Solution Structure of the Circular γ-Domain Analog from the Wheat Metallothionein Ec-1

Abstract
:
1. Introduction
2. Results and Discussion
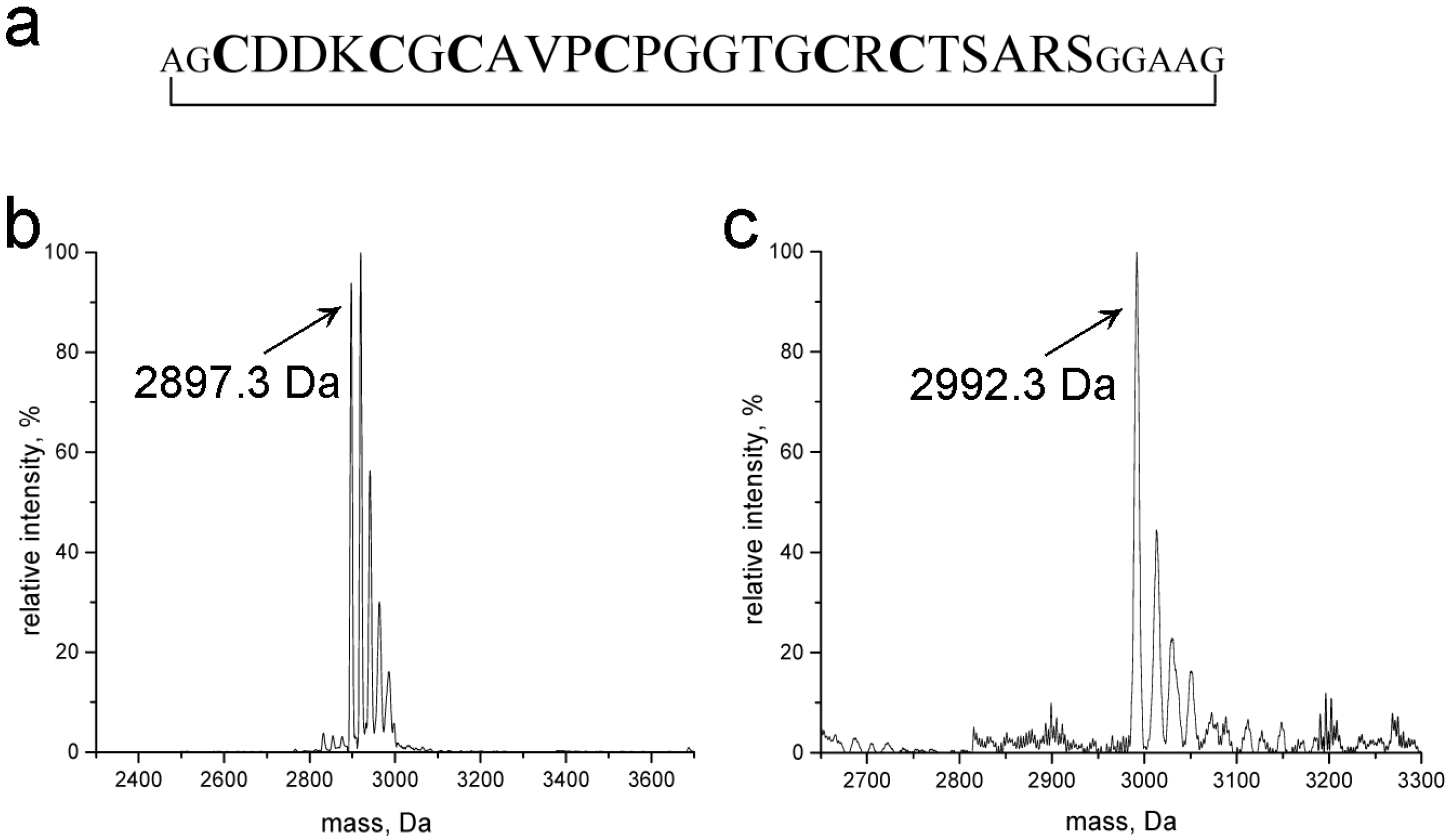
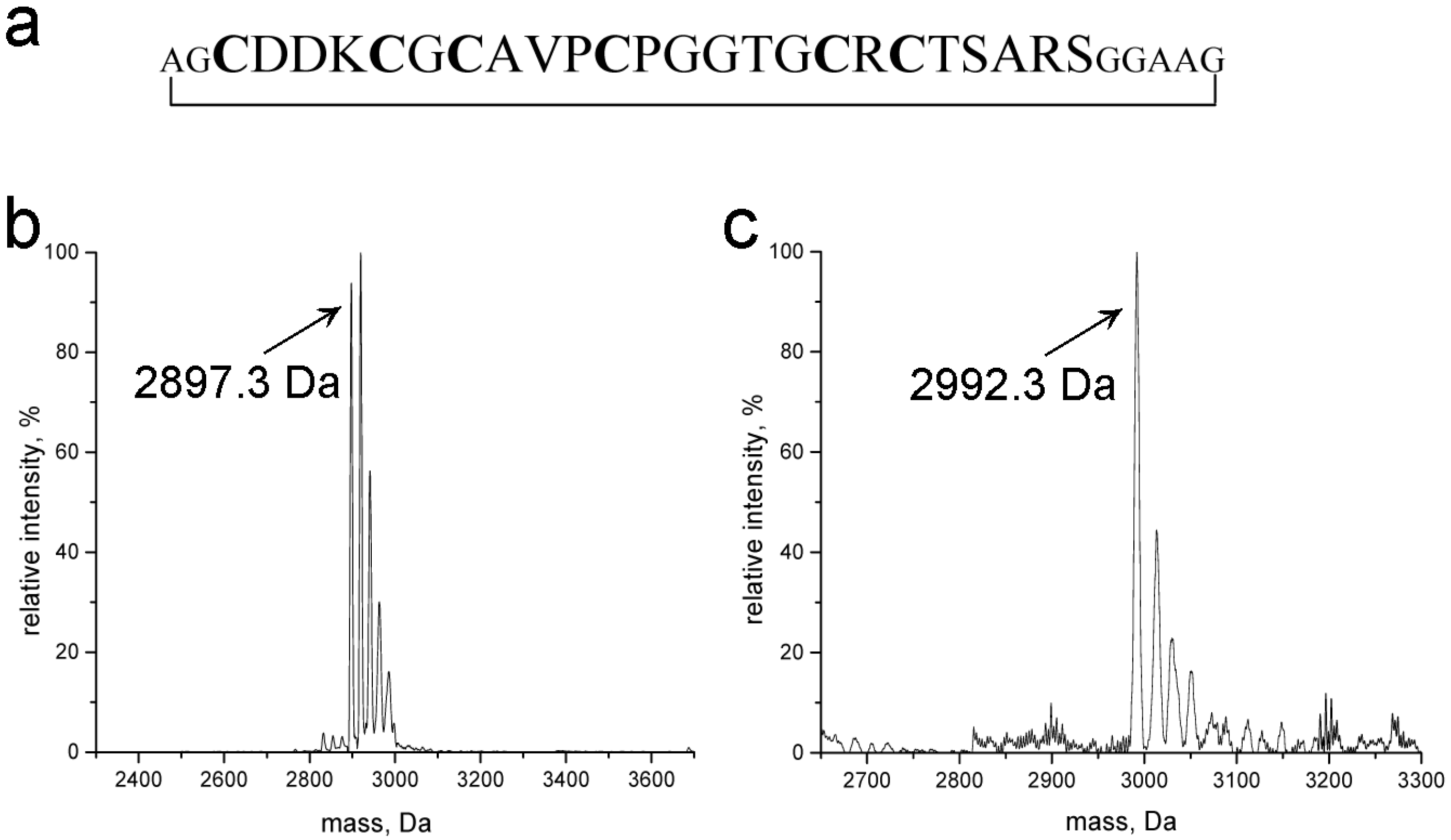
2.1. Design, Purity, and Metal Ion Binding Abilities of the Circular γ-Ec-1 Analog
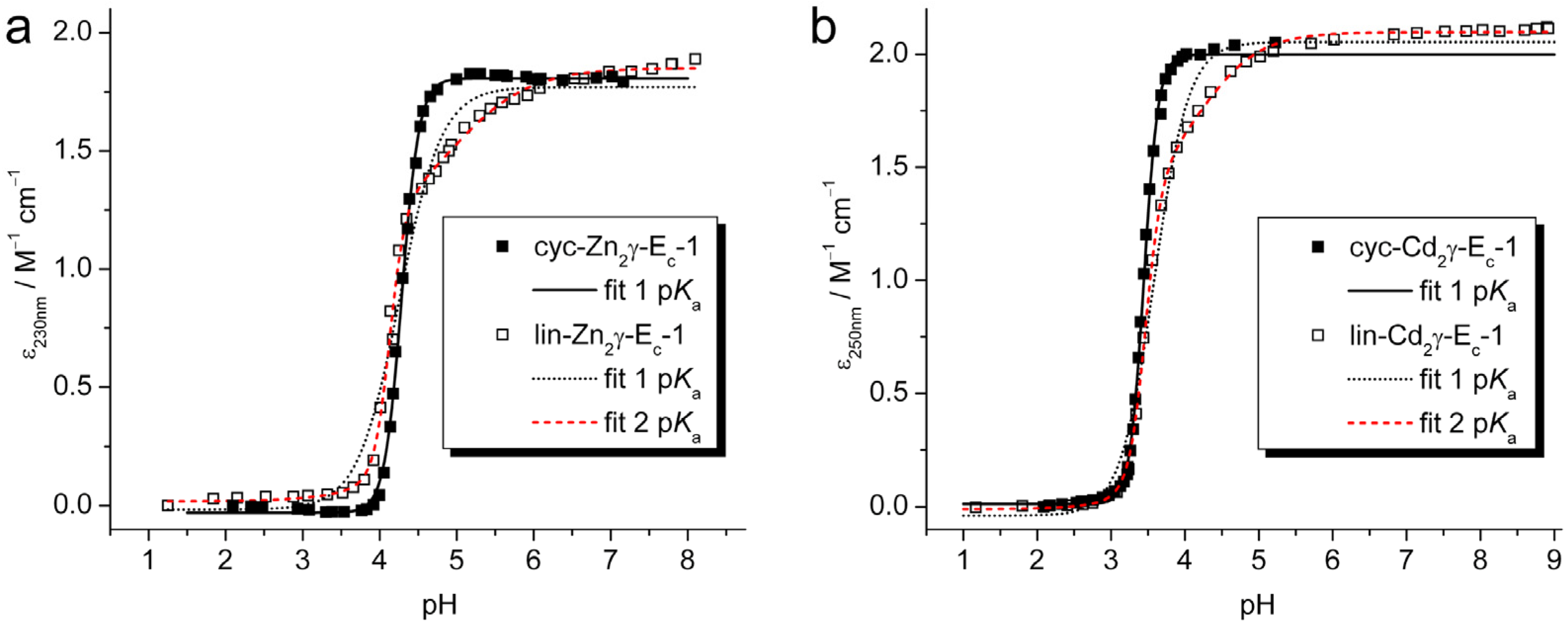
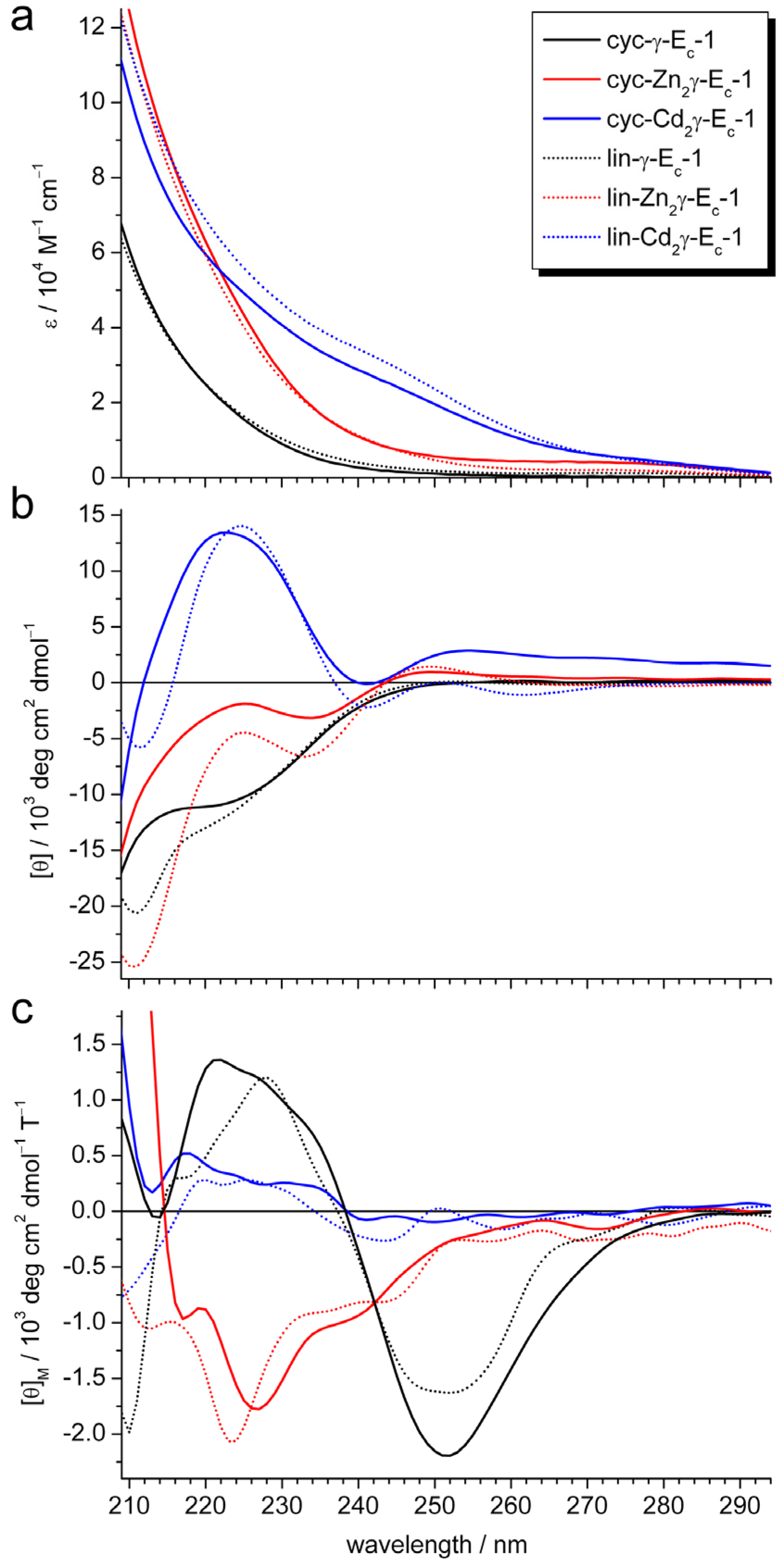
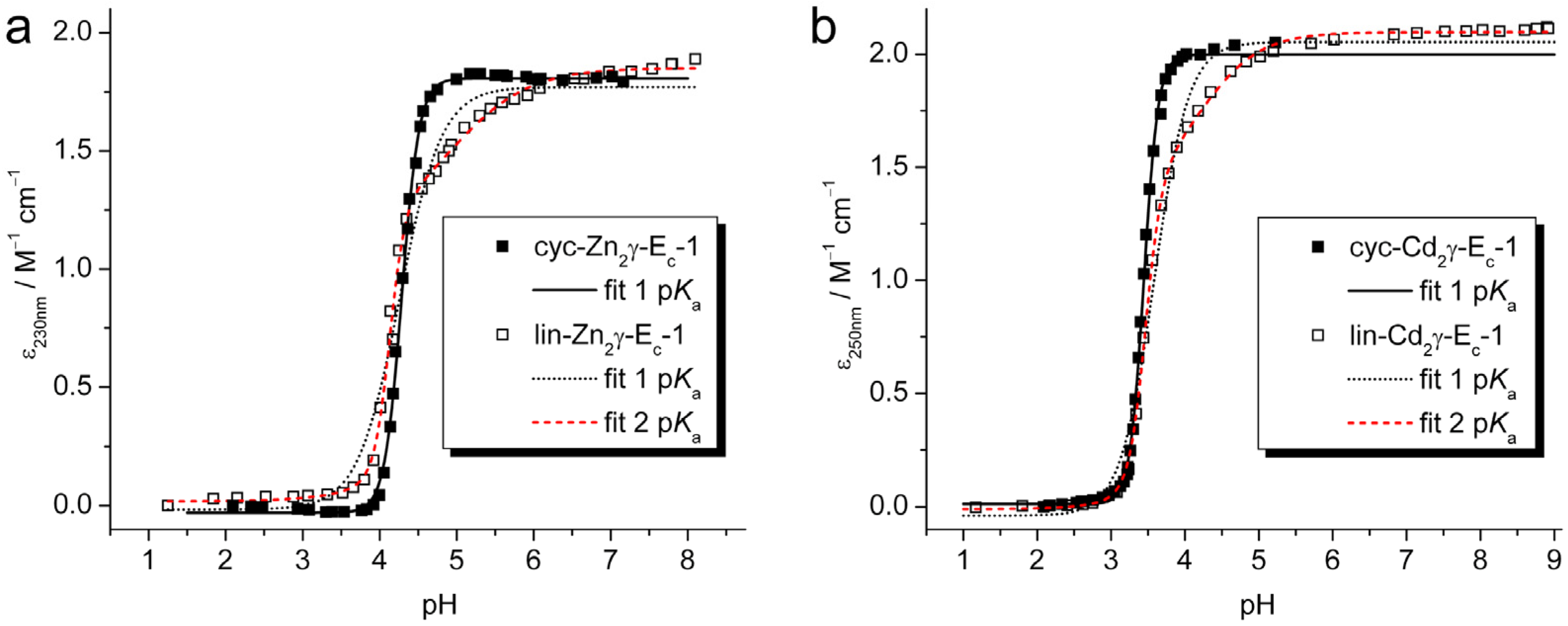
2.2. pH Stability of the Metal-Thiolate Clusters

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Equation 1 | cyc-Zn2γ-Ec-1 | lin-Zn2γ-Ec-1 | cyc-Cd2γ-Ec-1 | lin-Cd2γ-Ec-1 |
|---|---|---|---|---|
| AMT | 18062 ± 67 | 17696 ± 261 | 19981 ± 109 | 20539 ± 200 |
| AMTHn | −295 ± 82 | −161 ± 372 | 116 ± 101 | −395 ± 361 |
| pK | 4.287 ± 0.003 | 4.26 ± 0.03 | 3.444 ± 0.003 | 3.59 ± 0.02 |
| n | 3.85 ± 0.10 | 1.6 ± 0.1 | 4.5 ± 0.1 | 1.9 ± 0.1 |
| Equation 2 | ||||
| AMT | 18504 ± 141 | 20979 ± 61 | ||
| A MTHm | 11518 ± 1061 | 13634 ± 1174 | ||
| A MTHn+m | 176 ± 172 | −109 ± 124 | ||
| pK1 | 4.129 ± 0.010 | 3.466 ± 0.007 | ||
| pK2 | 4.9 ± 0.2 | 4.2 ± 0.1 | ||
| n | 4.3 ± 0.5 | 3.9 ± 0.3 | ||
| m | 0.9 ± 0.1 | 1.1 ± 0.1 |
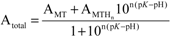 (1);[b]
(1);[b] 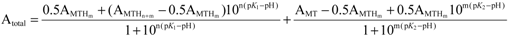 (2).
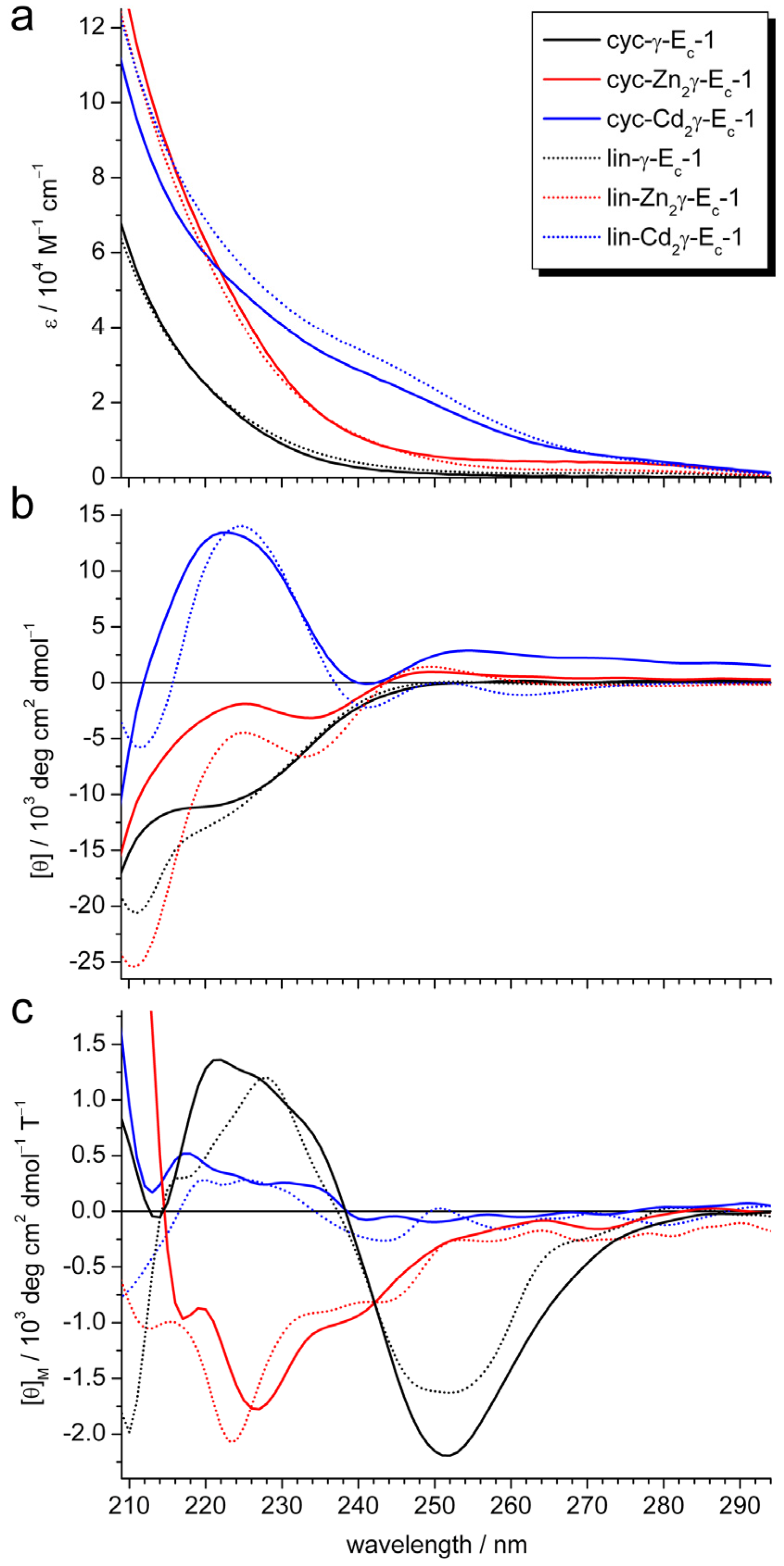
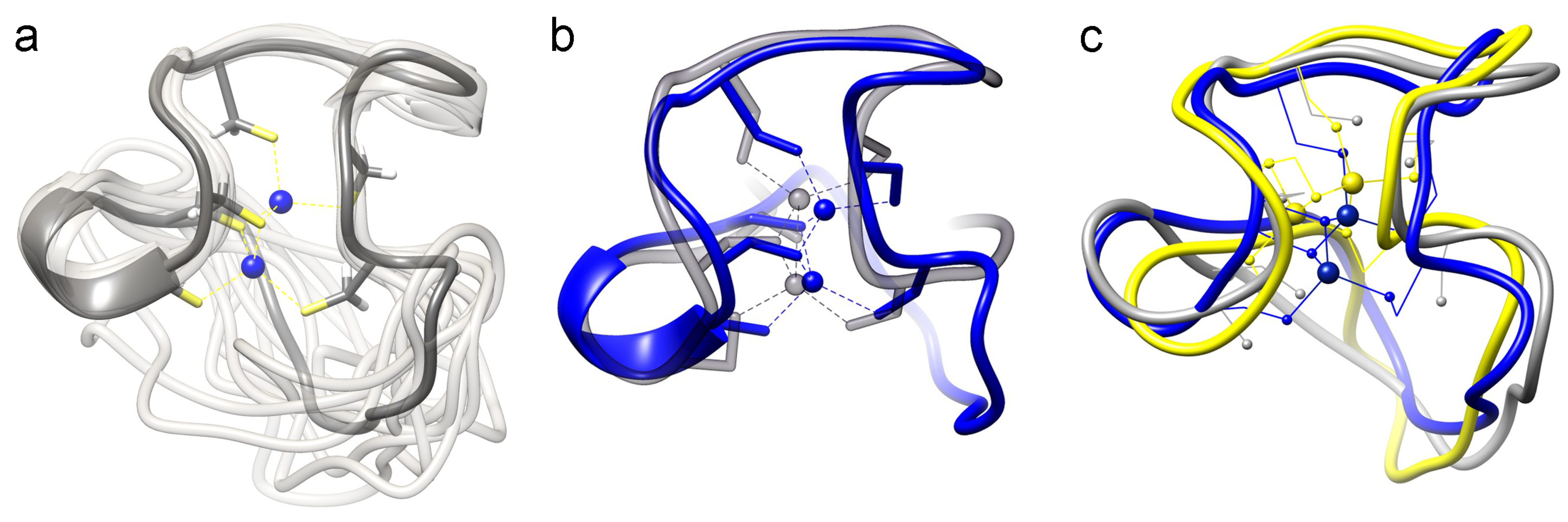
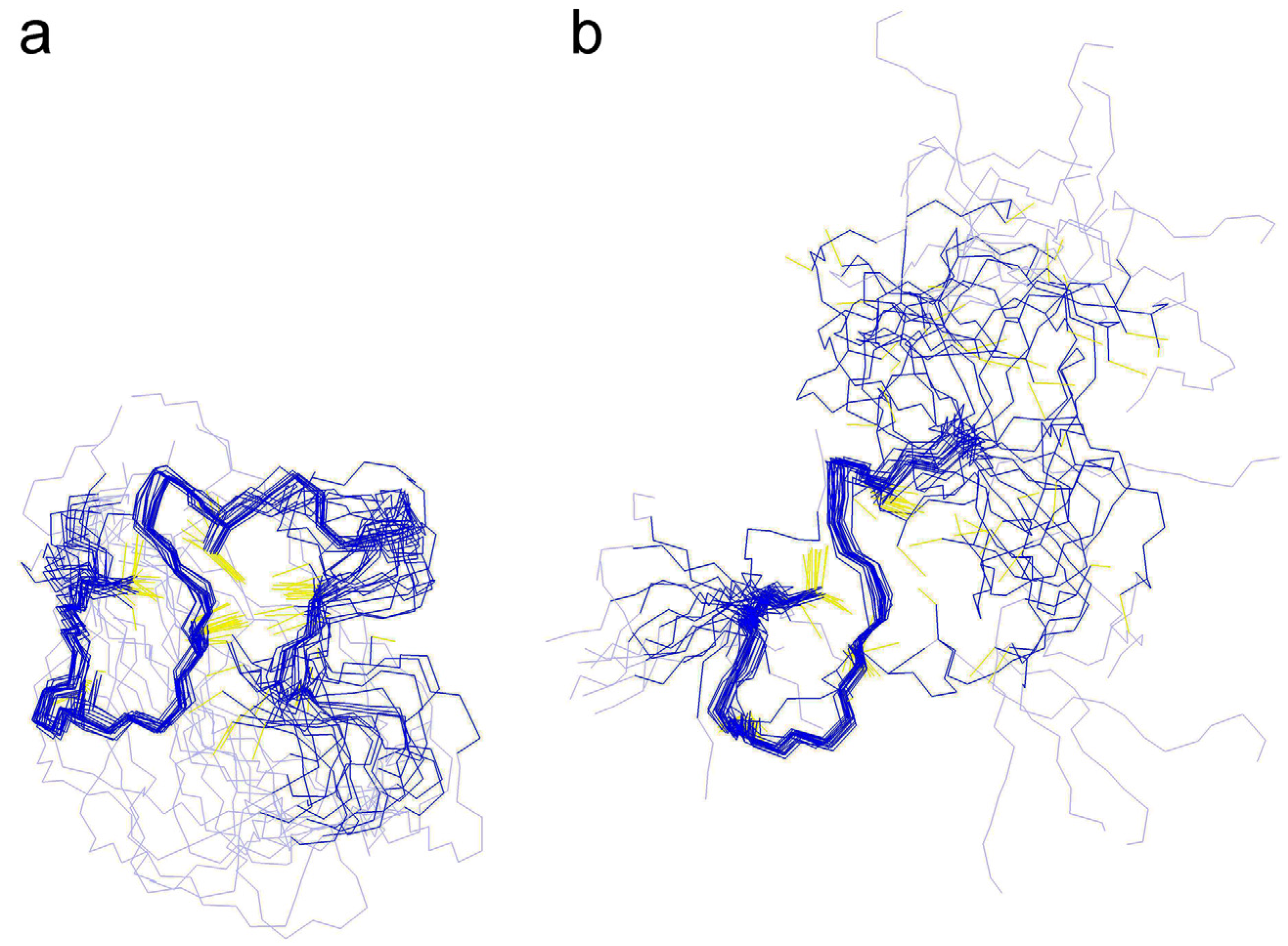
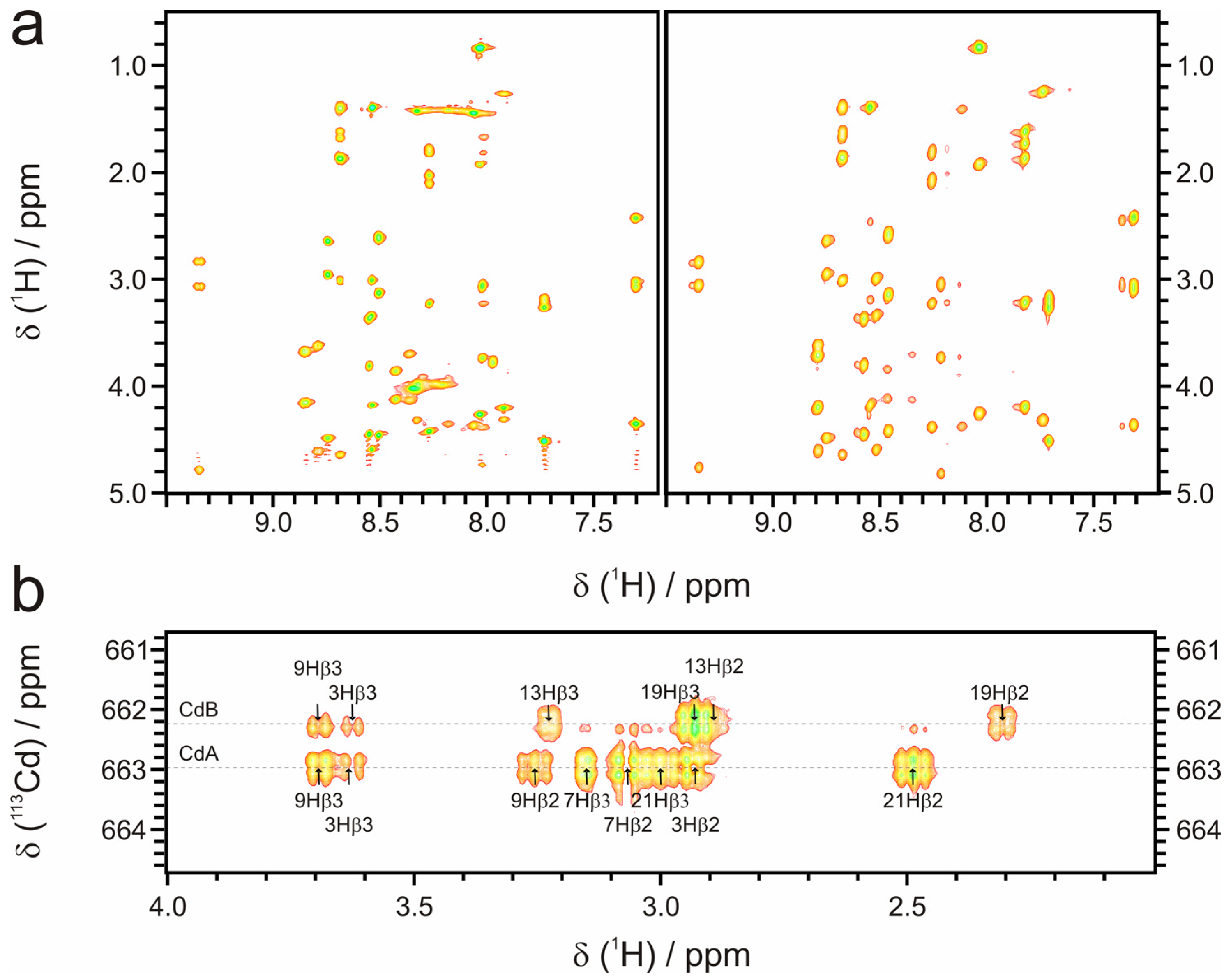
(2).2.3. Structural Analysis with NMR Spectroscopy
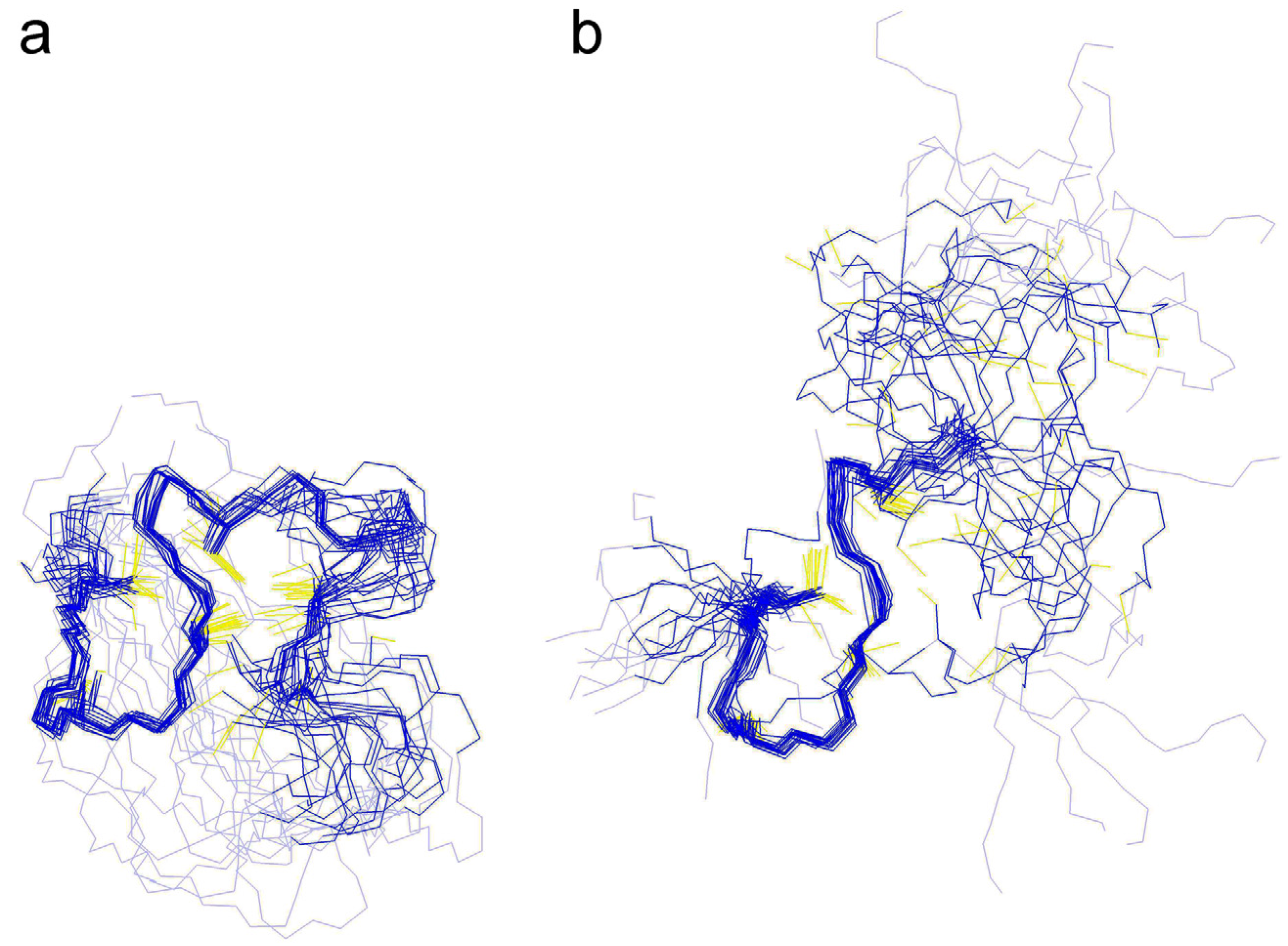
2.3.1. Conformational Constraints and Structure Calculation
| NOE restraints | ||
|---|---|---|
| Total | 201 | |
| Sequential (|i-j| < 1) | 142 | |
| Medium (1 < |i-j| <5) | 38 | |
| Long range (|i-j| <5) | 21 | |
| RMSD to average structure, Å (res. 2-23) Before water refinement | After water refinement | |
| Backbone (N,Cα,C) | 0.71 | 0.80 |
| All heavy atoms | 1.03 | 1.10 |
| Number of close contacts(within 1.6 Å for H atoms, 2.2 Å for heavy atoms) | 18 | 0 |
| RMS deviation for bond angles (°) | 0.2 | 1.4 |
| RMS deviation for bond lengths(Å) | 0.001 | 0.016 |
| Ordered residues | 3A-14A | 2A-23A |
| Procheck-NMR (residues 2-23) [30] | ||
| In most favored regions (%) | 58,3 | 73.9 |
| In additional allowed (%) | 41.7 | 26.1 |
| In generously allowed (%) | 0 | 0 |
| In disallowed regions (%) | 0 | 0 |
| Verify3D (residues 2-23) [31] | ||
| Raw score | 0.43 | 0.5 |
| Z-score | −0.48 | 0.64 |
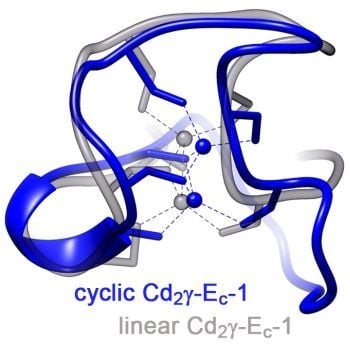
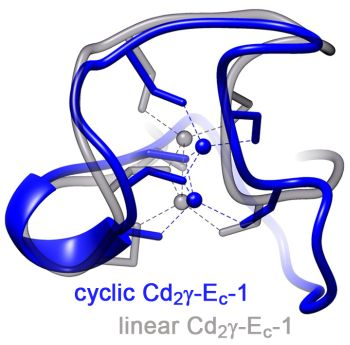
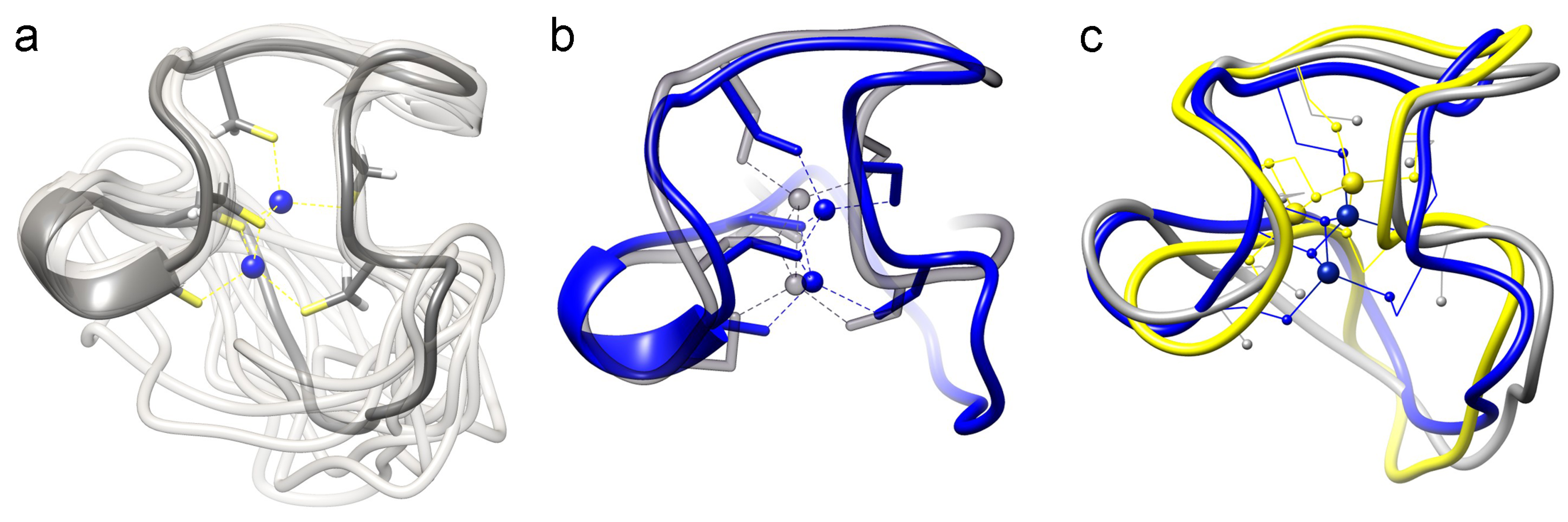
2.3.2. Comparison of the cyc-γ-Ec-1 Structure with its Linear Analog
3. Experimental
3.1. Chemicals and Solutions
3.2. Expression and Purification of cyc-γ-Ec-1
3.3. Preparation of cyc-Zn2γ−Ec-1 and cyc-Cd2γ−Ec-1
3.4. Mass Spectrometry
3.5. UV/Vis, CD, and MCD Spectroscopy
3.6. pH Titrations
3.7. NMR Spectroscopy
3.8. Structure Calculation
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Vašák, M.; Kägi, J.H.R.; Holmquist, B.; Vallee, B.L. Spectral studies of cobalt(II)- and nickel(II)-metallothionein. Biochemistry 1981, 20, 6659–6664. [Google Scholar]
- Cobbett, C.; Goldsbrough, P. Phytochelatins and metallothioneins: Roles in heavy metal detoxification and homeostasis. Annu. Rev. Plant Biol. 2002, 53, 159–182. [Google Scholar] [CrossRef]
- Freisinger, E. Plant MTs—Long neglected members of the metallothionein superfamily. Dalton Trans. 2008, 2008, 6663–6675. [Google Scholar] [CrossRef]
- Jacob, C.; Maret, W.; Vallee, B.L. Control of zinc transfer between thionein, metallothionein, and zinc proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 3489–3494. [Google Scholar] [CrossRef]
- Maret, W. Redox biochemistry of mammalian metallothioneins. J. Biol. Inorg. Chem. 2011, 16, 1079–1086. [Google Scholar] [CrossRef]
- Palmiter, R.D. The elusive function of metallothioneins. Proc. Natl. Acad. Sci. USA 1998, 95, 8428–8430. [Google Scholar] [CrossRef]
- Vašák, M.; Hasler, D.W. Metallothioneins: new functional and structural insights. Curr. Opin. Chem. Biol. 2000, 4, 177–183. [Google Scholar] [CrossRef]
- Braun, W.; Vašák, M.; Robbins, A.H.; Stout, C.D.; Wagner, G.; Kägi, J.H.R.; Wüthrich, K. Comparison of the NMR solution structure and the X-ray crystal-structure of rat metallothionein-2. Proc. Natl. Acad. Sci. USA 1992, 89, 10124–10128. [Google Scholar] [CrossRef]
- Narula, S.S.; Brouwer, M.; Hua, Y.; Armitage, I.M. Three-dimensional solution structure of Callinectes sapidus metallothionein-1 determined by homonuclear and heteronuclear magnetic resonance spectroscopy. Biochemistry 1995, 34, 620–631. [Google Scholar] [CrossRef]
- Riek, R.; Prêcheur, B.; Wang, Y.; Mackay, E.A.; Wider, G.; Güntert, P.; Liu, A.; Kägi, J.H.R.; Wüthrich, K. NMR structure of the sea urchin (Strongylocentrotus purpuratus) metallothionein MTA. J. Mol. Biol. 1999, 291, 417–428. [Google Scholar] [CrossRef]
- Schultze, P.; Wörgötter, E.; Braun, W.; Wagner, G.; Vašák, M.; Kägi, J.H.R.; Wüthrich, K. Conformation of [Cd7]-metallothionein-2 from rat liver in aqueous solution determined by nuclear magnetic resonance spectroscopy. J. Mol. Biol. 1988, 203, 251–268. [Google Scholar] [CrossRef]
- Zangger, K.; Öz, G.; Otvos, J.D.; Armitage, I.M. Three-dimensional solution structure of mouse [Cd7]-metallothionein-1 by homonuclear and heteronuclear NMR spectroscopy. Protein Sci. 1999, 8, 2630–2638. [Google Scholar]
- Loebus, J.; Peroza, E.A.; Blüthgen, N.; Fox, T.; Meyer-Klaucke, W.; Zerbe, O.; Freisinger, E. Protein and metal cluster structure of the wheat metallothionein domain γ-Ec-1. The second part of the puzzle. J. Biol. Inorg. Chem. 2011, 16, 683–694. [Google Scholar] [CrossRef]
- Binz, P.-A.; Kägi, J.H.R. Metallothionein: Molecular evolution and classification. In Metallothionein IV; Klaassen, C., Ed.; Birkhäuser Verlag: Basel, Switzerland, 1999; pp. 7–13. [Google Scholar]
- Hanley-Bowdoin, L.; Lane, B.G. A novel protein programmed by the mRNA conserved in dry wheat embryos. The principal site of cysteine incorporation during early germination. Eur. J. Biochem. 1983, 135, 9–15. [Google Scholar] [CrossRef]
- Peroza, E.A.; Freisinger, E. Metal ion binding properties of Triticum aestivum Ec-1 metallothionein: Evidence supporting two separate metal-thiolate clusters. J. Biol. Inorg. Chem. 2007, 12, 377–391. [Google Scholar] [CrossRef]
- Peroza, E.A.; Schmucki, R.; Güntert, P.; Freisinger, E.; Zerbe, O. The βE-domain of the wheat Ec-1 metallothionein: A metal-binding domain with a distinctive structure. J. Mol. Biol. 2009, 387, 207–218. [Google Scholar] [CrossRef]
- Marmorstein, R.; Carey, M.; Ptashne, M.; Harrison, S.C. DNA Recognition by Gal4—Structure of a Protein DNA Complex. Nature 1992, 356, 408–414. [Google Scholar] [CrossRef]
- Craik, D.J.; Daly, N.L.; Mulvenna, J.; Plan, M.R.; Trabi, M. Discovery, structure and biological activities of the cyclotides. Curr. Protein Pept. Sci. 2004, 5, 297–315. [Google Scholar] [CrossRef]
- Rosengren, K.J.; Daly, N.L.; Plan, M.R.; Waine, C.; Craik, D.J. Twists, knots, and rings in proteins—Structural definition of the cyclotide framework. J. Biol. Chem. 2003, 278, 8606–8616. [Google Scholar] [CrossRef]
- Saether, O.; Craik, D.J.; Campbell, I.D.; Sletten, K.; Juul, J.; Norman, D.G. Elucidation of the primary and three-dimensional structure of the uterotonic polypeptide kalata B1. Biochemistry 1995, 34, 4147–4158. [Google Scholar] [CrossRef]
- Iwai, H.; Plückthun, A. Circular beta-lactamase: Stability enhancement by cyclizing the backbone. FEBS Lett. 1999, 459, 166–172. [Google Scholar] [CrossRef]
- Rizo, J.; Gierasch, L.M. Constrained peptides: Models of bioactive peptides and protein substructures. Ann. Rev. Biochem. 1992, 61, 387–418. [Google Scholar] [CrossRef]
- Tarasava, K.; Freisinger, E. Institute of Inorganic Chemistry, University of Zurich, Zurich, Switzerland. Unpublished work, 2013. [Google Scholar]
- Pedersen, A.O.; Jacobsen, J. Reactivity of the thiol-group in human and bovine albumin at pH 3–9, as measured by exchange with 2,2'-dithiodipyridine. Eur. J. Biochem. 1980, 106, 291–295. [Google Scholar] [CrossRef]
- Freisinger, E.; Vašák, M. Cadmium in metallothioneins. Met. Ions Life Sci. 2013, 11, 339–371. [Google Scholar] [CrossRef]
- Hill, A.V. The possible effects of the aggregation of the molecules of haemoglobin on its oxygen dissociation curve. J. Physiol. (London) 1910, 40, 4–7. [Google Scholar]
- Parella, T. NMRGuide3.5; BRUKER Biospin: Billerica, MA, USA, 1998. [Google Scholar]
- Linge, J.P.; Williams, M.A.; Spronk, C.A.E.M.; Bonvin, A.M.J.J.; Nilges, M. Refinement of protein structures in explicit solvent. Proteins 2003, 50, 496–506. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Macarthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck: A program to check the stereochemical quality of protein structures. J. Appl. Chrystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [CrossRef]
- Öz, G.L.; Pountney, D.L.; Armitage, I.M. NMR spectroscopic studies of I = 1/2 metal ions in biological systems. Biochem. Cell Biol. 1998, 76, 223–234. [Google Scholar]
- Freisinger, E. Spectroscopic characterization of a fruit-specific metallothionein: M acuminata MT3. Inorg. Chim. Acta 2007, 360, 369–380. [Google Scholar] [CrossRef]
- Markley, J.L.; Bax, A.; Arata, Y.; Hilbers, C.W.; Kaptein, R.; Sykes, B.D.; Wright, P.E.; Wüthrich, K. Recommendations for the presentation of NMR structures of proteins and nucleic acids. J. Mol. Biol. 1998, 280, 933–952. [Google Scholar] [CrossRef]
- Harris, R.K.; Mann, B.E. NMR and the Periodic Table; Academic Press: New York, NY, USA, 1978; pp. 262–265. [Google Scholar]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; Wiley-Interscience: New York, NY, USA, 1986; pp. 1–292. [Google Scholar]
- Keller, R.L.J. Computer Aided Resonance Assignment Tutorial; CANTINA Verlag: Goldau, Switzerland, 2004; pp. 1–81. [Google Scholar]
- Bartels, C.; Xia, T.H.; Billeter, M.; Güntert, P.; Wüthrich, K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 1995, 6, 1–10. [Google Scholar]
- Goddard, T.D.; Kneller, D.G. SPARKY 3; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Güntert, P.; Mumenthaler, C.; Wüthrich, K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997, 273, 283–298. [Google Scholar] [CrossRef]
- Herrmann, T.; Güntert, P.; Wüthrich, K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 2002, 319, 209–227. [Google Scholar] [CrossRef]
- Güntert, P. Automated NMR protein structure calculation. Progr. Nucl. Magn. Reson. Spectrosc. 2003, 43, 105–125. [Google Scholar] [CrossRef]
- Schwieters, C.D.; Kuszewski, J.J.; Tjandra, N.; Clore, G.M. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003, 160, 65–73. [Google Scholar] [CrossRef]
- Schwieters, C.D.; Kuszewski, J.J.; Clore, G.M. Using Xplor-NIH for NMR molecular structure determination. Progr. Nucl. Magn. Reson. Spectrosc. 2006, 48, 47–62. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Koradi, R.; Billeter, M.; Wüthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics 1996, 14, 51–55. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tarasava, K.; Johannsen, S.; Freisinger, E. Solution Structure of the Circular γ-Domain Analog from the Wheat Metallothionein Ec-1. Molecules 2013, 18, 14414-14429. https://doi.org/10.3390/molecules181114414
Tarasava K, Johannsen S, Freisinger E. Solution Structure of the Circular γ-Domain Analog from the Wheat Metallothionein Ec-1. Molecules. 2013; 18(11):14414-14429. https://doi.org/10.3390/molecules181114414
Chicago/Turabian StyleTarasava, Katsiaryna, Silke Johannsen, and Eva Freisinger. 2013. "Solution Structure of the Circular γ-Domain Analog from the Wheat Metallothionein Ec-1" Molecules 18, no. 11: 14414-14429. https://doi.org/10.3390/molecules181114414
APA StyleTarasava, K., Johannsen, S., & Freisinger, E. (2013). Solution Structure of the Circular γ-Domain Analog from the Wheat Metallothionein Ec-1. Molecules, 18(11), 14414-14429. https://doi.org/10.3390/molecules181114414