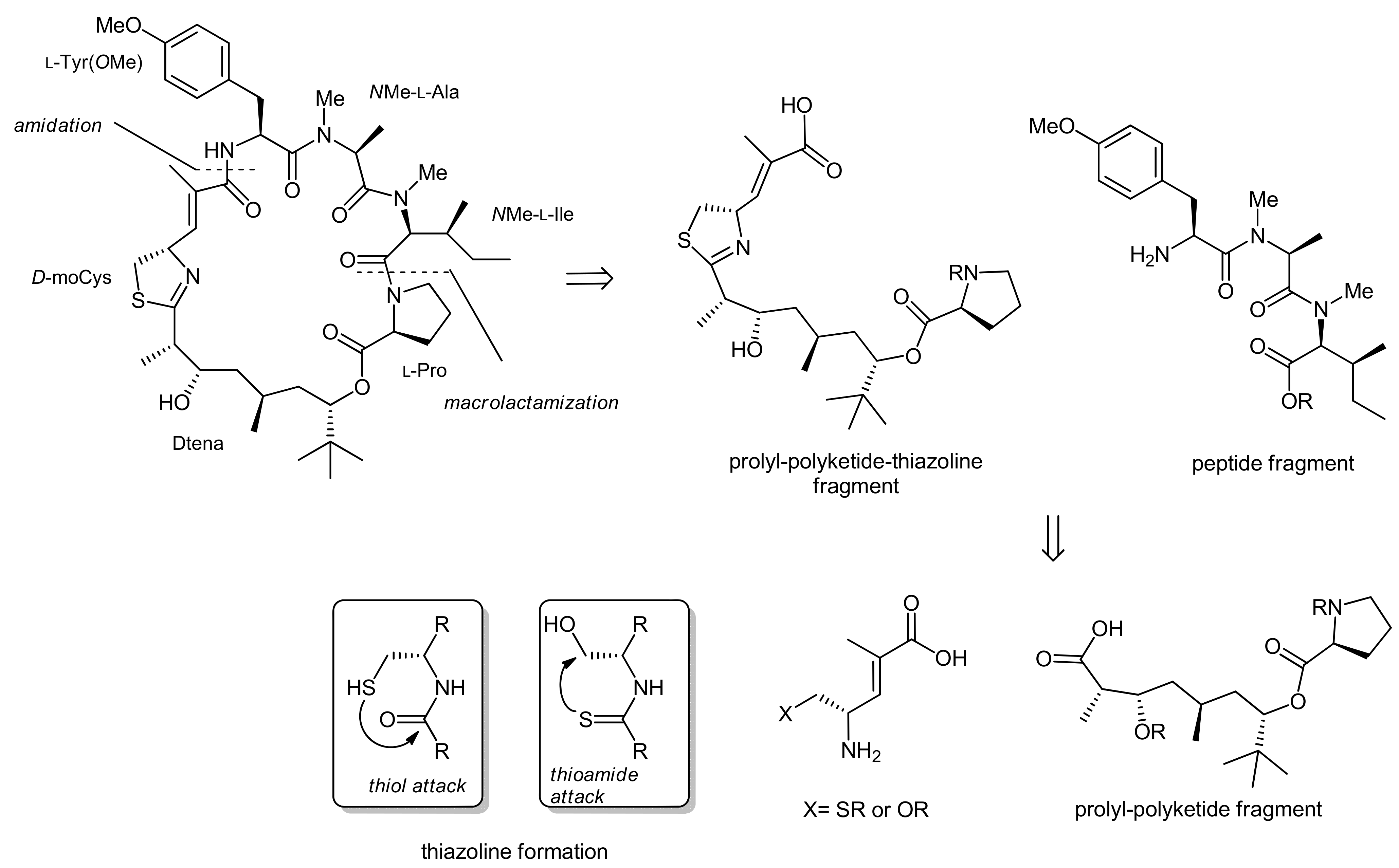
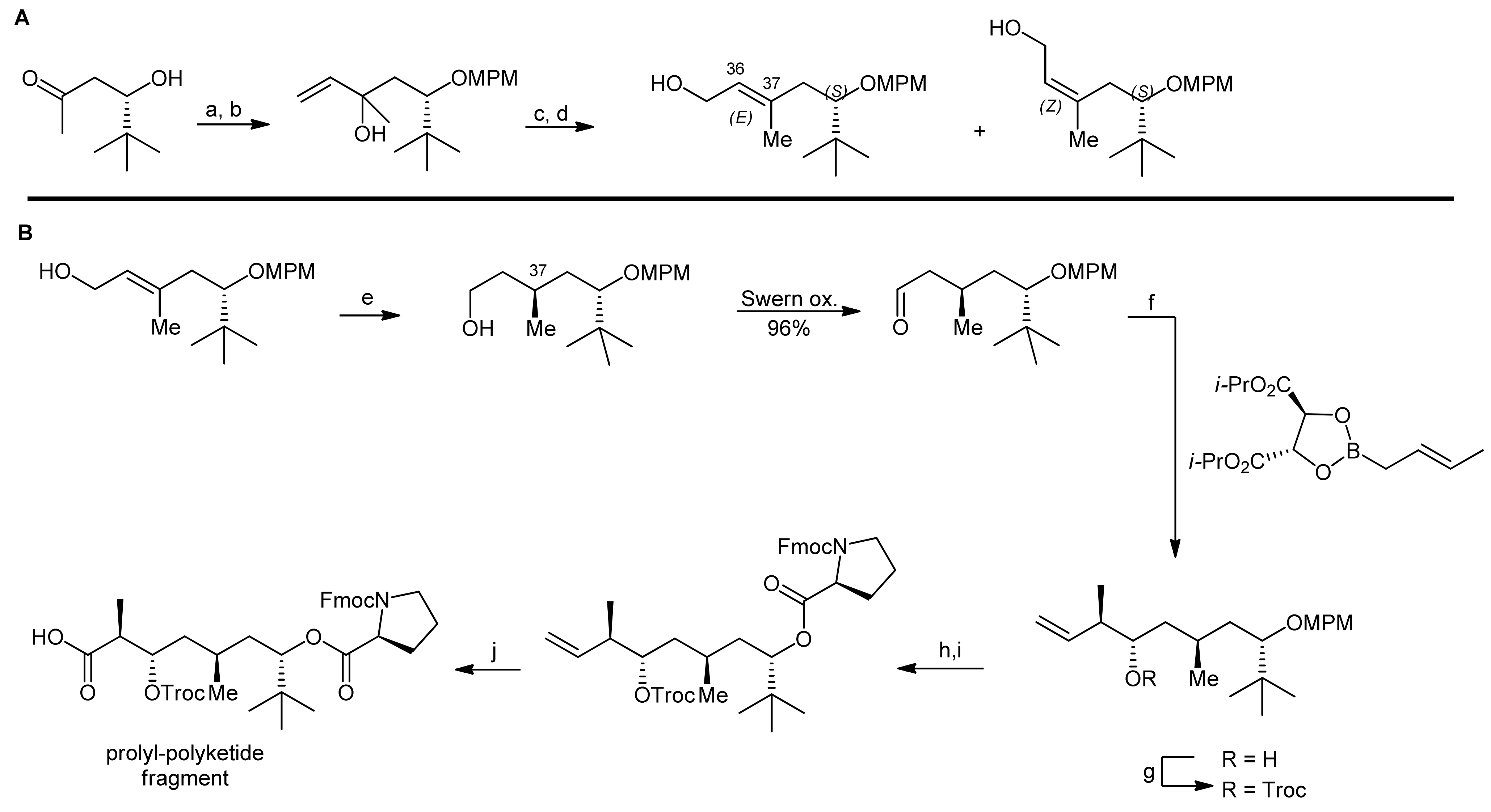
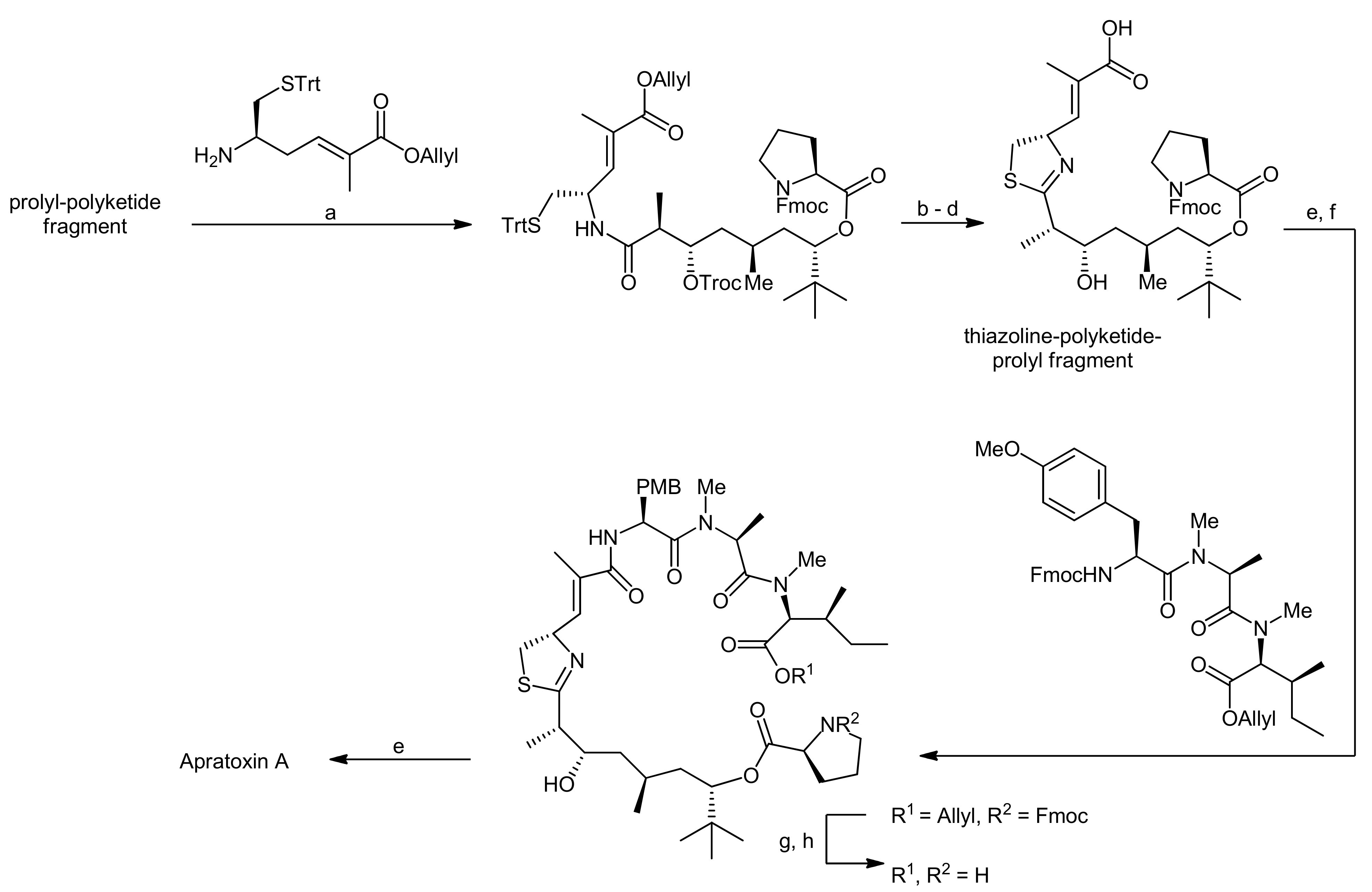
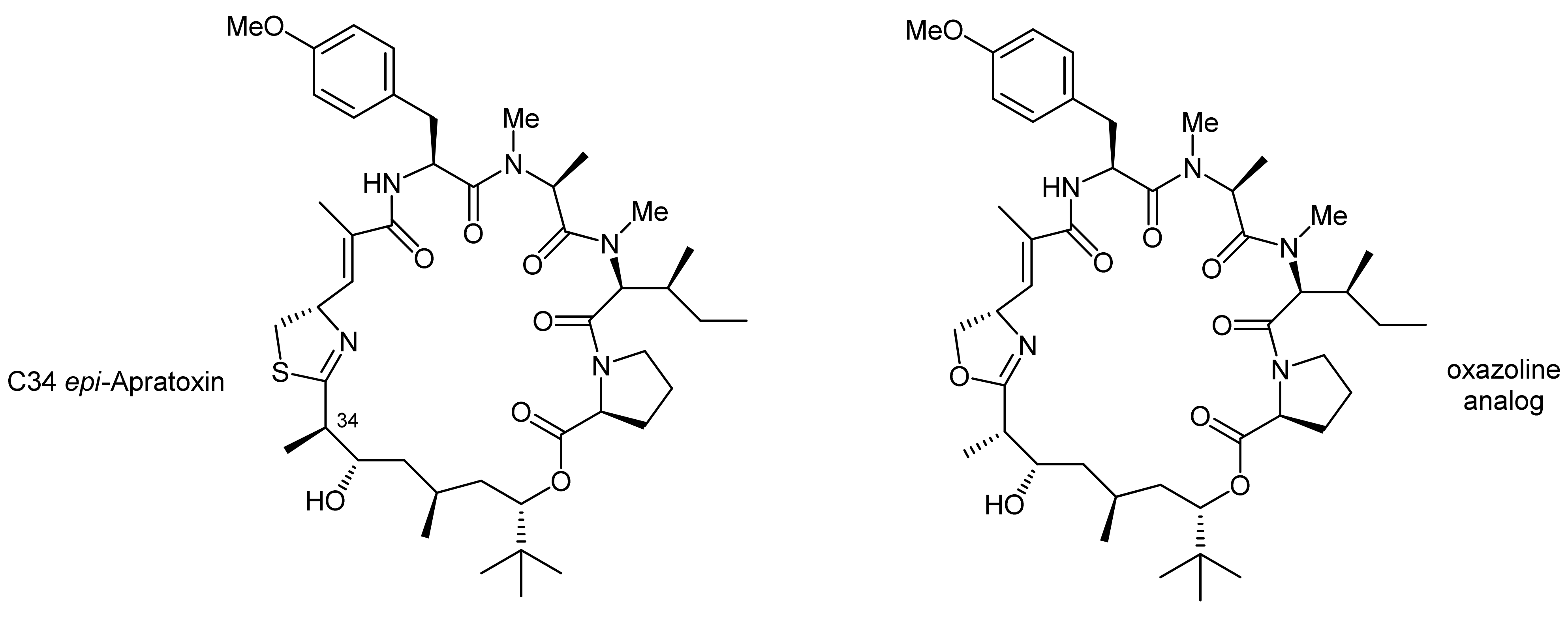
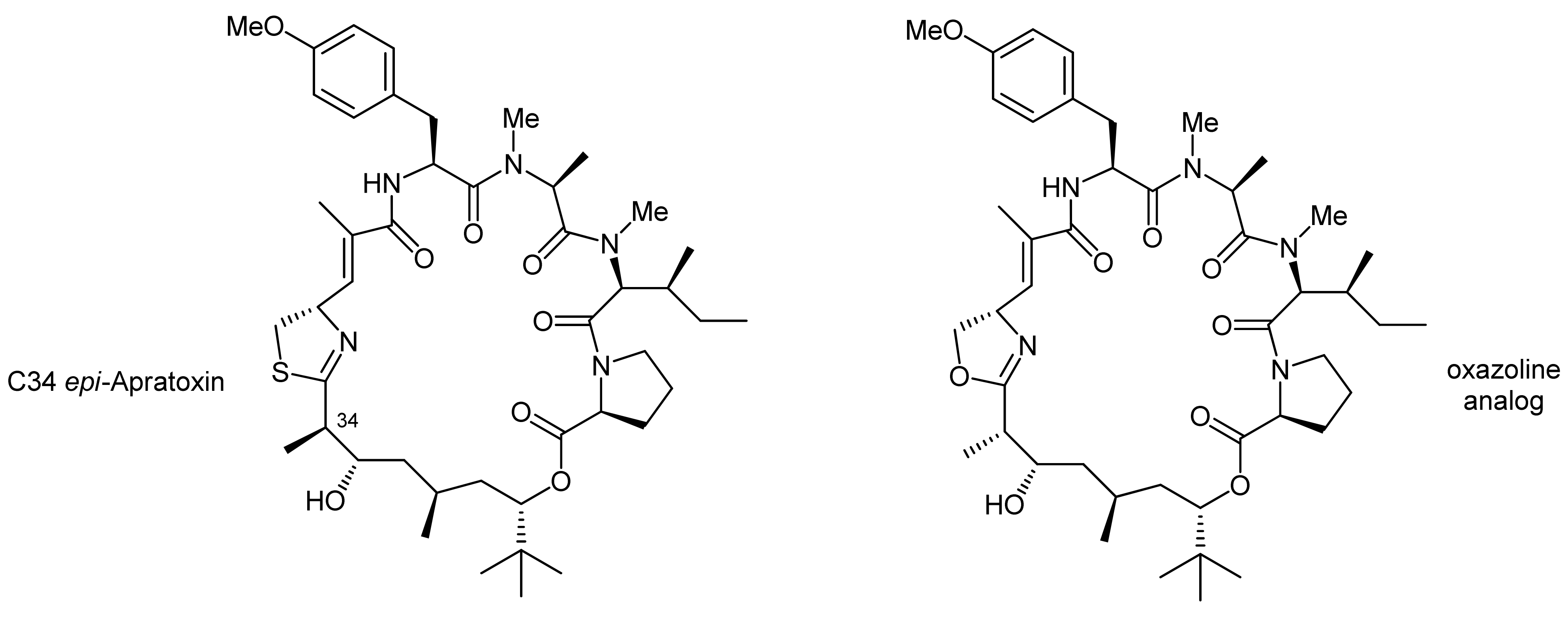
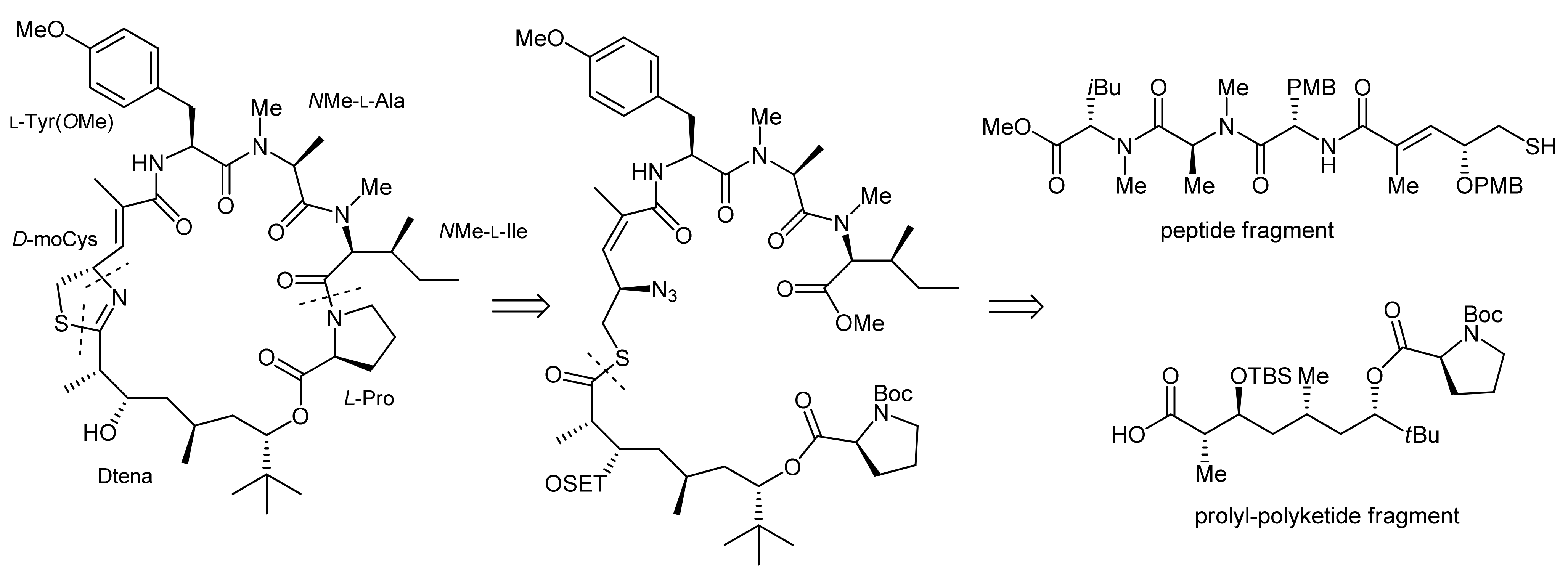
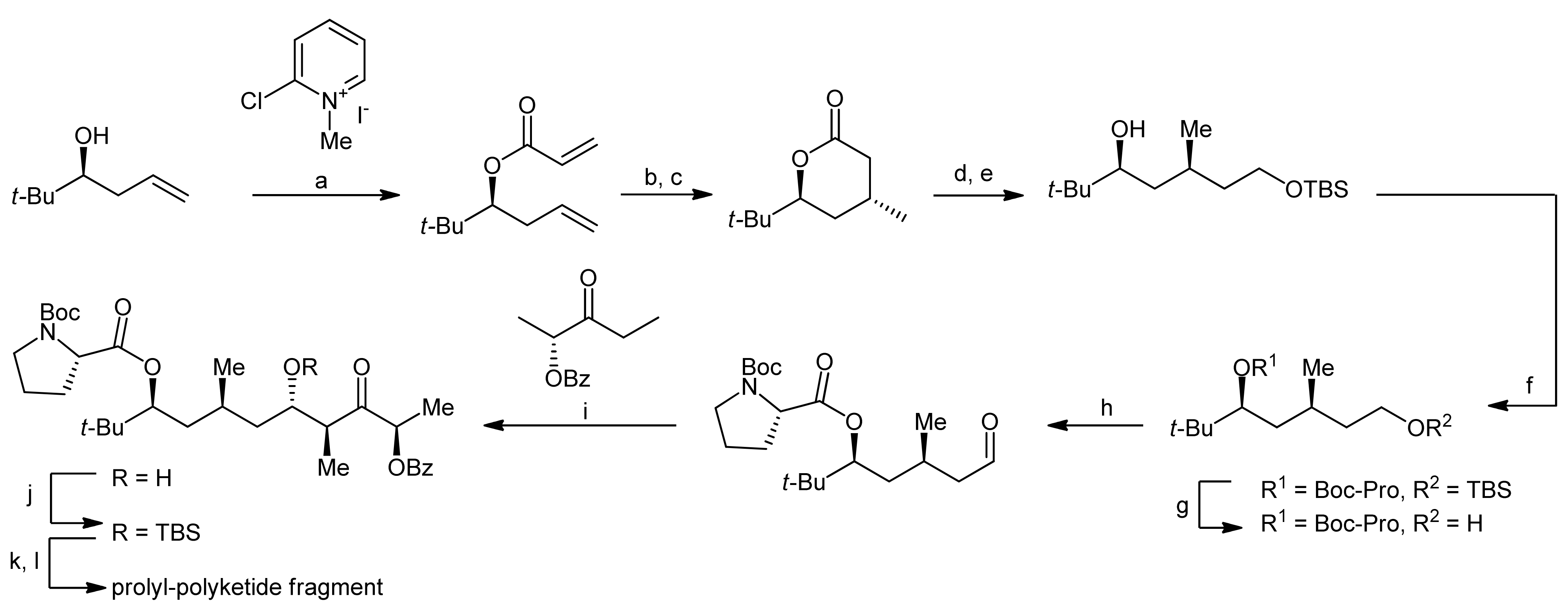
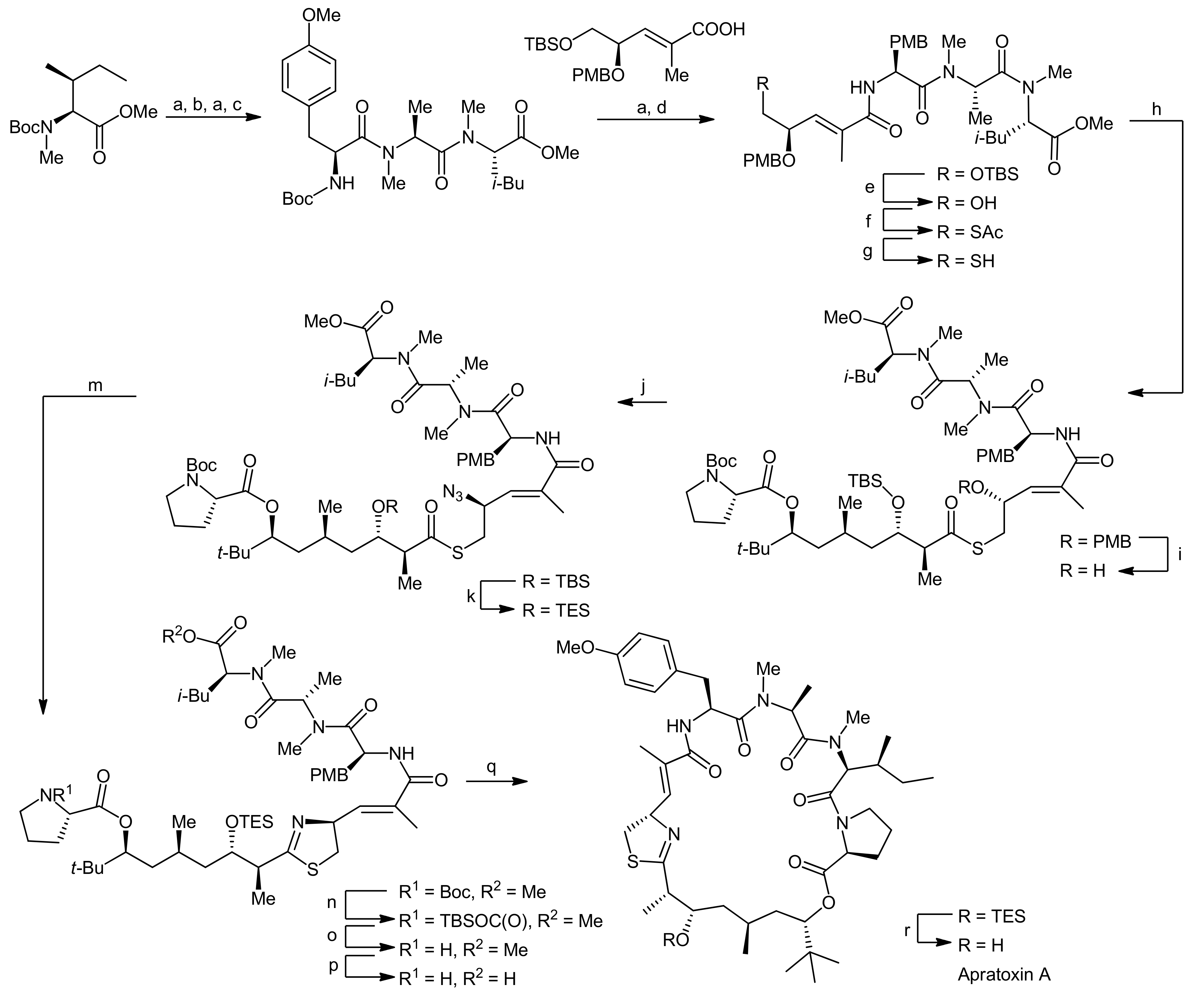
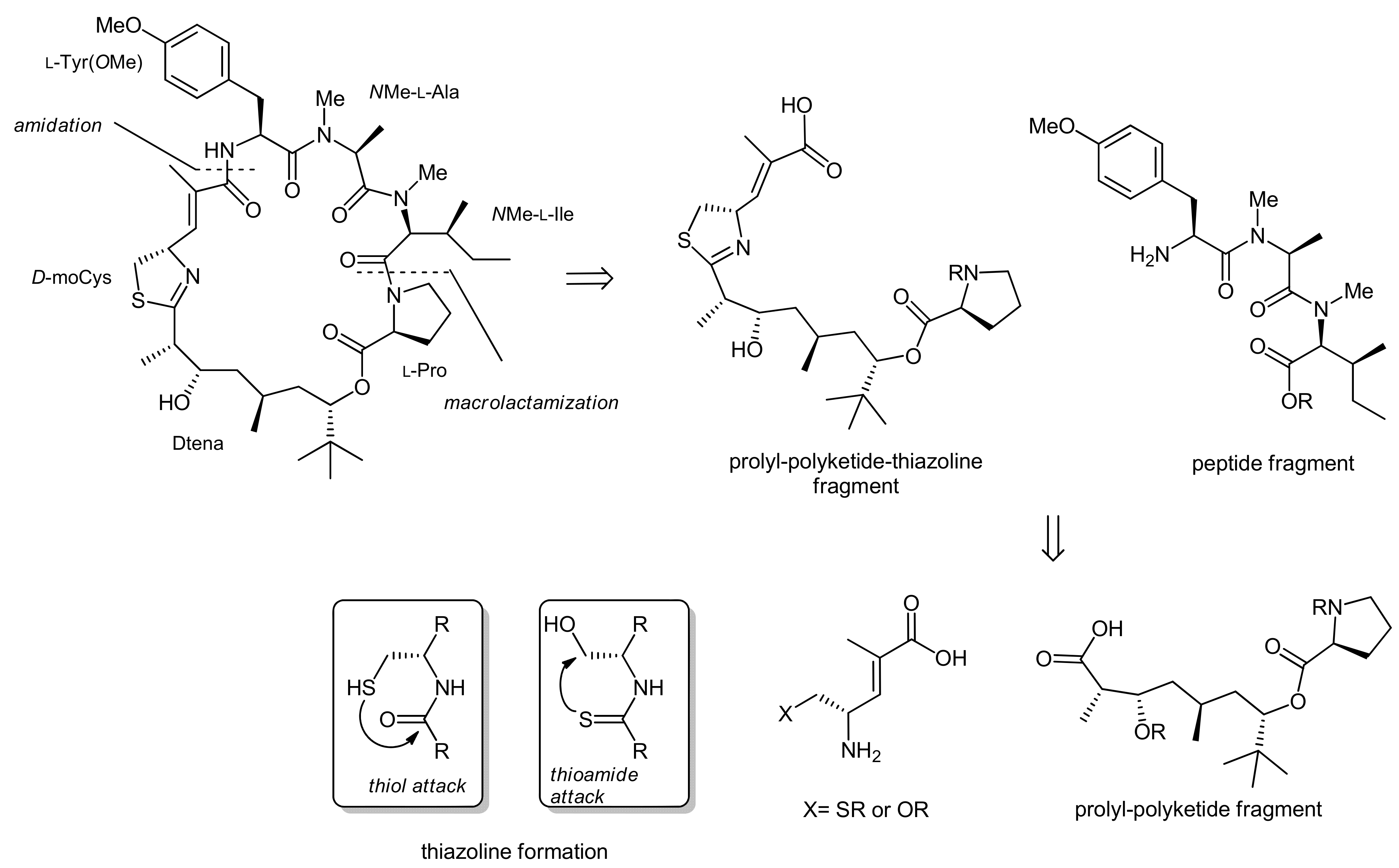
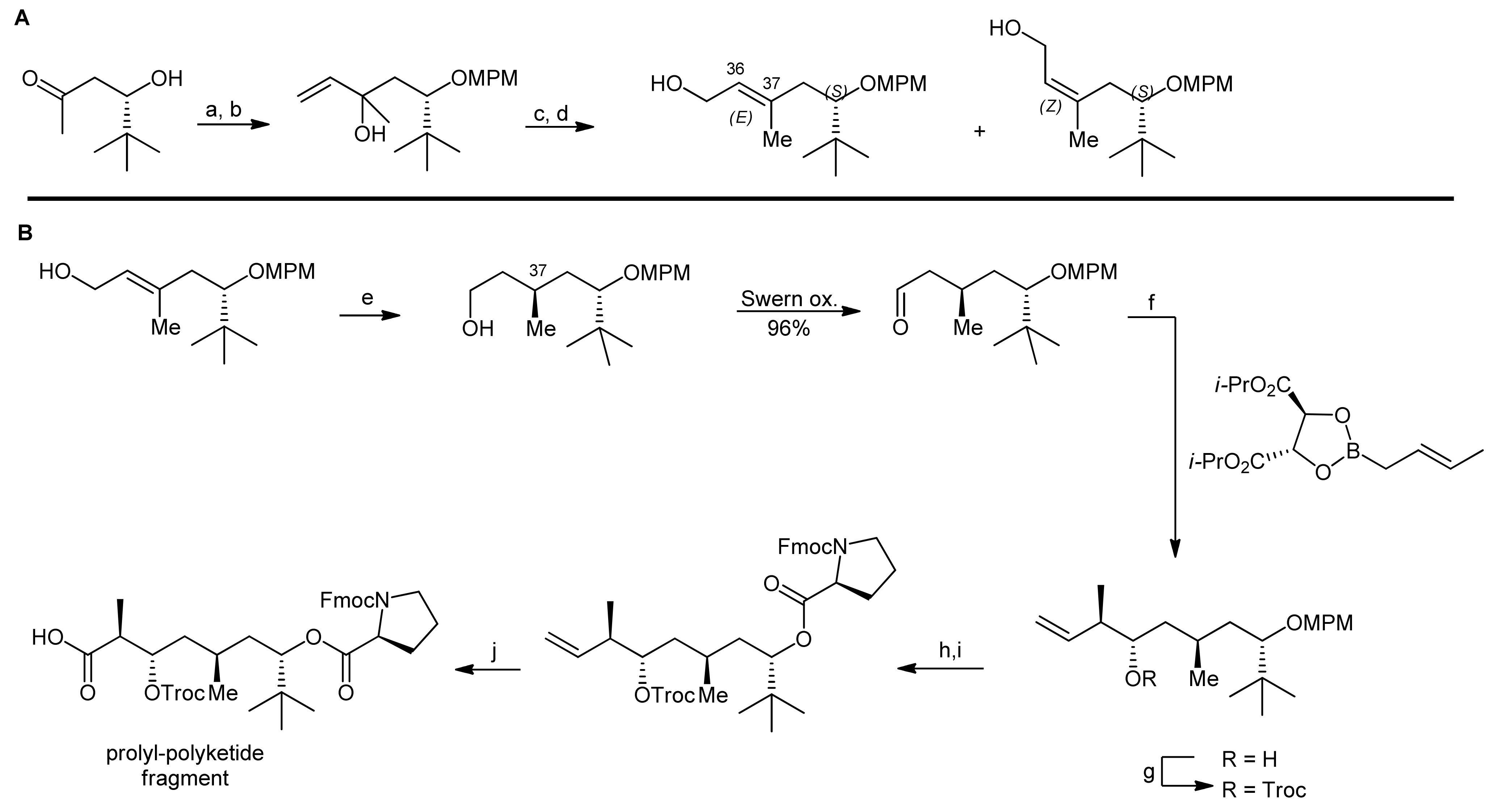
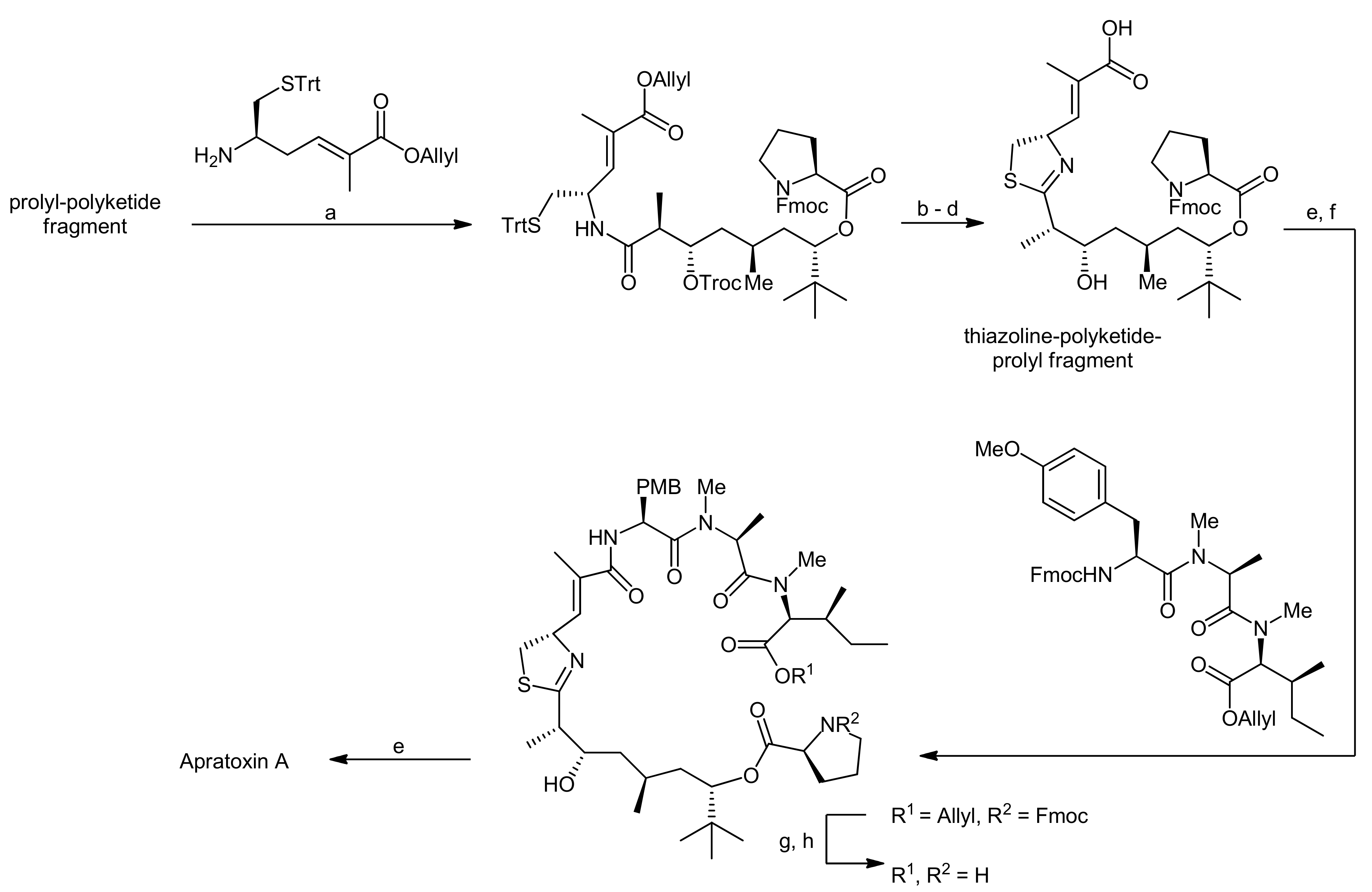
2. Apratoxin A—The Challenge of Efficient Incorporation of Heterocylic Building Blocks
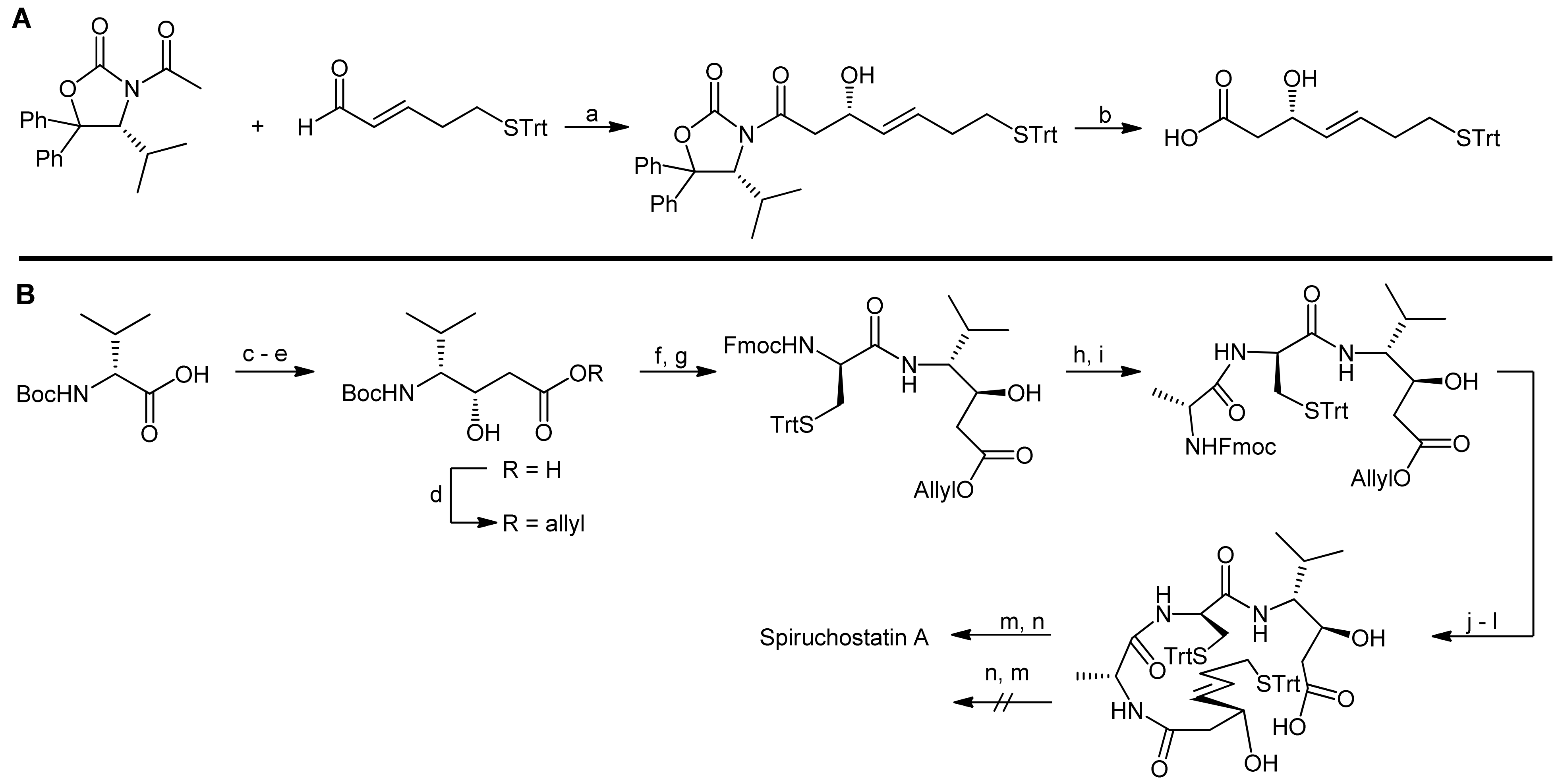
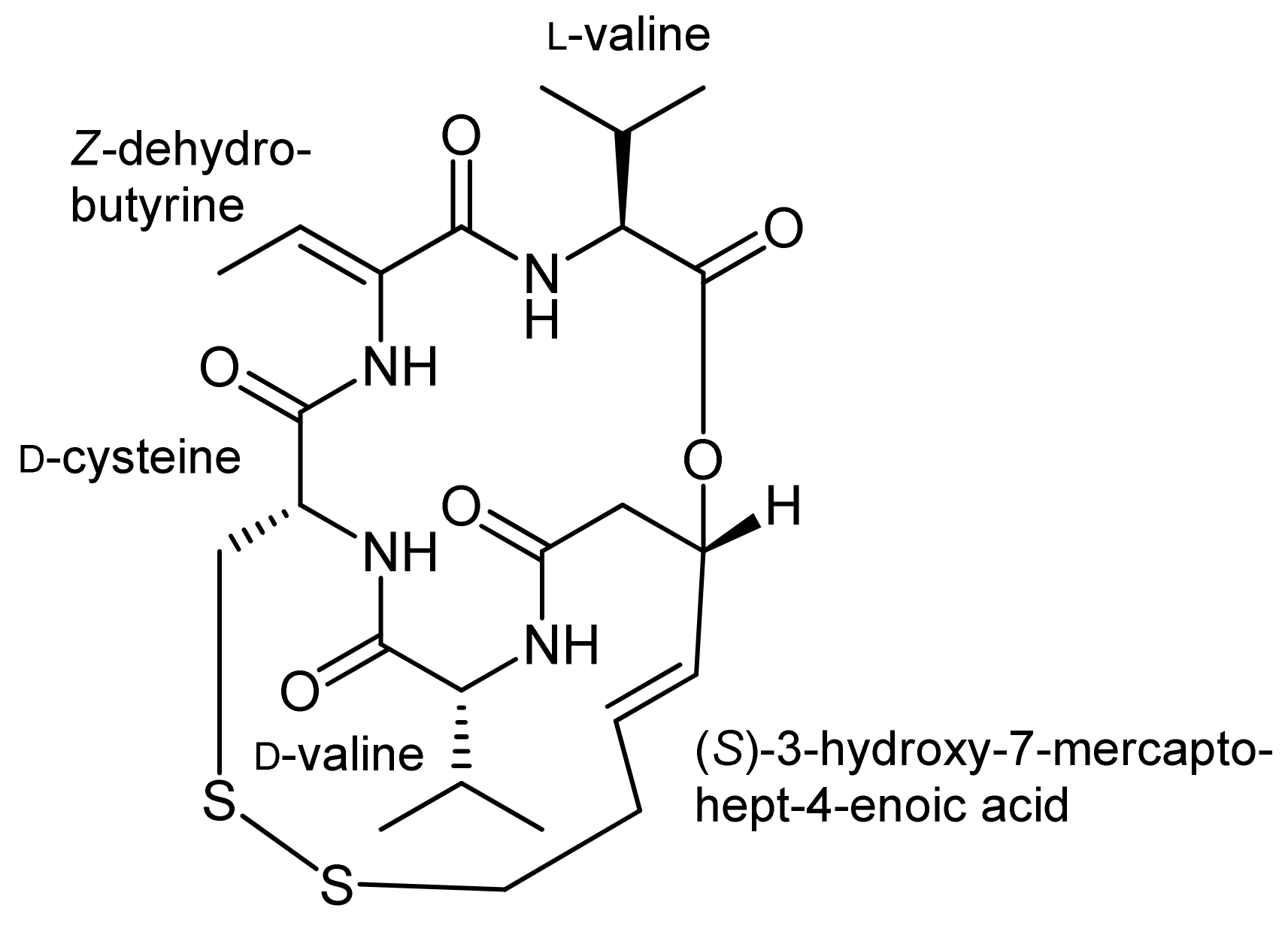
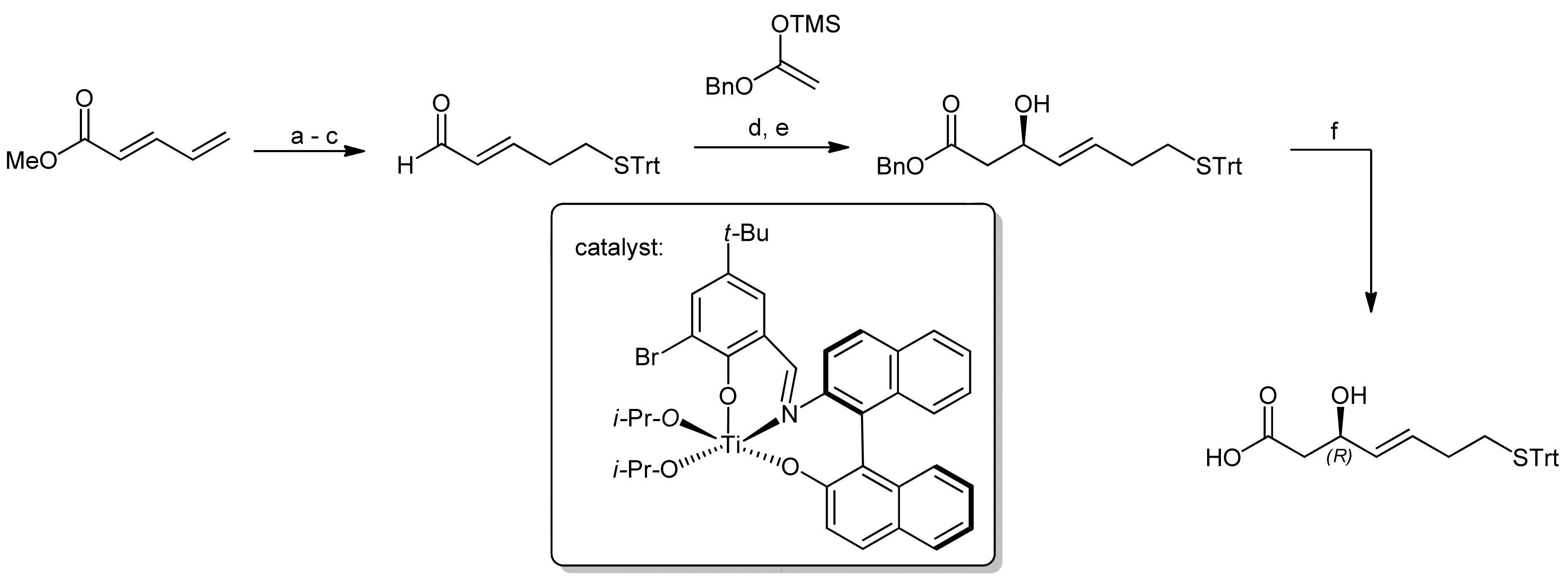
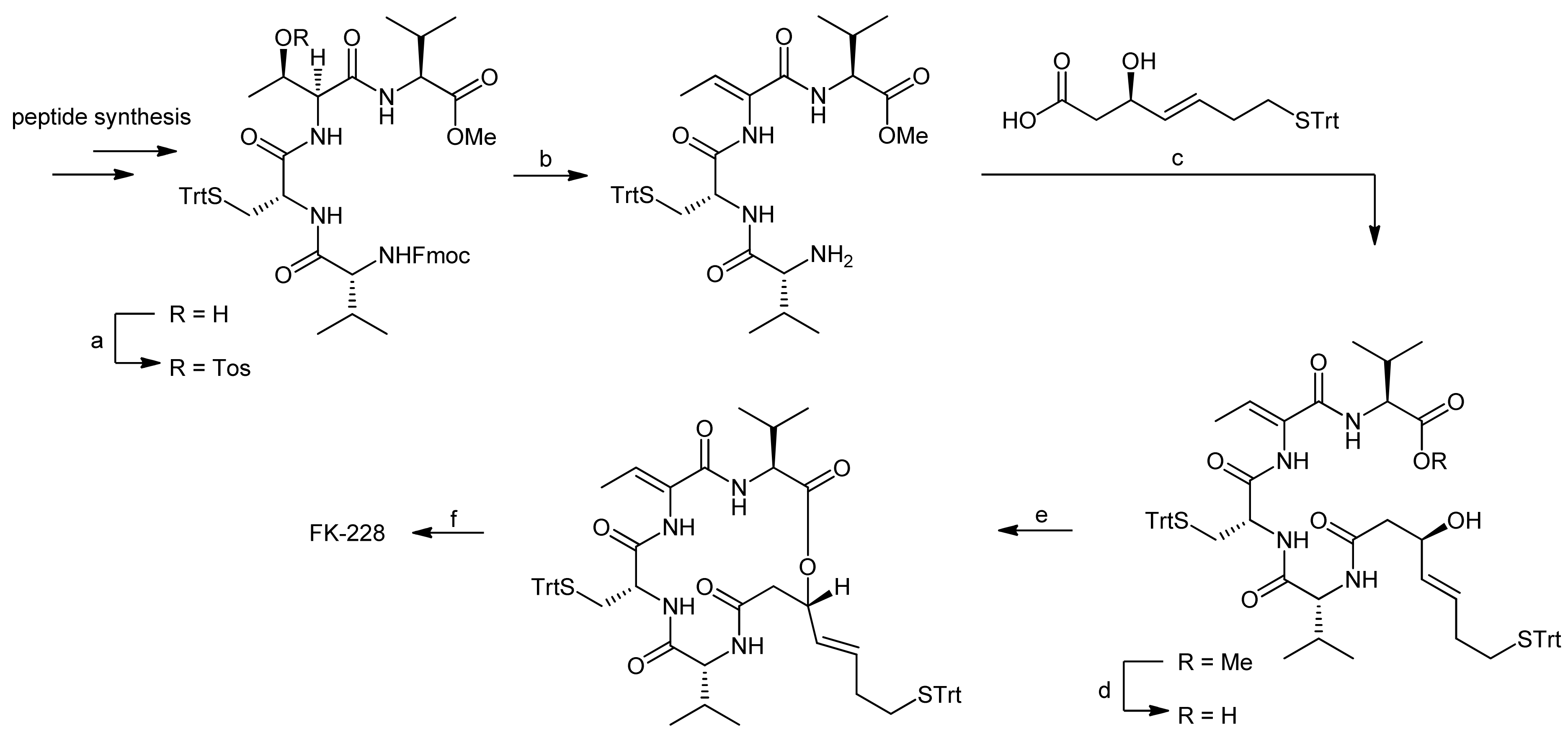
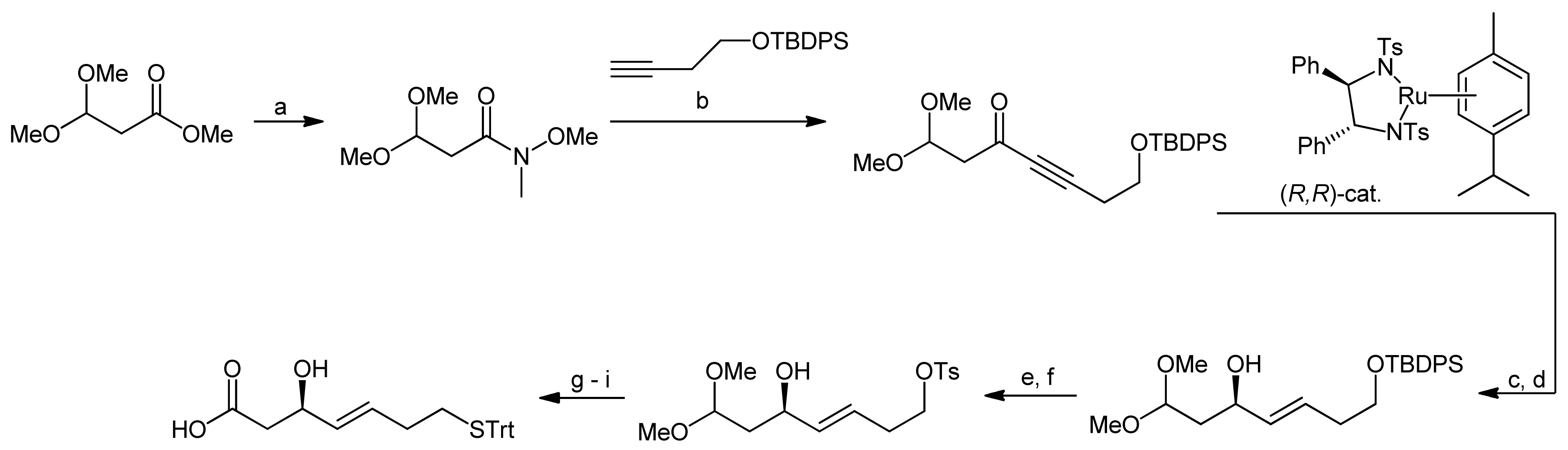
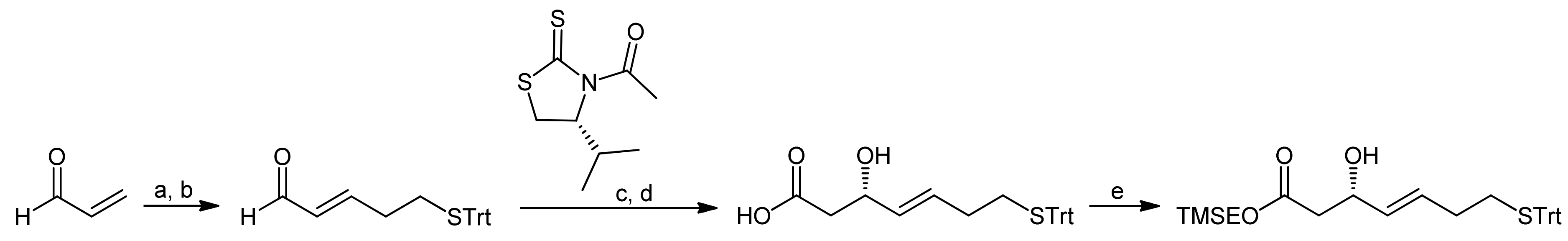
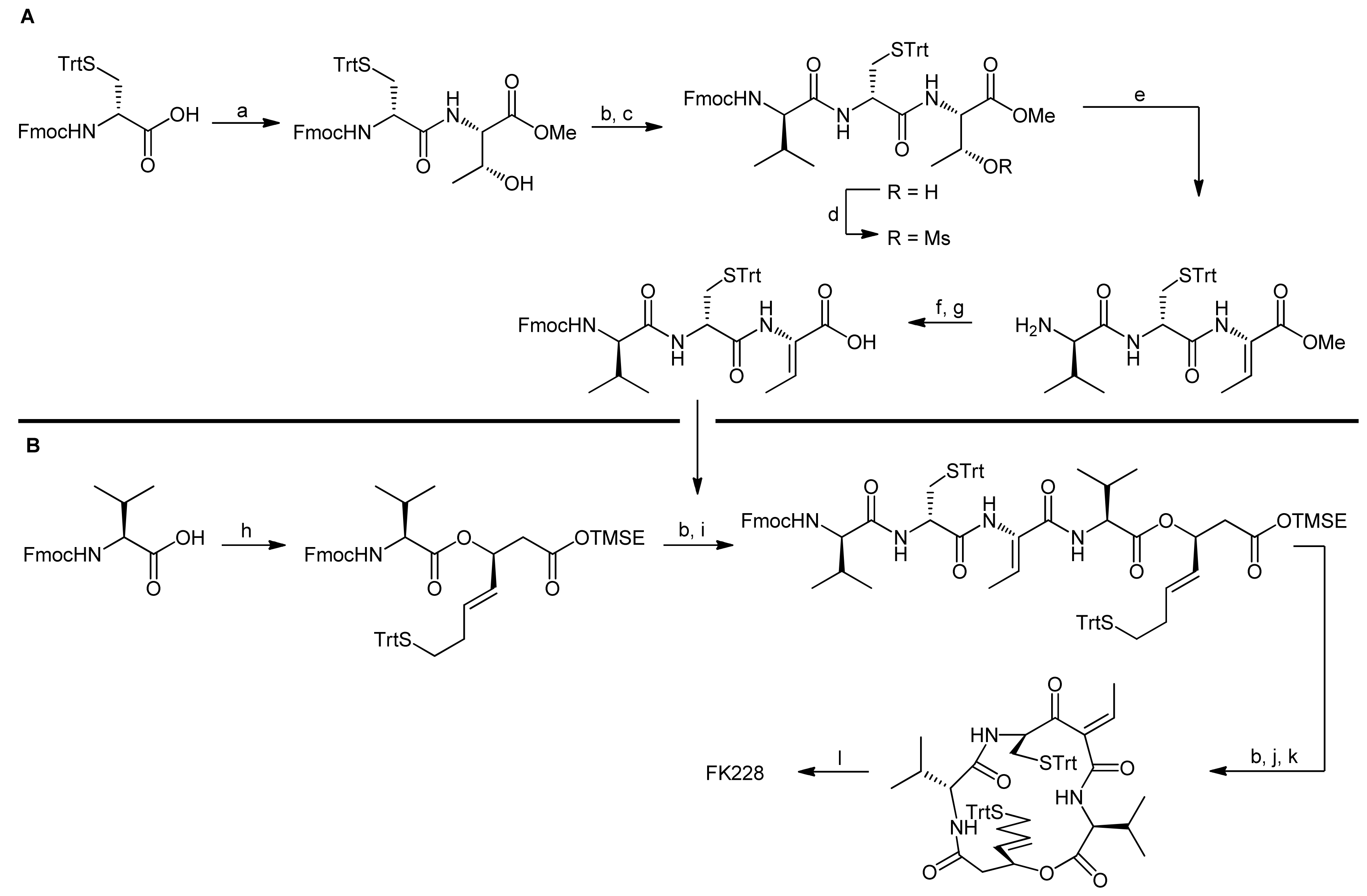
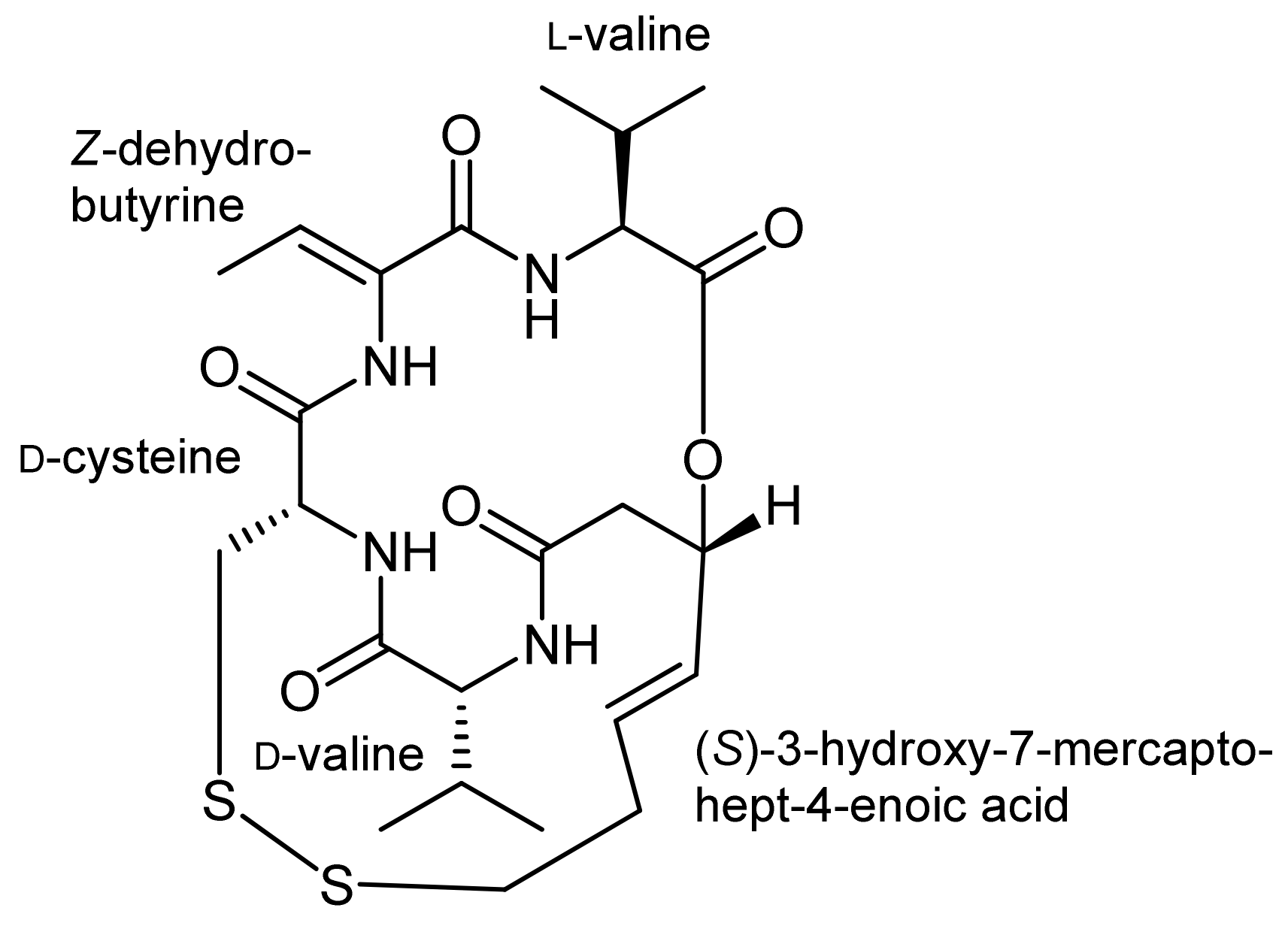
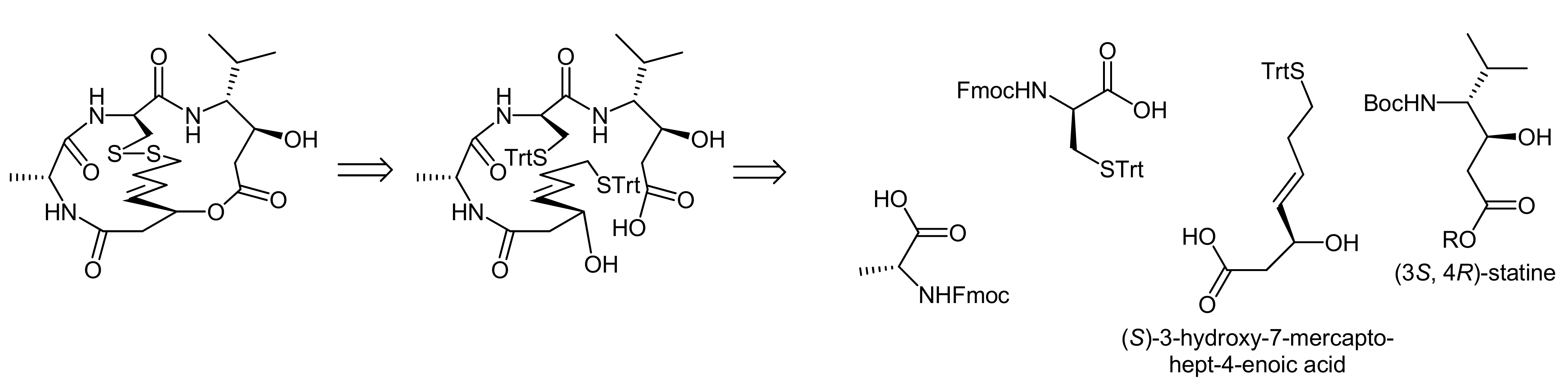
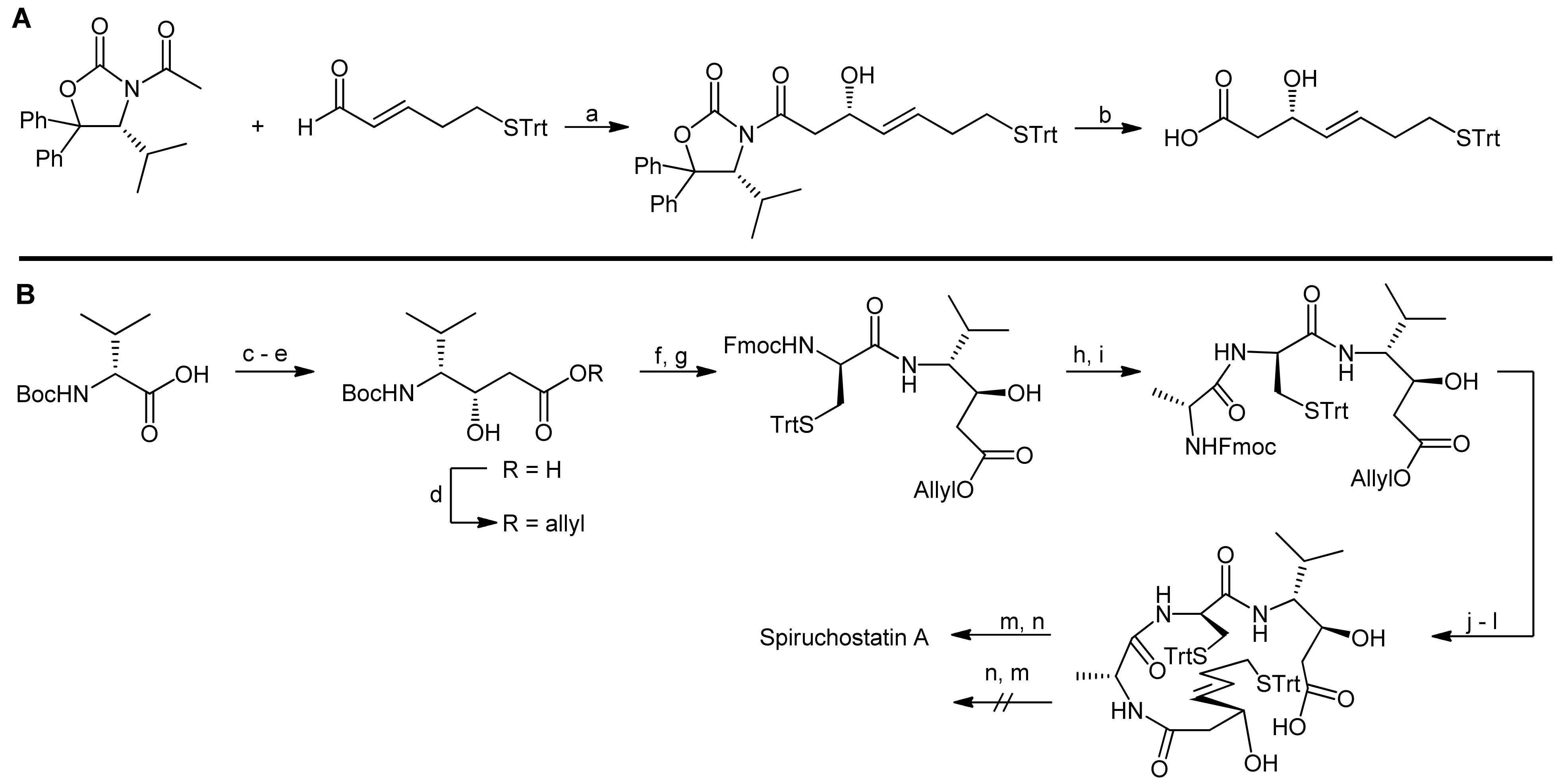
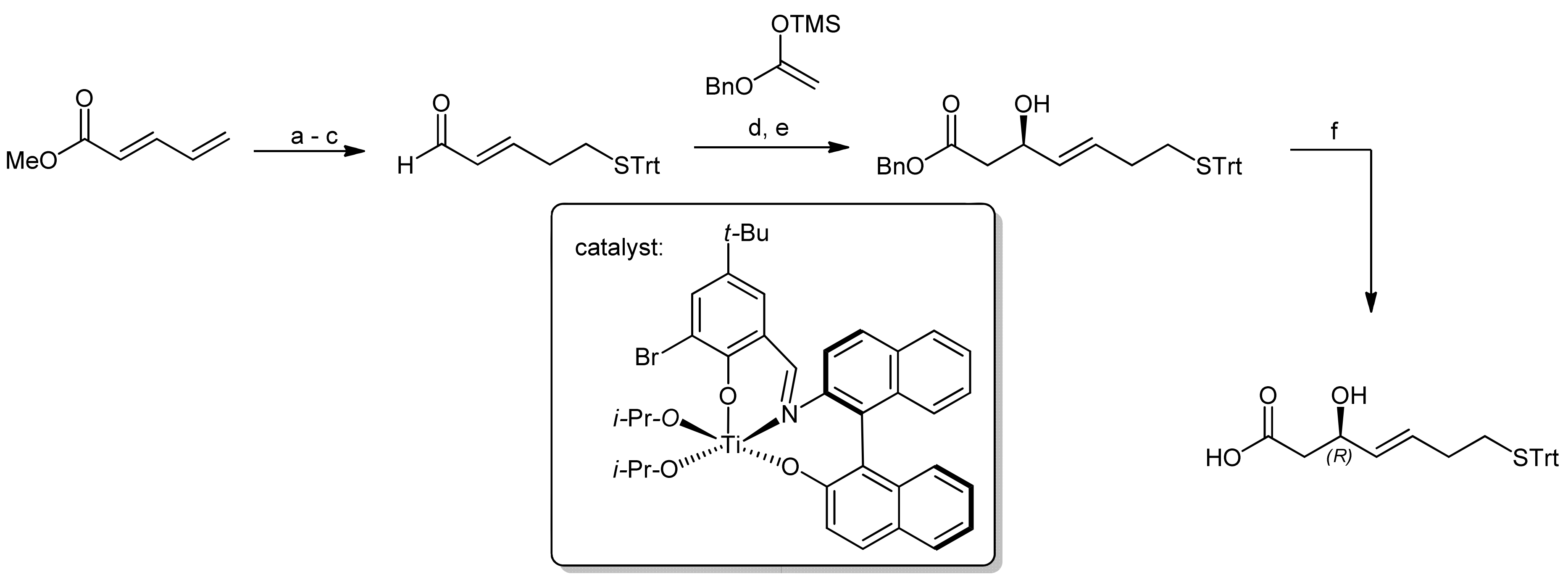
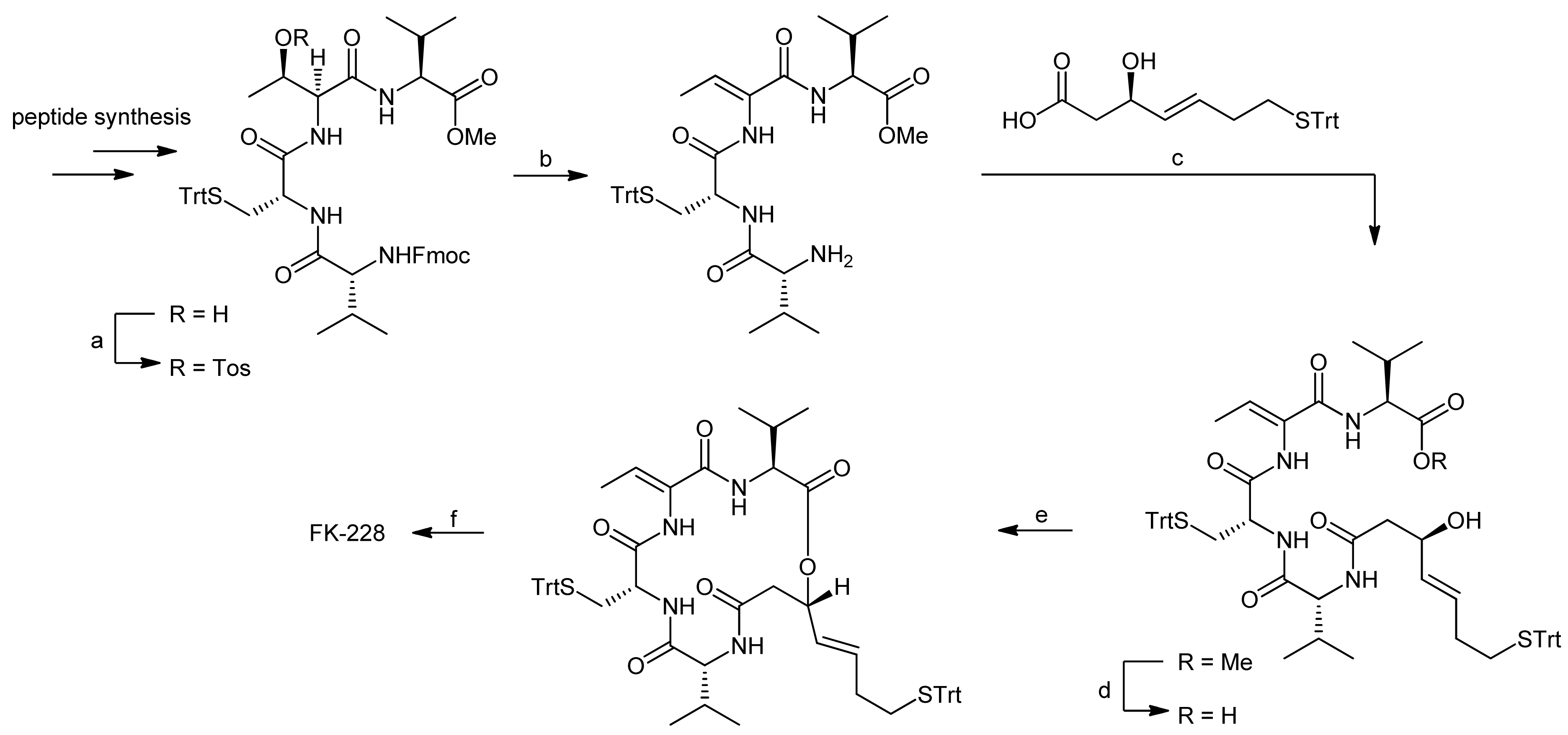
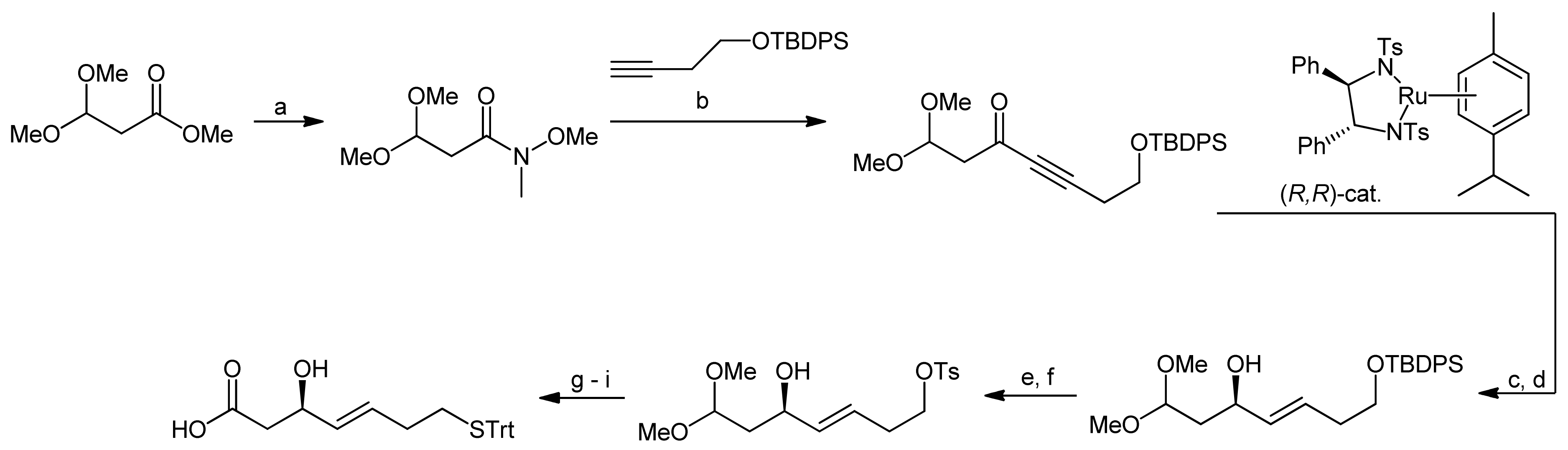
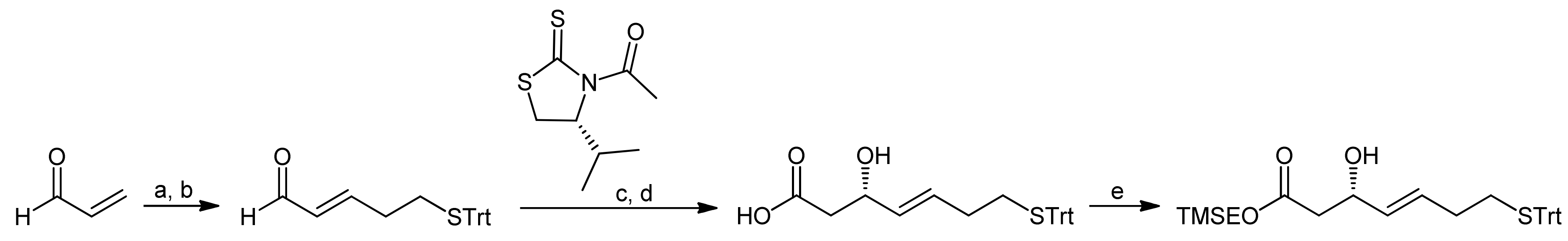
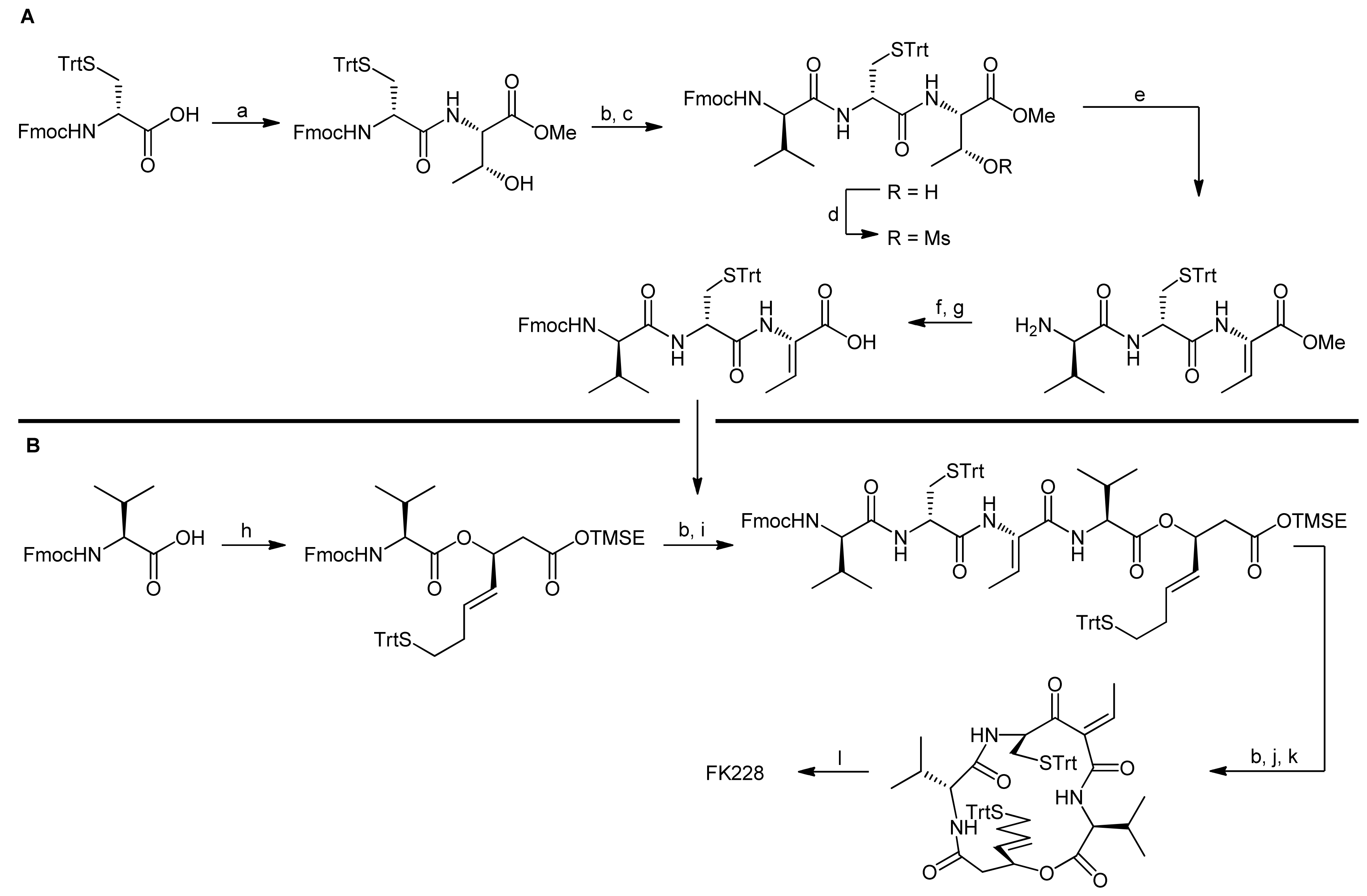
3. Spiruchostatin A and FK288: The Challenges to Find Efficient Macrocylization Routes
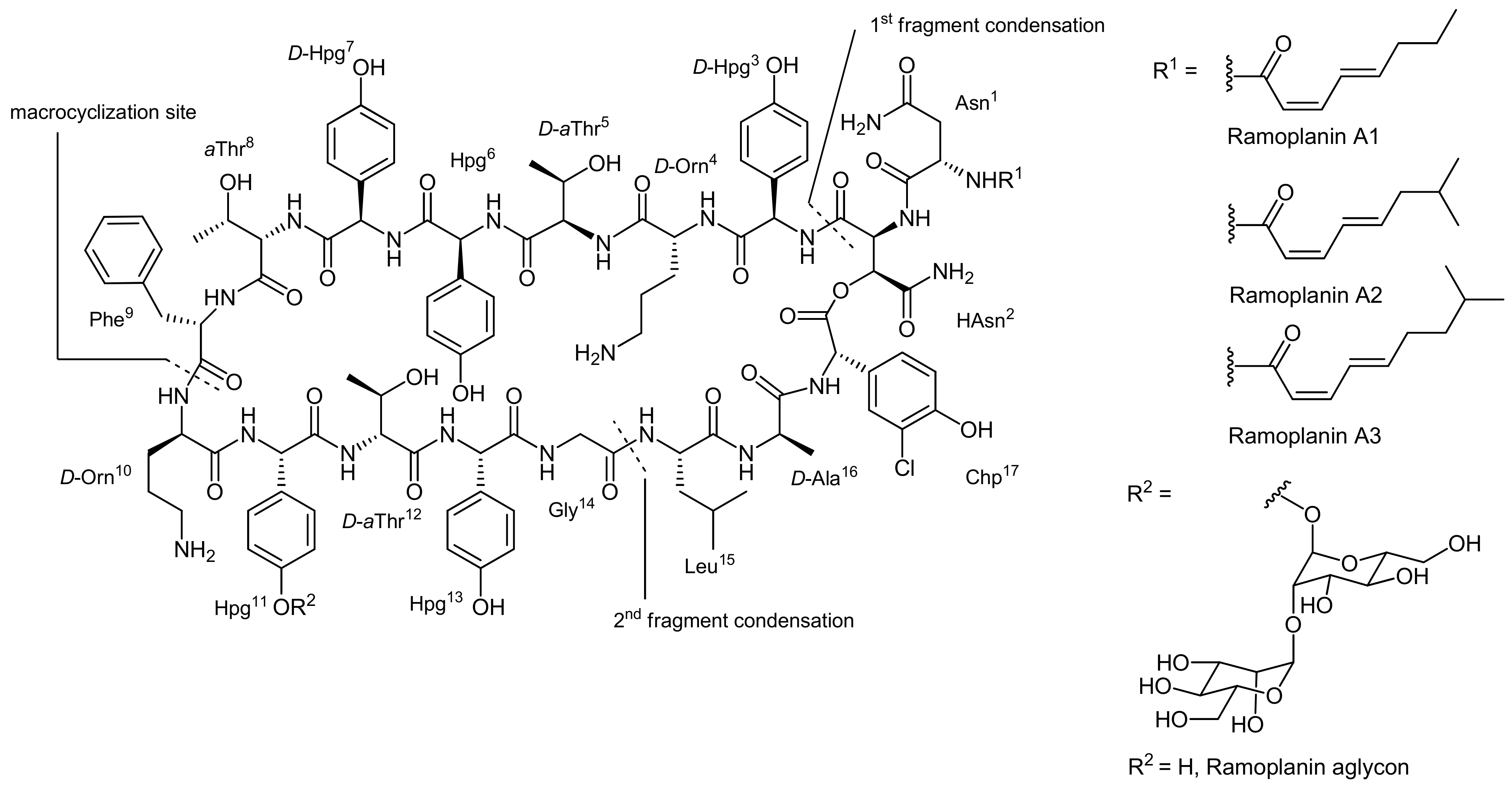
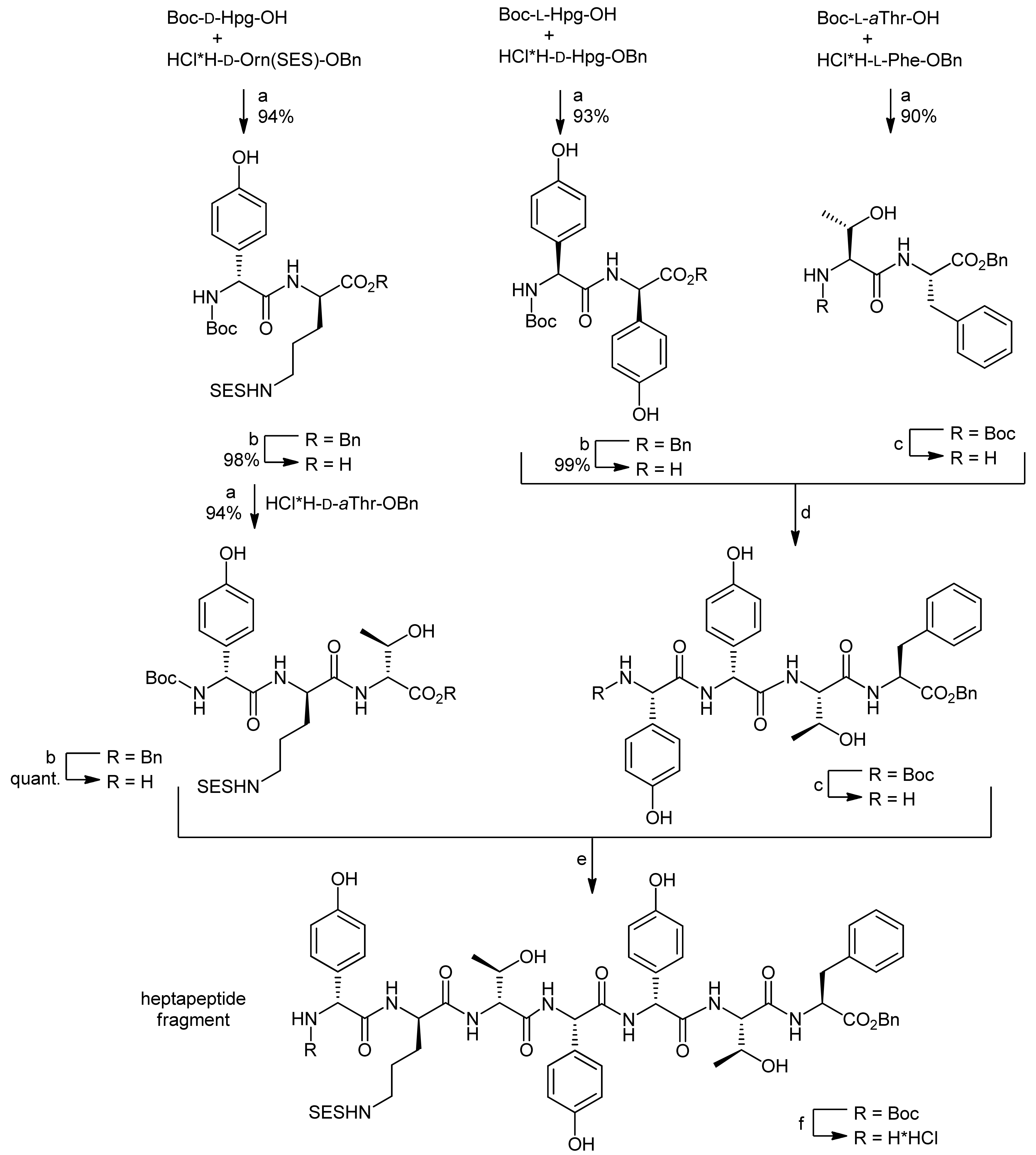
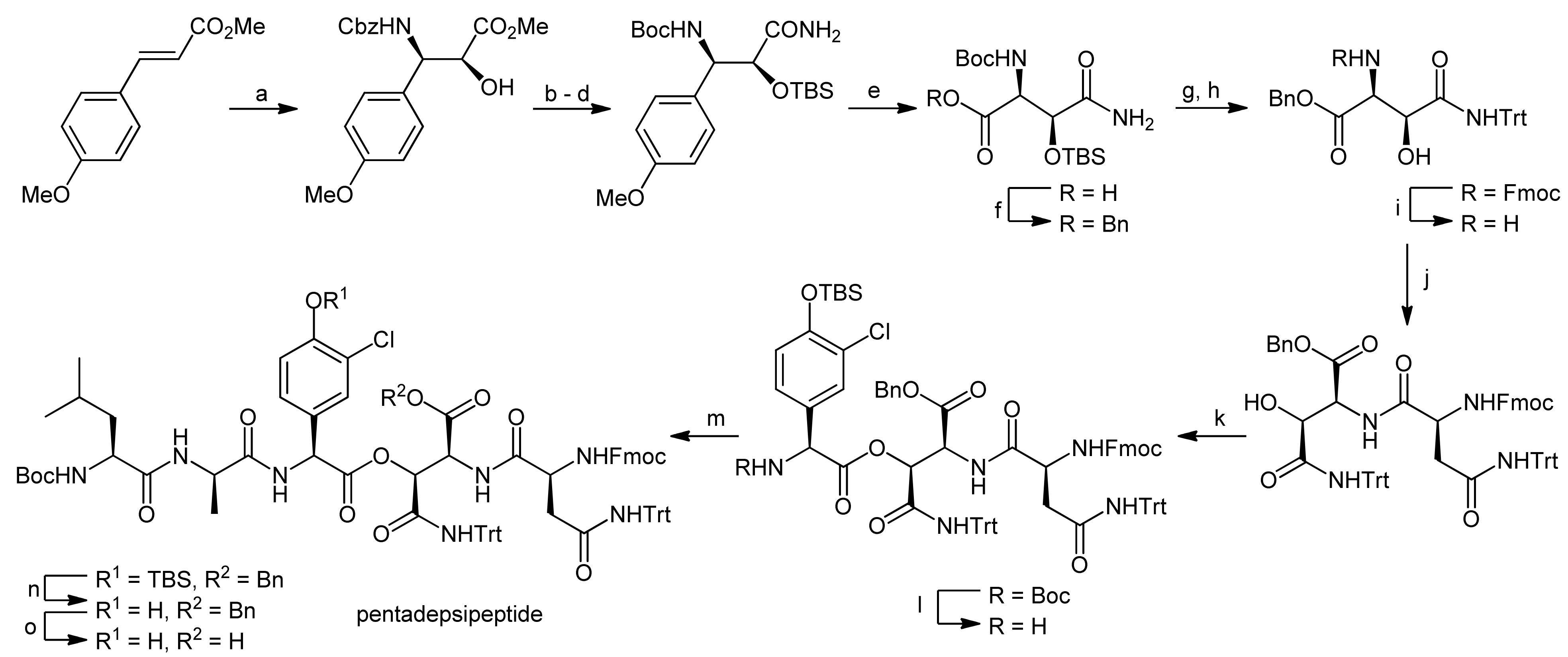
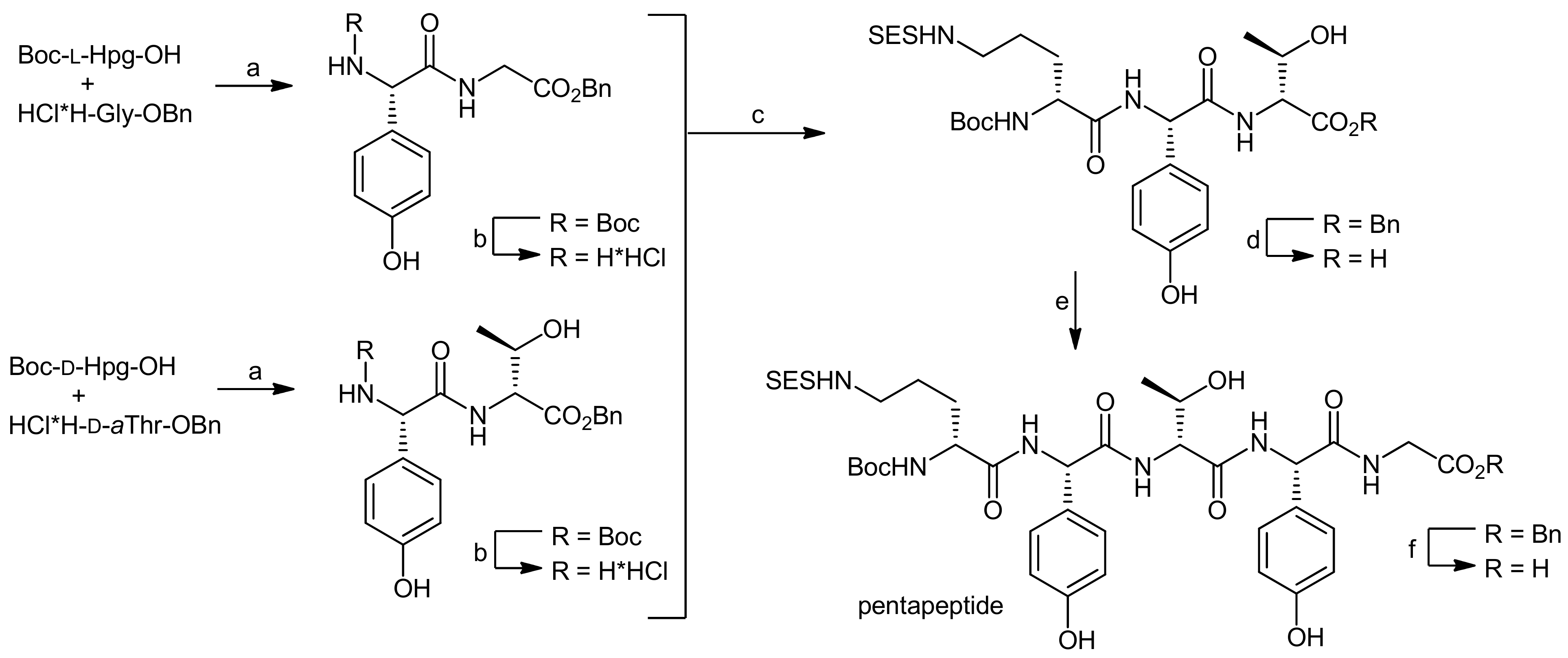
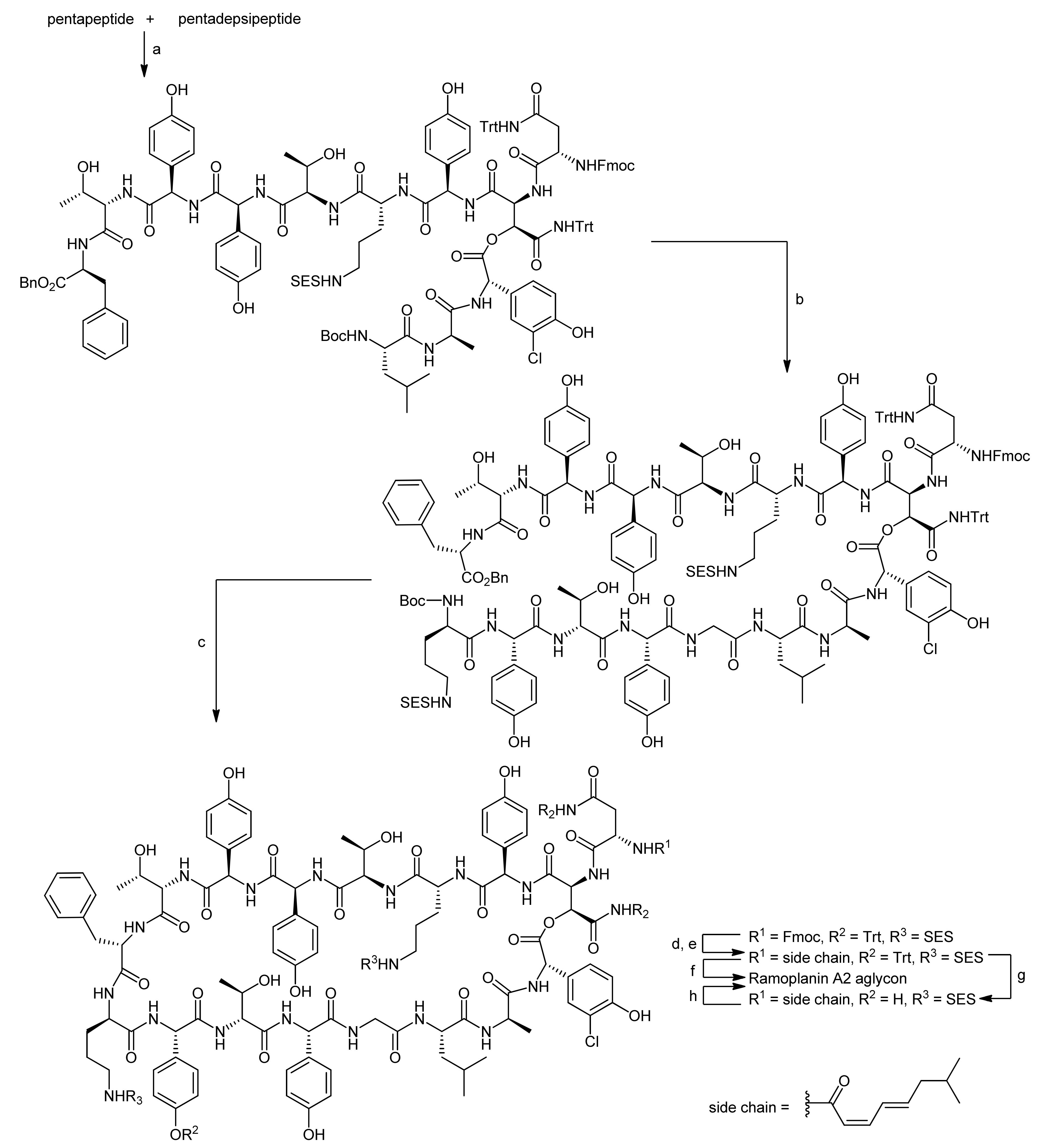
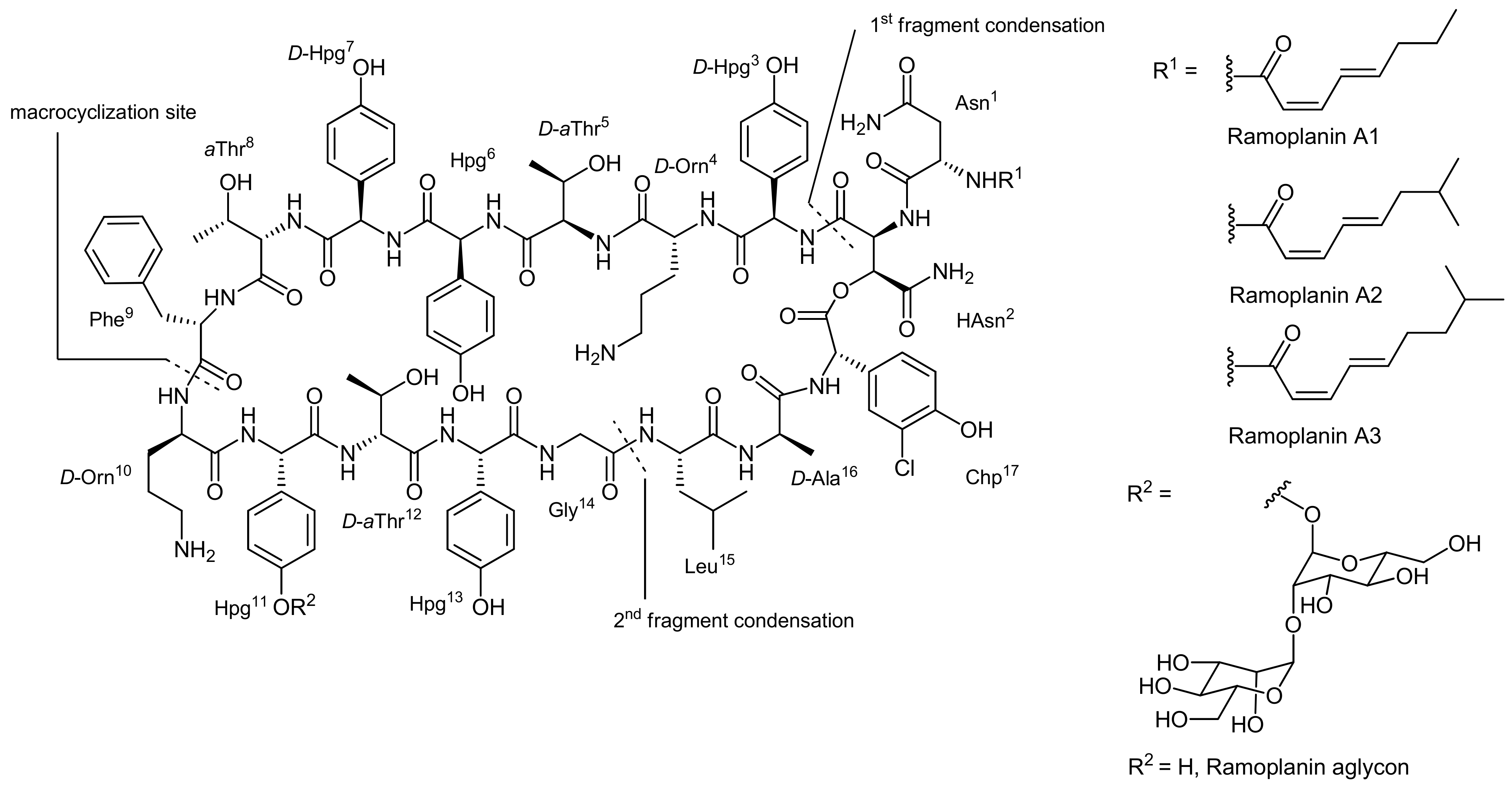
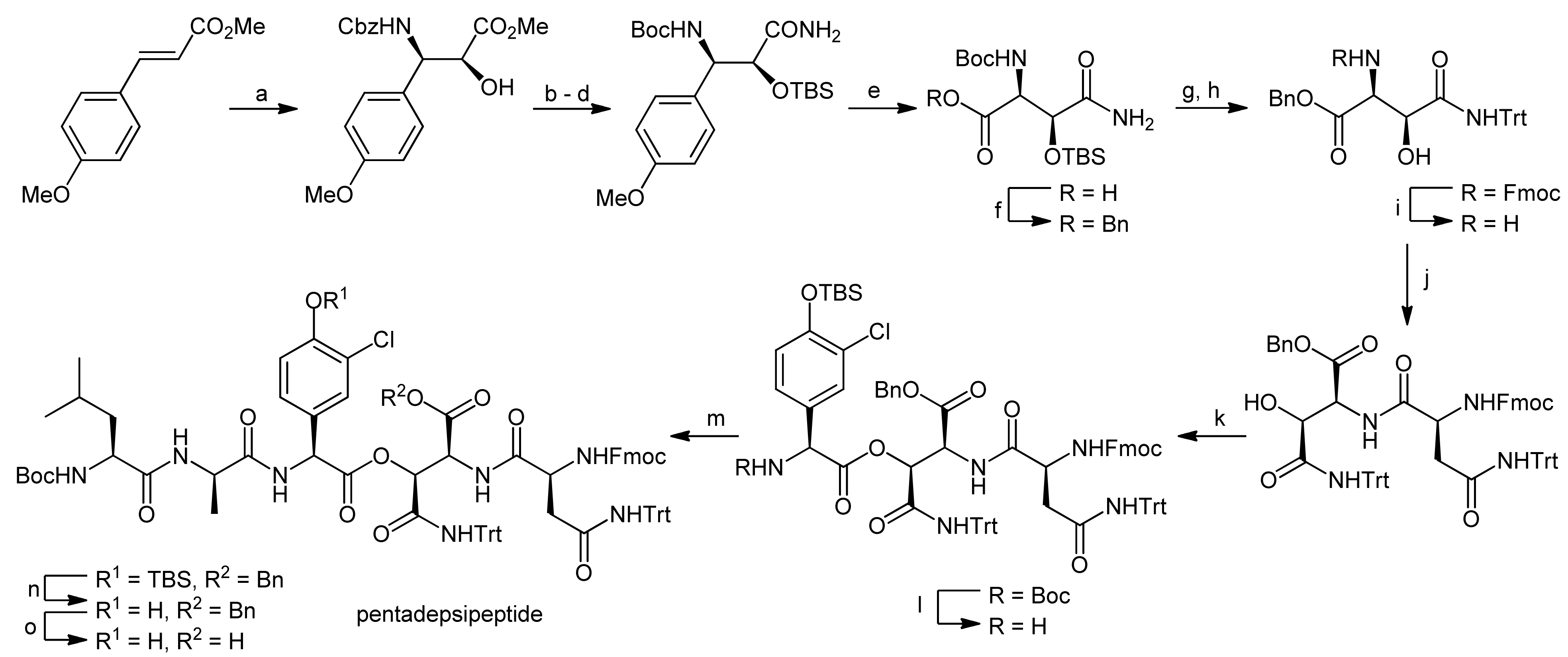
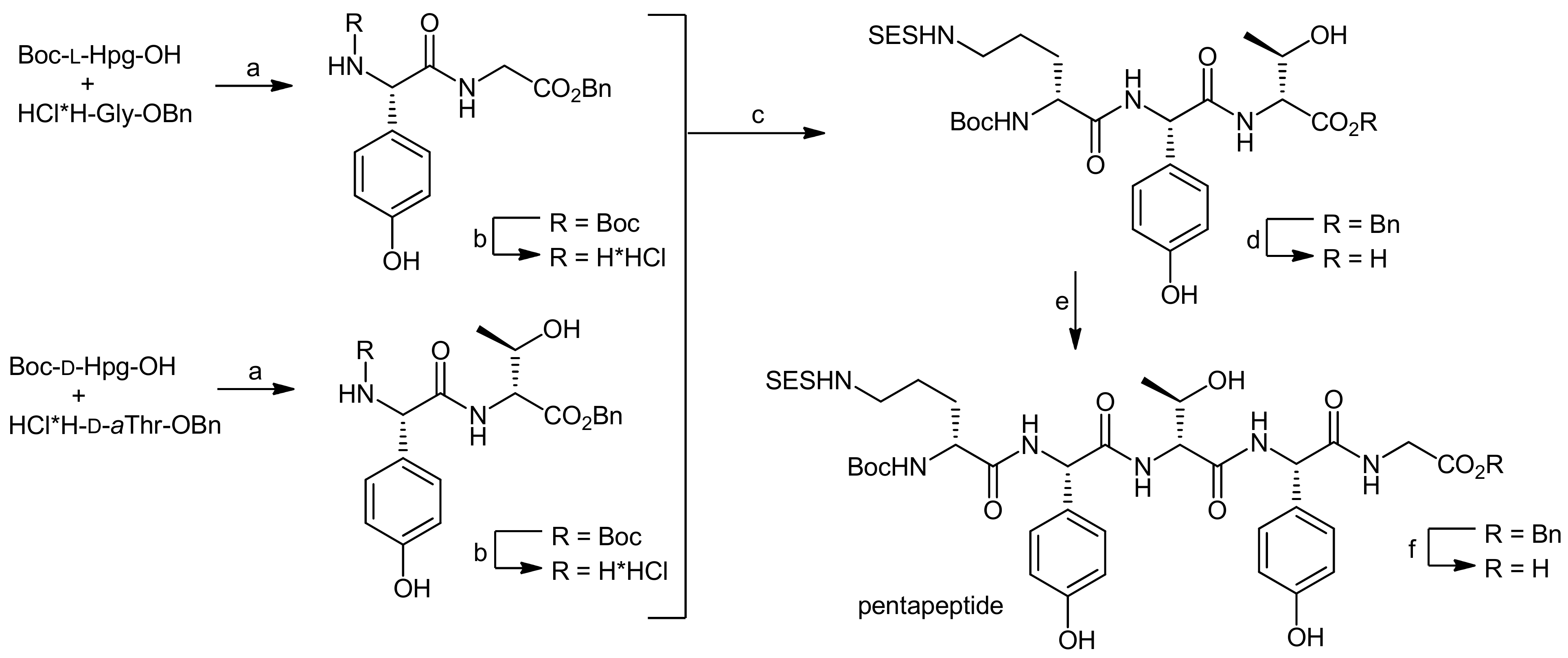
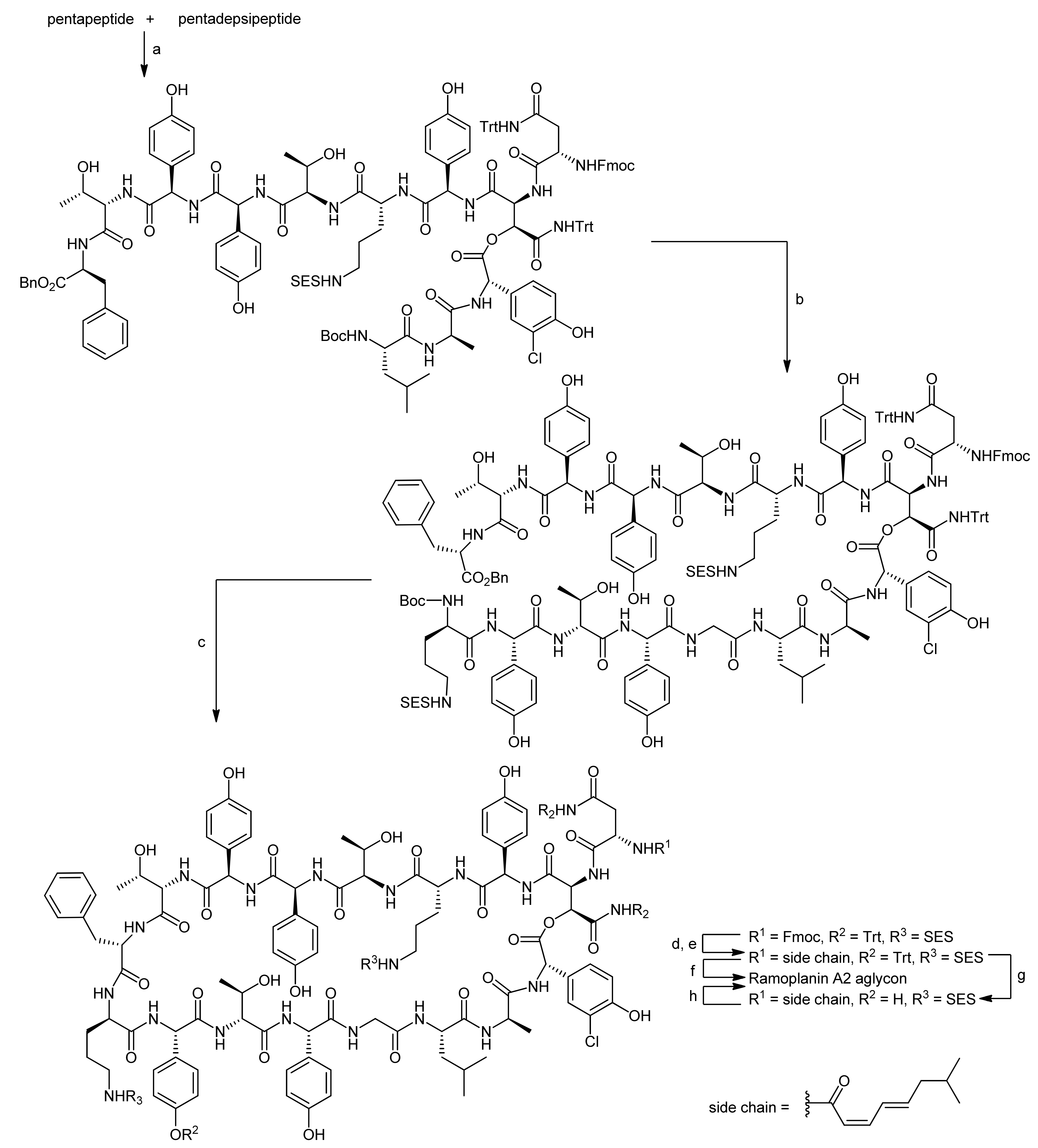
4. Ramoplanin Aglycons: The Challenges to Properly Assemble Cyclodepsipeptides from Smaller Fragments
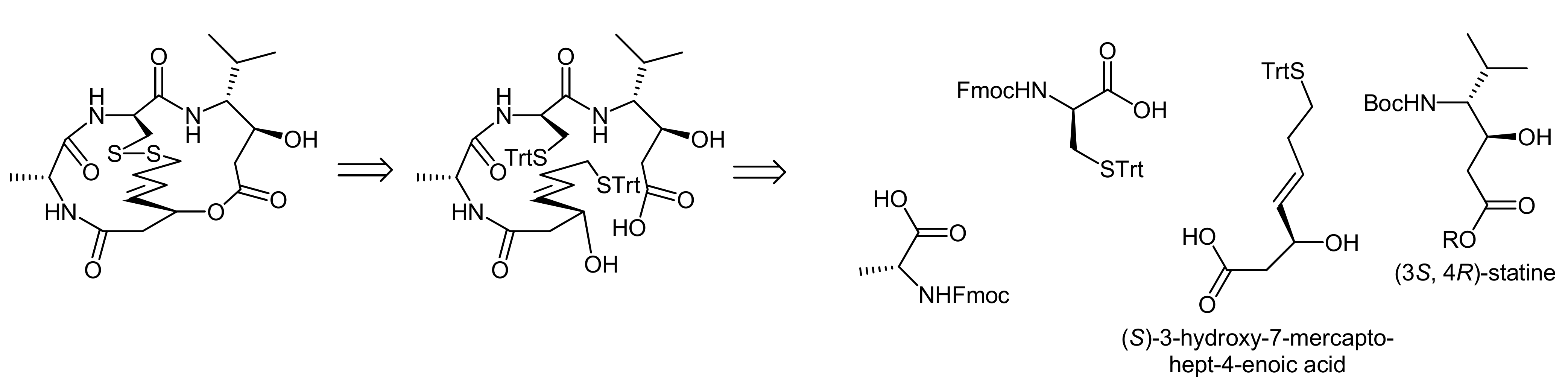
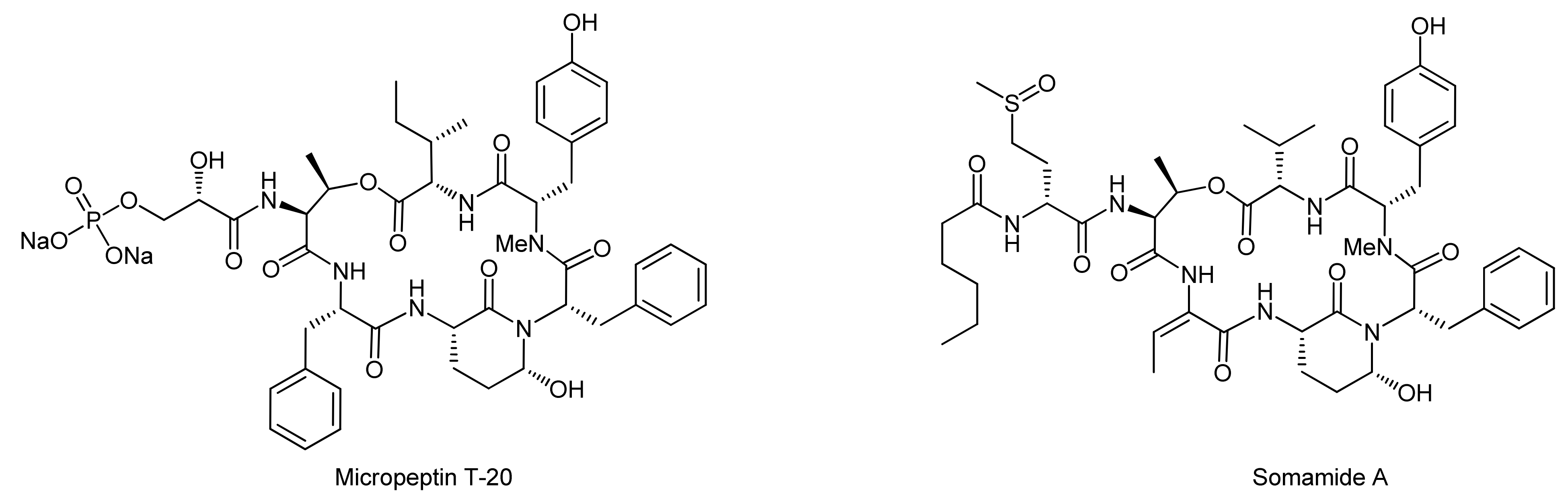
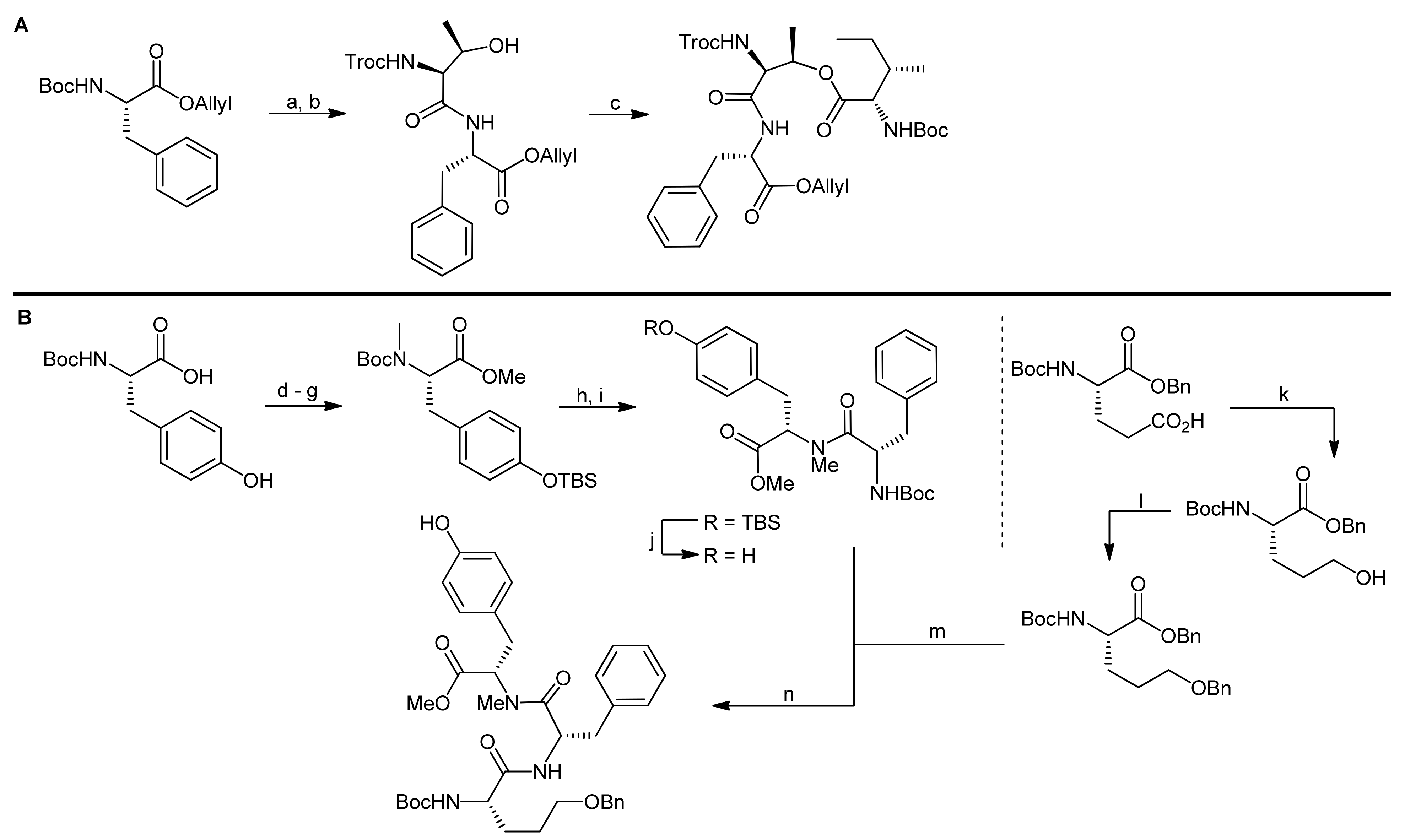
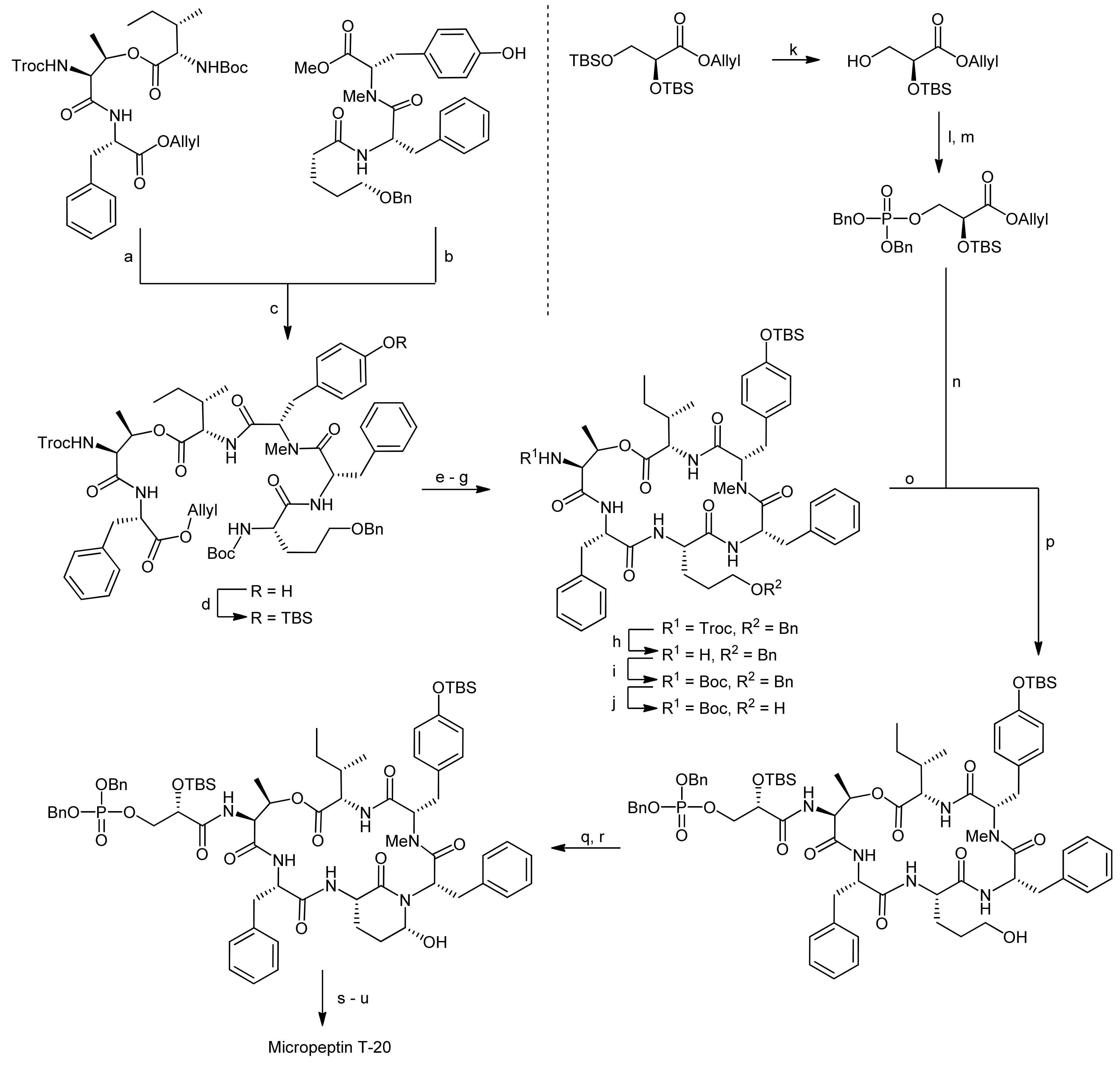
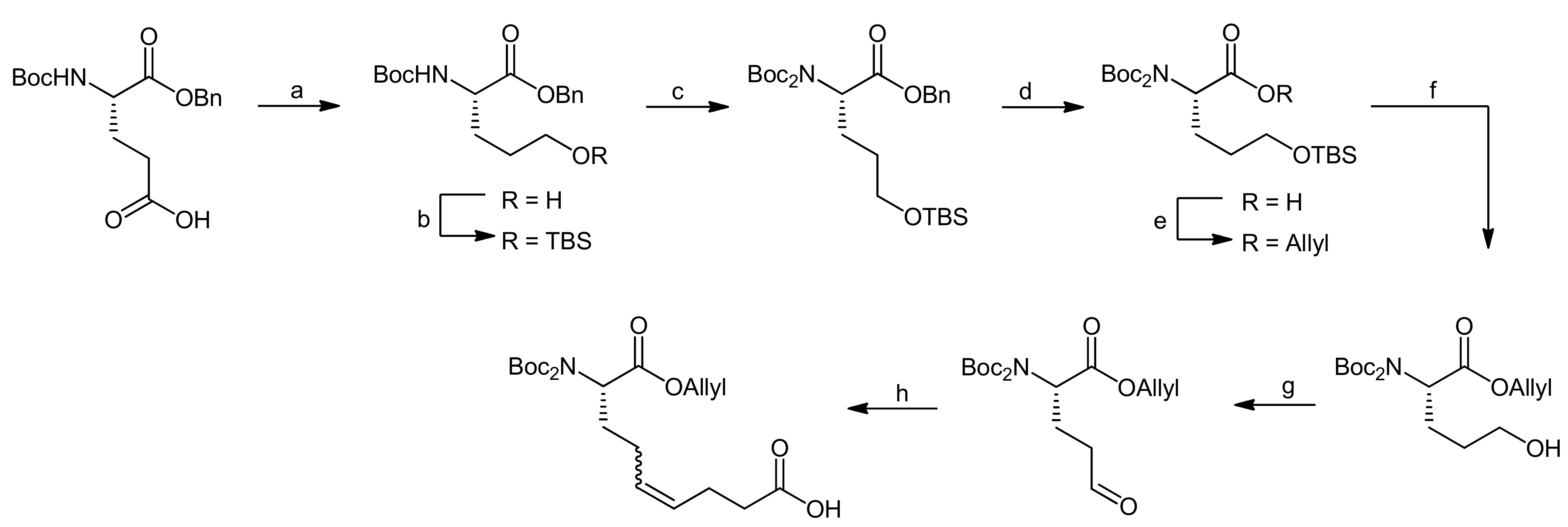
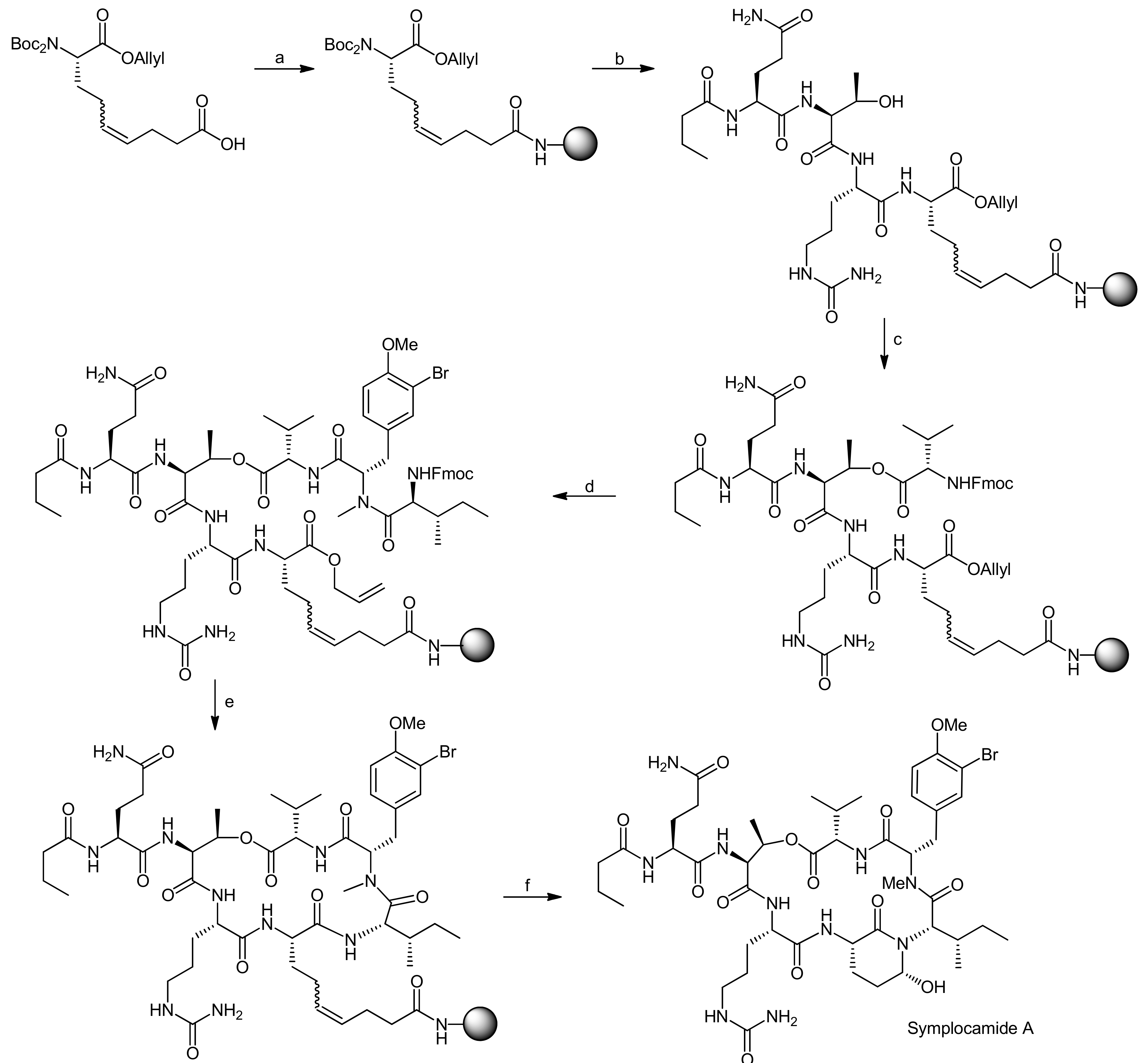
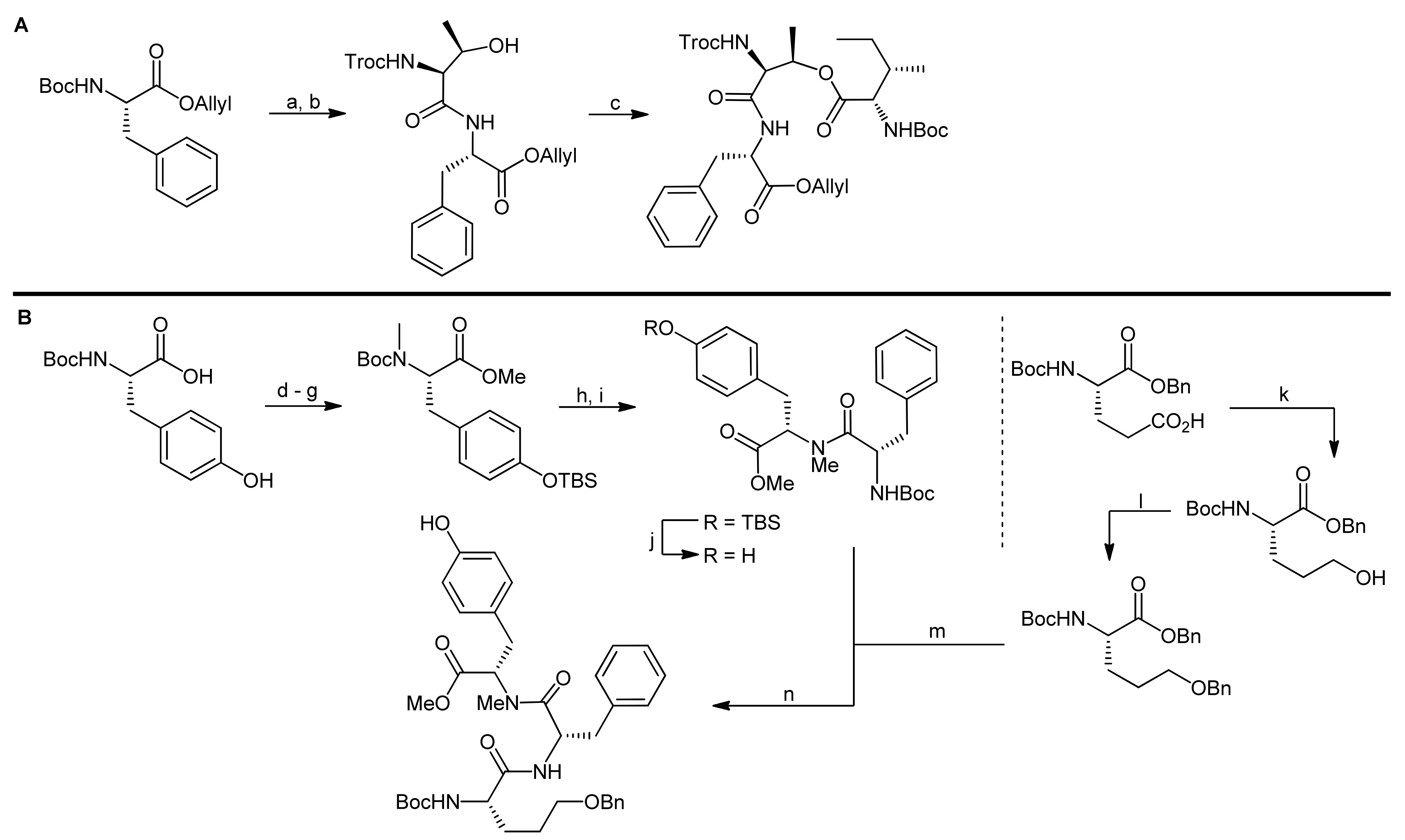
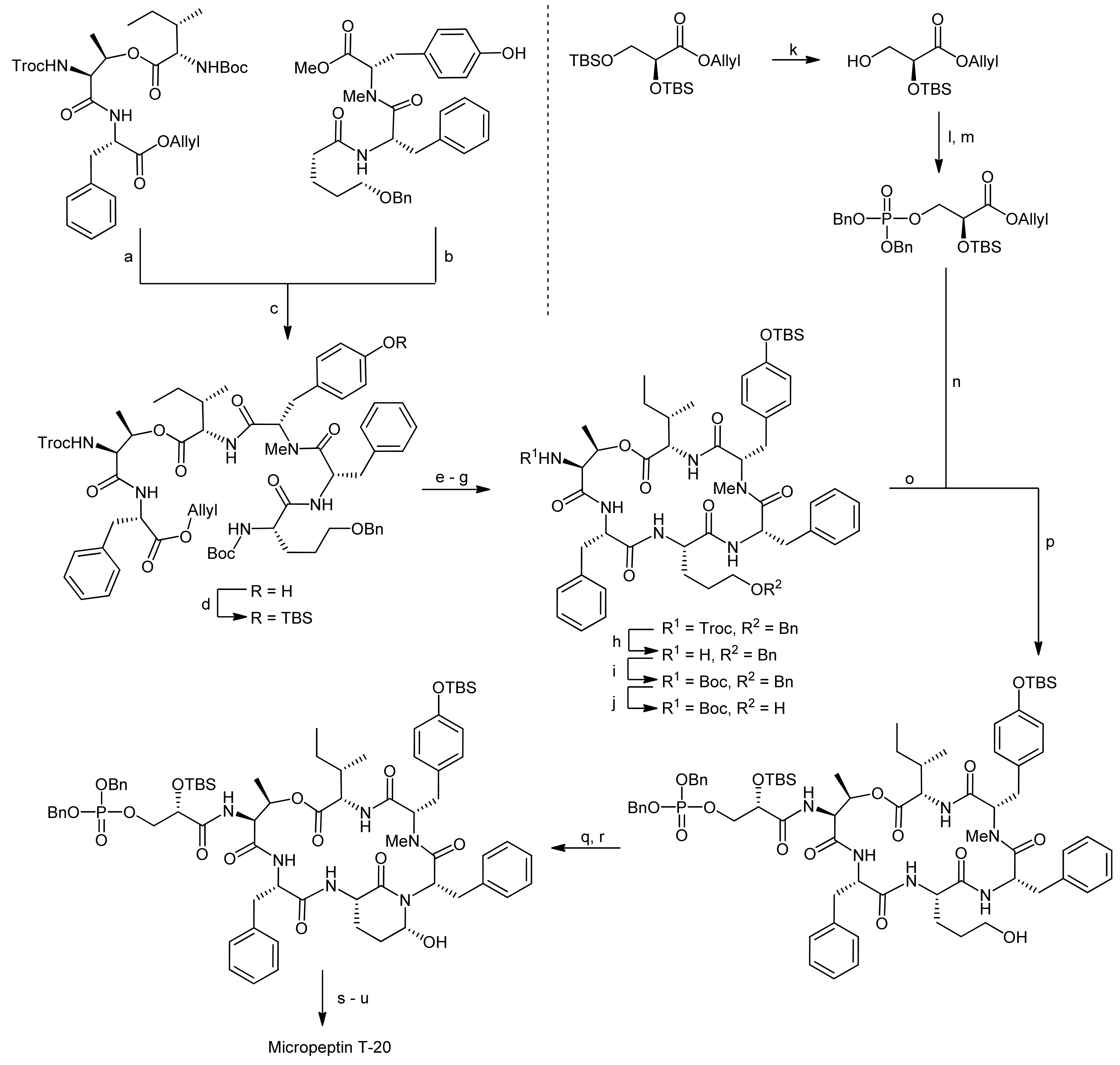
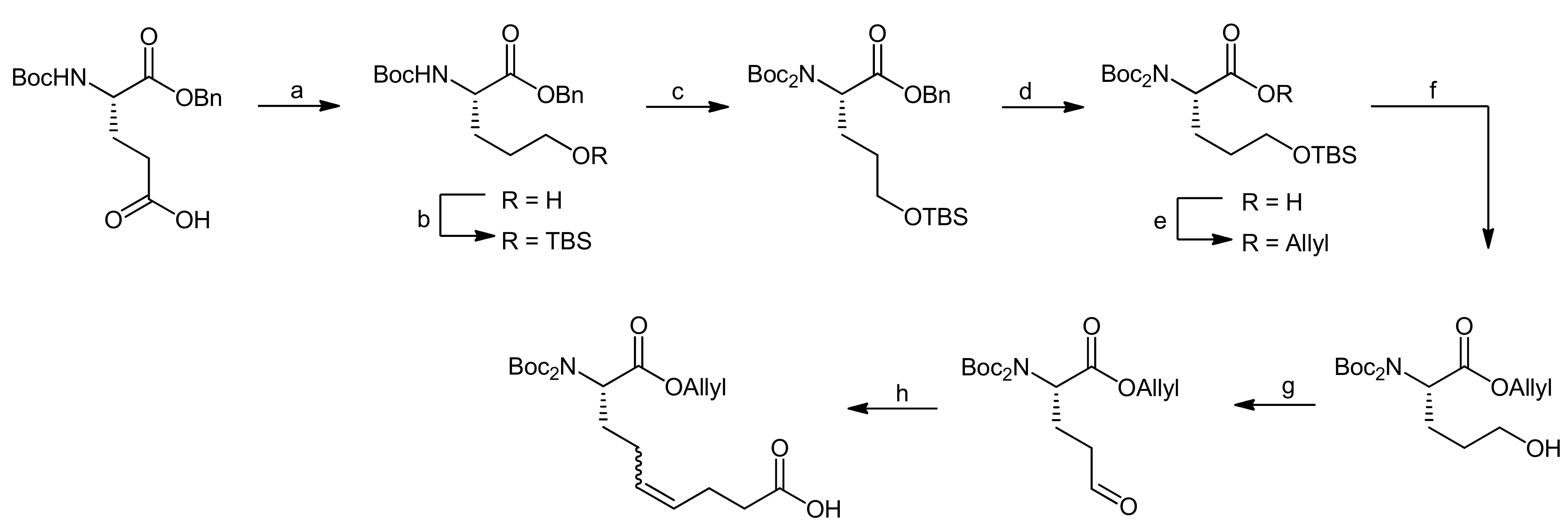
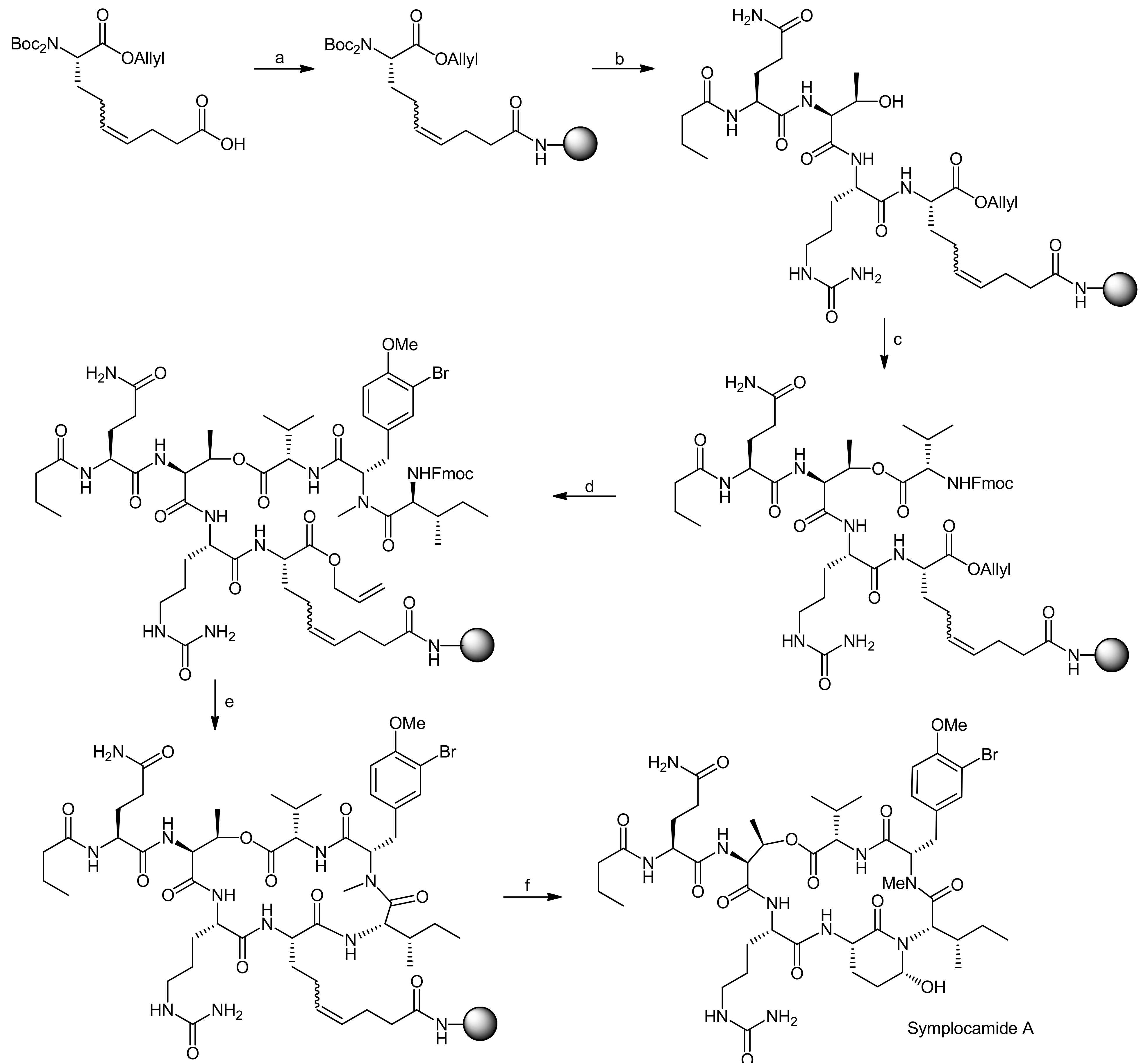
5. Syntheses of 3-Amino-6-hydroxy-2-piperidone (Ahp) Cyclodepsipeptides: The Challenge of Synthesizing Cyclodepsipeptides with Chemically Instable Elements
6. Conclusions
Acknowledgments
References
- Sarabia, F.; Chammaa, S.; Ruiz, A.S.; Ortiz, L.M.; Herrera, F.J. Chemistry and biology of cyclic depsipeptides of medicinal and biological interest. Curr. Med. Chem. 2004, 11, 1309–1332. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Walsh, C.T. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem. Rev. 2006, 106, 3468–3496. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Donia, M.S.; Schmidt, E.W. Ribosomal peptide natural products: Bridging the ribosomal and nonribosomal worlds. Nat. Prod. Rep. 2009, 26, 537–559. [Google Scholar] [CrossRef] [PubMed]
- Breinbauer, R.; Vetter, I.R.; Waldmann, H. From protein domains to drug candidates—Natural products as guiding principles in the design and synthesis of compound libraries. Angew. Chem. Int. Ed. 2002, 41, 2878–2890. [Google Scholar] [CrossRef]
- Groner, B. Peptides as Drugs: Discovery and Development. In Peptides as Drugs: Discovery and Development; Groner, B., Ed.; Wiley-VCH: Weinheim, Germany, 2009; pp. 1–8. [Google Scholar]
- Lemmens-Gruber, R.; Kamyar, M.R.; Dornetshuber, R. Cyclodepsipeptides—Potential drugs and lead compounds in the drug development process. Curr. Med. Chem. 2009, 16, 1122–1137. [Google Scholar] [CrossRef] [PubMed]
- Vera, M.D.; Joullié, M.M. Natural products as probes of cell biology: 20 years of didemnin research. Med. Res. Rev. 2002, 22, 102–145. [Google Scholar] [CrossRef] [PubMed]
- Taori, K.; Paul, V.J.; Luesch, H. Structure and activity of largazole, a potent antiproliferative agent from the floridian marine cyanobacterium symploca sp. J. Am. Chem. Soc. 2008, 130, 1806–1807. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yao, H.; Xu, J.; Jiang, S. Synthetic routes and biological evaluation of largazole and its analogues as potent histone deacetylase inhibitors. Molecules 2011, 16, 4681–4694. [Google Scholar] [CrossRef] [PubMed]
- Bowers, A.; West, N.; Taunton, J.; Schreiber, S.L.; Bradner, J.E.; Williams, R.M. Total synthesis and biological mode of action of largazole: A potent class i histone deacetylase inhibitor. J. Am. Chem. Soc. 2008, 130, 11219–11222. [Google Scholar] [CrossRef] [PubMed]
- Bowers, A.A.; Greshock, T.J.; West, N.; Estiu, G.; Schreiber, S.L.; Wiest, O.; Williams, R.M.; Bradner, J.E. Synthesis and conformation-activity relationships of the peptide isosteres of fk228 and largazole. J. Am. Chem. Soc. 2009, 131, 2900–2905. [Google Scholar] [CrossRef] [PubMed]
- Eggen, M.; Georg, G.I. The cryptophycins: Their synthesis and anticancer activity. Med. Res. Rev. 2002, 22, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Hamann, M.T. Chemistry and biology of kahalalides. Chem. Rev. 2011, 111, 3208–3235. [Google Scholar] [CrossRef] [PubMed]
- Hamann, M.T.; Scheuer, P.J. Kahalalide f: A bioactive depsipeptide from the sacoglossan mollusk elysia rufescens and the green alga bryopsis sp. J. Am. Chem. Soc. 1993, 115, 5825–5826. [Google Scholar] [CrossRef]
- López-Macià, À.; Jiménez, J.C.; Royo, M.; Giralt, E.; Albericio, F. Synthesis and structure determination of kahalalide f. J. Am. Chem. Soc. 2001, 123, 11398–11401. [Google Scholar] [CrossRef] [PubMed]
- Stolze, S.C.; Kaiser, M. Challenges in the syntheses of peptidic natural products. Synthesis 2012, 44, 1755–1777. [Google Scholar]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. New apratoxins of marine cyanobacterial origin from guam and palau. Bioorg. Med. Chem. 2002, 10, 1973–1978. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Corbett, T.H. Total structure determination of apratoxin a, a potent novel cytotoxin from the marine cyanobacterium lyngbya majuscula. J. Am. Chem. Soc. 2001, 123, 5418–5423. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Forsyth, C.J. Total synthesis of apratoxin a. J. Am. Chem. Soc. 2003, 125, 8734–8735. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Forsyth, C.J. Synthesis of the apratoxin 2,4-disubstituted thiazoline via an intramolecular aza-wittig reaction. Org. Lett. 2003, 5, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Forsyth, C.J. Total synthesis of the marine cyanobacterial cyclodepsipeptide apratoxin A. Proc. Natl. Acad. Sci. USA 2004, 101, 12067–12072. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Numajiri, Y.; Munakata, A.; Takahashi, T. Total synthesis of apratoxin A. Org. Lett. 2006, 8, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Numajiri, Y.; Takahashi, T.; Doi, T. Total synthesis of (−)-apratoxin a, 34-epimer, and its oxazoline analogue. Chem.-Asian J. 2009, 4, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Zou, B.; Cai, G.; Hu, X.; Liu, J.O. Total synthesis of the cyclodepsipeptide apratoxin a and its analogues and assessment of their biological activities. Chemistry 2006, 12, 7615–7626. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Liu, Y.; Luesch, H. Systematic chemical mutagenesis identifies a potent novel apratoxin A/E hybrid with improved in vivo antitumor activity. ACS Med. Chem. Lett. 2011, 2, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.; Suyama, T.L.; Engene, N.; Wingerd, J.S.; Matainaho, T.; Gerwick, W.H. Apratoxin d, a potent cytotoxic cyclodepsipeptide from papua new guinea collections of the marine cyanobacteria lyngbya majuscula and lyngbya sordida. J. Nat. Prod. 2008, 71, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Robertson, B.D.; Wengryniuk, S.E.; Coltart, D.M. Asymmetric total synthesis of apratoxin D. Org. Lett. 2012, 14, 5192–5195. [Google Scholar] [CrossRef] [PubMed]
- Masuoka, Y.; Nagai, A.; Shin-ya, K.; Furihata, K.; Nagai, K.; Suzuki, K.-I.; Hayakawa, Y.; Seto, H. Spiruchostatins A and B, novel gene expression-enhancing substances produced by pseudomonas sp. Tetrahedron Lett. 2001, 42, 41–44. [Google Scholar] [CrossRef]
- Doi, T.; Iijima, Y.; Kazuo, S.Y.; Ganesan, A.; Takahashi, T. A total synthesis of spiruchostatin A. Tetrahedron Lett. 2006, 47, 1177–1180. [Google Scholar] [CrossRef]
- Shiina, I.; Kubota, M.; Ibuka, R. A novel and efficient macrolactonization of omega-hydroxycarboxylic acids using 2-methyl-6-nitrobenzoic anhydride (mnba). Tetrahedron Lett. 2002, 43, 7535–7539. [Google Scholar] [CrossRef]
- Doi, T.; Iijima, Y.; Munakata, A.; Shin-ya, K.; Ganesan, A.; Takahashi, T. A solid-phase total synthesis of the cyclic depsipeptide hdac inhibitor spiruchostatin A. Tetrahedron Lett. 2009, 50, 2970–2972. [Google Scholar]
- Fuse, S.; Okada, K.; Iijima, Y.; Munakata, A.; Machida, K.; Takahashi, T.; Takagi, M.; Shin-ya, K.; Doi, T. Total synthesis of spiruchostatin b aided by an automated synthesizer. Org. Biomol. Chem. 2011, 9, 3825–3833. [Google Scholar] [CrossRef] [PubMed]
- Yurek-George, A.; Habens, F.; Brimmell, M.; Packham, G.; Ganesan, A. Total synthesis of spiruchostatin a, a potent histone deacetylase inhibitor. J. Am. Chem. Soc. 2004, 126, 1030–1031. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, T.; Watanabe, K.; Narita, K.; Oguchi, T.; Abe, H.; Katoh, T. Total synthesis of spiruchostatin B, a potent histone deacetylase inhibitor, from a microorganism. Chem. Commun. 2008, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Narita, K.; Kikuchi, T.; Watanabe, K.; Takizawa, T.; Oguchi, T.; Kudo, K.; Matsuhara, K.; Abe, H.; Yamori, T.; Yoshida, M.; et al. Total synthesis of the bicyclic depsipeptide hdac inhibitors spiruchostatins A and B, 5''-epi-spiruchostatin B, fk228 (fr901228) and preliminary evaluation of their biological activity. Chemistry 2009, 15, 11174–11186. [Google Scholar] [CrossRef] [PubMed]
- Calandra, N.A.; Cheng, Y.L.; Kocak, K.A.; Miller, J.S. Total synthesis of spiruchostatin a via chemoselective macrocyclization using an accessible enantiomerically pure latent thioester. Org. Lett. 2009, 11, 1971–1974. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Manda, T.; Matsumoto, S.; Mukumoto, S.; Nishigaki, F.; Kawamura, I.; Shimomura, K. Fr901228, a novel antitumor bicyclic depsipeptide produced by chromobacterium violaceum No. 968. Iii. Antitumor activities on experimental tumors in mice. J. Antibiot. 1994, 47, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Nakajima, H.; Hori, Y.; Fujita, T.; Nishimura, M.; Goto, T.; Okuhara, M. Fr901228, a novel antitumor bicyclic depsipeptide produced by chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J. Antibiot. 1994, 47, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, N.; Ueda, H.; Takase, S.; Tanaka, H.; Yamamoto, K.; Tada, T. Fr901228, a novel antitumor bicyclic depsipeptide produced by chromobacterium violaceum No. 968. II. Structure determination. J. Antibiot. 1994, 47, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Li, K.W.; Wu, J.; Xing, W.N.; Simon, J.A. Total synthesis of the antitumor depsipeptide fr-901,228. J. Am. Chem. Soc. 1996, 118, 7237–7238. [Google Scholar] [CrossRef]
- Greshock, T.J.; Johns, D.M.; Noguchi, Y.; Williams, R.M. Improved total synthesis of the potent hdac inhibitor fk228 (fr-901228). Org. Lett. 2008, 10, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Di Maro, S.; Pong, R.C.; Hsieh, J.T.; Ahn, J.M. Efficient solid-phase synthesis of fk228 analogues as potent antitumoral agents. J. Med. Chem. 2008, 51, 6639–6641. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Packham, G.; Ganesan, A. Macrolactamization versus macrolactonization: Total synthesis of fk228, the depsipeptide histone deacetylase inhibitor. J. Org. Chem. 2008, 73, 9353–9361. [Google Scholar] [CrossRef] [PubMed]
- Yurek-George, A.; Cecil, A.R.; Mo, A.H.; Wen, S.; Rogers, H.; Habens, F.; Maeda, S.; Yoshida, M.; Packham, G.; Ganesan, A. The first biologically active synthetic analogues of fk228, the depsipeptide histone deacetylase inhibitor. J. Med. Chem. 2007, 50, 5720–5726. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gambs, C.; Abe, Y.; Wentworth, P.; Janda, K.D. Total synthesis of the depsipeptide fr-901375. J. Org. Chem. 2003, 68, 8902–8905. [Google Scholar] [CrossRef] [PubMed]
- Nagao, Y.; Hagiwara, Y.; Kumagai, T.; Ochiai, M.; Inoue, T.; Hashimoto, K.; Fujita, E. New c-4-chiral 1,3-thiazolidine-2-thiones: Excellent chiral auxiliaries for highly diastereo-controlled aldol-type reactions of acetic acid and.Alpha..Beta.-unsaturated aldehydes. J. Org. Chem. 1986, 51, 2391–2393. [Google Scholar] [CrossRef]
- González, Á.; Aiguadé, J.; Urpí, F.; Vilarrasa, J. Asymmetric acetate aldol reactions in connection with an enantioselective total synthesis of macrolactin A. Tetrahedron Lett. 1996, 37, 8949–8952. [Google Scholar] [CrossRef]
- Walker, S.; Chen, L.; Hu, Y.; Rew, Y.; Shin, D.; Boger, D.L. Chemistry and biology of ramoplanin: A lipoglycodepsipeptide with potent antibiotic activity. Chem. Rev. 2005, 105, 449–476. [Google Scholar] [CrossRef] [PubMed]
- Skelton, N.J.; Harding, M.M.; Mortishire-Smith, R.J.; Rahman, S.K.; Williams, D.H.; Rance, M.J.; Ruddock, J.C. Structure elucidation and solution conformation of the glycopeptide antibiotic ramoplanose (uk-71,903): A cyclic depsipeptide containing an antiparallel.Beta.-sheet and a.Beta.-bulge. J. Am. Chem. Soc. 1991, 113, 7522–7530. [Google Scholar] [CrossRef]
- Jiang, W.L.; Wanner, J.; Lee, R.J.; Bounaud, P.Y.; Boger, D.L. Total synthesis of the ramoplanin a2 and ramoplanose aglycon. J. Am. Chem. Soc. 2002, 124, 5288–5290. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.L.; Wanner, J.; Lee, R.J.; Bounaud, P.Y.; Boger, D.L. Total synthesis of the ramoplanin a2 and ramoplanose aglycon. J. Am. Chem. Soc. 2003, 125, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L.; Lee, R.J.; Bounaud, P.-Y.; Meier, P. Asymmetric synthesis of orthogonally protected l-threo-β-hydroxyasparagine. J. Org. Chem. 2000, 65, 6770–6772. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Rew, Y.; Boger, D.L. Total synthesis and structure of the ramoplanin A1 and A3 aglycons: Two minor components of the ramoplanin complex. Proc. Natl. Acad. Sci. USA 2004, 101, 11977–11979. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Smitka, T.A.; Bonjouklian, R.; Clardy, J. Atomic structure of the trypsin-a90720a complex: A unified approach to structure and function. Chem. Biol. 1994, 1, 113–117. [Google Scholar] [CrossRef]
- Matern, U.; Schleberger, C.; Jelakovic, S.; Weckesser, J.; Schulz, G.E. Binding structure of elastase inhibitor scyptolin a. Chem. Biol. 2003, 10, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- McDonough, M.A.; Schofield, C.J. New structural insights into the inhibition of serine proteases by cyclic peptides from bacteria. Chem. Biol. 2003, 10, 898–900. [Google Scholar] [CrossRef] [PubMed]
- Radau, G. Cyanopeptides: A new and nearly inexhaustible natural resource for the design and structure-activity relationship studies of new inhibitors of trypsin-like serine proteases. Curr. Enzyme Inhib. 2005, 1, 295–307. [Google Scholar] [CrossRef]
- Zainuddin, E.N.; Mentel, R.; Wray, V.; Jansen, R.; Nimtz, M.; Lalk, M.; Mundt, S. Cyclic depsipeptides, ichthyopeptins A and B, from microcystis ichthyoblabe. J. Nat. Prod. 2007, 70, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- von Ehlert, E.; Oberer, L.; Merkel, P.; Huhn, T.; Blom, J.F. Cyanopeptolin 954, a chlorine-containing chymotrypsin inhibitor of microcystis aeruginosa niva cya 43. J. Nat. Prod. 2005, 68, 1324–1327. [Google Scholar] [CrossRef] [PubMed]
- Zafrir, E.; Carmeli, S. Micropeptins from an israeli fishpond water bloom of the cyanobacterium microcystis sp. J. Nat. Prod. 2009, 73, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Blom, J.F.; Bister, B.; Bischoff, D.; Nicholson, G.; Jung, G.; Süssmuth, R.D.; Jüttner, F. Oscillapeptin J, a new grazer toxin of the freshwater cyanobacterium planktothrix rubescens. J. Nat. Prod. 2003, 66, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Matern, U.; Oberer, L.; Falchetto, R.A.; Erhard, M.; König, W.A.; Herdman, M.; Weckesser, J. Scyptolin A and B, cyclic depsipeptides from axenic cultures of scytonema hofmanni pcc 7110. Phytochemistry 2001, 58, 1087–1095. [Google Scholar] [CrossRef]
- Okano, T.; Sano, T.; Kaya, K. Micropeptin t-20, a novel phosphate-containing cyclic depsipeptide from the cyanobacterium microcystis aeruginosa. Tetrahedron Lett. 1999, 40, 2379–2382. [Google Scholar] [CrossRef]
- Nogle, L.M.; Williamson, R.T.; Gerwick, W.H. Somamides a and b, two new depsipeptide analogues of dolastatin 13 from a fijian cyanobacterial assemblage of lyngbya majuscula and schizothrix species. J. Nat. Prod. 2001, 64, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Yokokawa, F.; Inaizumi, A.; Shioiri, T. Synthetic studies of micropeptin t-20, a novel 3-amino-6-hydroxy-2-piperidone (ahp)-containing cyclic depsipeptide. Tetrahedron Lett. 2001, 42, 5903–5908. [Google Scholar] [CrossRef]
- Yokokawa, F.; Inaizumi, A.; Shioiri, T. Synthetic studies of the cyclic depsipeptides bearing the 3-amino-6-hydroxy-2-piperidone (ahp) unit. Total synthesis of the proposed structure of micropeptin t-20. Tetrahedron 2005, 61, 1459–1480. [Google Scholar] [CrossRef]
- Yokokawa, F.; Shioiri, T. Total synthesis of somamide A, an ahp (3-amino-6-hydroxy-2-piperidone)-containing cyclic depsipeptide. Tetrahedron Lett. 2002, 43, 8673–8677. [Google Scholar] [CrossRef]
- Stolze, S.C.; Meltzer, M.; Ehrmann, M.; Kaiser, M. Solid phase total synthesis of the 3-amino-6-hydroxy-2-piperidone (ahp) cyclodepsipeptide and protease inhibitor symplocamide A. Chem. Commun. 2010, 46, 8857–8859. [Google Scholar] [CrossRef] [PubMed]
- Stolze, S.C.; Meltzer, M.; Ehrmann, M.; Kaiser, M. Development of a solid-phase approach to the natural product class of ahp-containing cyclodepsipeptides. Eur. J. Org. Chem. 2012, 2012, 1616–1625. [Google Scholar] [CrossRef]
- Linington, R.G.; Edwards, D.J.; Shuman, C.F.; McPhail, K.L.; Matainaho, T.; Gerwick, W.H. Symplocamide a, a potent cytotoxin and chymotrypsin inhibitor from the marine cyanobacterium symploca sp. J. Nat. Prod. 2007, 71, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.E.; Le Quement, S.T.; Meldal, M. Solid-phase synthesis of carboxylic and oxamic acids via oso4/naio4/hmta-mediated oxidative cleavage of acetylenic peptides. Org. Lett. 2007, 9, 2469–2472. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.E.; Meldal, M. Highly efficient solid-phase oxidative cleavage of olefins by oso4-naio4 in the intramolecular n-acyliminium pictet-spengler reaction. Org. Lett. 2005, 7, 2695–2698. [Google Scholar] [CrossRef] [PubMed]
- Le Quement, S.T.; Nielsen, T.E.; Meldal, M. Scaffold diversity through intramolecular cascade reactions of solid-supported cyclic n-acyliminium intermediates. J. Comb. Chem. 2007, 9, 1060–1072. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2013 by the authors. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stolze, S.C.; Kaiser, M. Case Studies of the Synthesis of Bioactive Cyclodepsipeptide Natural Products. Molecules 2013, 18, 1337-1367. https://doi.org/10.3390/molecules18021337
Stolze SC, Kaiser M. Case Studies of the Synthesis of Bioactive Cyclodepsipeptide Natural Products. Molecules. 2013; 18(2):1337-1367. https://doi.org/10.3390/molecules18021337
Chicago/Turabian StyleStolze, Sara C., and Markus Kaiser. 2013. "Case Studies of the Synthesis of Bioactive Cyclodepsipeptide Natural Products" Molecules 18, no. 2: 1337-1367. https://doi.org/10.3390/molecules18021337
APA StyleStolze, S. C., & Kaiser, M. (2013). Case Studies of the Synthesis of Bioactive Cyclodepsipeptide Natural Products. Molecules, 18(2), 1337-1367. https://doi.org/10.3390/molecules18021337