Three New Germacrane-Type Sesquiterpenes with NGF-Potentiating Activity from Valeriana officinalis var. latiofolia

Abstract
:1. Introduction
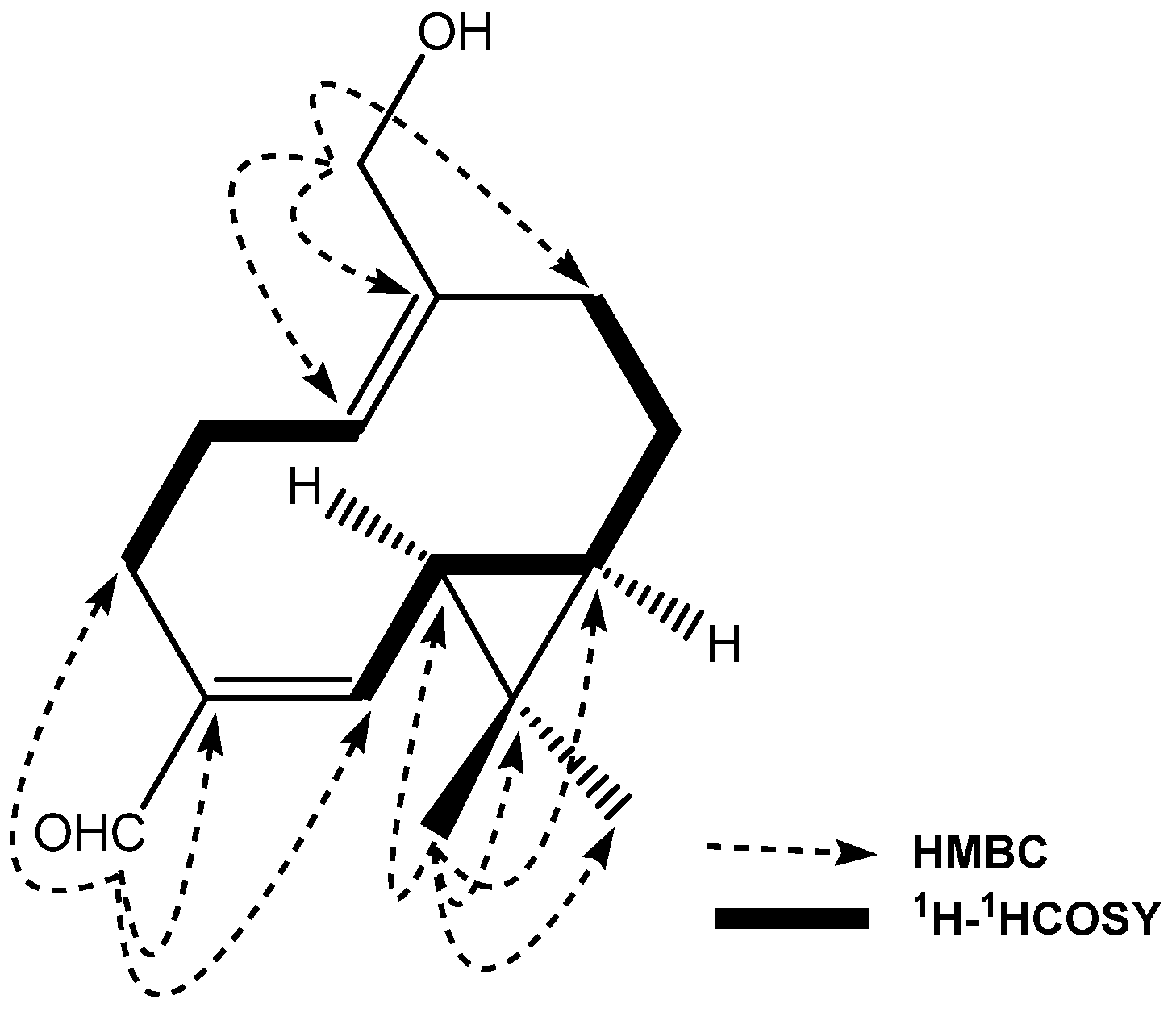
2. Results and Discussion

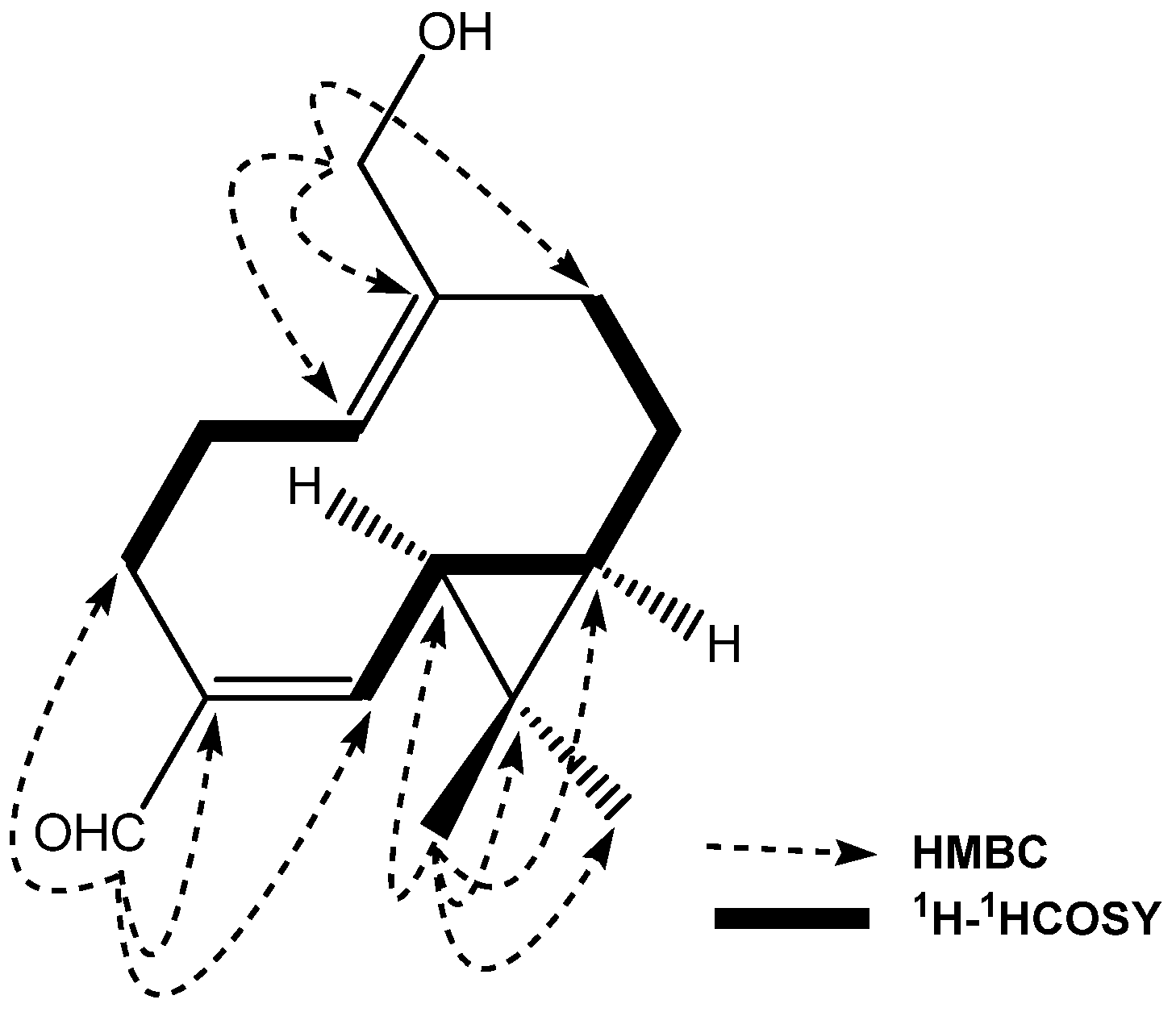{kind=link}
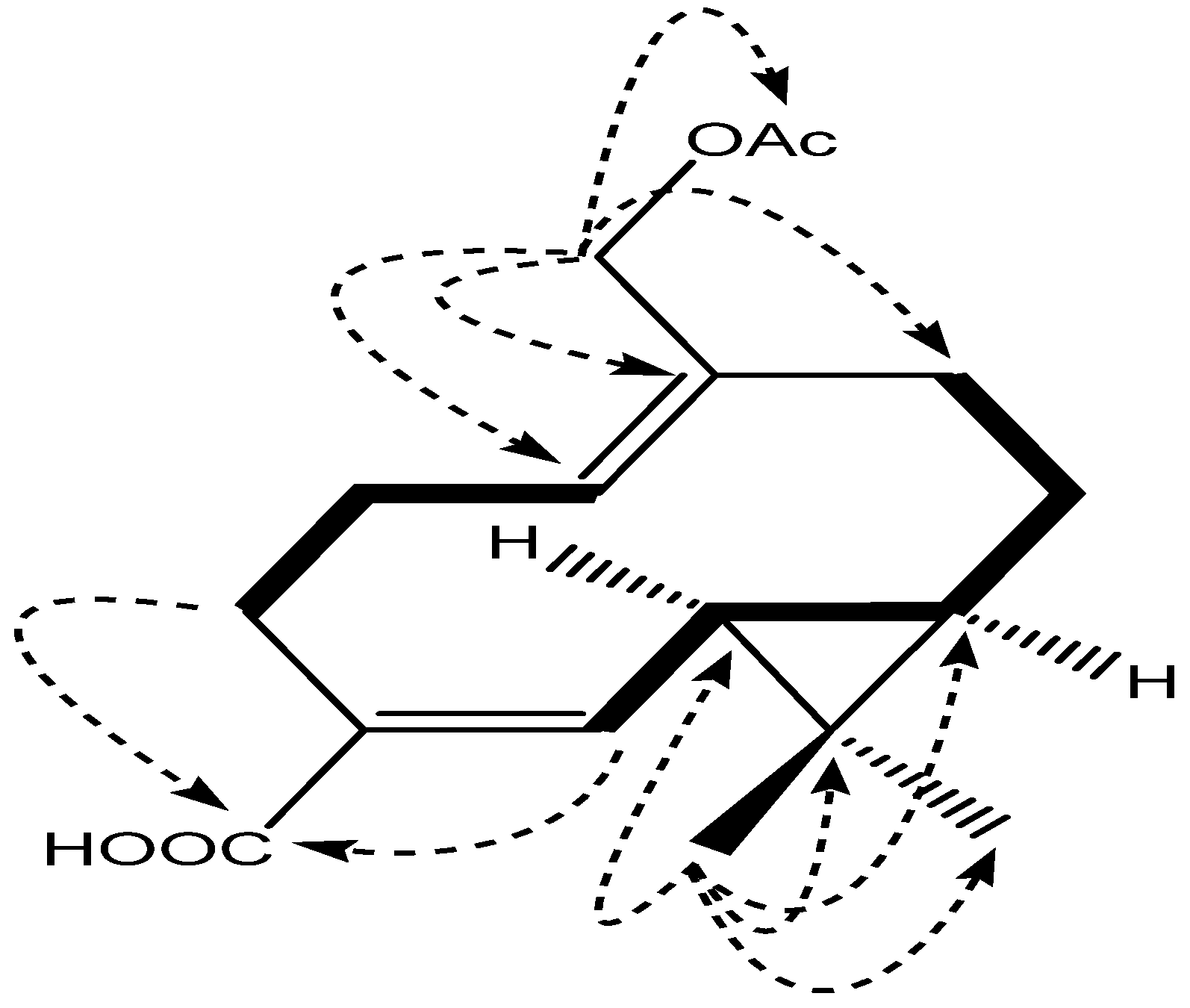{kind=link}
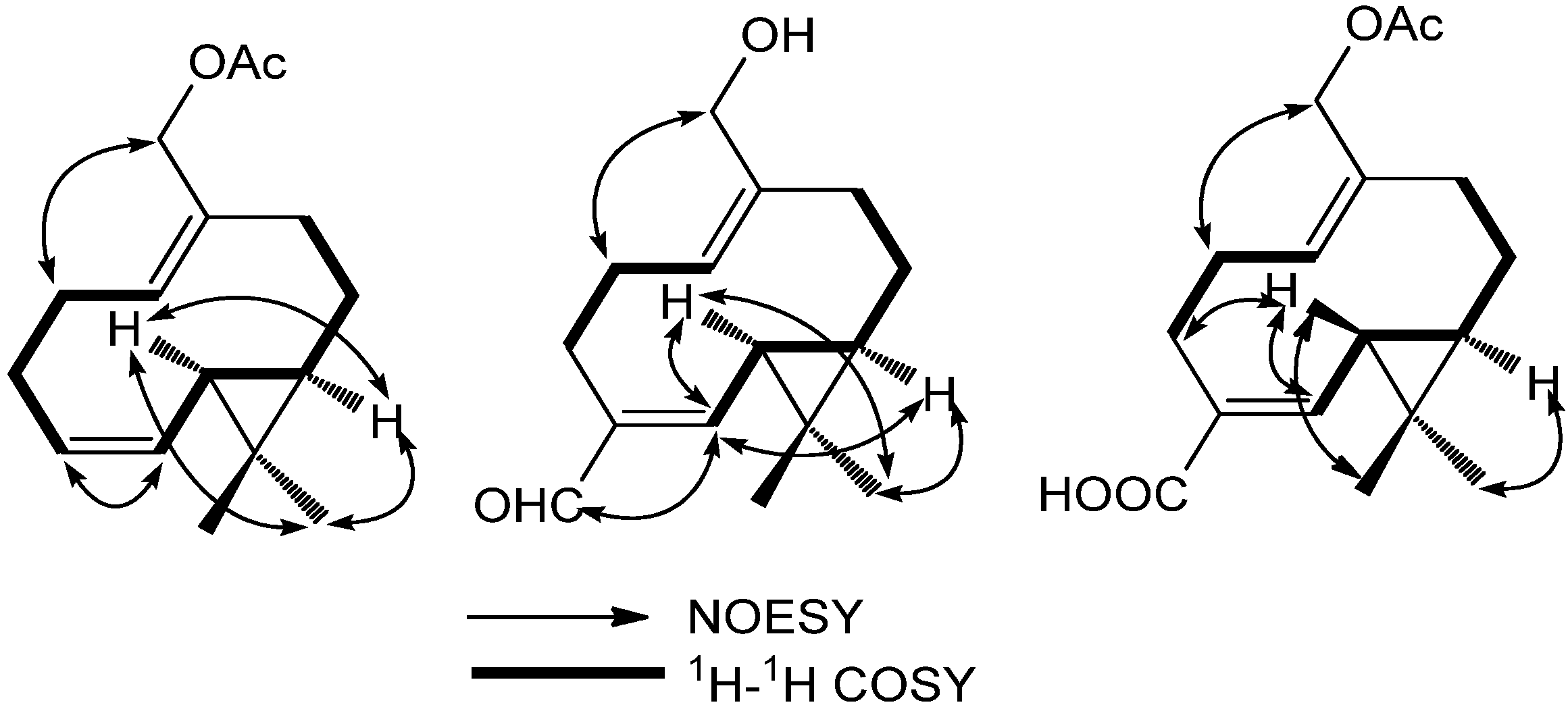{kind=link}
{kind=link}
{kind=link}
| NO. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | |
| 1 | 128.8 | 5.27 dd (5.4, 10.2) | 128.2 | 5.22 dd (5.4, 11.4) | 130.7 | 5.33 dd (5.4, 11.4) |
| 2a | 28.2 | 2.23 m H-α | 28.3 | 2.22 m (H-α) | 29.2 | 2.38 m (H-β) |
| 2b | 2.11 m H-β | 2.15 m (H-β) | 2.26 m (H-α) | |||
| 3a | 26.8 | 2.14 m H-β | 24.0 | 2.71 dt (12.0, 4.2, H-α) | 27.1 | 2.71 dt (12.6, 4.0, H-α) |
| 3b | 2.08 m H-α | 2.05 td (12.6, 4.2, H-β) | 2.17 td (12.6, 4.0, H-β) | |||
| 4 | 128.3 | 5.34 ddd (9.0, 9.0, 3.2) | 143.9 | 132.1 | ||
| 5 | 129.6 | 5.21 dd (9.0, 9.0) | 158.9 | 6.48 d (9.0) | 144.8 | 6.72 d (9.6) |
| 6 | 26.4 | 1.14 t (9.0, H-α) | 31.2 | 1.52 t (13.2, 9.0, H-α) | 30.0 | 1.27 dd (9.6, 4.0, H-α) |
| 7 | 31.9 | 0.47 m (H-α) | 38.8 | 1.02 m (H-α) | 36.7 | 0.82 m (H-α) |
| 8a | 23.0 | 1.73 m (H-β) | 24.6 | 1.86 m (H-β) | 24.7 | 1.81 m (H-β) |
| 8b | 0.76 m (H-α) | 0.90 m (H-α) | 0.89 m (H-α) | |||
| 9a | 35.3 | 2.39 m (H-β) | 35.5 | 2.60 m (H-β) | 36.2 | 2.36 m (H-β) |
| 9b | 1.87 t (12.6, α) | 1.88 m (H-α) | 2.25 m (H-α) | |||
| 10 | 133.1 | 139.1 | 134.3 | |||
| 11 | 17.2 | 22.4 | 21.5 | |||
| 12 | 28.9 | 1.04 s | 28.7 | 1.16 s | 28.9 | 1.12 s |
| 13 | 15.6 | 0.98 s | 16.0 | 1.20 s | 16.1 | 1.14 s |
| 14a | 61.7 | 4.42 d (12.0) | 196.4 | 9.20 s | 172.9 | |
| 14b | 4.21 d (12.0) | |||||
| 15 | 171.3 | 59.1 | 3.69 d (12.0) | 62.8 | 4.32 d (12.0) | |
| 3.43 d (12.0) | 4.15 d (12.0) | |||||
| 16 | 21.1 | 2.01 s | 172.0 | |||
| 17 | 20.8 | 1.97 s | ||||
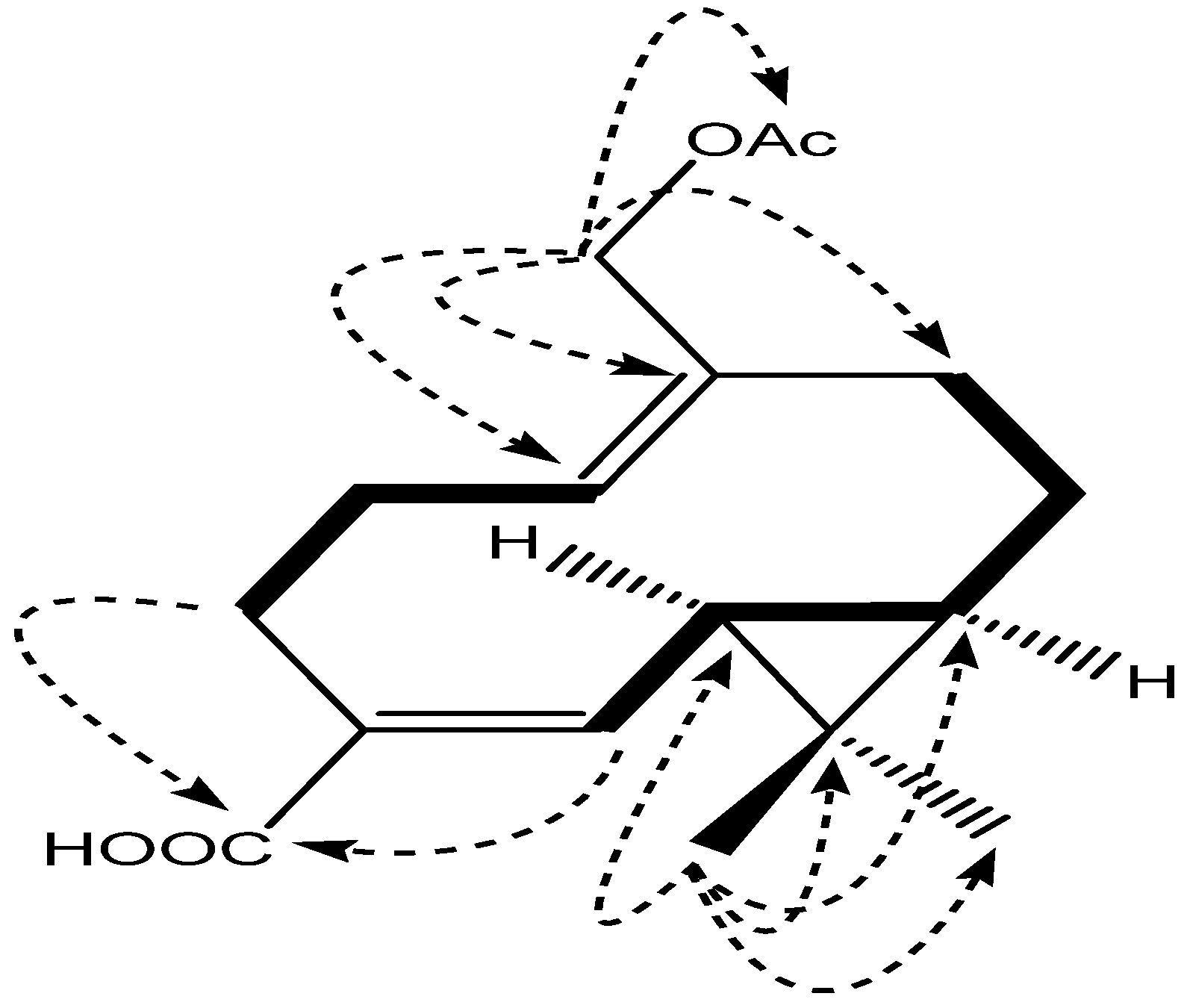
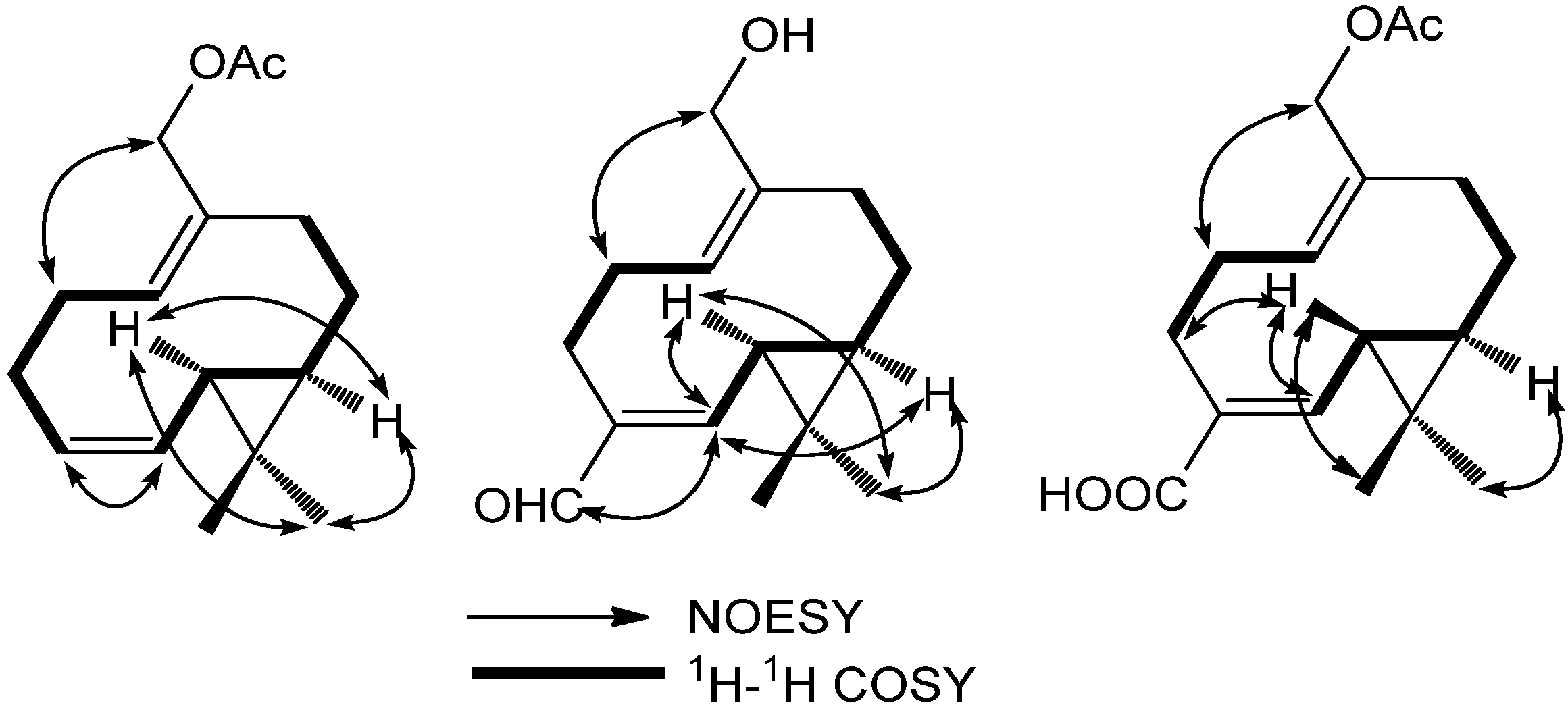
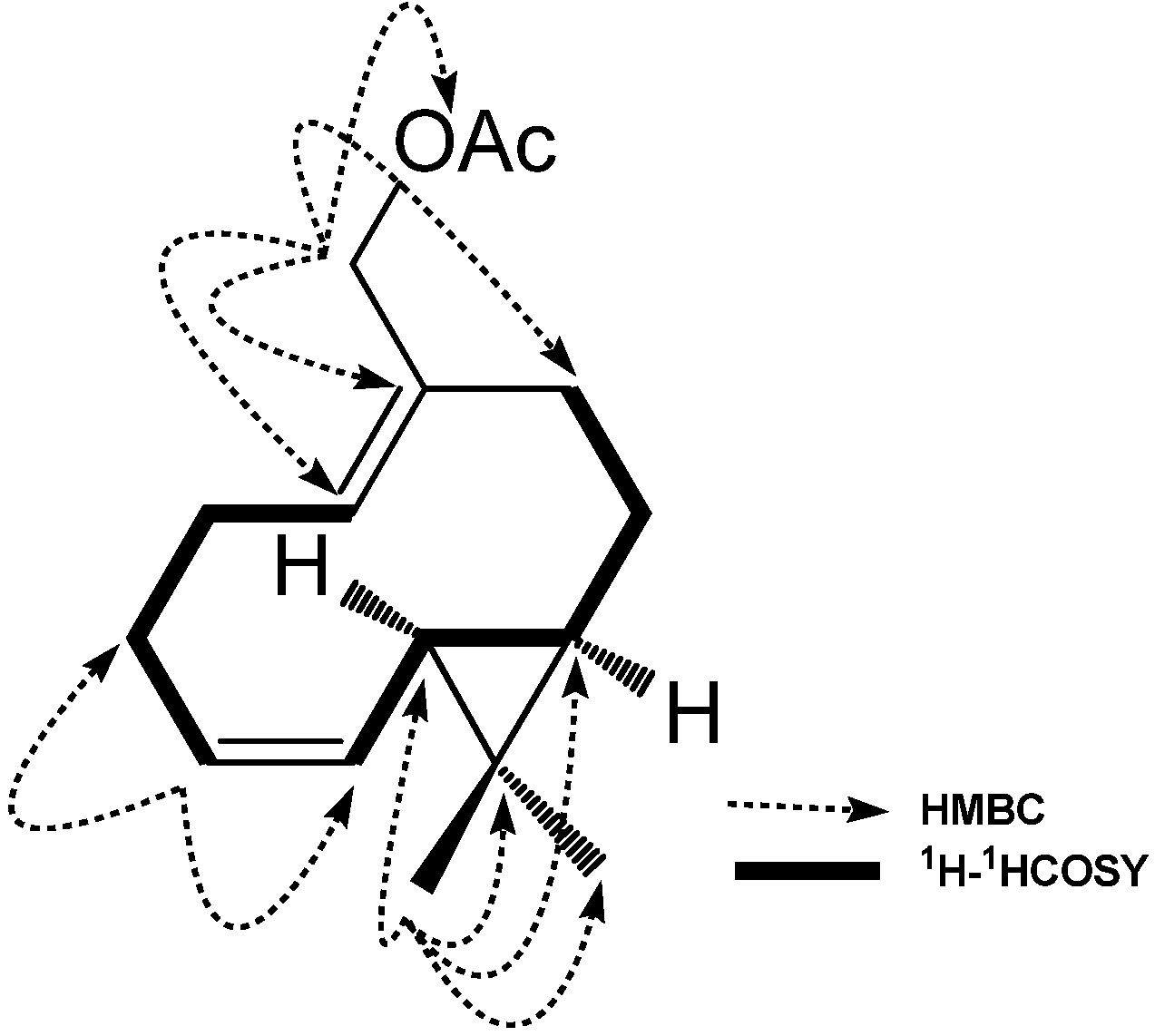
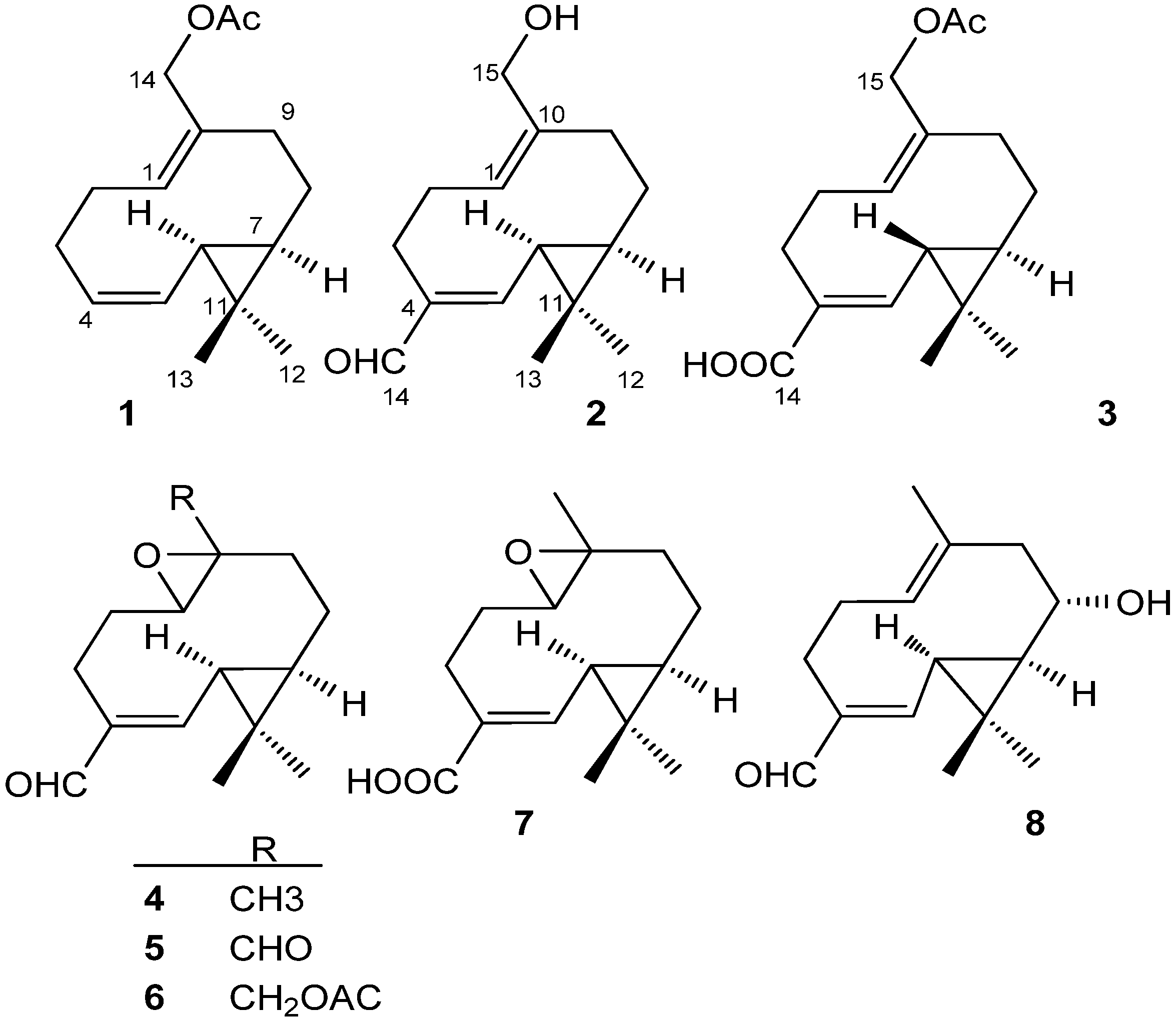
| Compound | NGF(ng/mL) | Cell viability (%) | ||
|---|---|---|---|---|
| 10 µmol | 30 µmol | 100 µmol | ||
| 1 | 2 | 24.85 ± 0.98 | 33.97 ± 1.77 b | 43.61 ± 2.11 c |
| 2 | 2 | 24.42 ± 1.12 | 35.34 ± 1.48 b | 44.30 ± 1.85 c |
| 3 | 2 | 26.48 ± 0.89 a | 37.51 ± 1.66 b | 50.15 ± 2.23 c |
| 4 | 2 | 23.50 ± 1.26 | 29.33 ± 0.88 b | 33.87 ± 1.63 c |
| 5 | 2 | 24.37 ± 1.01 | 30.12 ± 1.97 b | 40.79 ± 1.17 c |
| 6 | 2 | 24.74 ± 1.47 | 34.84 ± 2.35 b | 44.15 ± 2.19 c |
| 7 | 2 | 24.45 ± 1.01 | 33.96 ± 1.13 b | 46.69 ± 2.14 c |
| 8 | 2 | 24.26 ± 0.73 | 33.79 ± 1.17 b | 49.25 ± 1.25 c |
| 2 | 23.08% ± 1.28 | |||
| 50 | 100% | |||
| 0 | 3.12% ± 0.88 | |||
3. Experimental
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Compound Characterization
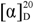 +20.0 (c 0.8, CHCl3); UV (CHCl3) λmax 237 nm; IR (film) νmax 3445, 3171, 2960, 2924, 2852, 1730, 1627, 1261, 1095, 1024 cm−1; 1H-NMR (CHCl3, 600 MHz) data, see Table 1; 13C-NMR (CHCl3, 150 MHz) data, see Table 1; HR-ESI-MS m/z 271.1678 [M+Na]+ (calcd. for C16H24O2Na, 271.1669). +53.9 (c 0.1, MeOH); UV (CHCl3) λmax 264 nm; IR (film) νmax 3382, 2933, 2864, 1618, 1298, 1190, 1072, 921, 793 cm−1; 1H-NMR (CHCl3, 600 MHz) data, see Table 1; 13C-NMR (CHCl3, 150 MHz) data, see Table 1; HR-ESI-MS m/z 235.1696 [M+H]+ (calcd. for C15H23O2, 235.1693). +8.5 (c 0.47, MeOH); UV (CHCl3) λmax 240 and 271 nm; IR (film) νmax 3384, 3245, 2931, 2862, 1238, 1118, 1027, 862, 768 cm−1; 1H-NMR (CHCl3, 600 MHz) data, see Table 1; 13C-NMR (CHCl3, 150 MHz) data, see Table 1; HR-ESI-MS m/z 293.1744 [M+H]+ (calcd. for C17H25O2, 293.1747).
+20.0 (c 0.8, CHCl3); UV (CHCl3) λmax 237 nm; IR (film) νmax 3445, 3171, 2960, 2924, 2852, 1730, 1627, 1261, 1095, 1024 cm−1; 1H-NMR (CHCl3, 600 MHz) data, see Table 1; 13C-NMR (CHCl3, 150 MHz) data, see Table 1; HR-ESI-MS m/z 271.1678 [M+Na]+ (calcd. for C16H24O2Na, 271.1669). +53.9 (c 0.1, MeOH); UV (CHCl3) λmax 264 nm; IR (film) νmax 3382, 2933, 2864, 1618, 1298, 1190, 1072, 921, 793 cm−1; 1H-NMR (CHCl3, 600 MHz) data, see Table 1; 13C-NMR (CHCl3, 150 MHz) data, see Table 1; HR-ESI-MS m/z 235.1696 [M+H]+ (calcd. for C15H23O2, 235.1693). +8.5 (c 0.47, MeOH); UV (CHCl3) λmax 240 and 271 nm; IR (film) νmax 3384, 3245, 2931, 2862, 1238, 1118, 1027, 862, 768 cm−1; 1H-NMR (CHCl3, 600 MHz) data, see Table 1; 13C-NMR (CHCl3, 150 MHz) data, see Table 1; HR-ESI-MS m/z 293.1744 [M+H]+ (calcd. for C17H25O2, 293.1747).3.5. Activity Screening in Vitro
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Chen, H.B.; Cheng, J.R. Taxonomic revision of the relative species of Valeriana officinalis Linn. from China. Bull. Bot. Res. 1991, 3, 29–40. [Google Scholar]
- Chen, H.B.; Cheng, J.R. Studies on the Medicinal Plants of Valerianaceae in China. China J. Chin. Mater. Med. 1994, 2, 67–70. [Google Scholar]
- Houghton, P.J. The biological activity of valerian and related plants. J. Ethnopharmacol. 1988, 22, 121–142. [Google Scholar] [CrossRef]
- Houghton, P.J. The scientific basis for the reputed activity of Valerian. J. Pharm. Pharmacol. 1999, 51, 505–512. [Google Scholar] [CrossRef]
- Valeriana officinalis (monograph). Altern. Med. Rev. 2004, 9, pp. 438–441. Available online: http://www.thorne.com/altmedrev/.fulltext/9/4/438.pdf (accessed on 14 November 2003).
- Thies, P.W. Zur Konstitution der isovalerian saureester Valepotriat, Acetoxyvalepotriat und Dihydrovalepotriat. Tetrahedron Lett. 1966, 11, 1163–1170. [Google Scholar] [CrossRef]
- Hendriks, H.; Geertsma, H.J.; Malingre, T.M. The occurrence of valeranone and crytofauronol in the essential oil of Valeriana officinalis cinalis L. collected in the northern part of the Netherlands. Pharm. Weekbl. 1981, 116, 1316–1320. [Google Scholar] [CrossRef]
- Leathwood, P.D.; Chauffard, F.; Heck, E.; Munoz-Box, R. Aqueous extract of valerian root (Valeriana officinalis L.) improves sleep quality in man. Pharmacol. Biochem. Behav. 1982, 1, 65–71. [Google Scholar]
- Sakamoto, T.; Mltani, Y.; Nakajima, K. Psychotropic effects of Japanese Valerian Root extract. Chem. Pharm. Bull. 1992, 3, 758–761. [Google Scholar] [CrossRef]
- Santos, M.S.; Ferreira, F.; Faro, C.; Pires, E.; Carvalho, A.P.; Cunha, A.P.; Macedo, T. The amount of GABA Present in aqueous extracts of valerian is sufficient to account for [3H] GABA release in synaptosomes. Planta Med. 1994, 5, 475–476. [Google Scholar]
- Zhang, Z.X.; Yao, X.S. The Advance of Chemical Study on the Medicinal Plant Valeriana officinalis L. Chin. J. Med. Chem. 2000, 3, 226–229. [Google Scholar]
- Liu, X.G.; Gao, P.Y.; Wang, G.S.; Song, S.J.; Li, L.Z.; Li, X.; Yao, X.S.; Zhang, Z.X. In vivo antidepressant activity of sesquiterpenes from the roots of Valeriana fauriei Briq. Fitoterapia 2012, 3, 599–603. [Google Scholar]
- Wang, Q.H.; Wang, C.F.; Zuo, Y.M.; Wang, Z.B.; Yang, B.Y.; Kuang, H.X. Compounds from the roots and rhizomes of Valeriana amurensis protect against neurotoxicity in PC12 cells. Molecules 2012, 12, 15013–15021. [Google Scholar]
- Letchamo, W.; Ward, W.; Heard, B.; Heard, D. Essential oil of Valeriana officinalis L. cultivars and their antimicrobial activity as influenced by harvesting time under commercial organic cultivation. J. Agric. Food Chem. 2004, 12, 3915–3919. [Google Scholar] [CrossRef]
- Huang, B.K.; Qin, L.P.; Chu, Q.C.; Zhang, Q.Y.; Gao, L.H.; Zheng, H.C. Comparison of headspace spme with hydrodistillation and sfe for analysis of the volatile components of the roots of Valeriana officinalis var. latifolia. Chromatographia 2009, 69, 489–496. [Google Scholar] [CrossRef]
- Zhou, Y.; Fang, Y.; Gong, Z.F.; Duan, X.Y.; Liu, Y.W. Two New Terpenoids from Valeriana officinalis. Chin. J. Nat. Med. 2009, 72, 270–273. [Google Scholar]
- Wang, P.C.; Hu, J.M.; Ran, X.H.; Chen, Z.Q.; Jiang, H.Z.; Liu, Y.Q.; Zhou, J.; Zhao, Y.X. Iridoids and sesquiterpenoids from the roots of Valeriana officinalis. J. Nat. Prod. 2009, 72, 1682–1685. [Google Scholar] [CrossRef]
- Wang, P.C.; Ran, X.H.; Chen, R.; Luo, H.R.; Liu, Y.Q.; Zhou, J.; Zhao, Y.X. Germacranetype sesquiterpenoids from the roots of Valeriana officinalis var. latifolia. J. Nat. Prod. 2010, 73, 1563–1567. [Google Scholar] [CrossRef]
- Wang, P.C.; Ran, X.H.; Luo, H.R.; Hu, J.M.; Chen, R.; Ma, Q.Y.; Dai, H.F.; Liu, Y.Q.; Xie, M.J.; Zhou, J. Volvalerelactones A and B, two new sesquiterpenoid lactones with an unprecedented skeleton from Valeriana officinalis var. latifolia. Org. Lett. 2011, 12, 3036–3039. [Google Scholar]
- Han, Z.Z.; Yan, Z.H.; Liu, Q.X.; Hu, X.Q.; Ye, J.; Li, H.L.; Zhang, W.D. Acylated iridoids from the roots of Valeriana officinalis var. latifolia. Planta Med. 2012, 15, 1645–1650. [Google Scholar]
- Muller, N.F.; Dessing, R.P. European Society of Chemical Pharmacy. In Europaea Drug Index, 2nd ed.; Elsevier: New York, NY, USA, 1992; p. 1260. [Google Scholar]
- Xu, L. United States Pharmacopeia/National Formulary(24): Valerian. World Phytomed. World Notes Plant Med. 2002, 3, 126–127. [Google Scholar]
- Blumenthal, M. Herbs continue slide in mainstream market: Sales down 14 percent. Herbal. Gram. 2003, 58, 71. [Google Scholar]
- Flora of China Editorial Committee of Chinese Academy of Sciences. Flora Repubulicae Popularis Sinicae; Science Press: Beijing, China, 1986; Volume 1, pp. 32–34. [Google Scholar]
- Wang, Y.W.; Chen, Q. The survey of medicinal efficacy of the Valeriana officinalis Linn. var. latifolia miq. of China. Asia Pac. Tradit. Med. 2005, 3, 69–71. [Google Scholar]
- Wu, T.S.; Chan, Y.Y.; Leu, Y.L. Sesquiterpenes from the root and stem of Aristolochia cucurbitafolia. J. Nat. Prod. 1998, 4, 511–544. [Google Scholar]
- Wu, T.S.; Leu, Y.L.; Chan, Y.Y. Constituents from the stem and root of Aristolochia kaempferi. Biol. Pharm. Bull. 2000, 10, 1216–1219. [Google Scholar]
- Guo, Y.Q.; Xu, J.; Li, Y.S.; Yamakuni, T.; Ohizumi, Y. Three-membered ring sesquiterpenoids with NGF-potentiating activity from the roots of Valeriana fauriei. Planta Med. 2006, 4, 373–375. [Google Scholar]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–8 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, H.-W.; Chen, L.; Li, B.; Yin, H.-L.; Tian, Y.; Wang, Q.; Xiao, Y.-H.; Dong, J.-X. Three New Germacrane-Type Sesquiterpenes with NGF-Potentiating Activity from Valeriana officinalis var. latiofolia. Molecules 2013, 18, 14138-14147. https://doi.org/10.3390/molecules181114138
Chen H-W, Chen L, Li B, Yin H-L, Tian Y, Wang Q, Xiao Y-H, Dong J-X. Three New Germacrane-Type Sesquiterpenes with NGF-Potentiating Activity from Valeriana officinalis var. latiofolia. Molecules. 2013; 18(11):14138-14147. https://doi.org/10.3390/molecules181114138
Chicago/Turabian StyleChen, Heng-Wen, Li Chen, Bin Li, Hai-Long Yin, Ying Tian, Qiong Wang, Yan-Hua Xiao, and Jun-Xing Dong. 2013. "Three New Germacrane-Type Sesquiterpenes with NGF-Potentiating Activity from Valeriana officinalis var. latiofolia" Molecules 18, no. 11: 14138-14147. https://doi.org/10.3390/molecules181114138
APA StyleChen, H.-W., Chen, L., Li, B., Yin, H.-L., Tian, Y., Wang, Q., Xiao, Y.-H., & Dong, J.-X. (2013). Three New Germacrane-Type Sesquiterpenes with NGF-Potentiating Activity from Valeriana officinalis var. latiofolia. Molecules, 18(11), 14138-14147. https://doi.org/10.3390/molecules181114138