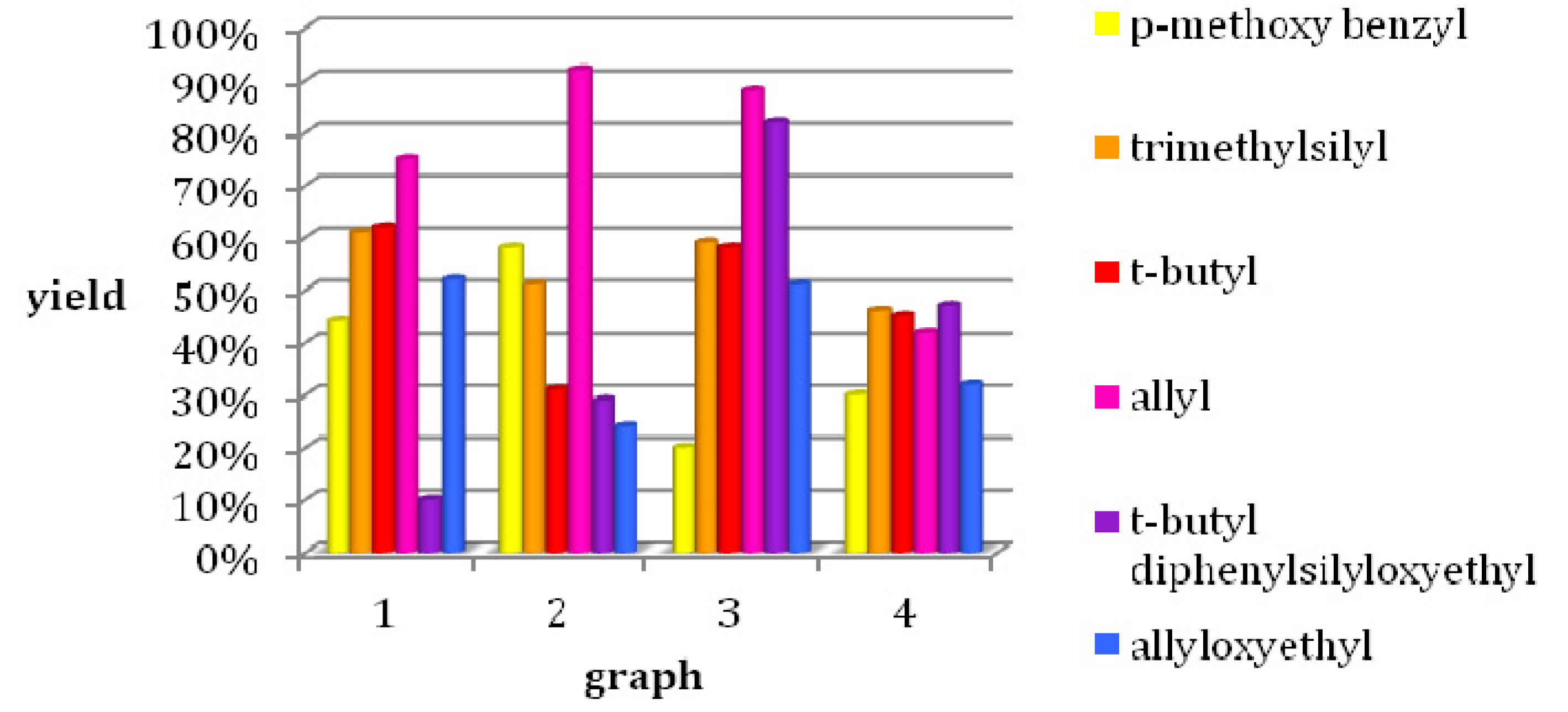
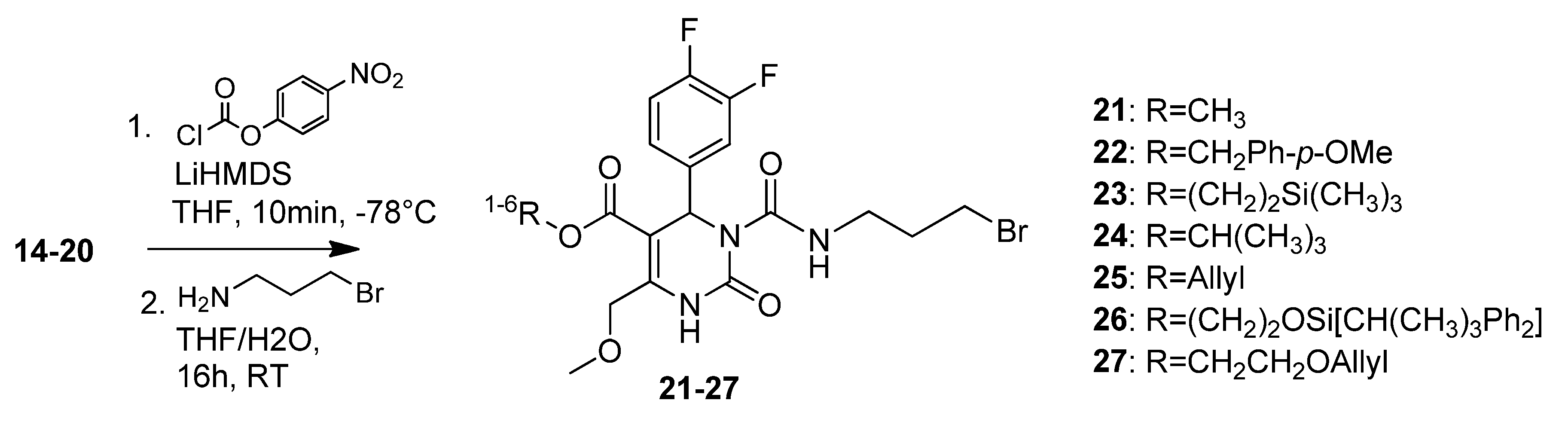
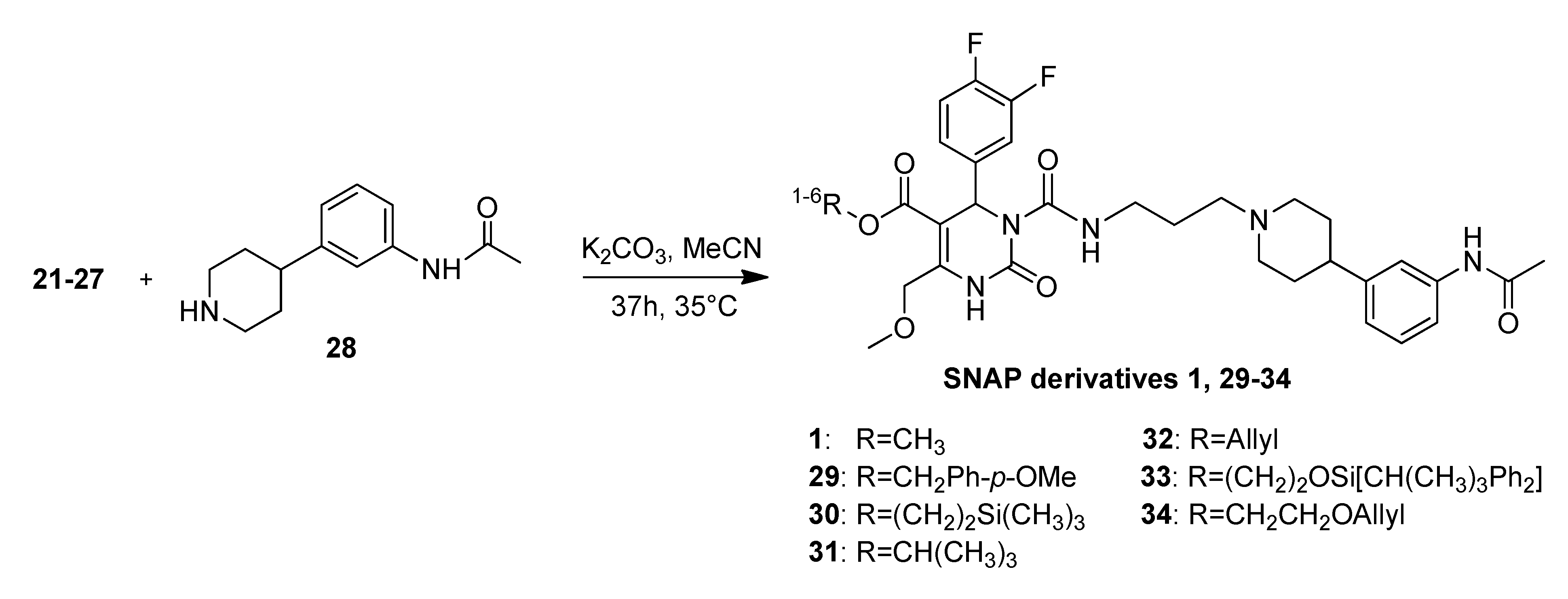
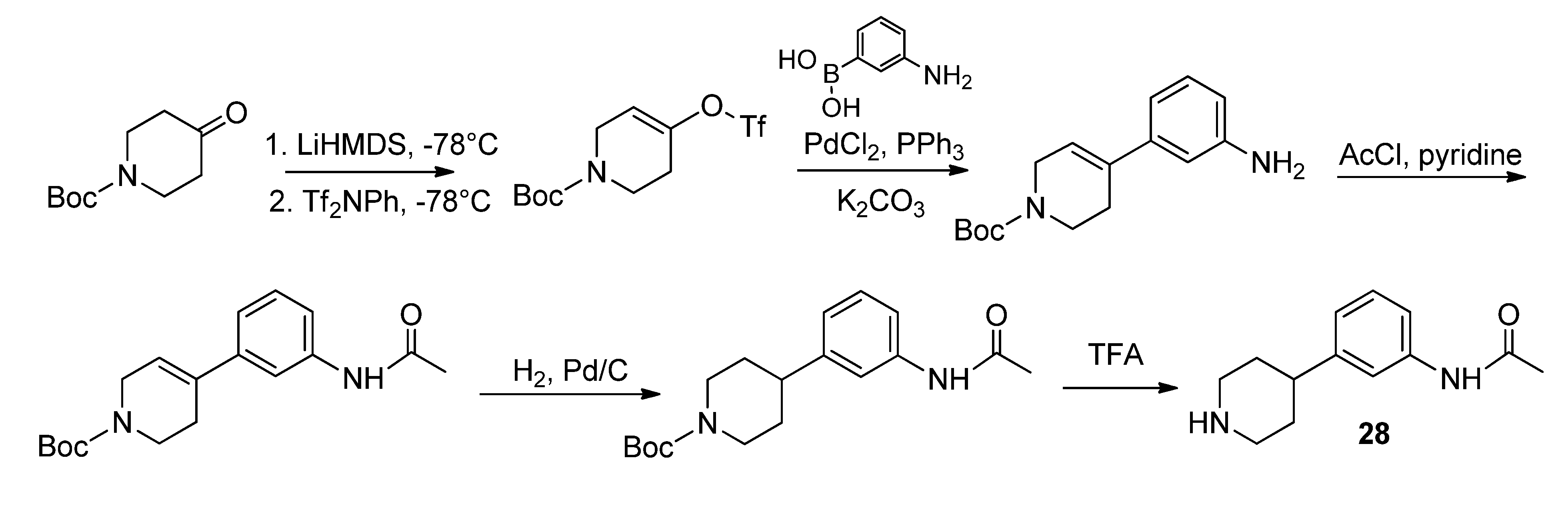
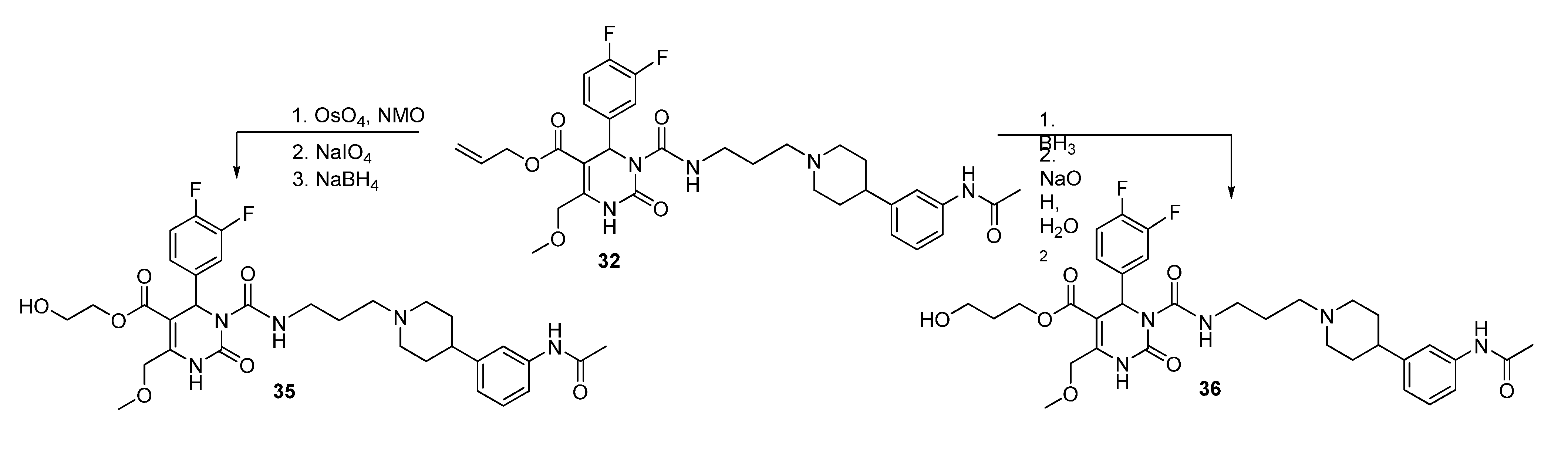
3.2. Syntheses
Synthesis of compounds
1,
14,
21 and
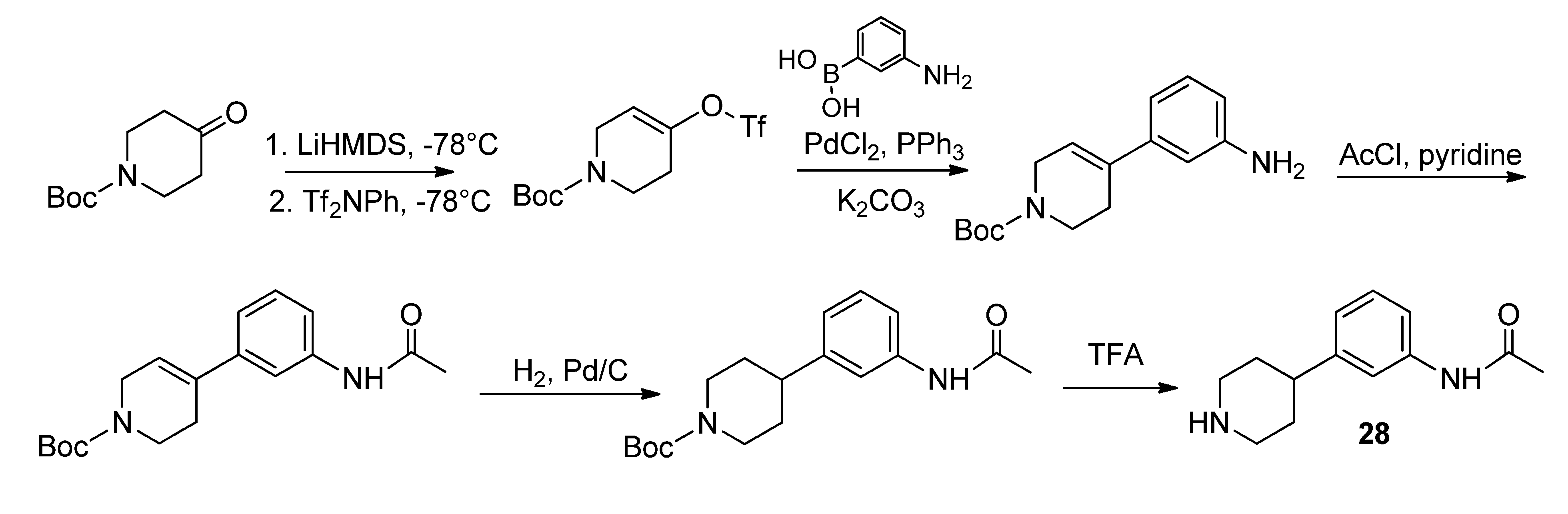
28 was conducted according to Schönberger [
17]. Synthesis of compounds
2,
7,
11,
18,
25 and
32 was conducted according to Philippe
et al. [
15]. Synthesis of compounds
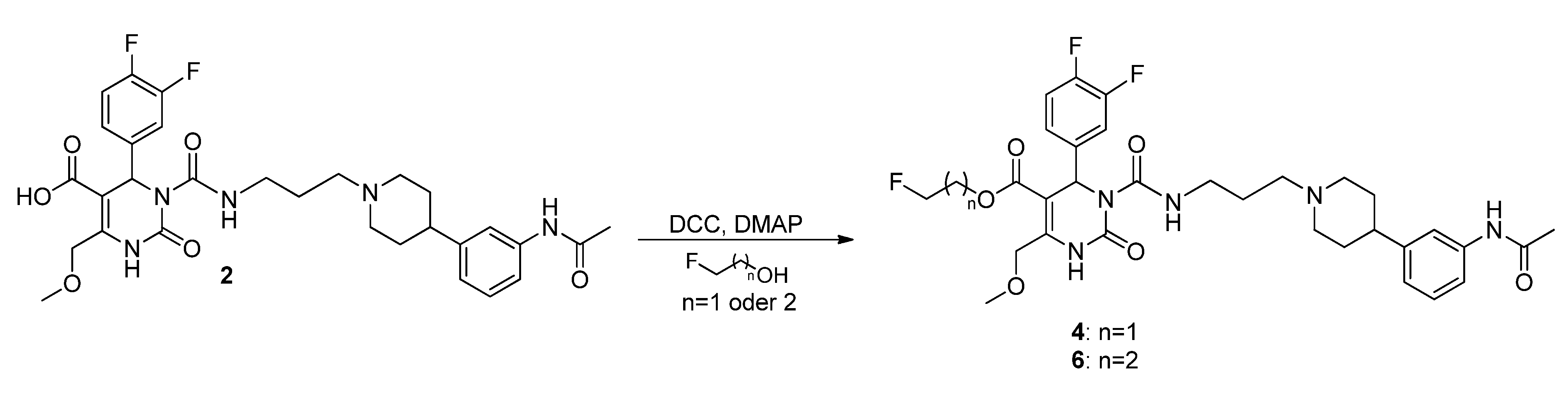
3 and
4 was conducted according to Philippe
et al. [
16].
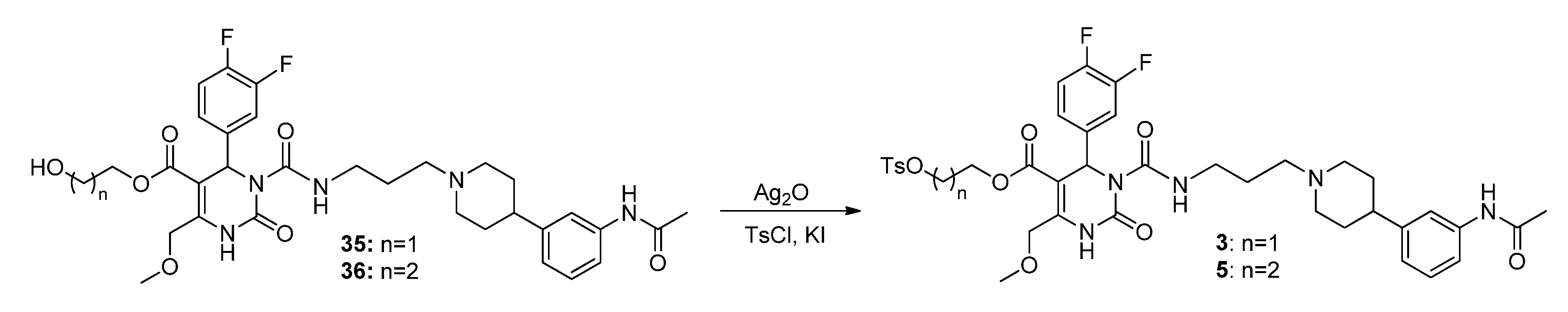
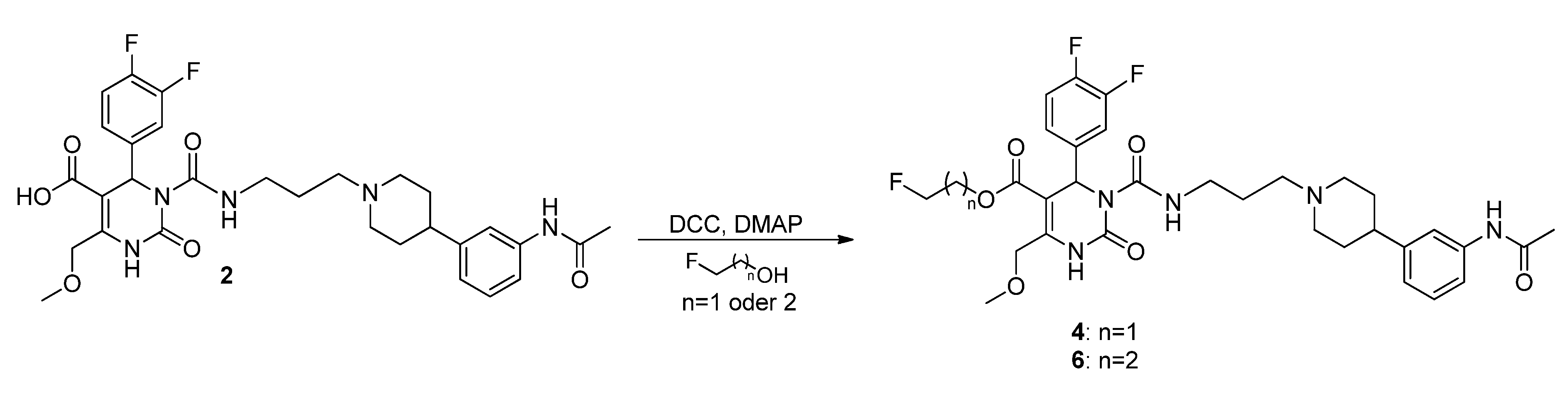
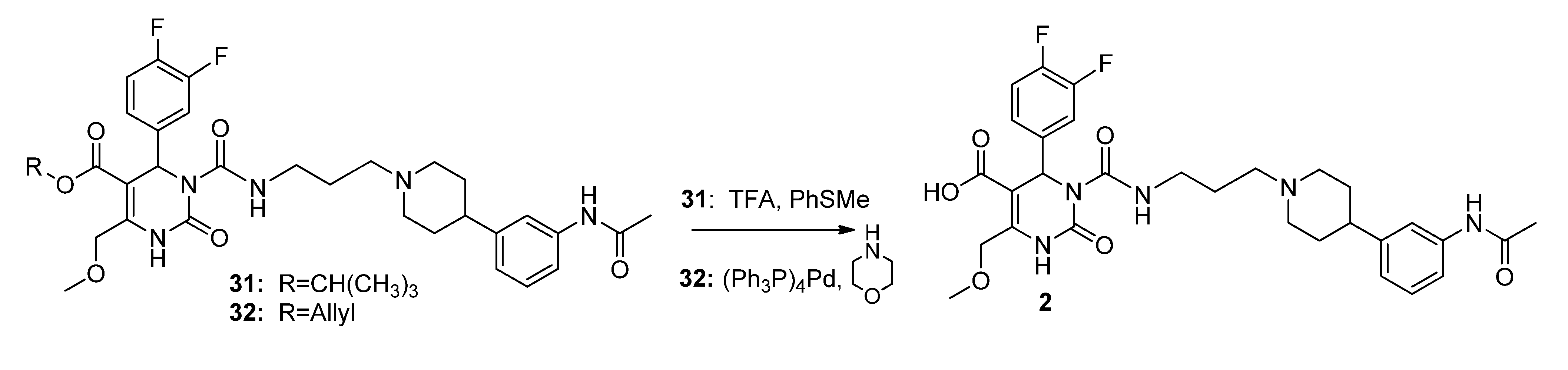
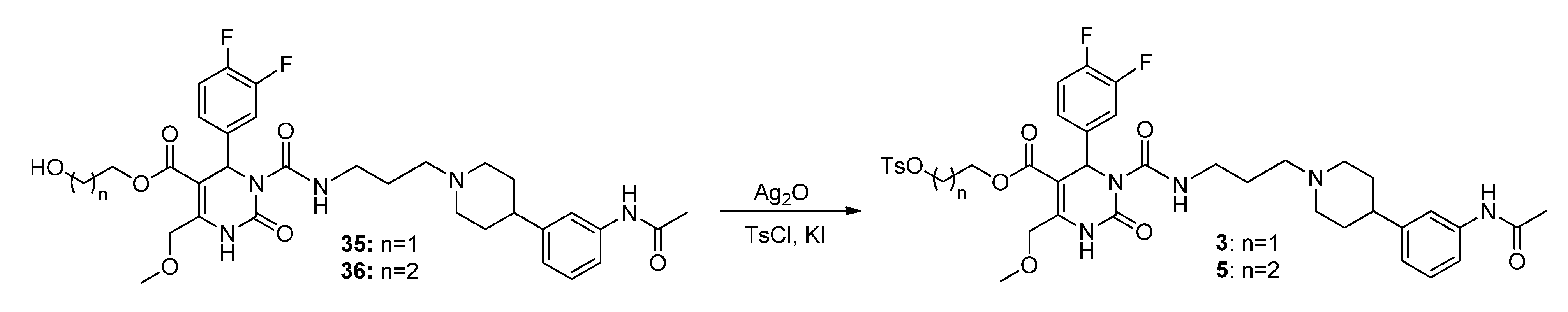
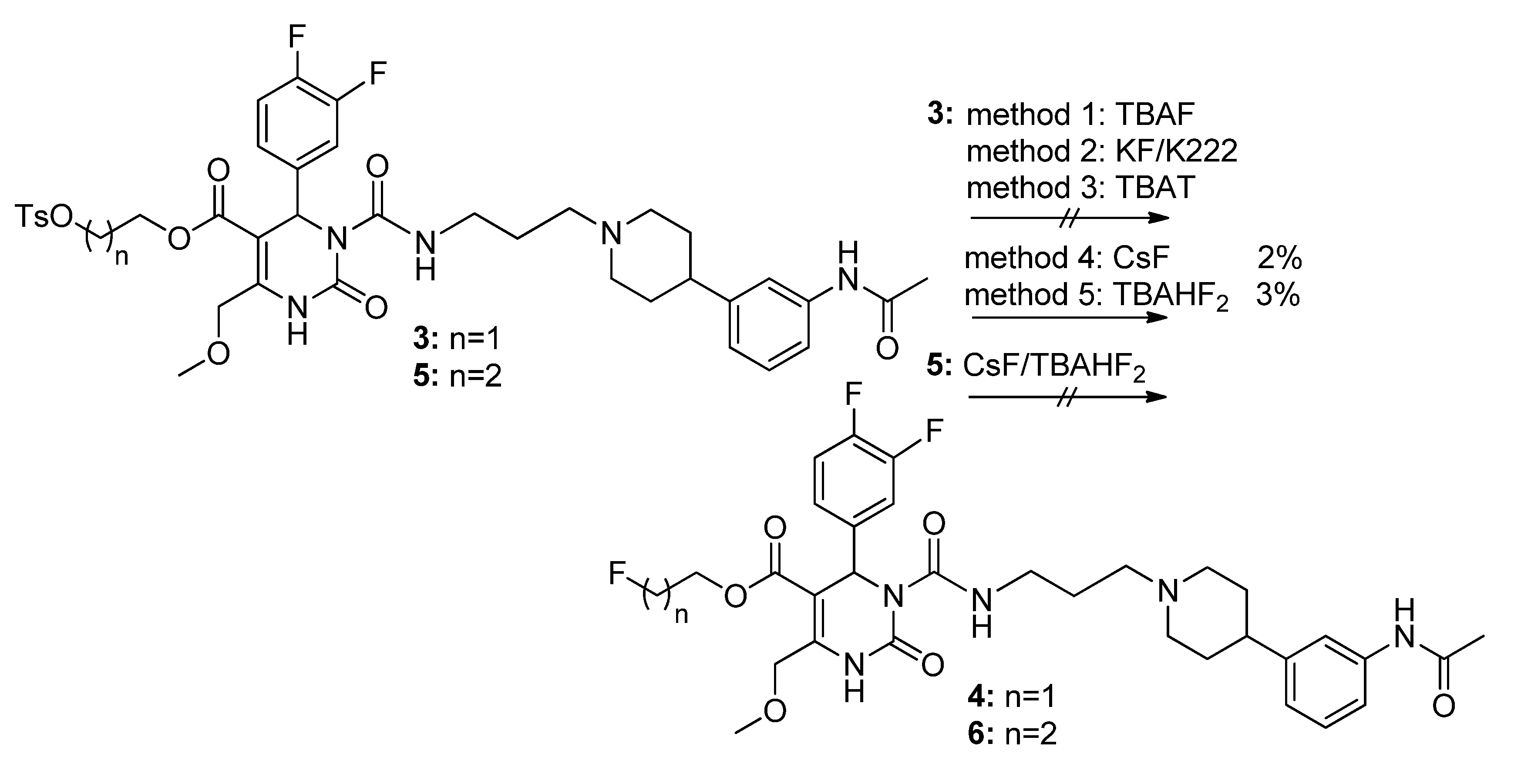
3-(Tosyloxy)propyl-3-(3-(4-(3-acetamidophenyl)piperidin-1-yl)propyl-carbamoyl)-4-(3,4-difluoro-phenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (5). To a stirred solution of alcohol 36 (116 mg, 0.18 mmol) in CH2Cl2 (1.0 mL), freshly produced Ag2O (83 mg, 0.36 mmol), tosyl chloride (69 mg, 0.36 mmol) and KI (60 mg, 0.36 mmol) were added. The mixture was stirred at 40 °C until completion of the reaction (TLC-monitoring). Thereafter, the reaction mixture was filtered and the solvent evaporated in vacuo. The residue was purified via column chromatography (silica gel, eluent: CH2Cl2/MeOH 9:1) to give 23 mg (26.1%) of product 5. 1H-NMR (200 MHz, CDCl3): δ (ppm) 1.87–1.97 (m, 8H, 9b-CH2, 19-CH2, 22,22′-(CH2)2), 2.05–2.08 and 3.08–3.15 (m, 4H, 21,21′-(CH2)2), 2.15 (s, 3H, 32-CH3), 2.42–2.51 (m, 6H, Tos-CH3, 20-CH2, 23-CH), 3.37–3.40 (m, 2H, 18-CH2), 3.45 (s, 3H, 7-OCH3), 3.94–4.04 (m, 2H, 9c-OCH2), 4.10–4.18 (m, 2H, 9a-OCH2), 4.65 (s, 2H, 6-OCH2), 6.58 (s, 1H, 3-CH), 6.92 (d, 1H, J = 7.2 Hz, 29-CH), 7.04–7.26 (m, 5H, 11-CH, 14-CH, 15-CH, 27-CH, 28-CH), 7.30-7.34 (m, 2H, 3′,3′′-(CH)2), 7.44 (s, 1H, 30-NH), 7.72–7.76 (m, 2H, 2′,2′′-(CH)2), 7.99 (s, 1H, 1-NH), 8.96 (t, 1H, J = 5.2 Hz, 17-NH); 13C-NMR (50 MHz, CDCl3): δ (ppm) 21.6 (Tos-CH3), 24.5 (32-CH3), 26.0 (19-CH2), 29.6 (9b-CH2), 32.4 (22,22′-(CH2)2), 39.4 (18-CH2), 42.2 (23-CH), 52.9 (3-CH), 54.1 (21,21′-(CH2)2), 56.4 (20-CH2), 59.1 (7-OCH3), 60.4 (9c-CH2OH), 66.6 (9a-OCH2), 68.0 (6-OCH2), 101.2 (4-C), 116.0/116.3 (11-CH), 117.2/117.5 (14-CH), 117.7 (27-CH), 118.1 (25-CH), 122.7 (29-CH), 122.9/123.0/123.1/123.2 (15-CH), 127.8 (2′,2′′-(CH)2), 128.9/129.0 (28-CH), 129.9 (3′,3′′-(CH)2), 132.7 (1′-C), 137.5 (10-C), 138.3 (26-C), 144.9 (4′-C), 146.6 (5-C), 146.8 (24-C), 152.1 (2-CO), 153.2 (16-CO), 163.8 (8-COO), 168.6 (31-CON); MS: m/z (%) 812 (1), 371 (46), 286 (56), 231 (43), 71 (29), 70 (100), 65 (28), 57 (38), 56 (55); HRMS: Calcd. for C40H48F2N5O9S [M + H]+: 812.3141. Found: 812.3148.
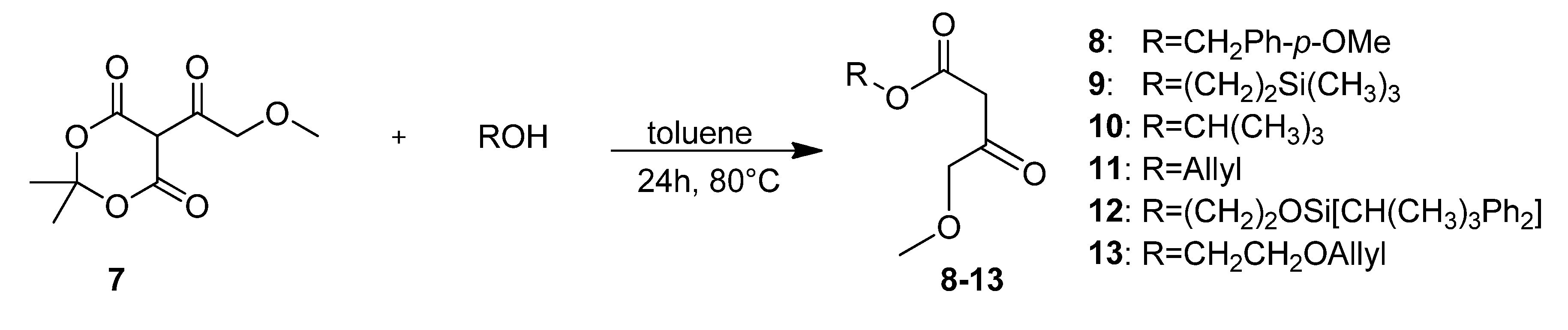
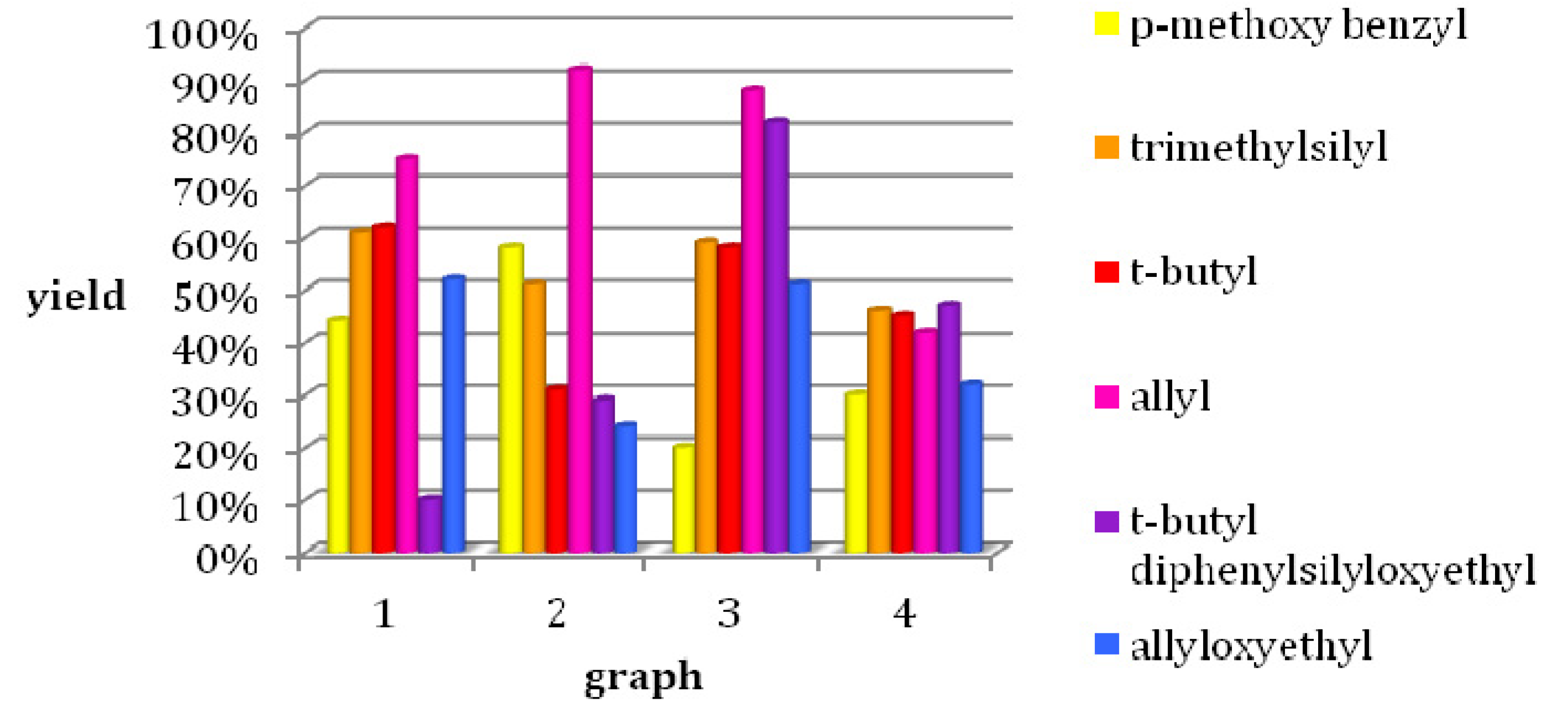
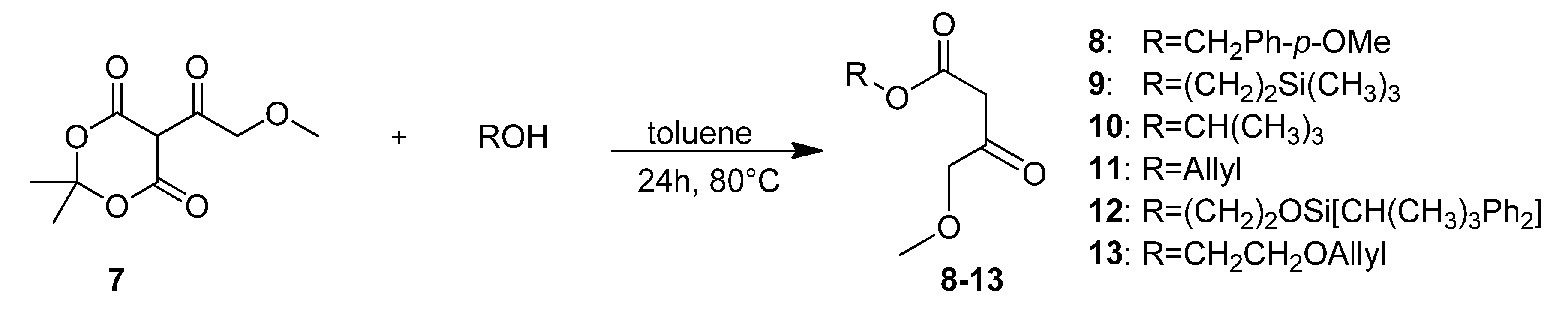
4-Methoxybenzyl 4-methoxy-3-oxobutanotate (8). 5-(2-Methoxyacetyl)-2,2-dimethyl-1,3-dioxane-4,6-dione (7, 724 mg, 3.35 mmol) and (4-methoxyphenyl)methanol (925 mg, 6.69 mmol) in toluene (10.0 mL) were heated to 80 °C for 24 h. After cooling to room temperature the solvent was removed in vacuo and the residue was partly purified by column chromatography (silica gel, eluent: petroleum ether/EtOAc 3:1) to give 372 mg (44.0%) of 8 as a brown oil. The crude product was reacted without further purification in the next step.
2-(Trimethylsilyl)ethyl 4-methoxy-3-oxobutanoate (9). A mixture of 5-(2-methoxyacetyl)-2,2-dimethyl-1,3-dioxane-4,6-dione (7, 3.60 g, 16.65 mmol) and 2-(trimethylsilyl)ethanol (4.8 mL, 3.94 g, 33.32 mmol) in toluene (50.0 mL) was heated to 80 °C for 24 h. After cooling to room temperature the solvent was removed in vacuo and the residue was purified by column chromatography (silica gel, eluent: petroleum ether/EtOAc 9:1) to give 2.35 g (61.0%) of 9 as a light brown oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) −0.01 (s, 9H, Si(CH3)3), 0.91–1.00 (m, 2H, SiCH2), 3.36 (s, 3H, OCH3), 3.43 (s, 2H, 2-CH2), 4.03 (s, 2H, 4-OCH2), 4.12–4.21 (m, 2H, OCH2); 13C-NMR (50 MHz, CDCl3): δ (ppm) −1.7 (Si(CH3)3), 17.1 (SiCH2), 45.8 (2-CH2), 59.2 (OCH3), 63.6 (OCH2), 77.2 (4-OCH2), 167.0 (1-COO), 201.5 (3-CO); MS m/z (%): 232 (1), 147 (8), 117 (8), 75 (64), 74 (12), 73 (100), 72 (9), 59 (9), 45 (12); HRMS: Calcd. for C8H16O4Si [M – C2H4]: 204.0818. Found: 204.0813.
t-Butyl 4-methoxy-3-oxobutanoate (10). 5-(2-Methoxyacetyl)-2,2-dimethyl-1,3-dioxane-4,6-dione (7, 6.40 g, 19.60 mmol) and 2-methylpropan-2-ol (9.00 g, 121.42 mmol) were dissolved in toluene (90.0 mL) and the mixture was stirred for 24 h at 80 °C. After cooling to room temperature, the solvent and excess alcohol were removed in vacuo and the residue was purified by column chromatography (RP silica gel, eluent: ACN/H2O 9:1) and Kugelrohr distillation (b.p. ca 260 °C) to give compound 10 as a brown oil (3.44 g, 62.0%). 1H-NMR (200 MHz, CDCl3): δ (ppm) 1.40 (s, 9H, t-but-(CH3)3), 3.34 (s, 2H, 2-CH2), 3.36 (s, 3H, 5-OCH3), 4.02 (s, 2H, 4-CH2); 13C-NMR (50 MHz, CDCl3): δ (ppm) 27.8 (t-but-(CH3)3), 47.0 (2-CH2), 59.1 (OCH3), 77.1 (4-OCH2), 81.9 (t-but-C), 166.0 (1-COO), 201.8 (3-CO); MS: m/z (%) 132 (5), 115 (6), 87 (4), 69 (5), 59 (33), 58 (8), 57 (100), 56 (13), 45 (51), 43 (16), 42 (11), 41 (26); HRMS: Calcd. for C5H8O4 [M – C4H8]: 132.0423. Found: 132.0424.
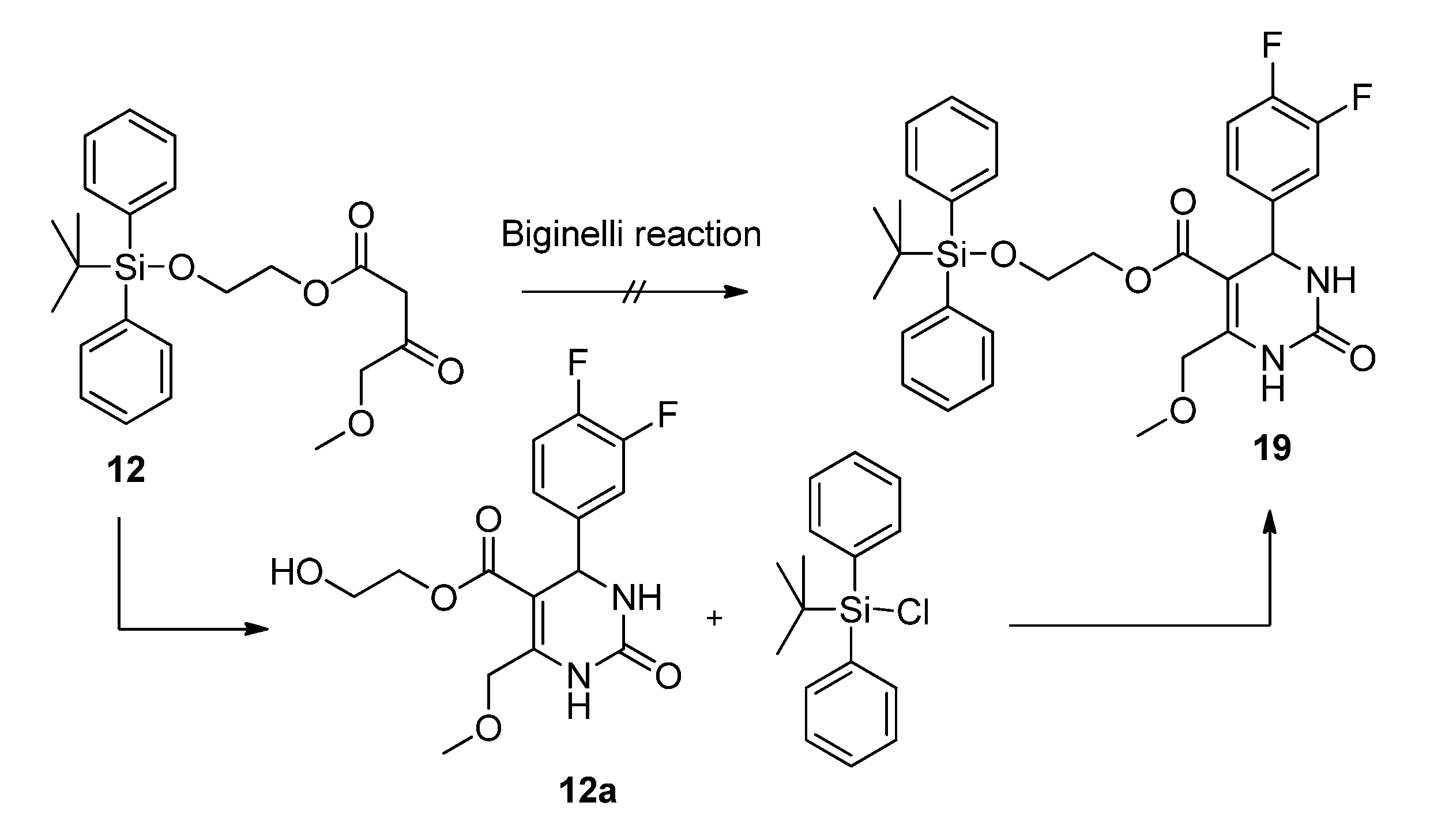
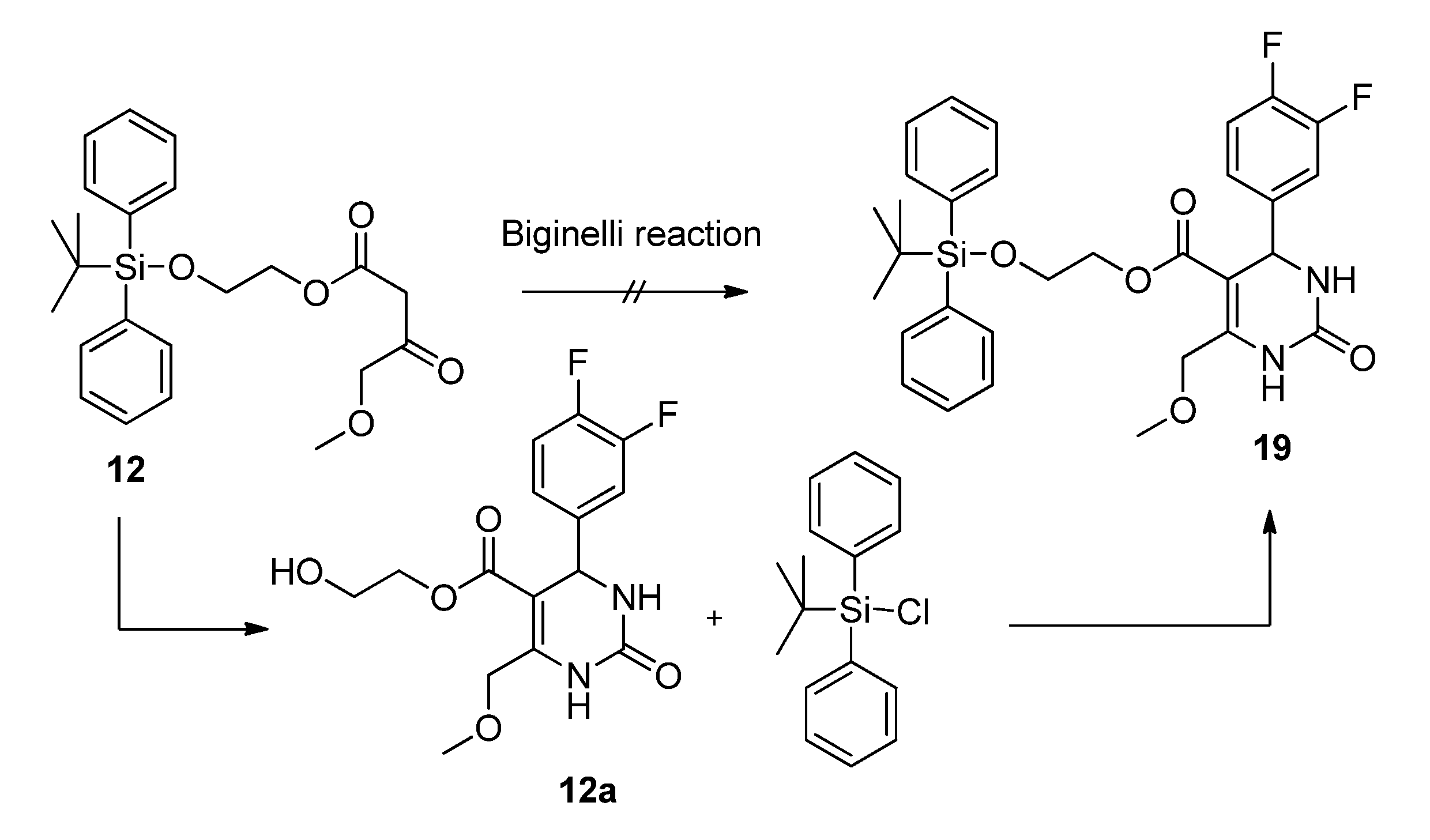
2-(t-Butyldiphenylsilyloxy)ethyl 4-methoxy-3-oxobutanoate (12). First, 2-(t-butyldiphenylsilyloxy)-ethanol was freshly prepared. To a mixture of ethylene glycol (5.59 g, 90.00 mmol), imidazole (6.13 g, 90.00 mmol), and absolute CH2Cl2 (140 mL) t-butylchlorodiphenylsilane (23.03 mL, 24.74 g, 90.00 mmol) dissolved in absolute CH2Cl2 was added dropwise at 0 °C. After stirring for 24 h at room temperature, the reaction mixture was extracted with water to remove unreacted ethylene glycol. The solvent was removed under reduced pressure to give 21.81 g (81.1%) of 2-(t-butyldiphenyl-silyloxy)ethanol as a colorless oil which crystallized upon cooling to afford colorless crystals. The crude product (21.69 g, 72.19 mmol) was used for the next step without further purification and heated to 80 °C for 24 h with 5.23 g (24.21 mmol) of 5-(2-methoxyacetyl)-2,2-dimethyl-1,3-dioxane-4,6-dione (7) in toluene (73.0 mL). After cooling to room temperature the solvent was removed in vacuo and the residue was purified by column chromatography (reversed-phase silica gel, eluent: acetonitrile/H2O 4:1→1:0) to give 3.10 g (10.4%) of 12 as reddish brown oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) 1.07 (s, 9H, t-but-(CH3)3), 3.40 (s, 3H, OCH3), 3.49 (s, 2H, 2-CH2), 3.85–3.90 (m, 2H, 9b-OCH2), 4.08 (s, 2H, 4-OCH2), 4.24–4.29 (m, 2H, 9a-OCH2), 7.37–7.45 (m, 6H, 2′-(CH)2, 4′-(CH)2, 6′-(CH)2), 7.66–7.70 (m, 4H, 3′-(CH)2, 5′-(CH)2); 13C-NMR (50 MHz, CDCl3): δ (ppm) 19.1 (t-but-C), 26.7 (t-but-(CH3)3), 45.6 (2-CH2), 59.3 (OCH3), 61.7 (9b-OCH2), 66.3 (9a-OCH2), 77.2 (4-OCH2), 127.7 (3′-(CH)2, 5′-(CH)2), 129.7 (4′-(CH)2), 133.2 (1′-(C)2), 135.5 (2′-(CH)2, 6′-(CH)2), 166.9 (1-COO), 201.3 (3-CO); MS: m/z (%) 383 (4), 349 (3), 243 (18), 199 (89), 185 (24), 184 (48), 165 (65), 154 (42), 139 (24), 111 (56), 94 (23), 69 (33), 45 (100); HRMS: m/z calcd. for C23H30O5SiNa [M + Na]+: 437.1760. Found: 437.1760.
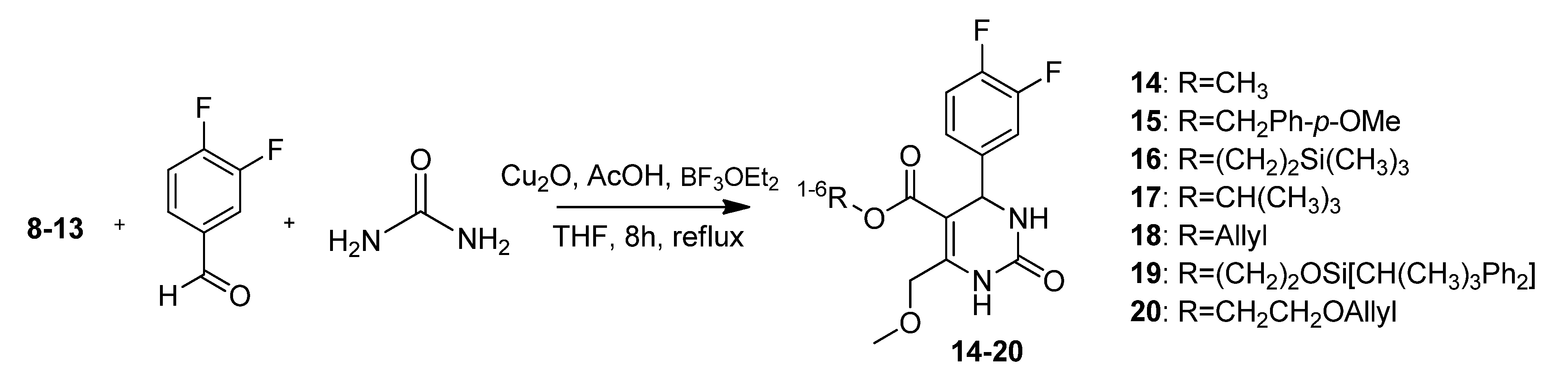
2-Hydroxyethyl 4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (12a). To a well-stirred solution of allyloxyethyl 4-methoxy-3-oxobutanoate (12, 3.00 g, 7.24 mmol), 3,4-difluorobenzaldehyde (1.20 g, 8.44 mmol) and urea (0.73 g, 12.15 mmol) in THF (7.0 mL), Cu2O (117 mg, 0.82 mmol), glacial acetic acid (47 µL) and boron trifluoride diethyl etherate (1.29 mL, 1.46 g, 10.32 mmol) were added. The resulting reaction mixture was heated under reflux for 8 h. After cooling to room temperature the mixture was poured onto a mixture of ice (12 g) and NaHCO3 (2 g). The resulting cloudy solution was filtered over Celite and washed with CH2Cl2 (10 mL). The biphasic solution was separated in a separatory funnel and the aqueous phase was washed with CH2Cl2 (3 × 7 mL). The combined organic layers were dried over Na2SO4, filtered and evaporated in vacuo. The crude product was purified by column chromatography (silica gel, eluent: CH2Cl2/MeOH 9:1) to give 0.90 g (36.3%) of 12a as yellow oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) 3.44 (s, 3H, 7-OCH3), 3.71 (m, 2H, 9b-CH2OH), 4.12–4.20 (m, 2H, 9a-OCH2), 4.63 (s, 2H, 6-OCH2), 5.37 (d, J = 2.4 Hz, 3-CH), 6.77 (s, 1H, 2a-CH), 7.02–7.18 (m, 3H, 11-CH, 14-CH, 15-CH), 7.75 (s, 1H, 1-NH); 13C-NMR (50 MHz, CDCl3): δ (ppm) 54.3 (3-CH), 59.1 (7-OCH3), 60.9 (9b-CH2OH), 65.9 (9a-OCH2), 68.5 (6-OCH2), 98.2 (4-C), 115.4/115.8 (11-CH), 117.3/117.7 (14-CH), 122.4/122.5 (15-CH), 140.5 (10-C), 148.2 (5-C), 152.3 (2-CO), 164.8 (8-COO); MS: m/z (%) 342 (22), 310 (14), 280 (33), 267 (74), 253 (27), 229 (100), 221 (24), 167 (50), 153 (38), 45 (40); HRMS: m/z calcd. for C15H16F2N2O5Na [M + Na]+: 365.0925. Found: 365.0932.
Allyloxyethyl 4-methoxy-3-oxobutanoate (13). A mixture of 5-(2-methoxyacetyl)-2,2-dimethyl-1,3-dioxane-4,6-dione (7, 38.74 g, 179.20 mmol) and allyl alcohol (57.4 mL, 537.60 mmol) in toluene (10.0 mL) was heated to 80 °C for 24 h. After cooling to room temperature the solvent was removed in vacuo and the residue was purified by bulb-to-bulb distillation to give 20.04 g (51.7%) of 13 as a yellow oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) 3.36 (s, 2H), 3.65 (m, 2H), 3.96–3.99 (m, 2H), 4.04 (s, 2H), 4.22–4.27 (m, 2H), 5.27 (d, J = 19.1, 2H), 5.88 (m, 1H); 13C-NMR (50 MHz, CDCl3): δ (ppm) 45.6, 59.2, 64.3, 67.5, 71.9, 77.1, 117.1, 134.4, 166.9, 201.3; MS: m/z (%) 73 (23), 69 (37), 60 (23), 57 (41), 55 (60), 43 (100), 42 (17), 41 (87); HRMS: m/z calcd. for C10H16O5: 216.0998. Found: 216.1003.
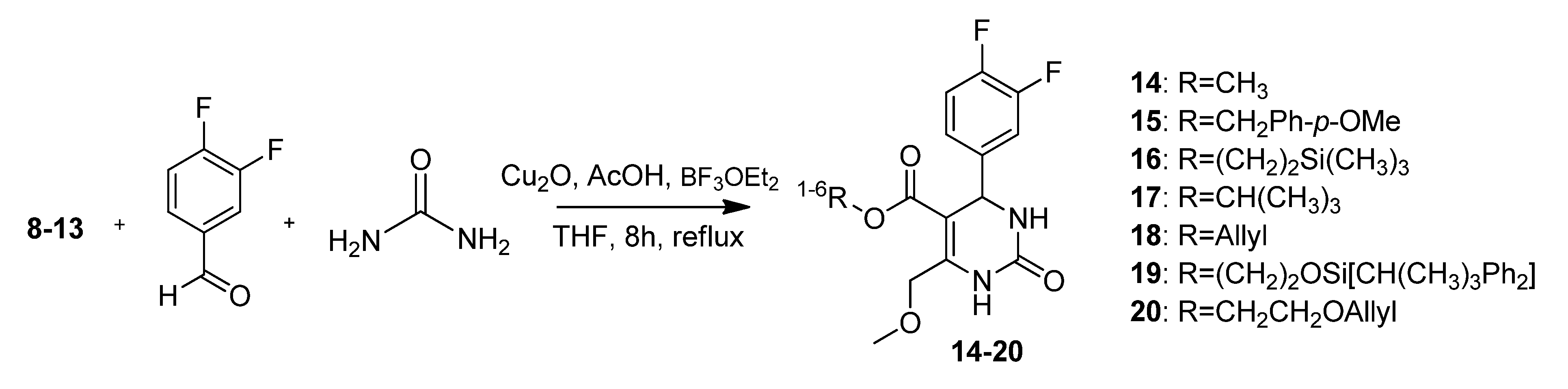
4-Methoxybenzyl 4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (15). To a well-stirred solution of 4-methoxybenzyl 4-methoxy-3-oxobutanoate (8, 330 mg, 1.31 mmol), 3,4-difluorobenzaldehyde (192 mg, 1.35 mmol) and urea (118 mg, 1.96 mmol) in THF (1.2 mL), Cu2O (19 mg, 0.13 mmol), glacial acetic acid (7.6 µL) and boron trifluoride diethyl etherate (0.2 mL, 240 mg, 1.69 mmol) were added. The resulting reaction mixture was heated under reflux for 8 h. After cooling to room temperature the mixture was poured onto ice (2 g) and NaHCO3 (200 mg). The resulting cloudy solution was filtered over Celite and washed with CH2Cl2 (5 mL). The biphasic solution was separated in a separatory funnel and the aqueous phase was washed with CH2Cl2 (3 × 30 mL). The combined organic layers were dried over Na2SO4, filtered and evaporated in vacuo. The product was partly purified by column chromatography (silica gel, eluent: EtOAc/MeOH 4:1) to give 315 mg (57.5%) of 15 as a yellow oil. The crude product was reacted without further purification in the next step.
2-(Trimethylsilyl)ethyl 4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (16). To a well-stirred solution of 2-(trimethylsilyl)ethyl 4-methoxy-3-oxobutanoate (9, 2.35 g, 10.11 mmol), 3,4-difluorobenzaldehyde (1.48 mg, 10.41 mmol) and urea (910 mg, 15.17 mmol) in THF (8.7 mL), Cu2O (146 mg, 1.02 mmol), glacial acetic acid (59 µL) and boron trifluoride diethyl etherate (1.6 mL, 1.82 g, 12.80 mmol) were added. The resulting reaction mixture was heated under reflux for 8 h (TLC monitoring EtOAc/hexane 1:1). After cooling to room temperature the mixture was poured onto ice (15 g) and NaHCO3 (3 g). The resulting cloudy solution was filtered over Celite and washed with CH2Cl2 (12 mL). The biphasic solution was separated in a separatory funnel and the aqueous phase was washed with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over Na2SO4, filtered and evaporated in vacuo. The crude product was purified by column chromatography (silica gel, eluent: CH2Cl2/MeOH 9:1) to give 2.05 mg (51.0%) of 16 as yellow oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) −0.01 (s, 9H, 9c-Si(CH3)3), 0.84–0.93 (m, 2H, 9b-SiCH2), 3.42 (s, 3H, 7-OCH3), 4.05–4.14 (m, 2H, 9a-OCH2), 4.62 (s, 2H, 6-OCH2), 5.31 (s, 2H, 2a-NH), 6.96 (s, 1H, 3-CH), 7.01–7.15 (m, 3H, 11-CH, 14-CH, 15-CH), 7.65 (s, 1H, 1-NH); 13C-NMR (50 MHz, CDCl3): δ (ppm) −1.7 (9c-Si(CH3)3), 17.4 (9b-SiCH2), 54.3 (3-CH), 59.0 (7-OCH3), 62.3 (9a-OCH2), 68.5 (6-OCH2), 98.8 (4-C), 115.4/115.8 (11-CH), 117.1/117.6 (14-CH), 122.4/122.5 (15-CH), 140.5 (10-C), 147.1 (5-C), 152.5 (2-CO), 165.0 (8-COO); MS m/z (%): 398 (1), 370 (11), 355 (27), 323 (13), 293 (11), 281 (11), 257 (10), 253 (9), 225 (10), 185 (10), 167 (13), 84 (12), 75 (13), 73 (100), 45 (26); HRMS: Calcd. for C18H24F2N2O4SiNa [M + Na]+: 421.1371. Found: 421.1365.
t-Butyl-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (17). To a stirred mixture of t-butyl 4-methoxy-3-oxobutanoate 10 (3.00 g, 15.94 mmol), 3,4-difluorobenzaldehyde (1.8 mL, 2.33 g, 16.40 mmol) and urea (1.44 g, 23.98 mmol) in THF (14.0 mL), Cu2O (230 mg, 1.61 mmol) and CH3COOH were added at room temperature, followed by dropwise addition of boron trifluoride diethyl etherate (2.5 mL, 2.88 g, 20.20 mmol). The resulting mixture was stirred and refluxed for 8 h. After cooling to room temperature, the reaction mixture was poured into a mixture of ice (25 g) and NaHCO3 (5 g). The resulting mixture was filtered over Celite and washed with CH2Cl2 (20 mL). The organic phase was separated from the filtrate and the aqueous layer was extracted with CH2Cl2 (3 x 15 mL). The combined organic layers were dried over Na2SO4 and the solvent was evaporated in vacuo to give 4.92 g of raw product (a brown oil) that was purified via column chromatography (silica gel, eluent: CH2Cl2/MeOH 9:1) to give compound 17 as a yellow oil (1.74 g, 30.8%). 1H-NMR (200 MHz, CDCl3): δ (ppm) 1.32 (s, 9H, t-but-(CH3)3), 3.41 (s, 3H, 7-OCH3), 4.60 (s, 2H, 6-OCH2), 5.24 (s, 1H, 3-CH), 7.00–7.08 (m, 3H, 11-CH, 14-CH, 15-CH), 7.60 (s, 1H, NH); 13C-NMR (50 MHz, CDCl3): δ (ppm) 28.0 (t-but-(CH3)3), 54.7 (3-CH), 58.9 (7-OCH3), 68.5 (6-OCH2), 81.1 (t-but-C), 99.9 (4-C), 115.4/115.7 (11-CH), 117.1/117.4 (14-CH), 122.37/ 122.45/122.5/122.6 (15-CH), 140.7 (10-C), 146.3 (5-C), 152.5 (2-C), 164.03 (8-COO); MS: m/z (%) 354 (1), 298 (11), 265 (6), 221 (6), 185 (39), 121 (11), 71 (23), 70 (12), 69 (22), 57 (100), 55 (20), 43 (19), 41 (17); HRMS: Calcd. for C17H20F2N2O4Na [M + Na]+: 377.1289. Found: 377.1284.
2-(t-Butyldiphenylsilyloxy)ethyl 4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydro-pyrimidine-5-carboxylate (19). To a mixture of 2-hydroxyethyl 4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (12a, 0.78 g, 2.28 mmol), imidazole (155 mg, 2.28 mmol), and absolute CH2Cl2 (140 mL), tert-butylchlorodiphenylsilane (0.6 mL, 626 mg, 2.28mmol) dissolved in absolute CH2Cl2 was added dropwise at 0 °C. After stirring for 24 h at room temperature, the solvent was removed under reduced pressure and the residue was purified by column chromatography (silica gel, eluent: CH2Cl2/MeOH 9:1) to give 1.20 g (90.7%) of 19 as a colorless oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) 1.06 (s, 9H, t-but-(CH3)3), 3.42 (s, 3H, 7-OCH3), 3.79–3.83 (m, 2H, 9b-OCH2), 4.06–4.29 (m, 2H, 9a-OCH2), 4.63 (s, 2H, 6-OCH2), 5.27 (d, 1H, J = 2.9 Hz, 3-CH), 6.59 (s, 1H, 2a-NH), 6.96–7.15 (m, 3H, 11-CH, 14-CH, 15-CH), 7.36–7.46 (m, 6H, 2′-(CH)2, 4′-(CH)2, 6′-(CH)2), 7.63–7.76 (m, 5H, 1-NH, 3′-(CH)2, 5′-(CH)2); 13C-NMR (50 MHz, CDCl3): δ (ppm) 19.1 (t-but-C), 26.7 (t-but-(CH3)3), 54.4 (3-CH), 59.0 (7-OCH3), 62.0 (9b-OCH2), 65.4 (9a-OCH2), 68.5 (6-OCH2), 98.4 (4-C), 115.3/115.7 (11-CH), 117.2/117.5 (14-CH), 122.4/122.5/122.6 (15-CH), 127.7 (3′-(CH)2, 5′-(CH)2), 129.8 (4′-(CH)2), 133.1 (1′-(C)2), 135.4 (2′-(CH)2, 6′-(CH)2), 140.4 (10-C), 147.5 (5-C), 152.3 (2-CO), 164.6 (8-COO); MS: m/z (%) 580 (1), 523 (18), 493 (4), 282 (15), 281 (100), 251 (51), 238 (12), 235 (12), 199 (18), 165 (18), 140 (30), 135 (12), 45 (10); HRMS: m/z calcd. for C31H34F2N2O5SiNa [M + Na]+: 603.2103. Found: 603.2121.
Allyloxyethyl 4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (20). To a well-stirred solution of allyloxyethyl 4-methoxy-3-oxobutanoate (13, 20.04 g, 98.68 mmol), 3,4-difluorobenzaldehyde (9.8 mL, 12.55 g, 88.34 mmol) and urea (7.95 g, 132.51 mmol) in THF (113 mL), Cu2O (1.26 g, 8.83 mmol), glacial acetic acid (506 µL) and boron trifluoride diethyl etherate (114.8 mL, 130.17 g, 917.04 mmol) were added. The resulting reaction mixture was heated under reflux for 8 h. After cooling to room temperature the mixture was poured on a mixture of ice (125 g) and NaHCO3 (25 g). The resulting cloudy solution was filtered over Celite and washed with CH2Cl2 (5 mL). The biphasic solution was separated in a separatory funnel and the aqueous phase was washed with CH2Cl2 (3 × 40 mL). The combined organic layers were dried over Na2SO4, filtered and evaporated in vacuo. The crude product was purified by column chromatography (silica gel, eluent: EtOAc/petroleum ether 3:1) to give 9.00 g (24.4%) of 20 as yellow oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) 3.43 (s, 3H), 3.94–4.23 (m, 6H), 4.63 (s, 2H), 5.16–5.29 (m, 2H), 5.76–5.96 (m, 1H), 6.52 (s, 1H), 7.04–7.21 (m, 3H), 7.66 (s, 1H); 13C-NMR (50 MHz, CDCl3): δ (ppm) 54.5, 59.1, 63.3, 67.7, 68.5, 71.9, 98.4, 115.6/115.9, 117.2, 117.4/117.6, 134.2, 147.6, 152.2, 164.5; MS: m/z (%) 382 (14), 280 (51), 269 (28), 222 (31), 221 (39), 167 (51), 45 (35), 41 (100); HRMS: m/z calcd for C18H21O5F2N2: 383.1419. Found: 383.1426; CHN: calcd for C18H21O5F2N2·H2O: C, 55.20; H, 5.16; N, 7.15. Found: C, 54.60; H, 5.10; N, 6.82.
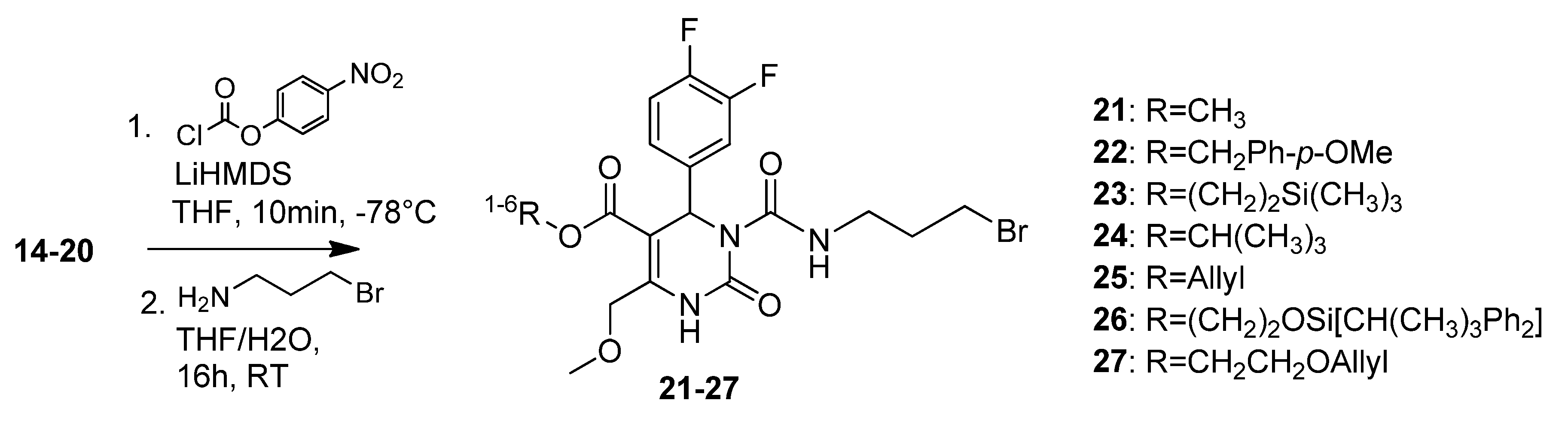
4-Methoxybenzyl 3-((3-bromopropyl)carbamoyl)-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (22). To a solution of 4-methoxybenzyl 4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (15, 252 mg, 0.60 mmol) and 4-nitrophenylchloroformate (425 mg, 2.11 mmol) in THF (7.5 mL), LiHMDS (1.7 mL, 284 mg, 1.70 mmol, 1 M in THF) was added dropwise at -78 °C. After 10 min the reaction was quenched with water (190 µL) and the mixture was allowed to warm to 0 °C. After addition of K2CO3 (333 mg, 2.41 mmol) and 3-aminopropylbromide hydrobromide (396 mg, 1.69 mmol) the reaction mixture was stirred at room temperature overnight. The yellow suspension was washed with saturated aqueous NaHCO3 and the biphasic solution was separated in a separatory funnel. The aqueous phase was extracted with Et2O (2 × 20 mL). The combined organic layers were dried over Na2SO4 and evaporated in vacuo. The crude product was purified by column chromatography (silica gel, eluent: CH2Cl2/MeOH 9:1) to give 70 mg (19.9%) of 22 as yellow oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) 1.20-1.25 (m, 2H, 20-CH2), 1.99–2.08 (m, 2H, 19-CH2), 3.39–3.41 (m, 2H, 18-CH2), 3.43 (s, 3H, 7-OCH3), 3.91 (s, 3H, PMB-OCH3), 4.63 (s, 2H, 6-OCH2), 5.46 (s, 2H, PMB-OCH2), 6.59 (s, 1H, 3-CH), 6.79–6.85 (m, 3H, 11-CH, 14-CH, 15-CH), 6.99–7.14 (m, 4H, PMB-2′-CH, 3′-CH, 5′-CH, 6′-CH), 7.72 (s, 1H, 1-NH), 8.86 (t, 1H, J = 4.4 Hz, 17-NH); 13C-NMR (50 MHz, CDCl3): δ (ppm) 30.4 (20-CH2), 32.1 (19-CH2), 39.1 (18-CH2), 53.2 (3-CH), 55.2 (PMB-OCH3), 59.2 (7-OCH3), 66.7 (PMB-OCH2), 68.0 (6-OCH2), 106.9 (4-C), 113.9 (PMB-3′-CH, 5′-CH), 116.0/116.4 (11-CH), 117.1/117.4 (14-CH), 123.1 (15-CH), 130.3 (PMB-2′-CH, 6′-CH), 145.7 (2-CO), 153.3 (16-CO), 167.6/167.7 (8-COO), 5-C and 10-C not found; MS m/z (%): 325 (57), 294 (81), 279 (31), 265 (29), 222 (20), 137 (28), 121 (100), 45 (37); HRMS: Calcd. for C25H26F2N3O6Br [M − H]-: 580.0903. Found: 580.0895.
2-(Trimethylsilyl)ethyl 3-((3-bromopropyl)carbamoyl)-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (23). To a solution of 2-(trimethylsilyl)ethyl 4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (16, 590 mg, 1.48 mmol) and 4-nitrophenylchloroformate (1.05 mg, 5.18 mmol) in THF (18.5 mL), LiHMDS (4.2 mL, 694 mg, 4.15 mmol, 1 M in THF) was added dropwise at ‒78 °C. After 10 min the reaction was quenched with water (460 µL) and the mixture was allowed to warm to 0 °C. After addition of K2CO3 (819 mg, 5.93 mmol) and 3-aminopropylbromide hydrobromide (973 mg, 4.44 mmol), the reaction mixture was stirred at room temperature overnight. The yellow suspension was washed with saturated aqueous NaHCO3 and the biphasic solution was separated in a separatory funnel. The aqueous phase was extracted with Et2O (2 × 20 mL). The combined organic layers were dried over Na2SO4 and evaporated in vacuo. The crude product was purified by column chromatography (silica gel, eluent: EtOAc/petroleum ether 1:1 and CH2Cl2/MeOH 9.5:0.5) to give 490 mg (59.0%) of 23 as yellow oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) 0.14 (s, 9H, 9c-Si(CH3)3), 1.05–1.15 (m, 2H, 9b-SiCH2), 1.34–1.41 (m, 2H, 20-CH2), 2.18–2.28 (m, 19-CH2), 3.49–3.56 (m, 18-CH2), 3.59 (s, 3H, 7-OCH3), 4.18–4.35 (m, 2H, 9a-OCH2), 4.81 (s, 2H, 6-OCH2), 6.78 (s, 1H, 3-CH), 7.12–7.39 (m, 3H, 11-CH, 14-CH, 15-CH), 7.87 (s, 1H, 1-NH), 9.02 (t, 1H, J = 5.6 Hz, 17-NH); 13C-NMR (50 MHz, CDCl3): δ (ppm) −1.7 (9c-Si(CH3)3), 17.5 (9b-SiCH2), 30.4 (20-CH2), 32.2 (19-CH2), 39.1 (18-CH2), 53.0 (3-CH), 59.1 (7-OCH3), 63.1 (9a-OCH2), 68.1 (6-OCH2), 101.9 (4-C), 116.0/116.4 (11-CH), 117.1/117.4 (14-CH), 123.0/123.1 (15-CH), 137.4/137.5 (10-C), 147.4/147.6 (5-C), 152.5 (2-CO), 153.3 (16-CO), 164.3 (8-COO); MS m/z (%): 536 (100), 508 (4), 436 (15), 421 (9), 378 (71), 350 (40), 328 (3), 306 (5), 278 (17), 234 (14); HRMS: Calcd. for C22H30O5F2N3BrSiNa [M + Na]+: 584.1004. Found: 584.1008.
t-Butyl-3-(3-bromopropylcarbamoyl)-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetra-hydropyrimidine-5-carboxylate (24). To a solution of pyrimidinone 17 (1.65 g, 4.66 mmol) and p-nitrophenylchloroformate (3.29 g, 16.32 mmol) in THF (60.0 mL), LiHMDS (13.0 mL, 2.18 g, 13.03 mmol, 1 M in THF) was slowly added at −78 °C. After 10 min, the reaction was completed by addition of H2O (1.5 mL), warmed to 0 °C and neutralised with K2CO3 (2.57 g, 18.60 mmol). Thereafter, 3-aminopropylbromide hydrobromide (3.06 g, 13.98 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. The resulting yellow suspension was washed with NaHCO3 twice, the layers were separated and the aqueous layer was extracted with Et2O. The combined organic layers were dried over Na2SO4. After purification of the raw product (5.23 g, brown oil) via column chromatography (silica gel, eluent: CH2Cl2/MeOH 9.5:0.5), the bromide 24 could be obtained as a yellow oil (1.41 g, 58.2%). 1H-NMR (200 MHz, CDCl3): δ (ppm) 1.23 (t, 2H, J = 7.1 Hz, 20-CH2), 1.39 (s, 9H, t-but-(CH3)3), 2.04–2.21 (m, 2H, 19-CH2), 3.35–3.42 (m, 2H, 18-CH2), 3.45 (s, 3H, 7-OCH3), 4.65 (s, 2H, 6-OCH2), 6.54 (s, 1H, 3-CH), 7.02–7.23 (m, 3H, 11-CH, 14-CH, 15-H), 7.73 (s, 1H, 1-NH), 8.91 (t, 1H, J = 5.6 Hz, 17-NH); 13C-NMR (50 MHz, CDCl3): δ (ppm) 28.1 (t-but-(CH3)3), 30.5 (20-CH2Br), 32.1 (19-CH2), 39.1 (18-CH2), 53.6 (3-CH), 59.0 (7-OCH3), 68.1 (6-OCH2), 81.7 (t-but-C), 103.1 (4-C), 116.1/116.4 (11-CH), 117.0/117.3 (14-CH), 123.1 (15-CH), 137.8 (10-C), 147.5 (5-C), 152.5 (2-CO), 153.4 (16-CO), 163.3 (8-COO); MS: m/z (%) 518 (1), 353 (4), 322 (44), 297 (73), 279 (34), 266 (100), 265 (37), 221 (16), 185 (14), 167 (10), 57 (33), 41 (30); HRMS: Calcd. for C21H26F2N3O5BrNa [M + Na]+: 540.0922. Found: 540.0904.
2-(t-Butyldiphenylsilyloxy)ethyl 3-((3-bromopropyl)carbamoyl)-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (26). To a solution of 2-(t-butyl-diphenylsilyloxy)ethyl 4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (19, 1.10 g, 1.89 mmol) and 4-nitrophenylchloroformate (1.34 g, 6.65 mmol) in THF (24.0 mL), LiHMDS (5.3 mL, 892 mg, 5.33 mmol, 1 M in THF) was added dropwise at −78 °C. After 10 min the reaction was quenched with water (9.0 mL) and the mixture was allowed to warm to 0 °C. After addition of K2CO3 (1.05 g, 7.60 mmol) and 3-aminopropylbromide hydrobromide (1.25 g, 5.71 mmol) the reaction mixture was stirred at room temperature overnight. The yellow suspension was washed with saturated aqueous NaHCO3 and the biphasic solution was separated in a separatory funnel. The aqueous phase was extracted with Et2O. The combined organic layers were dried over Na2SO4 and evaporated in vacuo. The crude product was purified by column chromatography (silica gel, eluent: EtOAc/petroleum ether 1:1) to give 1.16 g (82.2%) of 26 as a yellow oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) 0.96 (s, 9H, t-but-(CH3)3), 1.17–1.24 (m, 2H, 20-CH2), 2.01–2.11 (m, 2H, 19-CH2), 3.32–3.51 (m, 5H, 18-CH2, 7-OCH3), 3.74–3.79 (m, 2H, 9b-OCH2), 4.12–4.24 (m, 2H, 9a-OCH2), 4.61 (s, 2H, 6-OCH2), 6.64 (s, 1H, 3-CH), 6.78–7.20 (m, 3H, 11-CH, 14-CH, 15-CH), 7.28–7.37 (m, 6H, 2′-(CH)2, 4′-(CH)2, 6′-(CH)2), 7.54–7.58 (m, 4H, 3′-(CH)2, 5′-(CH)2), 7.73 (s, 1H, 1-NH), 8.86 (t, J = 5.7 Hz, 17-NH); 13C-NMR (50 MHz, CDCl3): δ (ppm) 19.0 (t-but-C), 26.6 (t-but-(CH3)3), 30.4 (20-CH2), 32.1 (19-CH2), 39.1 (18-CH2), 53.2 (3-CH), 59.1 (7-OCH3), 61.8 (9b-OCH2), 65.7 (9a-OCH2), 68.1 (6-OCH2), 101.6 (4-C), 115.5/115.8/116.2 (11-CH), 117.2/117.5 (14-CH), 123.0/123.1/ 123.2 (15-CH), 127.7 (3′-(CH)2, 5′-(CH)2), 129.8 (4′-(CH)2), 133.1 (1′-(C)2), 135.4 (2′-(CH)2, 6′-(CH)2), 137.4 (10-C), 145.9 (5-C), 152.5 (2-CO), 153.3 (16-CO), 164.6 (8-COO); MS: m/z (%) 768 (53), 686 (2), 603 (100), 560 (47), 540 (16), 460 (1), 238 (2); HRMS: m/z calcd for C35H40F2N3O6BrSiNa [M + Na]+: 766.1736. Found: 766.1728.
Allyloxyethyl 3-((3-bromopropyl)carbamoyl)-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (27). To a solution of allyloxyethyl 4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (20, 9.00 g, 23.56 mmol) and 4-nitrophenylchloroformate (16.65 g, 82.60 mmol) in THF (300.0 mL), LiHMDS (66.08 mL, 11.06 g, 66.08 mmol, 1 M in THF) was added dropwise at −78 °C. After 10 min the reaction was quenched with water (9.0 mL) and the mixture was allowed to warm to 0 °C. After addition of K2CO3 (13.03 g, 94.40 mmol) and 3-aminopropylbromide hydrobromide (15.50 g, 70.80 mmol) the reaction mixture was stirred at room temperature overnight. The yellow suspension was washed with saturated aqueous NaHCO3 and the biphasic solution was separated in a separatory funnel. The aqueous phase was extracted with Et2O. The combined organic layers were dried over Na2SO4 and evaporated in vacuo. The crude product was purified by column chromatography (silica gel, eluent: CH2Cl2/MeOH 10:1) to give 6.60 g (51.3%) of 27 as a yellow oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) 2.02–2.08 (m, 2H), 3.36–3.47 (m, 5H), 3.60–3.62 (m, 2H), 3.94–4.29 (m, 6H), 4.65 (s, 1H), 5.15–5.28 (m, 2H), 5.75–5.94 (m, 1H), 6.64 (s, 1H), 7.02–7.25 (m, 3H), 7.77 (s, 1H), 8.89 (t, J = 5.6 Hz, 1H); 13C-NMR (50 MHz, CDCl3): δ (ppm) 30.4, 32.1, 39.1, 59.1, 63.7, 67.6, 68.1, 71.9, 101.5, 116.0/116.4, 117.1, 117.3/117.4, 122.9, 134.2, 137.8, 146.0, 152.5, 153.2, 163.9; MS: m/z (%) 546 (2), 381 (64), 350 (52), 311 (30), 280 (48), 279 (100), 249 (33), 222 (47), 220 (32), 167 (29), 41 (91); HRMS: m/z calcd. for C22H26O6BrF2N3H: 548.1035. Found (M+1)+: 548.1044; CHN: Calcd. for C22H26O6BrF2N3H·H2O: C, 47.58; H, 4.73; N, 7.57. Found: C, 47.47; H, 4.81; N, 7.28.
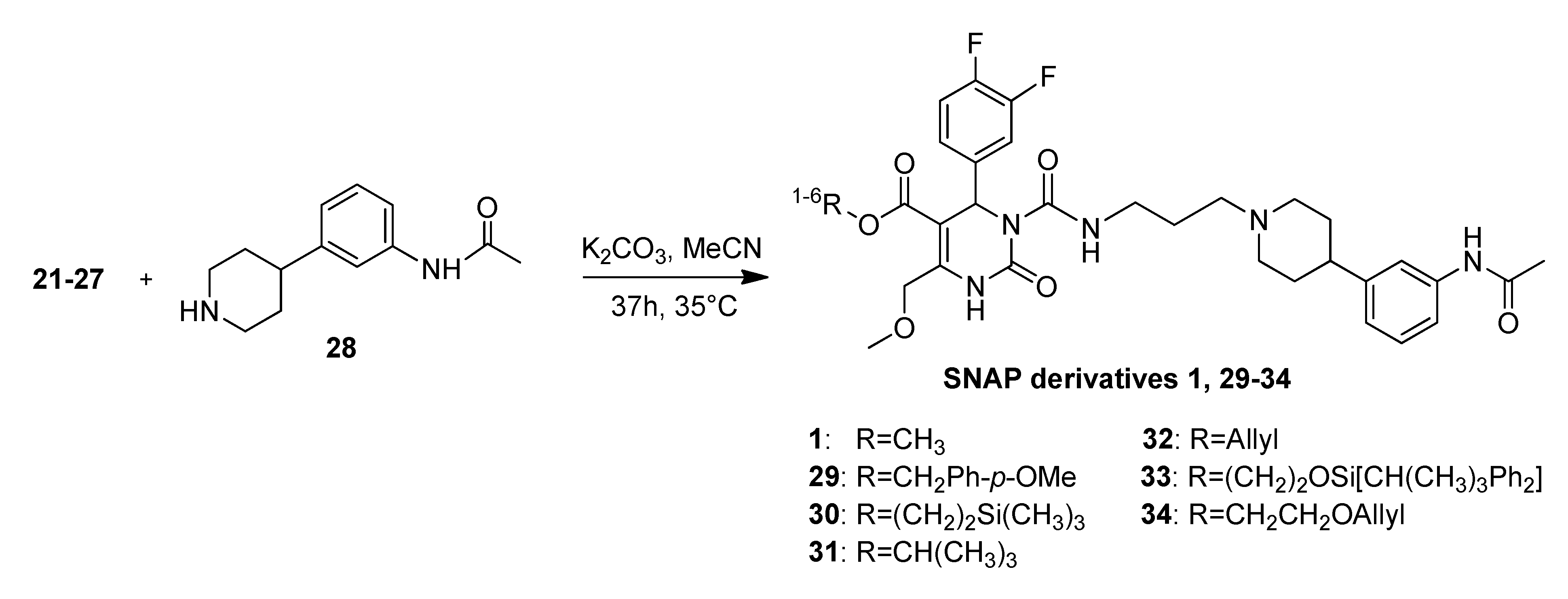
4-Methoxybenzyl 3-((3-(4-(3-acetamidophenyl)piperidin-1-yl)propyl)carbamoyl-4-(3,4-difluoro-phenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (29). Under an argon atmosphere a mixture of N-(3-(piperidin-4-yl)phenyl)acetamide (28, 109 mg, 0.50 mmol), 4-methoxybenzyl 3-((3-bromopropyl)carbamoyl)-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (22, 190 mg, 0.33 mmol) and K2CO3 (480 mg, 3.40 mmol) in ACN (12.6 mL) was stirred at 35 °C for 37 h. The resulting yellow suspension was filtered and the filtration residue was washed with EtOAc. After removal of the solvent in vacuo the obtained oily residue was dissolved in EtOAc (10 mL) and the organic phase was washed with saturated aqueous NaHCO3 (2 × 8 mL). The aqueous phase was washed with EtOAc (2 × 10 mL) and the combined organic layers were dried over Na2SO4, filtered and evaporated in vacuo. The obtained residue was purified by column chromatography (silica gel, eluent: CH2Cl2/MeOH 10:1) to give 75 mg (30.3%) of 29 as yellow oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) 1.67–1.79 (m, 6H, 19-CH2, 22,22′-(CH2)2), 2.00–2.13 and 3.08–3.13 (m, 7H, 21,21′-(CH2)2, 32-CH3), 2.48–2.51 (m, 3H, 20-CH2, 23-CH), 3.29–3.41 (m, 5H, 7-OCH3, 18-CH2), 3.78 (s, 3H, PMB-OCH3), 4.65 (d, 2H, J = 2.6 Hz, 6-OCH2), 6.62 (d, 1H, J = 6.2 Hz, 3-CH), 6.81–7.22 (m, 10H, 11-CH, 14-CH, 15-CH, 27-CH, 28-CH, 29-CH, 2′-CH, 3′-CH, 4′-CH, 5′-CH), 7.39 (25-CH), 7.97 (br s, 1H, 30-NH), 8.15 (s, 1H, 1-NH), 8.96–8.98 (m, 1H, 17-NH); 13C-NMR (50 MHz, CDCl3): δ (ppm) 24.3 (32-CH3), 25.8 (19-CH2), 32.2 (22,22′-(CH2)2), 39.2 (18-CH2), 42.0 (23-CH), 53.2 (3-CH), 53.9 (21,21′-(CH2)2), 55.2 (PMB-OCH3), 56.1 (20-CH2), 59.0 (7-OCH3), 66.3 (PMB-OCH2), 68.0 (6-OCH2), 101.9 (4-C), 113.9 (3′-CH, 5′-CH), 116.0/116.3 (11-CH), 117.0/117.4 (14-CH), 117.8 (27-CH), 118.0 (25-CH), 122.7 (29-CH), 123.0 (15-CH), 127.4 (1′-C), 128.8 (28-CH), 130.0 (2′-CCH, 6′-CH), 137.8 (10-C), 138.4 (26-C), 145.9 (5-C), 146.5 (24-C), 152.3 (2-CO), 153.2 (16-CO), 164.1 (8-COO), 168.8 (31-CON), 4’-C not found; MS m/z (%): 301 (8), 231 (82), 213 (17), 167 (8), 153 (10), 121 (100), 95 (12), 70 (16), 57 (20); HRMS: Calcd. for C38H43F2N5O7: 720.3209. Found: 720.3216.
2-(Trimethylsilyl)ethyl 3-((3-(4-(3-acetamidophenyl)piperidin-1-yl)propyl-carbamoyl)-4-(3,4-difluoro-phenyl)-6-methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (30). Under an argon atmosphere a mixture of N-(3-(piperidin-4-yl)phenyl)acetamide (28, 251 mg, 1.15 mmol), 2-(trimethylsilyl)ethyl 3-((3-bromopropyl)carbamoyl)-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (23, 420 mg, 0.75 mmol) and K2CO3 (1.09 g, 7.89 mmol) in ACN (28.0 mL) was stirred at 35 °C for 37 h. The resulting yellow suspension was filtered and the filtration residue was washed with EtOAc. After removal of the solvent in vacuo the obtained oily residue was dissolved in EtOAc (20 mL) and the organic phase was washed with saturated aqueous NaHCO3 (2 × 15 mL). The aqueous phase was washed with EtOAc (2 × 20 mL) and the combined organic layers were dried over Na2SO4, filtered and evaporated in vacuo. The obtained residue was purified by column chromatography (silica gel, eluent: CH2Cl2/MeOH 9:1 + 0.5% TEA) to give 240 mg (45.8%) of 30 as a yellow oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) −0.02 (s, 9H, 9c-Si(CH3)3), 0.93–1.02 (m, 2H, 9b-SiCH2), 1.76 (m, 6H, 19-CH2, 22,22′-(CH2)2), 1.96–2.08 and 2.98–3.03 (m, 4H, 21,21′-(CH2)2), 2.15 (s, 3H, 32-CH3), 2.43 (t, 3H, J = 6.8 Hz, 20-CH2, 23-CH), 3.31–3.41 (m, 2H, 18-CH2), 3.44 (s, 3H, 7-OCH3), 4.15–4.24 (m, 2H, 9a-OCH2), 4.68 (s, 2H, 6-OCH2), 6.69 (s, 1H, 3-CH), 6.93 (d, 1H, J = 7.6 Hz, 29-CH), 7.03–7.35 (m, 5H, 11-CH, 14-CH, 15-CH, 27-CH, 28-CH), 7.49 (s, 1H, 25-CH), 8.05 (s, 1H, 30-NH), 8.25 (s, 1H, 1-NH), 9.02 (t, 1H, J = 5.1 Hz, 17-NH); 13C-NMR (50 MHz, CDCl3): δ (ppm) −1.8 (9c-Si(CH3)3), 17.3 (9b-SiCH2), 24.2 (32-CH3), 26.2 (19-CH2), 32.7 (22,22′-(CH2)2), 39.4 (18-CH2), 42.4 (23-CH), 52.9 (3-CH), 54.1 (21,21′-(CH2)2), 56.5 (20-CH2), 58.9 (7-OCH3), 63.0 (9a-OCH2), 67.9 (6-OCH2), 102.0 (4-C), 115.9/116.2 (11-CH), 116.9/117.3 (14-CH), 117.6 (27-CH), 118.2 (25-CH), 122.5 (29-CH), 122.8/122.9 (15-CH), 128.6 (28-CH), 137.6 (10-C), 138.2 (26-C), 145.8 (5-C), 147.0 (24-C), 152.3 (2-CO), 153.1 (16-CO), 164.3 (8-COO), 168.7 (31-CON); MS m/z (%): 700 (26), 600 (2), 345 (8), 259 (4), 231 (4); HRMS: Calcd. for C35H48F2N5O6Si [M + H]+: 700.3342. Found: 700.3343.
t-Butyl-3-(3-(4-(3-acetamidophenyl)piperidin-1-yl)propylcarbamoyl)-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (31). To a solution of N-(3-(piperidin-4-yl)phenyl)acetamide (28, 140 mg, 0.64 mmol) in ACN (16.0 mL), bromide 24 (215 mg, 0.41 mmol) and K2CO3 (605 mg, 4.38 mmol) were added under argon atmosphere and the mixture was stirred at 35 °C for 37 h. The yellow slurry was filtered, washed with EtOAc and the filtrate was evaporated to dryness. The oily residue was dissolved in EtOAc and washed twice with saturated NaHCO3. Then the aqueous phase was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and evaporated in vacuo prior to purification by column chromatography (silica gel, eluent: CH2Cl2/MeOH 10:1). Product 31 was obtained as a yellow-brownish oil (133 mg, 49.4%). 1H-NMR (200 MHz, CDCl3): δ (ppm) 1.41 (t-but-(CH3)3), 1.74–1.78 (m, 6H, 19-CH2, 22,22′-(CH2)2), 1.94–2.07 and 2.98–3.03 (m, 4H, 21,21′-(CH2)2), 2.15 (s, 3H, 32-CH3), 2.37–2.44 (m, 3H, 20-CH2, 23-CH), 3.32–3.39 (m, 2H, 18-CH2), 3.44 (s, 3H, 7-CH3), 4.66 (s, 2H, 6-CH2), 6.58 (s, 1H, 3-CH), 6.94 (d, 1H, J = 7.3 Hz, 29-CH), 7.03–7.34 (m, 6H, 11-CH, 14-CH, 15-CH, 25-CH, 27-CH, 28-CH), 7.45 (s, 1H, 30-NH), 7.82 (s, 1H, 1-NH), 9.00 (t, 1H, J = 5.2 Hz, 17-NH); 13C-NMR (50 MHz, CDCl3): δ (ppm) 24.4 (32-CH3), 26.4 (19-CH2), 28.1 (t-but-(CH3)3), 32.9 (22,22′-(CH2)2), 39.5 (18-CH2), 42.5 (23-CH), 53.5 (3-CH), 54.3 (21,21′-(CH2)2), 56.5 (20-CH2), 59.0 (7-OCH3), 68.0 (6-OCH2), 103.3 (4-C), 116.0/116.4 (11-CH), 116.9/117.3 (14-CH), 117.6 (27-CH), 118.3 (25-CH), 122.7 (29-CH), 123.0/123.09/123.14/123.2 (15-CH), 128.8 (28-CH), 138.0 (10-C), 138.1 (26-C), 144.8 (5-C), 147.2 (24-C), 152.3 (2-CO), 153.3 (16-CO), 163.5 (8-COO), 168.5 (31-CON); MS: m/z (%) 679 (100, [M + Na]+), 657 (20, [M + H]+), 621 (10), 601 (17), 324 (23), 302 (49); HRMS: Calcd. for C34H44F2N5O6 [M + H]+: 656.3260. Found: 656.3277; IR: (ν) (cm−1) 3304, 2925, 1709, 1608, 1516, 1392, 1366, 1278, 1230, 1164, 1119, 1080, 772, 732, 701.
2-(t-Butyldiphenylsilyloxy)ethyl 3-((3-(4-(3-acetamidophenyl)piperidin-1-yl)propyl)carbamoyl-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (33). Under an argon atmosphere a mixture of N-(3-(piperidin-4-yl)phenyl)acetamide (28) (239 mg, 1.09 mmol), 2-(t-butyldiphenylsilyloxy)ethyl 3-((3-bromopropyl)carbamoyl)-4-(3,4-difluorophenyl)-6(methoxymethyl)- 2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (26, 775 mg, 1.04 mmol) and K2CO3 (1.52 g, 11.00 mmol) in ACN (40 mL) was stirred at 35 °C for 37 h. The resulting yellow suspension was filtered and the filtration residue was washed with EtOAc. After removal of the solvent in vacuo the obtained oily residue was dissolved in EtOAc and the organic phase was washed with saturated aqueous NaHCO3. The aqueous phase was washed with EtOAc and the combined organic layers were dried over Na2SO4, filtered and evaporated in vacuo. The obtained residue was purified by column chromatography (silica gel, eluent: CH2Cl2/MeOH 9:1) to give 428mg (47.7%) of 33 as a yellow oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) 1.01 (s, 9H, t-but-(CH3)3), 1.25–1.32 (m, 2H, 20-CH2), 1.74–1.94 (m, 6H, 19-CH2, 22,22′-(CH2)2), 2.01–2.09 and 3.09–3.14 (m, 4H, 21,21′-(CH2)2), 2.14 (s, 3H, 32-CH3), 2.48–2.60 (m, 3H, 20-CH2, 23-CH), 3.30-3.41 (m, 5H, 18-CH2, 7-OCH3), 3.79–3.84 (m, 2H, 9b-OCH2), 4.13–4.32 (m, 2H, 9a-OCH2), 4.65 (s, 2H, 6-OCH2), 6.69 (s, 1H, 3-CH), 6.90-7.26 (m, 7H, 11-CH, 14-CH, 15-CH, 25-CH, 27-CH, 28-CH, 29-CH), 7.33–7.42 (m, 6H, 2′-(CH)2, 4′-(CH)2, 6′-(CH)2), 7.59–7.63 (m, 5H, 3′-(CH)2, 5′-(CH)2, 30-NH), 8.15 (s, 1H, 1-NH), 8.86 (t, J = 5.7 Hz, 17-NH); 13C-NMR (50 MHz, CDCl3): δ (ppm) 19.0 (t-but-C), 24.3 (32-CH3), 25.8 (19-CH2), 26.6 (t-but-(CH3)3), 32.1 (22,22′-(CCH2)2), 39.2 (18-CH2), 42.0 (23-CH), 53.1 (3-CH), 53.8 (21,21′-(CH2)2), 56.0 (20-CH2), 59.0 (7-OCH3), 61.8 (9b-OCH2), 65.6 (9a-OCH2), 68.0 (6-OCH2), 101.7 (4-C), 115.8/116.2 (11-CH), 117.1/117.4 (14-CH), 117.8 (27-CH), 118.0 (25-CH), 122.9/ 122.8/123.0/123.1 (15-CH), 127.7 (3′-(CH)2, 5′-(CH)2), 128.8 (28-CH), 129.7 (4′-(CH)2), 133.0 (1′-(C)2), 135.4 (2′-(CH)2, 6′-(CH)2), 137.6 (10-C), 138.3 (26-C), 146.1 (5-C), 146.4 (24-C), 152.3 (2-CO), 153.1 (16-CO), 164.1 (8-COO), 168.7 (31-CON); MS: m/z (%) 883 (1), 523 (12), 281 (100), 231 (44), 199 (18), 168 (19), 140 (16), 70 (30), 57 (65), 56 (33), 45 (44), 43 (60), 41 (28); HRMS: m/z calcd. for C48H57F2N5O7Si: 882.4074. Found: 882.4087.
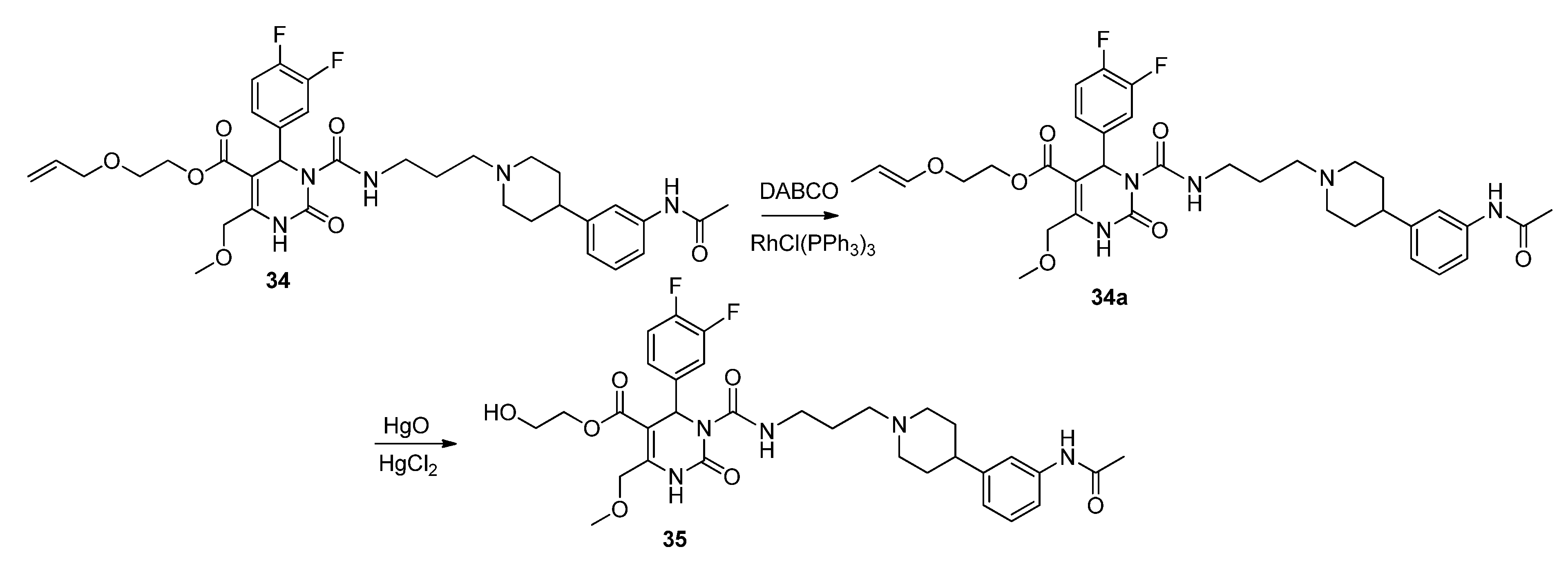
Allyloxyethyl 3-((3-(4-(3-acetamidophenyl)piperidin-1-yl)propyl)carbamoyl-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (34). Under an argon atmosphere a mixture of N-(3-(piperidin-4-yl)phenyl)acetamide (28, 4.04 mg, 18.47 µmol), allyloxyethyl 3-((3-bromopropyl)carbamoyl)-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (27, 6.60 g, 12.08 mmol) and K2CO3 (17.58 g, 127.20 mmol) in ACN (12.6 mL) was stirred at 35 °C for 37 h. The resulting yellow suspension was filtered and the filtration residue was washed with EtOAc. After removal of the solvent in vacuo the obtained oily residue was dissolved in EtOAc and the organic phase was washed with saturated aqueous NaHCO3. The aqueous phase was washed with EtOAc and the combined organic layers were dried over Na2SO4, filtered and evaporated in vacuo. The obtained residue was purified by column chromatography (silica gel, eluent: CH2Cl2/MeOH 9:1) to give 2.66 g (32.2%) of 34 as a yellow oil. 1H-NMR (200 MHz, CDCl3): δ (ppm) 1.72–1.81 (m, 6H), 1.99, 2.99 (m, 4H), 2.11 (s, 3H), 2.39 (m, 2H), 2.42 (m, 1H), 3.29, 3.39 (m, 2H), 3.37 (s, 3H), 3.55–3.60 (m, 2H), 3.92, 3.94 (d, J = 5.6 Hz, 2H), 4.17, 4.29 (m, 2H), 4.63 (m, 2H), 5.12–5.26 (m, 2H), 5.81 (m, 1H), 6.65 (s, 1H), 6.90 (d, J = 7.44, 1H), 6.98–7.19 (m, 5H), 7.25 (s, 1H), 7.44 (s, 1H), 7.95 (s, 1H), 8.98 (t, J = 5.3 Hz, 1H); 13C-NMR (50 MHz, CDCl3): δ (ppm) 24.4, 26.3, 32.8, 39.5, 42.5, 53.1, 54.2, 56.5, 58.9, 63.6, 67.6, 67.9, 71.9, 101.7, 115.9/116.3, 116.9, 117.2/117.3, 117.6, 118.2, 112.7, 122.8, 128.7, 134.2, 137.8, 138.2, 146.2, 147.16, 152.3, 153. 0, 164.0, 168.6; MS: m/z (%) 280 (33), 231 (100), 221 (21), 167 (34), 70 (26), 45 (21), 44 (21), 43 (54), 42 (31), 41 (57); HRMS: m/z calcd. for C35H43O7F2N5H: 684.3209. Found (M+1)+: 684.3195; CHN: calcd. for C35H43O7F2N5H•H2O: C, 59.90; H, 6.19; N, 9.98. Found: C, 59.09; H, 6.35; N, 9.69.
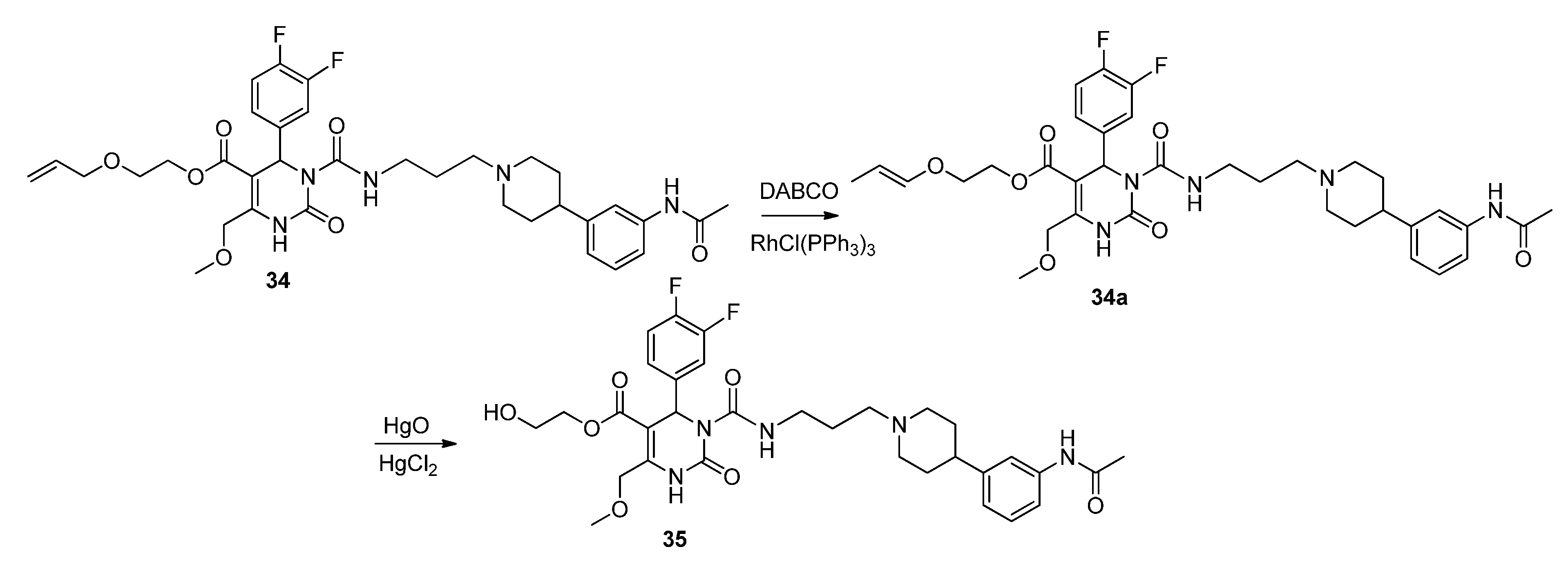
(E)-2-Prop-1-en-1-yloxy)ethyl 3-((3-(4-(3-acetamidophenyl)piperidin-1-yl)propyl)carbamoyl-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (34a). To a solution of allyloxyethyl 3-((3-(4-(3-acetamidophenyl)piperidin-1-yl)propyl)carbamoyl-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (34, 1.45 g, 2.12 mmol) and DABCO (1.00 g, 8.90 mmol) in EtOH (20 mL, 90%), RhCl(PPh3)3 (0.51 g, 0.55 mmol) was added. The reaction mixture was stirred for 15 min, cooled to room temperature, and then quenched with water. The aqueous phase was extracted with Et2O and dried with Na2SO4. After evaporation of the solvent, the crude product 34a was used for the next step without further purification.
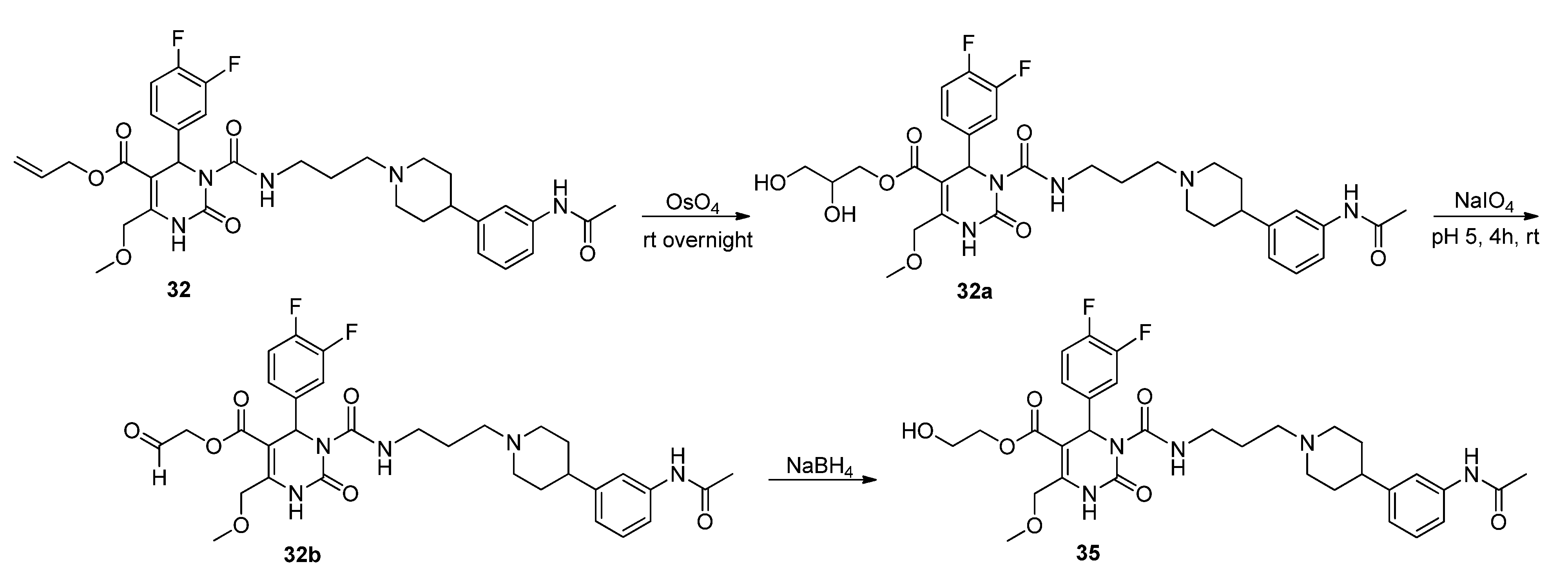
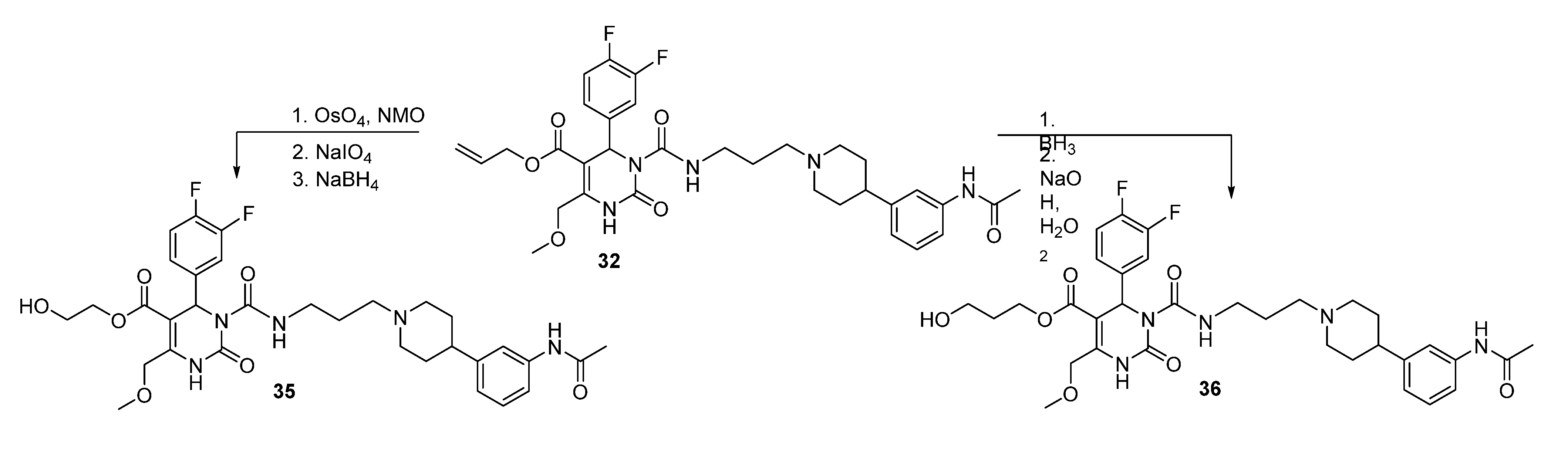
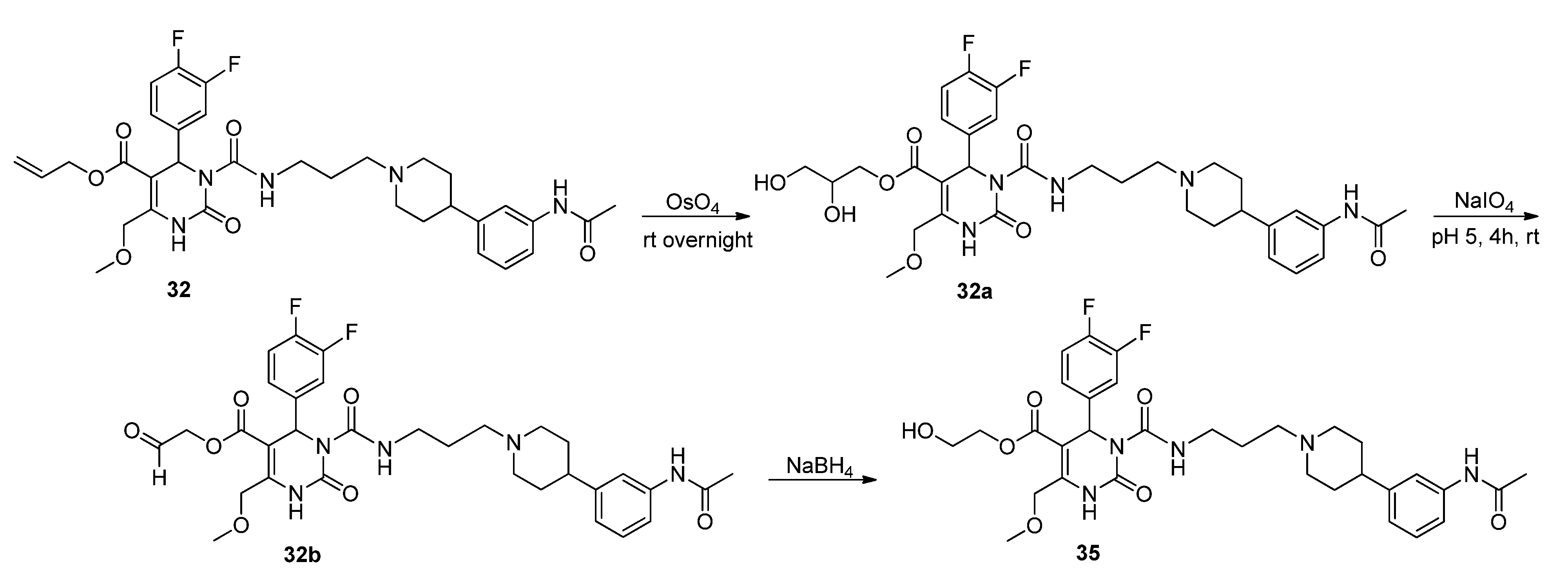
2-Hydroxyethyl 3-((3-(4-(3-acetamidophenyl)piperidine-1-yl)propyl)carbamoyl-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (35). Method 1(from 32): To a solution of OsO4 (2.5% in t-butanol, 0.5 mL, 0.04 mmol), N-methyl morpholine N-oxide monohydrate (46 mg, 0.39 mmol), H2O (0.8 mL), acetone (0.3 mL), and t-butanol (0.5 mL), allyl ester 32 (250 mg, 0.39 mmol) dissolved in dioxane (0.5 mL) was added dropwise. After stirring at room temperature overnight, the mixture was treated with celite (62 mg) and NaHSO3 (5 mg) and filtered over celite. After evaporation of the solvent, the crude 2,3-dihydroxypropyl ester 32a obtained was used for the next step.
To a solution of 32a (267 mg) in CH2Cl2 (0.6 mL), a solution of NaIO4 (93 mg, 0.44 mmol) in H2O (0.6 mL) was added. The two-layered mixture was stirred for 3–4 h, followed by separation of the organic layer. After evaporation of the solvent and drying in vacuo, the crude residue 32b was used in the next step without any further purification.
To a solution of 32b (88 mg) in MeOH (3.0 mL), NaBH4 (6.0 mg, 0.15 mmol) was added in portions under stirring, followed by stirring for another 45 min. The reaction was quenched with water, and the mixture extracted three times with Et2O. The organic layer was washed with water. After evaporation of the solvent the crude product was purified via column chromatography (silica gel, eluent: CH2Cl2/MeOH 10:1) to give 12 mg of 35 (13.3%) as a yellow oil.
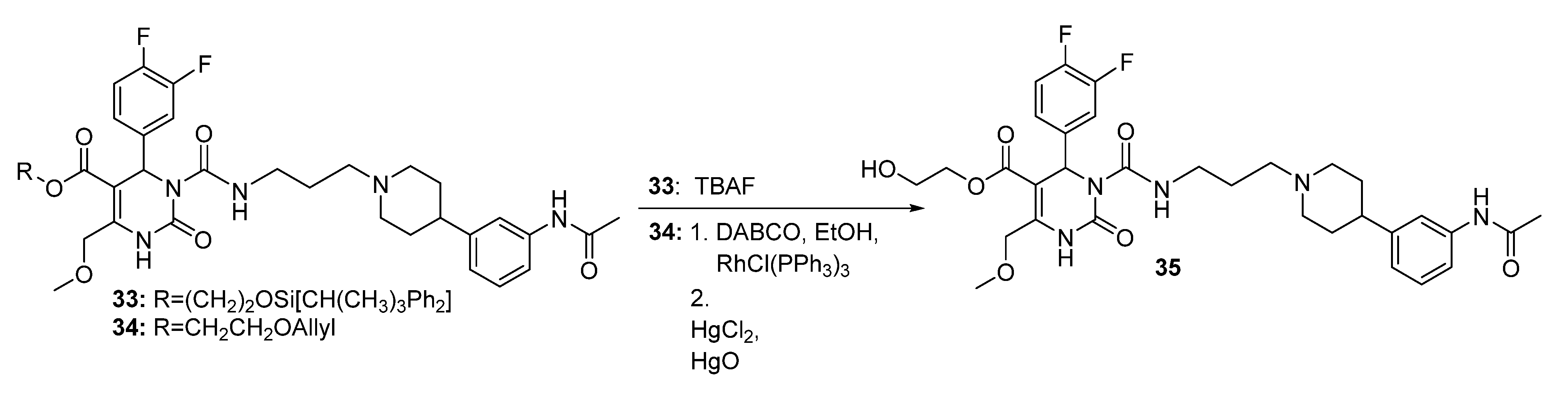
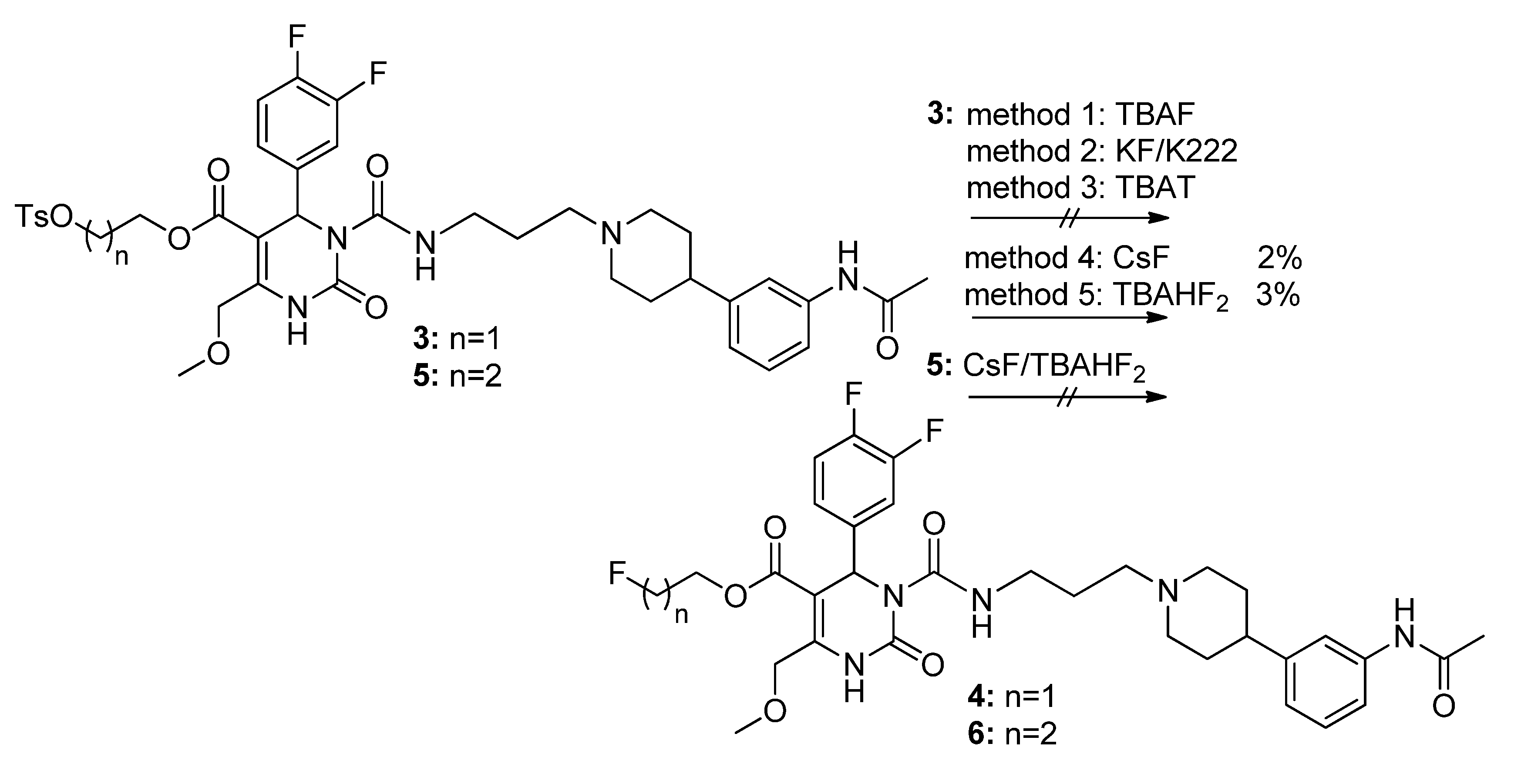
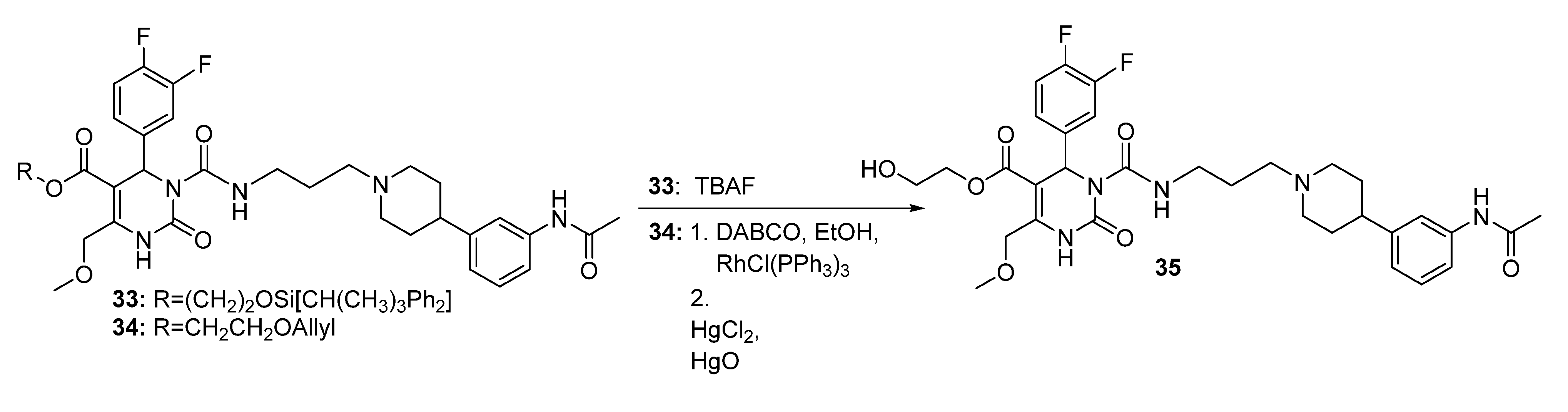
Method 2(from 33): To a solution of 2-(t-butyldiphenylsilyloxy)ethyl 3-((3-(4-(3-acetamidophenyl)piperidin-1-yl) propyl)carbamoyl-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (33) in THF (6.5 mL), TBAF (0.5 mL, 452 mg, 1.73, 1M in THF) was added dropwise. After stirring for 1.5 h at room temperature, the reaction was quenched with water (0.3 mL) and evaporated to dryness. The crude product was purified by column chromatography (reversed-phase silica gel, eluent: ACN/H2O 5:1 and silica gel, eluent: EtOAc/MeOH 10:1) to give 126 mg (40.3%) of 35 as a yellow oil.
Method 3(from 34a): A solution of HgCl2 (1.02 g, 3.76 mmol) in acetone/H2O (10:1, 10 mL) was added dropwise over a period of 3 min under stirring to a solution of (E)-2-(prop-1-en-1-yloxy)ethyl 3-((3-(4-(3-acetamidophenyl)piperidin-1-yl)propyl)carbamoyl)-4-(3,4-difluorophenyl)-6-(methoxy-methyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate 34a and HgO (1.02 g, 4.71 mmol) in acetone/H2O (10:1, 30 mL). After completion of the reaction (TLC-monitoring), HgO was removed by filtration over celite and the product was evaporated to dryness. The residue was purified by column chromatography (silica gel, eluent: CH2Cl2/MeOH 10:1) where part of the educt 34a could be recovered and used in another reaction again. The product was obtained in good yield (677 mg, 70.8%). 1H-NMR (200 MHz, CDCl3): δ (ppm) 1.74–1.85 (m, 6H, 19-CH2, 22,22′-(CH2)2), 2.02–2.09 and 3.01–3.06 (m, 4H, 21,21′-(CH2)2), 2.09 (s, 3H, 32-CH3), 2.42–2.45 (m, 3H, 20-CH2, 23-CH), 3.27–3.31 (m, 2H, 18-CH2), 3.39 (s, 3H, 7-OCH3), 3.74 (t, 2H, J = 4.4 Hz, 9b-CH2OH), 4.10-4.20 (m, 2H, 9a-OCH2), 4.62 (s, 2H, 6-OCH2), 6.62 (s, 1H, 3-CH), 6.86 (d, 1H, J = 7.5 Hz, 29-CH), 6.94–7.23 (m, 5H, 11-CH, 14-CH, 15-CH, 27-CH, 28-CH), 7.33–7.39 (m, 2H, 25-CH, 30-NH), 8.53 (s, 1H, 1-NH), 8.99 (t, 1H, J = 5.4 Hz, 17-NH); 13C-NMR (50 MHz, CDCl3): δ (ppm) 24.1 (32-CH3), 25.8 (19-CH2), 32.2 (22,22′-(CH2)2), 39.2 (18-CH2), 42.0 (23-CH), 53.0 (3-CH), 53.9 (21,21′-(CH2)2), 56.1 (20-CH2), 58.9 (7-OCH3), 60.3 (9b-CH2OH), 66.2 (9a-OCH2), 67.9 (6-OCH2), 101.6 (4-C), 115.8/116.1 (11-CH), 117.1/117.4 (14-CH), 117.8 (27-CH), 118.0 (25-CH), 122.6 (29-CH), 122.7/122.8 (15-CH), 128.7 (28-CH), 137.6 (10-C), 138.3 (26-C), 146.4 (5-C), 147.4 (24-C), 152.2 (2-CO), 153.2 (16-CO), 164.1 (8-COO), 169.0 (31-CON); MS: m/z (%) 644 (32), 345 (16), 302 (100), 259 (17), 231 (20), 160 (4), 114 (5); HRMS: Calcd. for C32H40F2N5O7 [M + H]+: 644.2896. Found: 644.2902.
3-Hydroxypropyl-3-(3-(4-(3-acetamidophenyl)piperidin-1-yl)propylcarbamoyl)-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (36). A solution of BH3-THF (1M, 0.31 mmol, 0.3 mL) was added dropwise to a solution of allyl ester 32 (131 mg, 0.21 mmol) in THF. The mixture was stirred at room temperature for 30 min prior to elimination of excess hydride ions via addition of water (0.4 mL). 3M NaOH solution (0.2 mL) and H2O2 solution (30%, 0.2 mL) were then added to the mixture. The stirred solution was heated to 60 °C for 2 h. Thereafter, the solution was cooled to room temperature and the solvent evaporated in vacuo. H2O was added and the reaction mixture was washed several times with CH2Cl2. The combined organic layers were dried over Na2SO4 and evaporated in vacuo. The intermediate product (108 mg) gave after purification by column chromatography (silica gel, eluent: CH2Cl2/MeOH 10:1) 43 mg of educt 32 and 23 mg of product 36 (26.1%). 1H-NMR (500 MHz, CDCl3): δ (ppm) 1.81–1.95 (22,22′-(CH2)2), 1.82 (m, 9b-CH2), 1.84 (m, 19-CH2), 2.16 and 3.13 (21,21′-(CH2)2), 2.17 (s, 32-CH3), 2.50 (m, 23-CH), 2.54 (m, 20-CH2), 3.33 and 3.41 (m, 18-CH2), 3.45 (s, 7-CH3), 3.52–3.64 (m, 9c-CH2), 4.22–4.31 (m, 9a-CH2), 4.67 (s, 6-CH2), 6.64 (s, 3-CH), 6.94 (d, 1H, J = 7.7 Hz, 29-CH), 7.08 (m, 14-CH), 7.09 (m, 15-CH), 7.18 (m, 11-CH), 7.21 (m, 28-CH), 7.39 (d, 1H, J = 7.9 Hz, 27-CH), 7.42 (s, 25-CH), 7.76 (s, 30-NH), 7.96 (s, 1-NH), 8.98 (t, 1H, J = 5.6 Hz, 17-NH); 13C-NMR (126 MHz, CDCl3): δ (ppm) 24.6 (32-CH3), 25.9 (19-CH2), 31.6 (9b-CH2), 32.2 (22,22′-(CH2)2), 39.3 (18-CH2), 42.0 (23-CH), 53.0 (3-CH), 54.1 (21,21′-(CH2)2), 56.3 (20-CH2), 59.1 (7-OCH3), 58.7 (9C-CH2OH), 61.5 (9a-OCH2), 68.1 (6-OCH2), 101.4 (4-C), 116.2 (d, J = 17.7, 11-CH), 117.4 (d, J = 17.3, 14-CH), 117.9 (27-CH), 118.1 (25-CH), 122.8 (29-CH), 123.1 (dd, J = 6.3, 3.6, 15-CH), 129.0 (28-CH), 137.6 (10-C), 138.3 (26-C), 146.5 (24-C), 146.6 (5-C), 150.0 (12- or 13-CF), 150.1 (12- or 13-CF), 152.2 (2-C), 153.3 (16-C), 164.5 (8-C), 168.6 (31-C); 19F-NMR (471 MHz, CDCl3): δ (ppm) -136.7 (m, 12- or 13-CF), -138.1 (m, 12- or 13-CF); MS: m/z (%) 658 (48), 345 (5), 302 (100), 259 (13), 231 (14), 160 (5), 113 (5); HRMS: Calcd. for C33H47F2N5O7Na [M + Na]+: 680.3271. Found: 680.2858.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}