Dibenzo[1,2,5]thiadiazepines Are Non-Competitive GABAA Receptor Antagonists

Abstract
: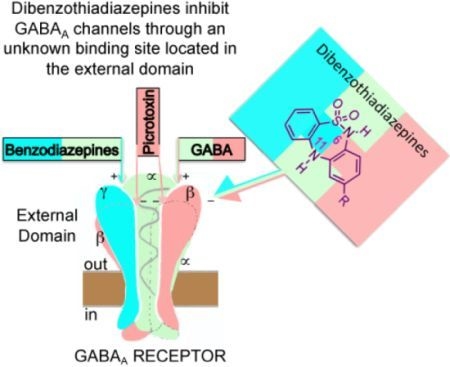
1. Introduction

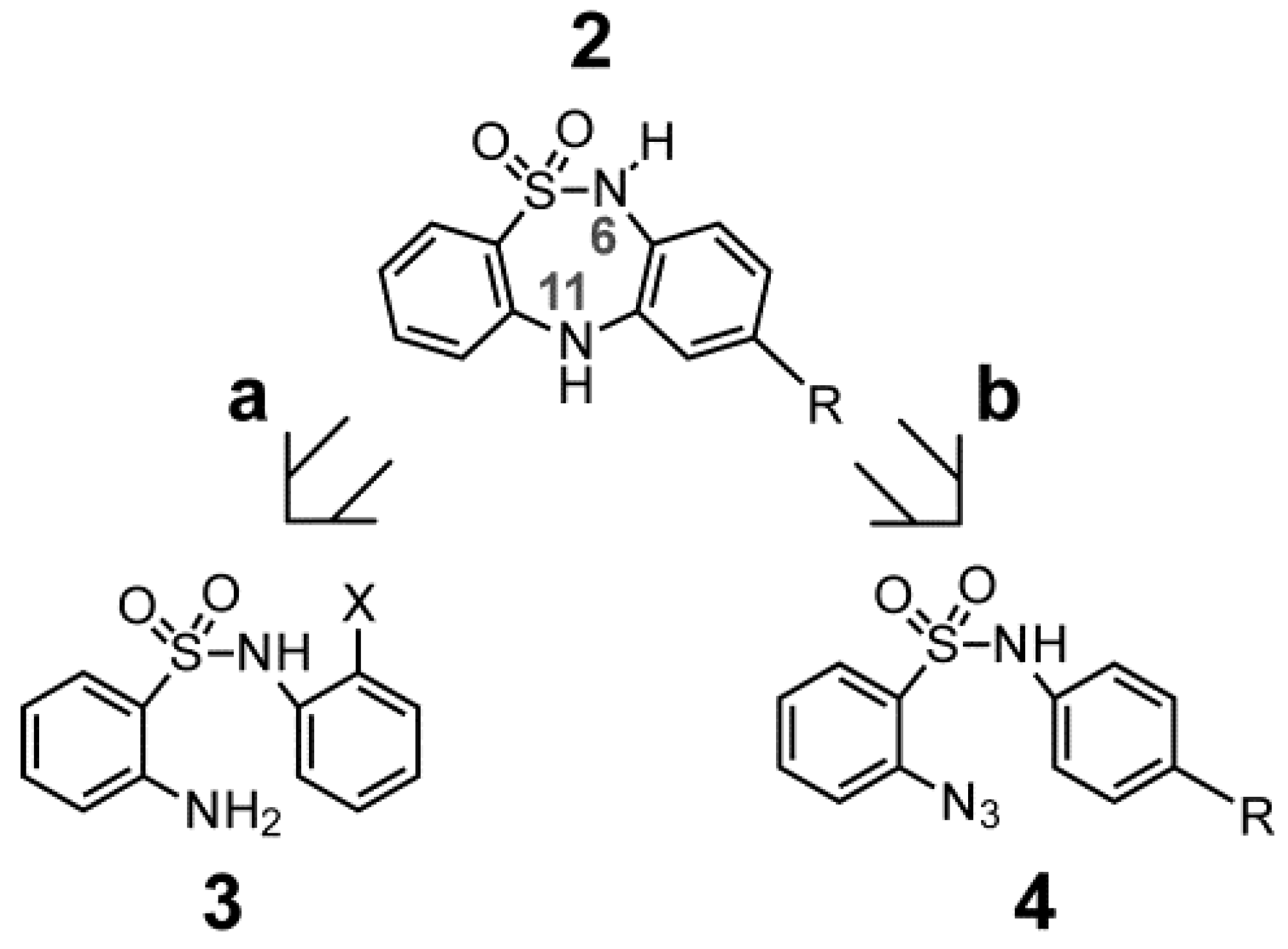
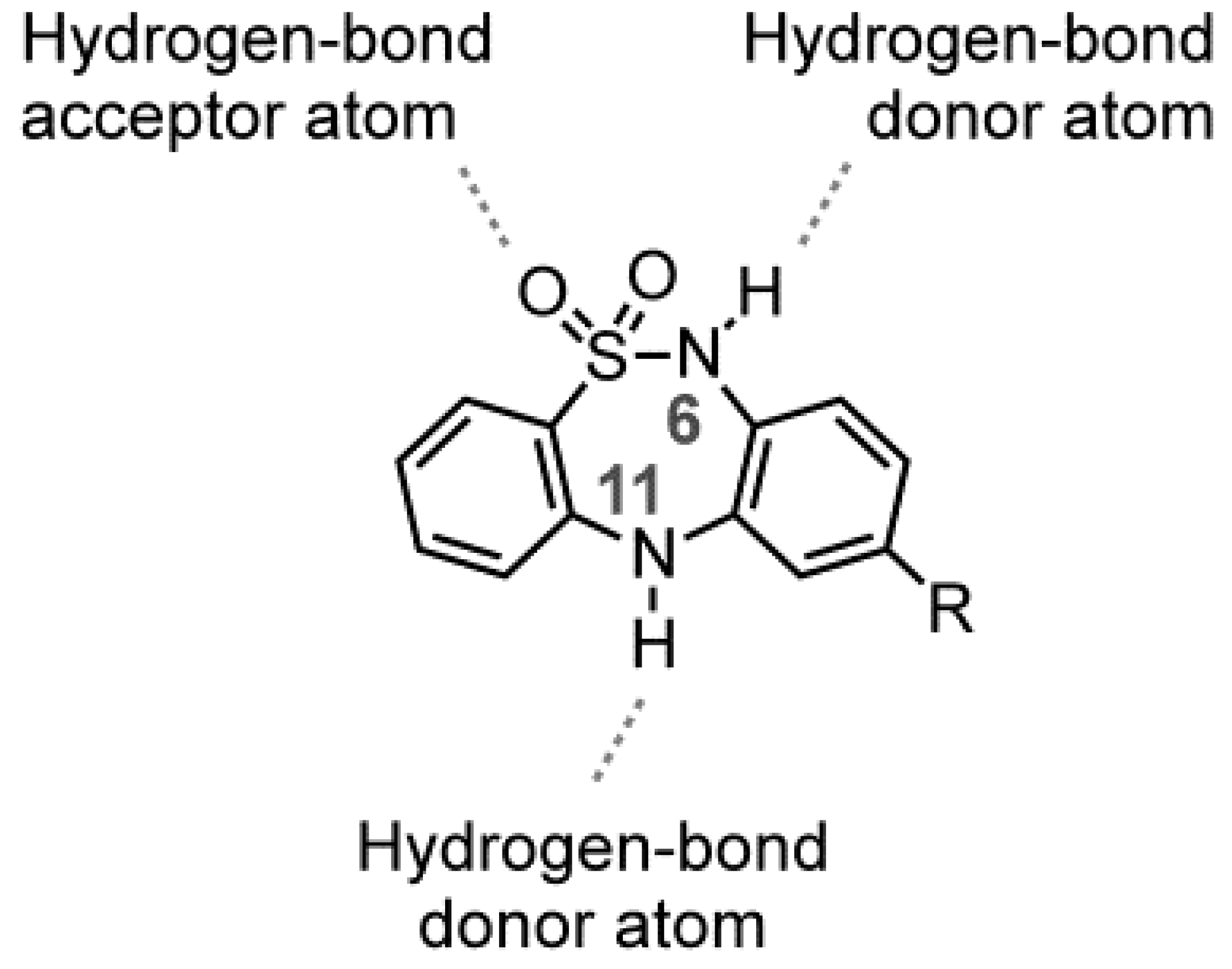
2. Results and Discussion
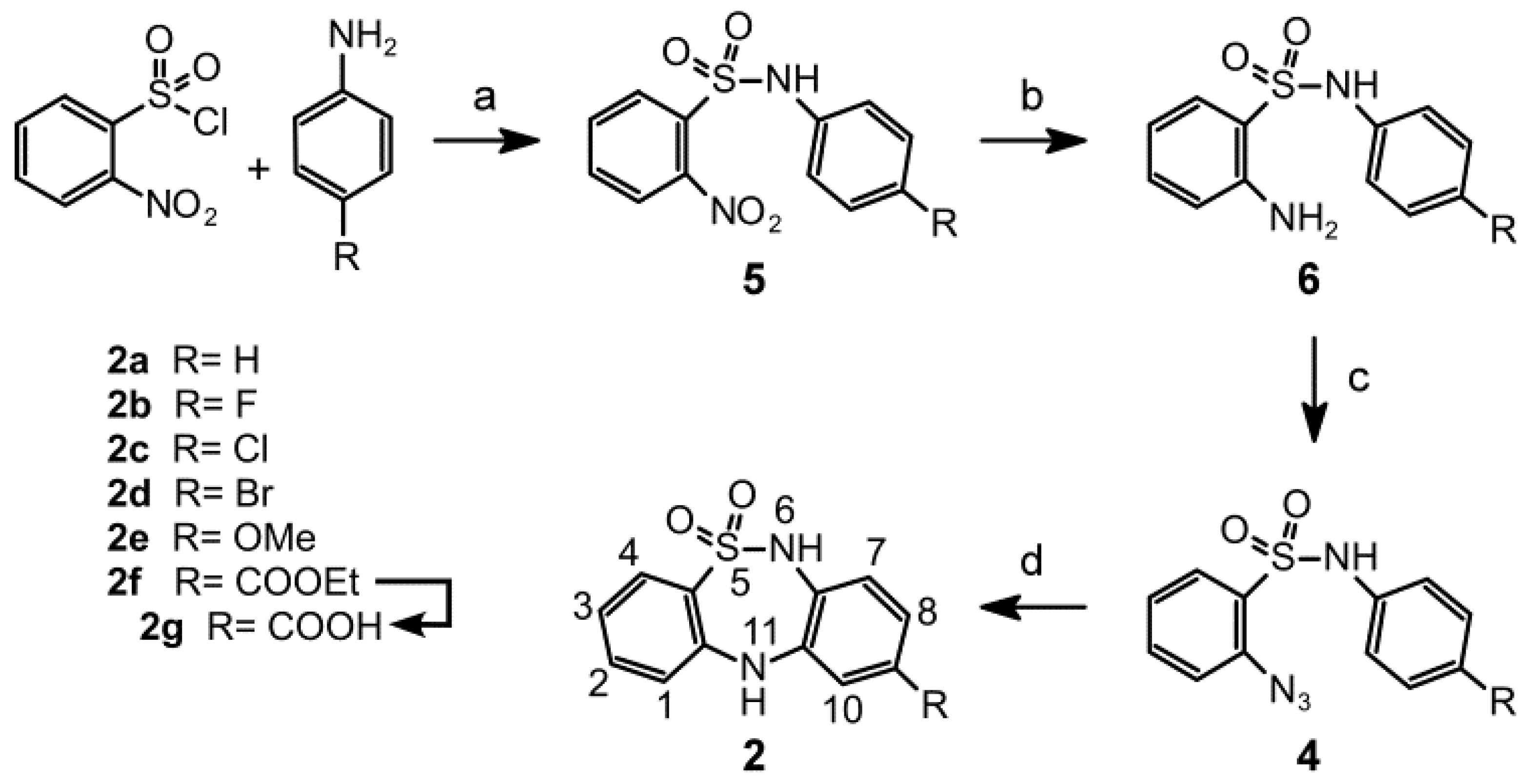
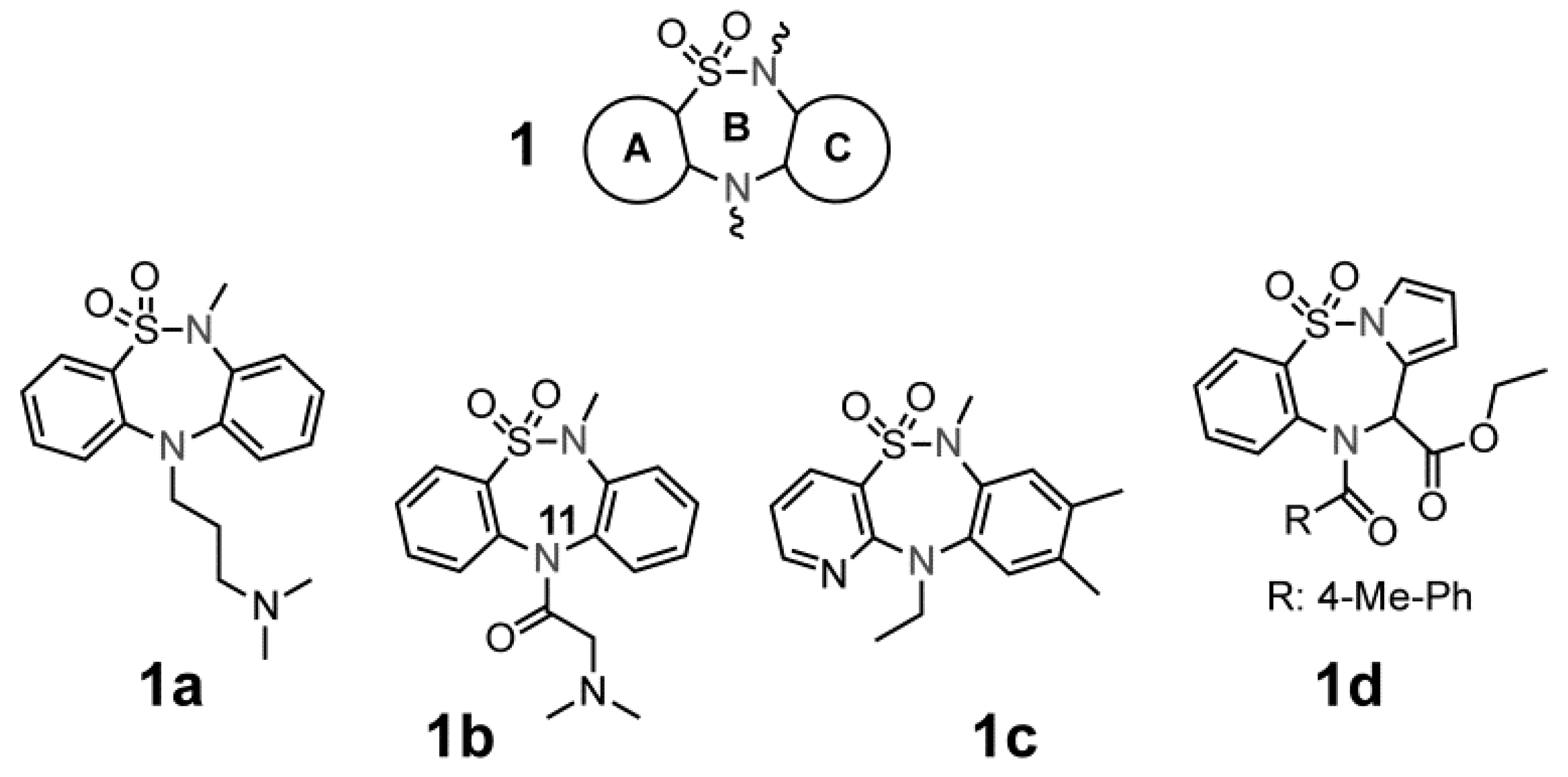
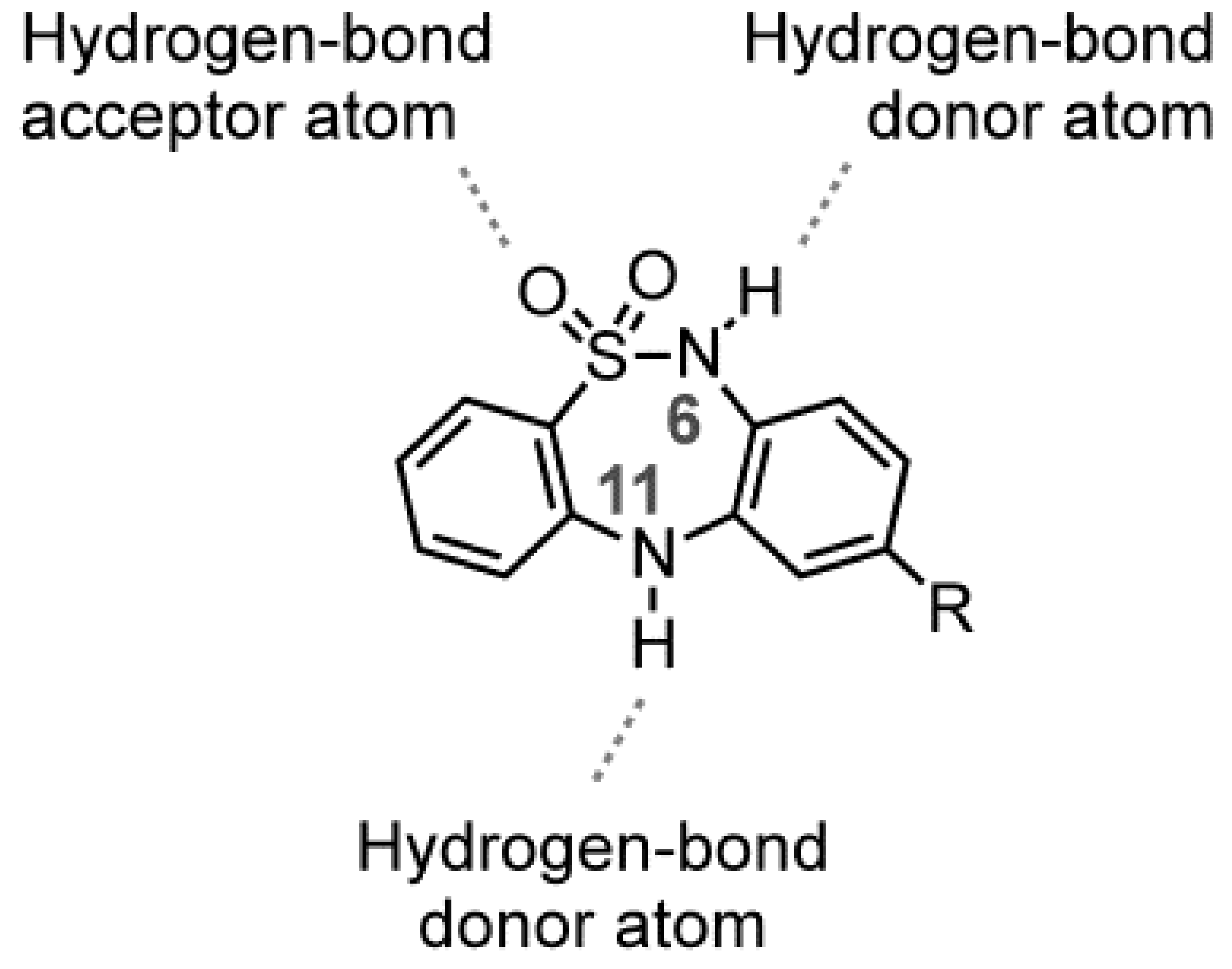
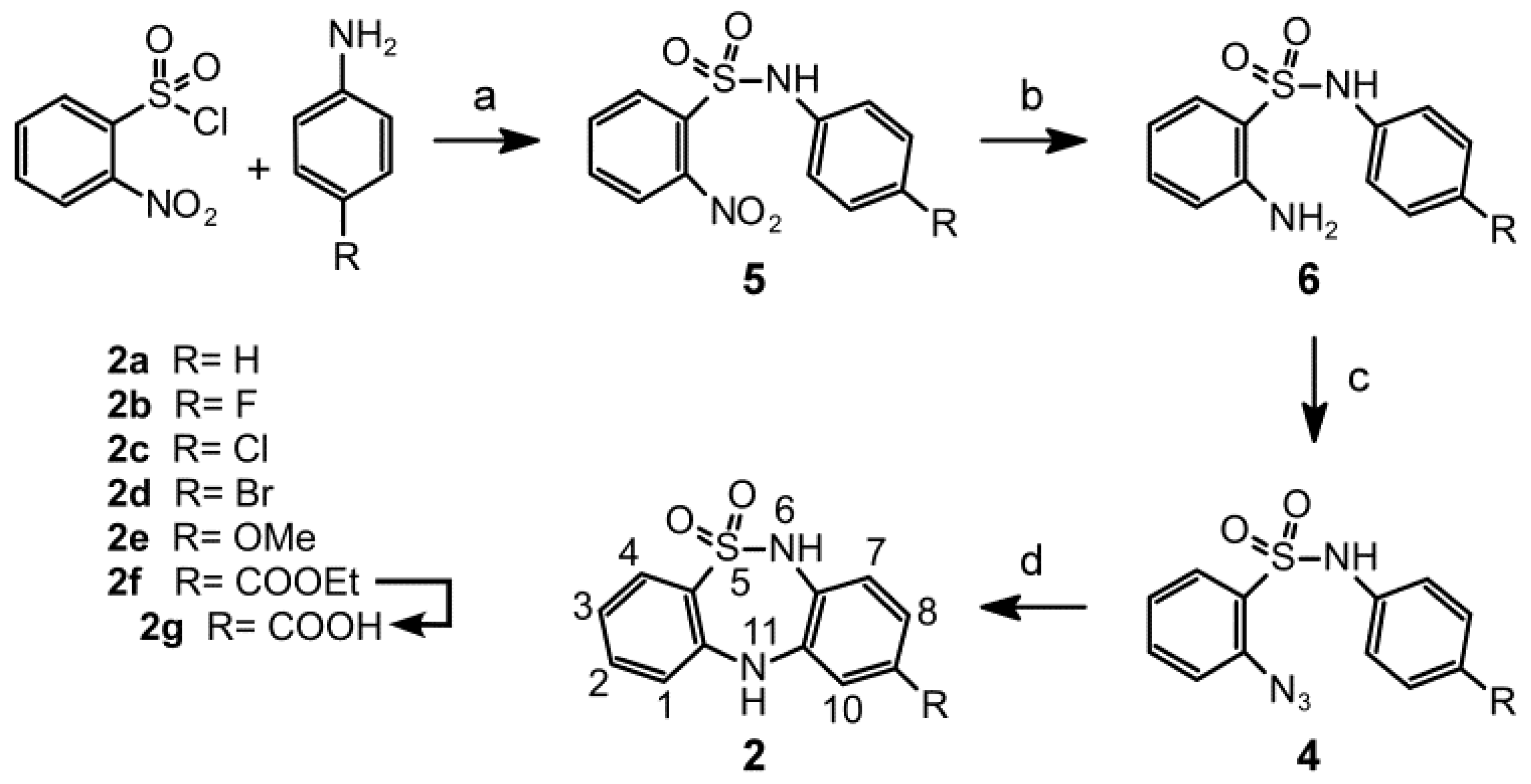
2.1. Chemistry
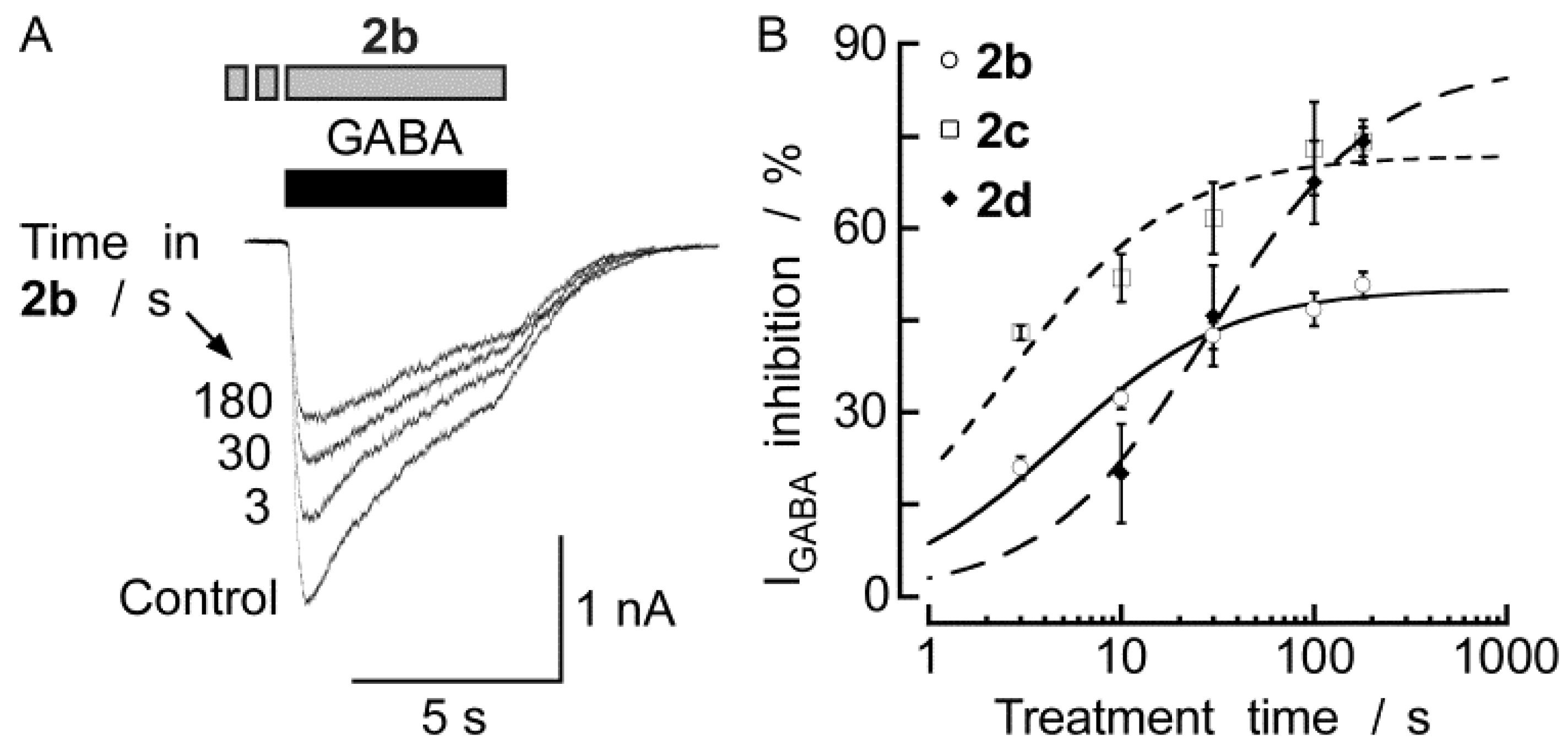
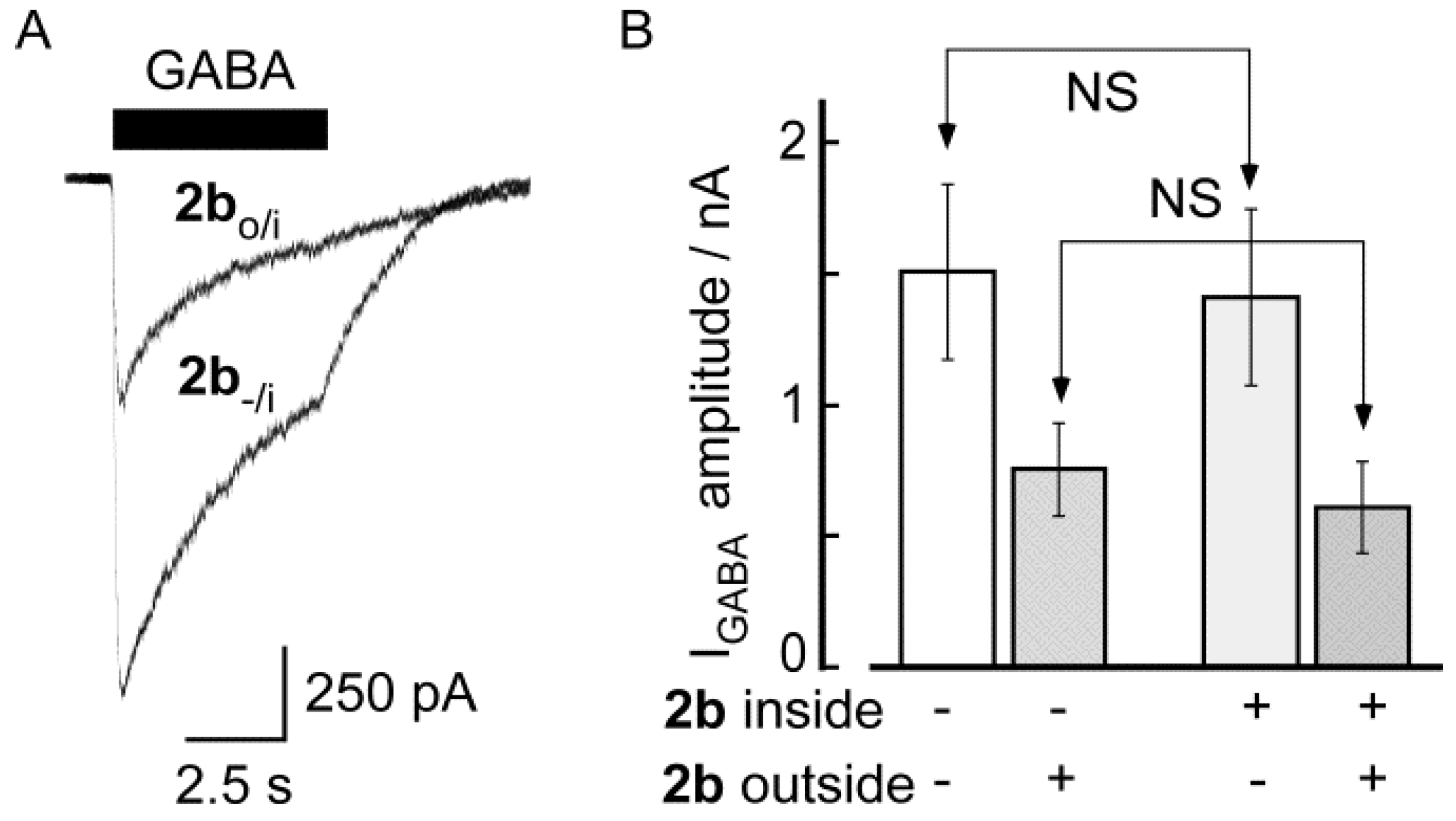
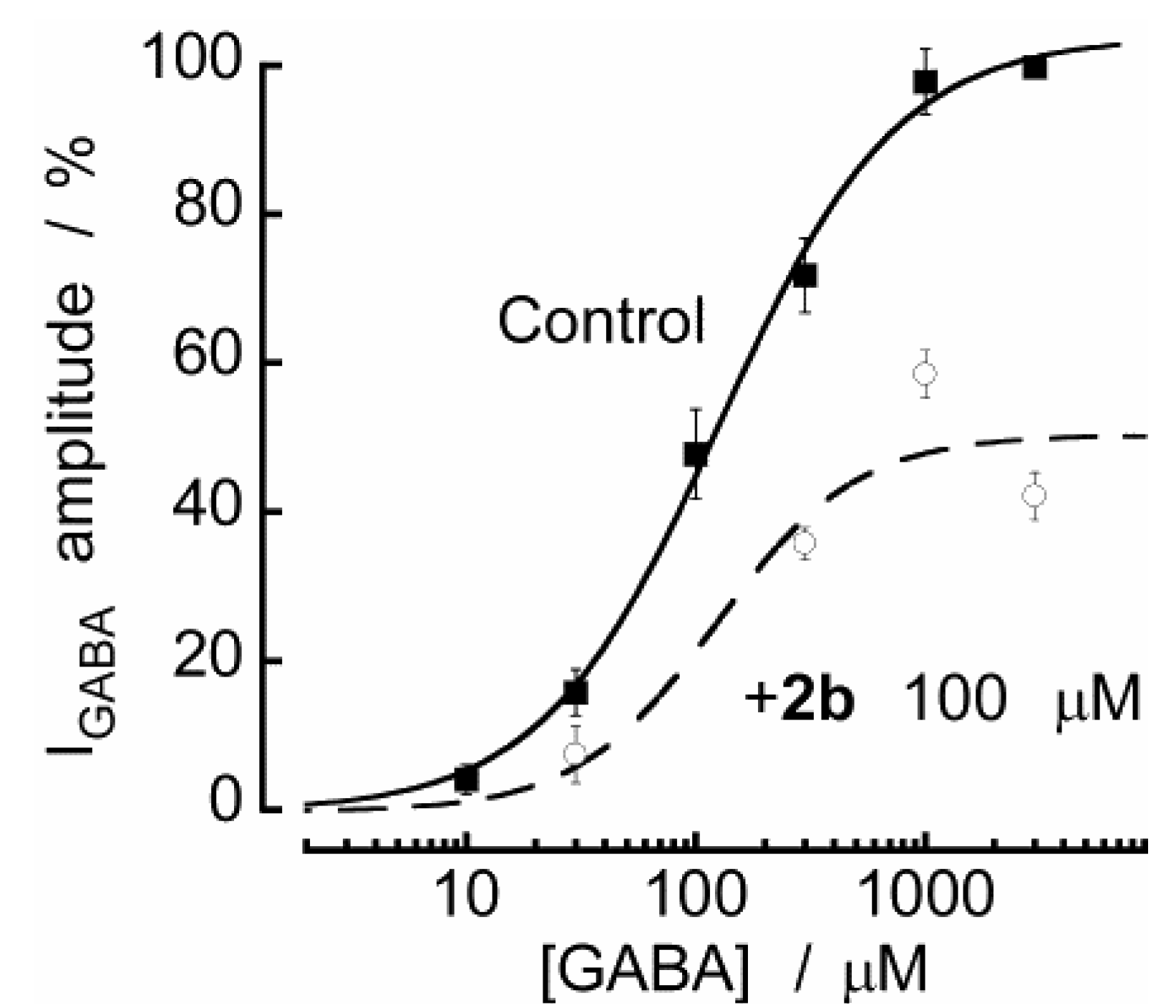
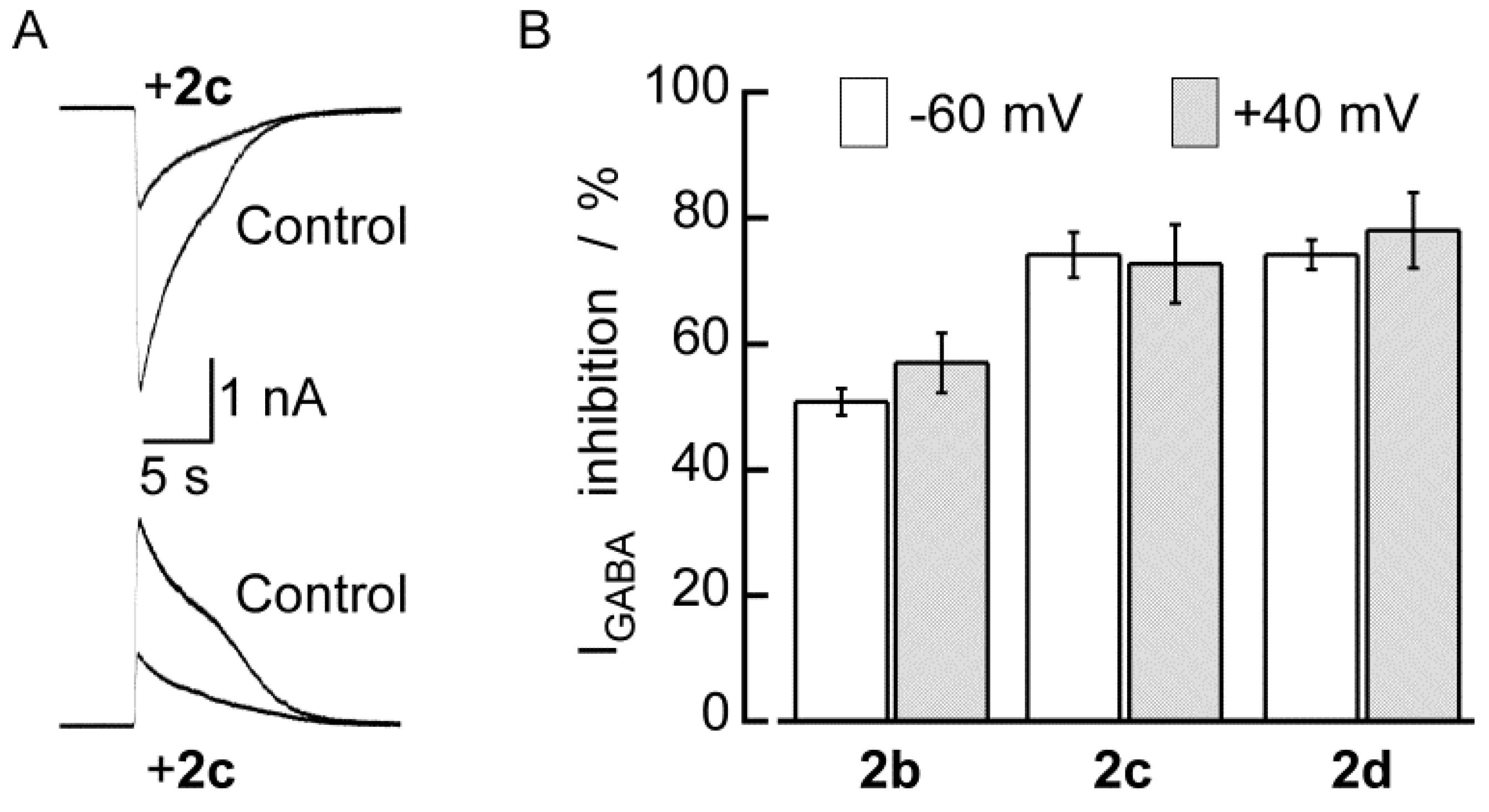
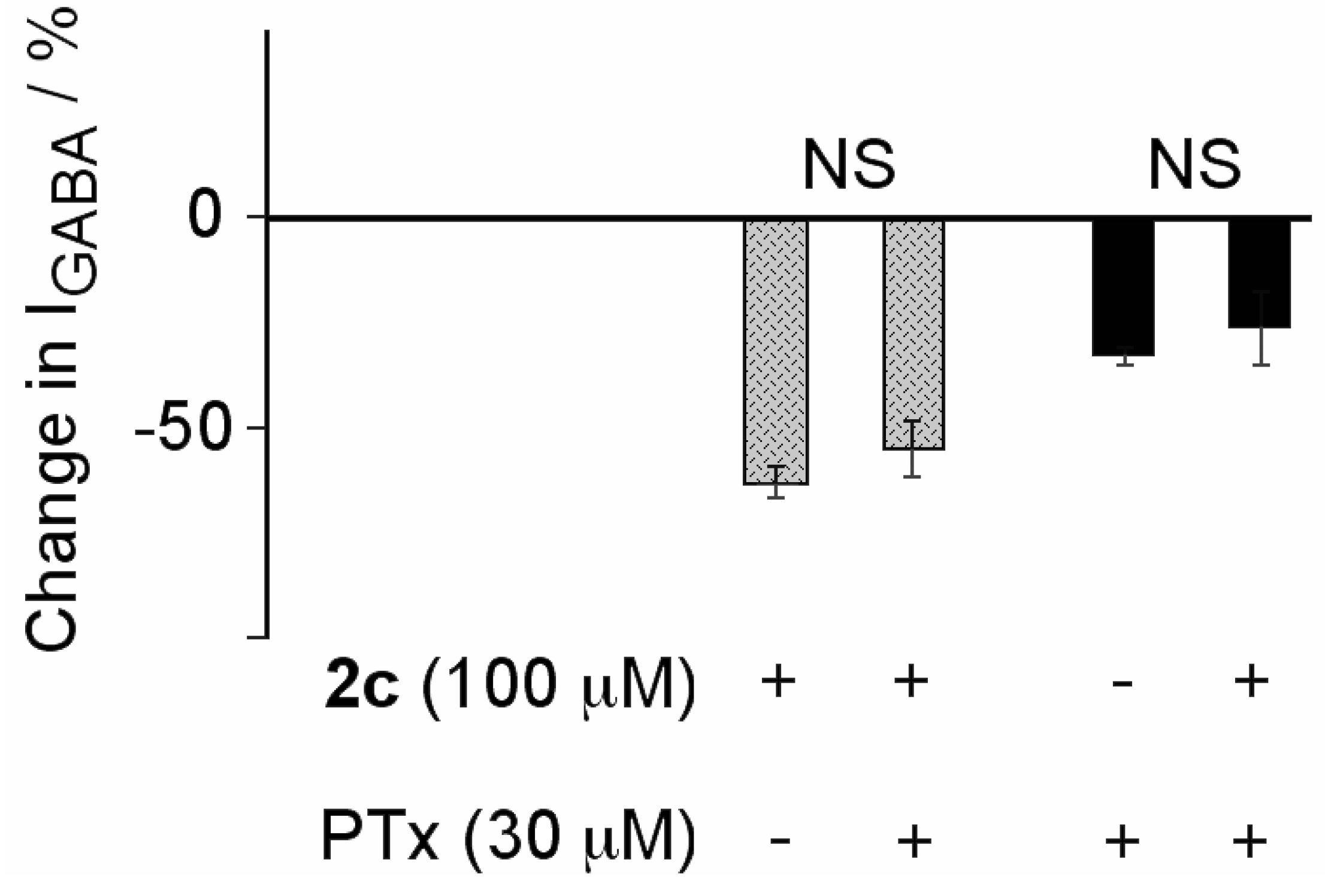
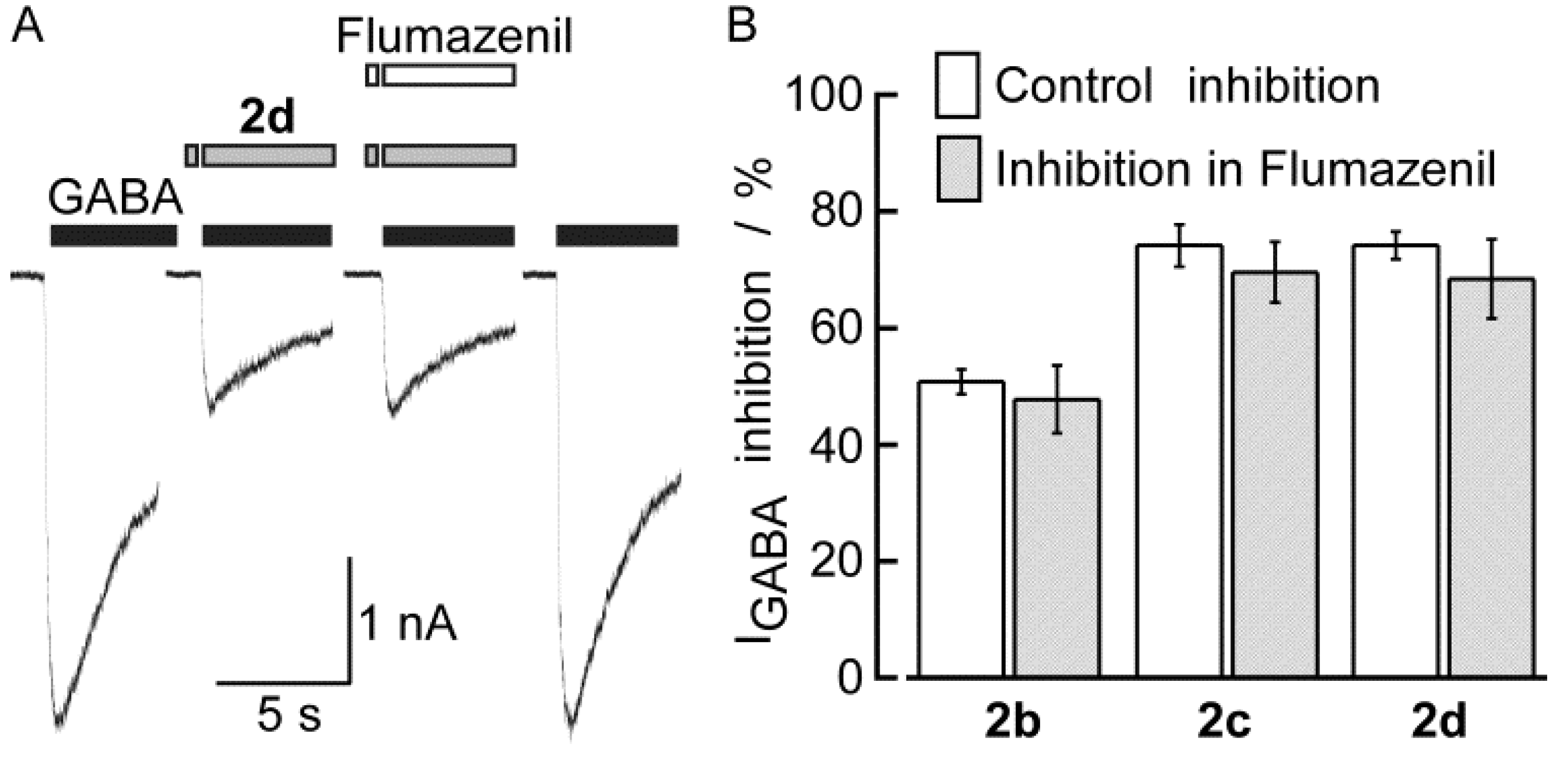
2.2. Biological Results
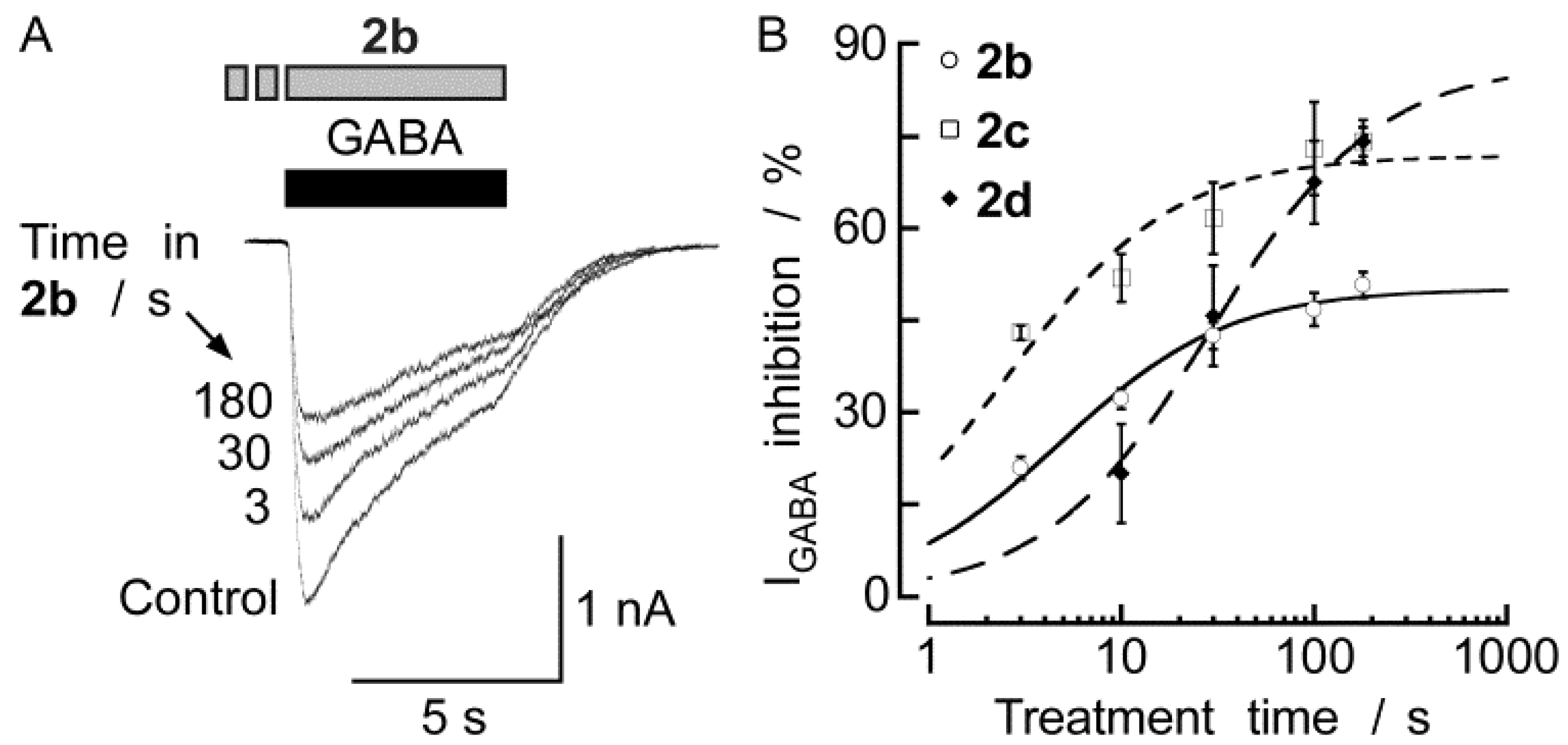

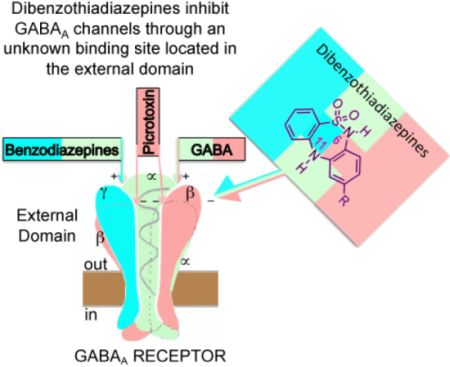{kind=link}
{kind=link}
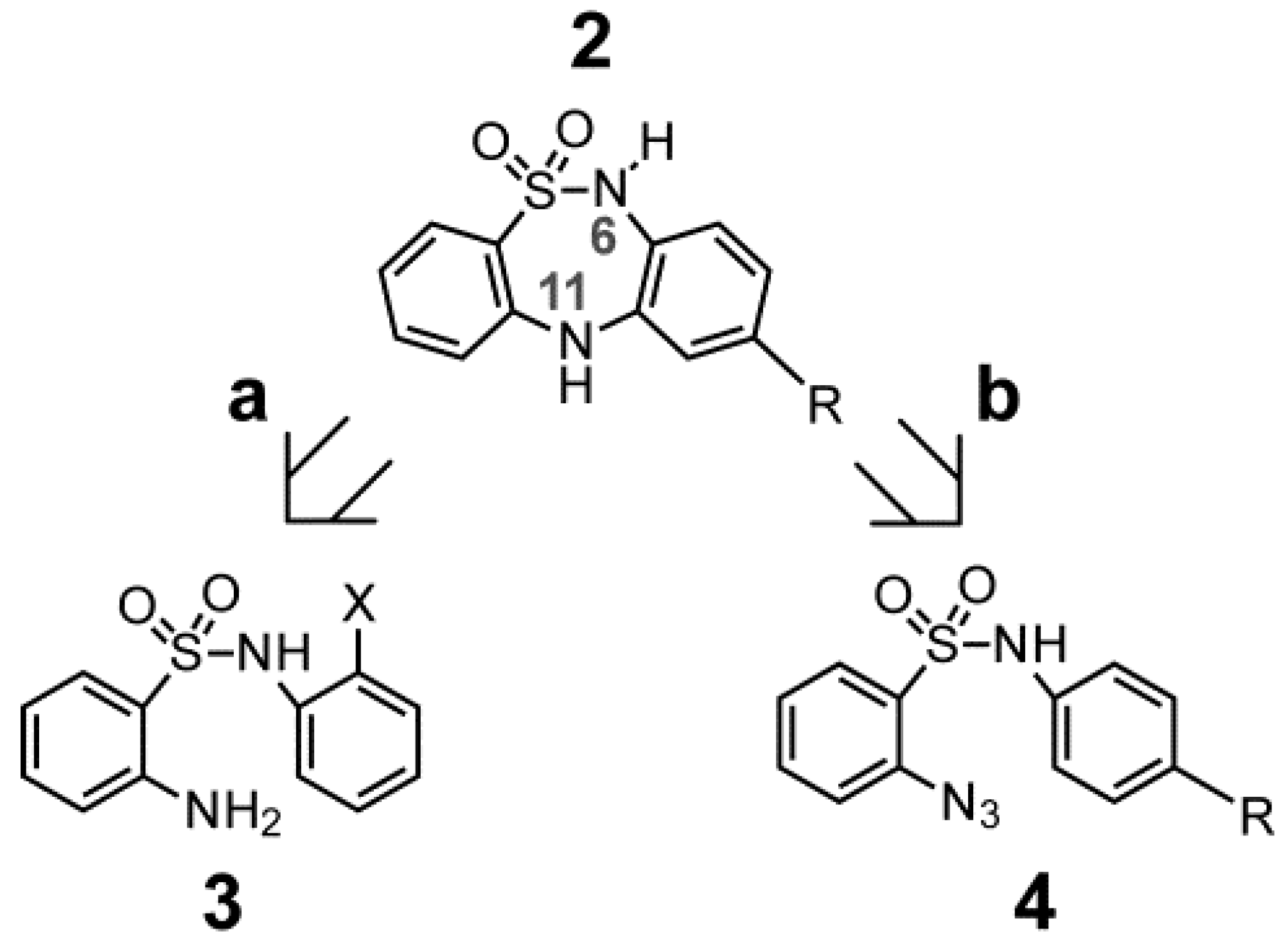{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Percentage of inhibition [a] | p IC50/M | cLog P [b] | Yield/% | mp/°C |
|---|---|---|---|---|---|
| 2a | 28.4 ± 1.4 (5) | ND | 2.18 | 69 | 198 |
| 2b | 50.8 ± 2.1 (12) | 3.98 | 1.50 | 70 | 202 |
| 2c | 74.2 ± 3.6 (6) | 4.30 | 2.69 | 79 | 248 |
| 2d | 74.2 ± 2.4 (10) | 4.32 | 2.97 | 85 | 250 |
| 2e | 43.7 ± 3.8 (3) | ND | 1.92 | 67 | 171 |
| 2f | 47.1 ± 3.7 (7) | ND | 2.25 | 12 | 227 |
| 2g | 16.0 ± 2.4 (6) | ND | 1.87 | 100 | 350 |
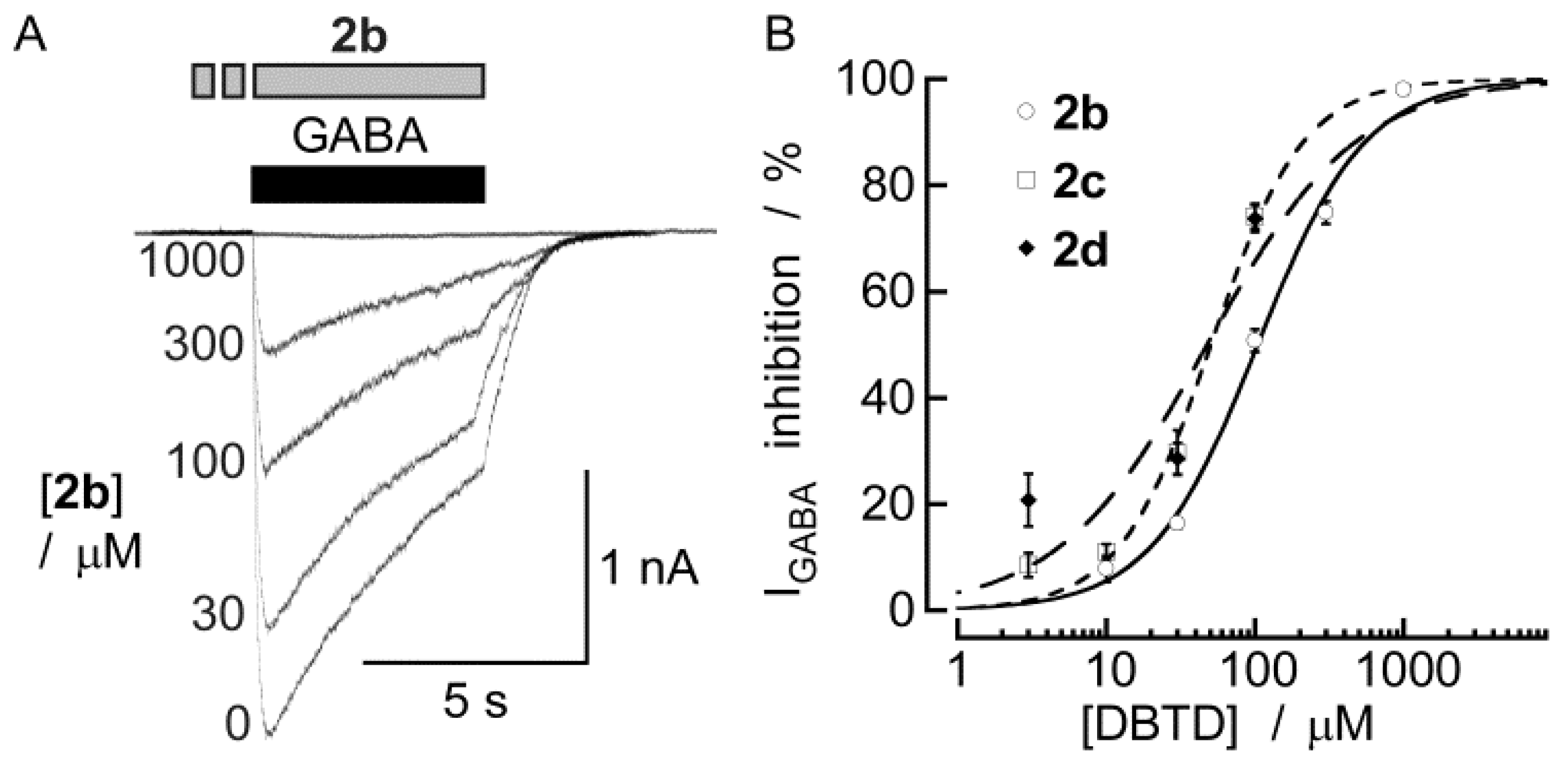
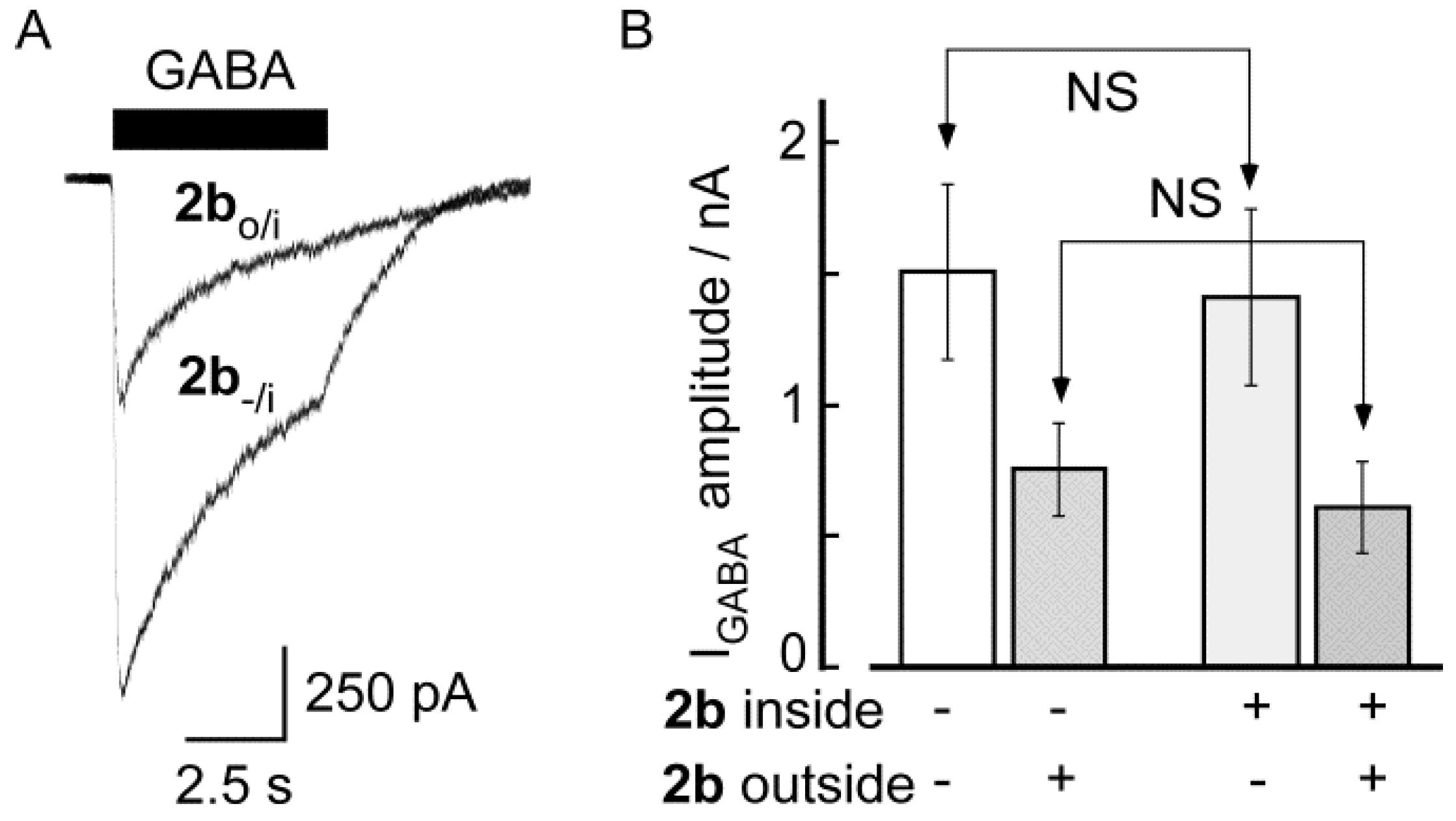
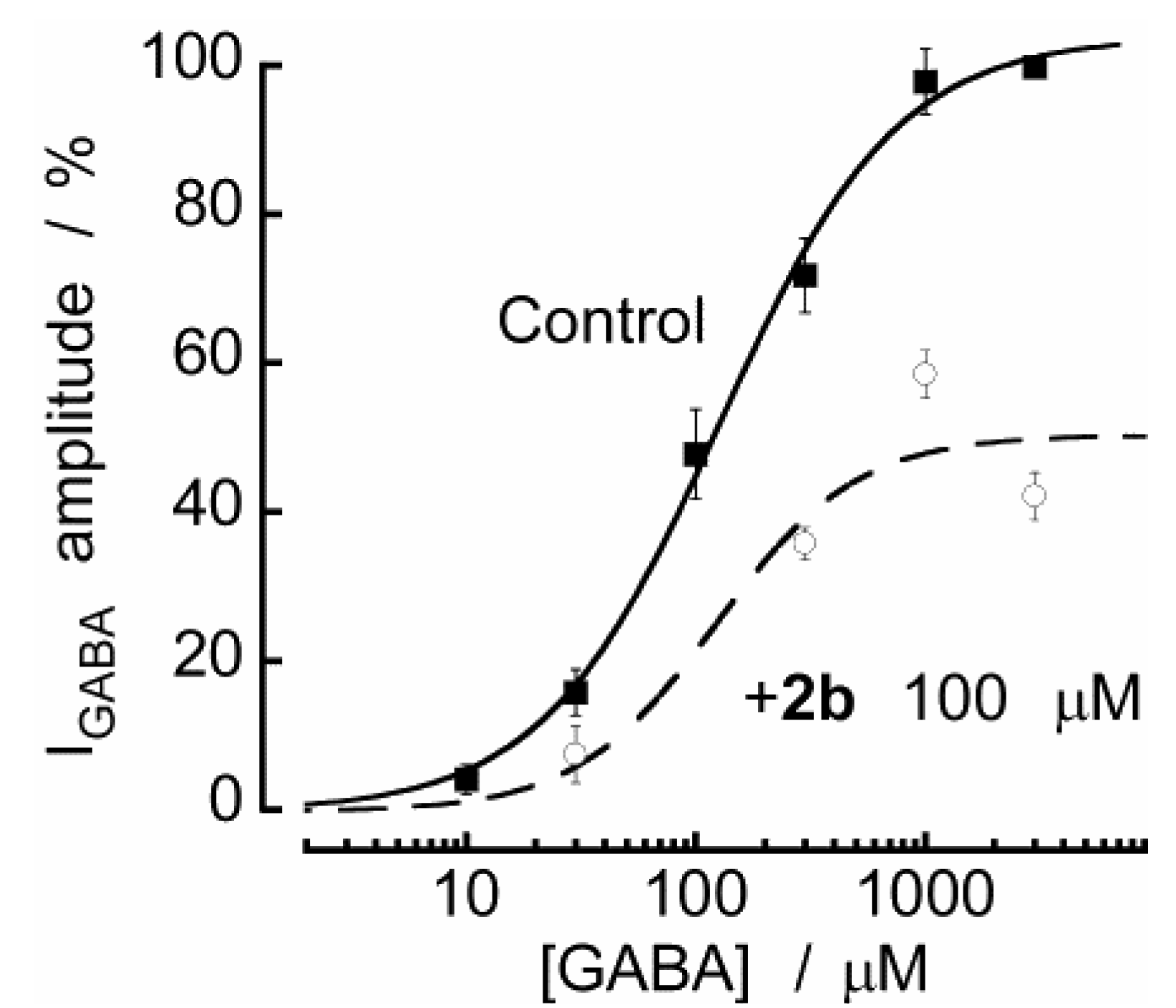
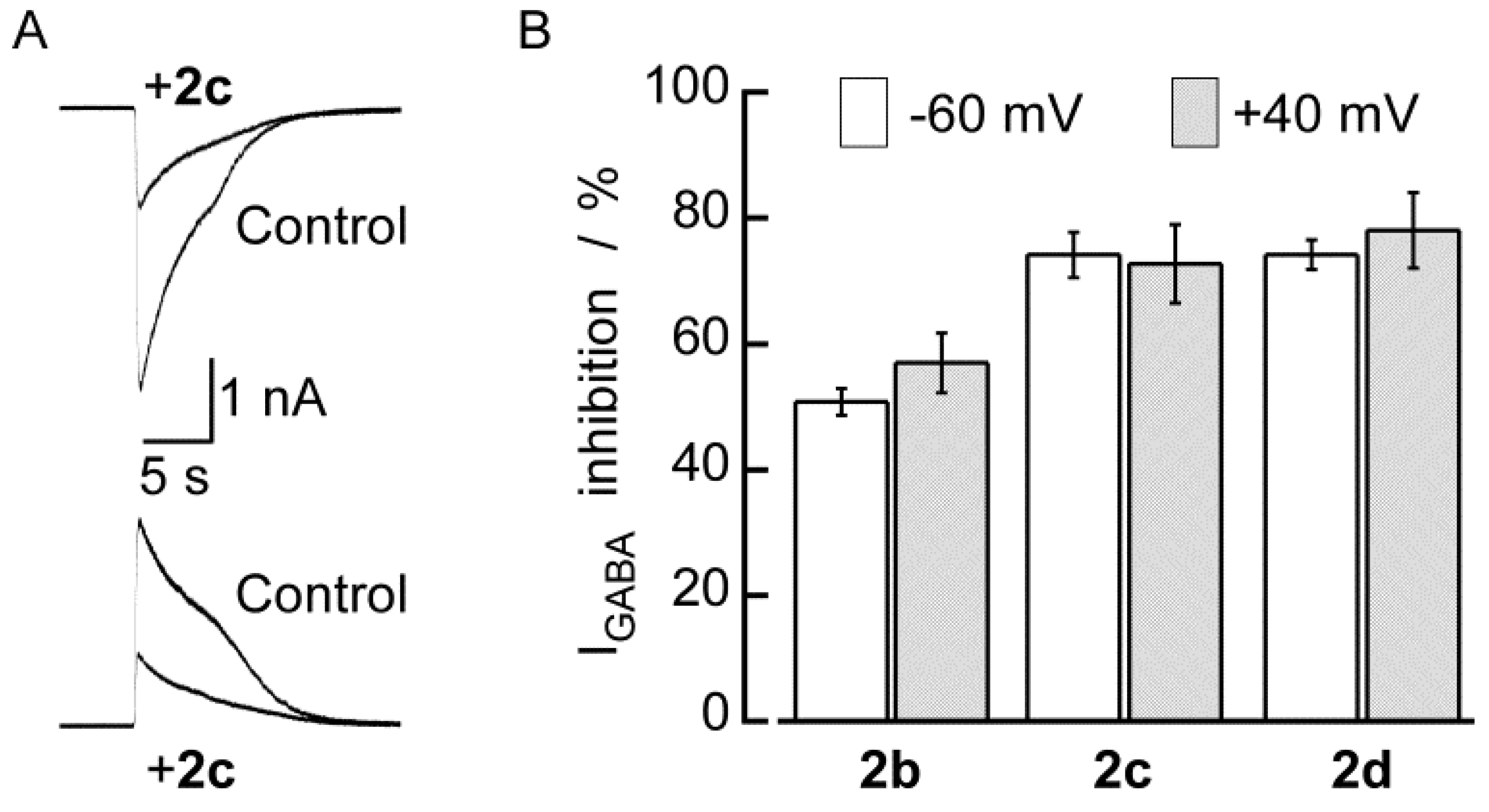
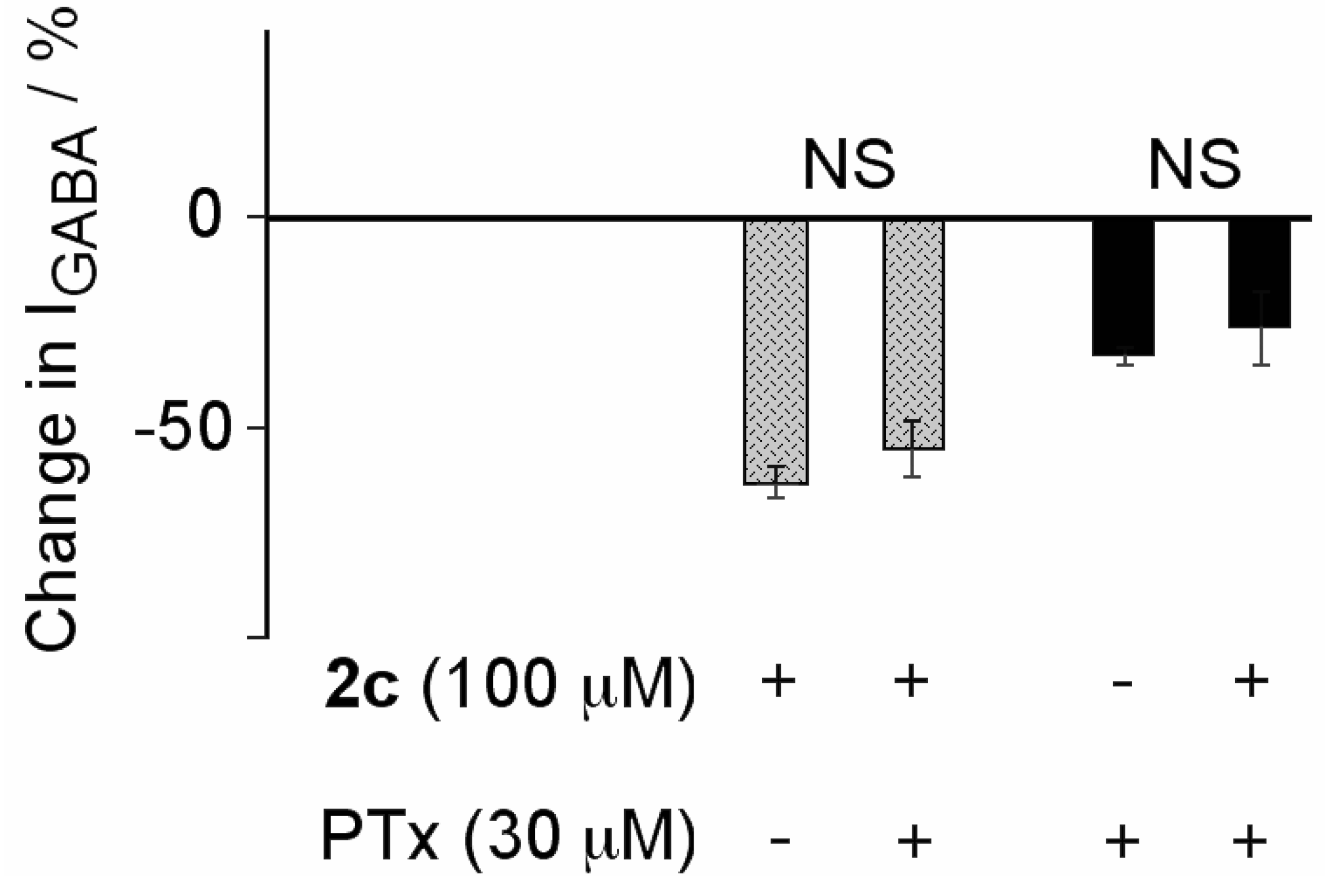
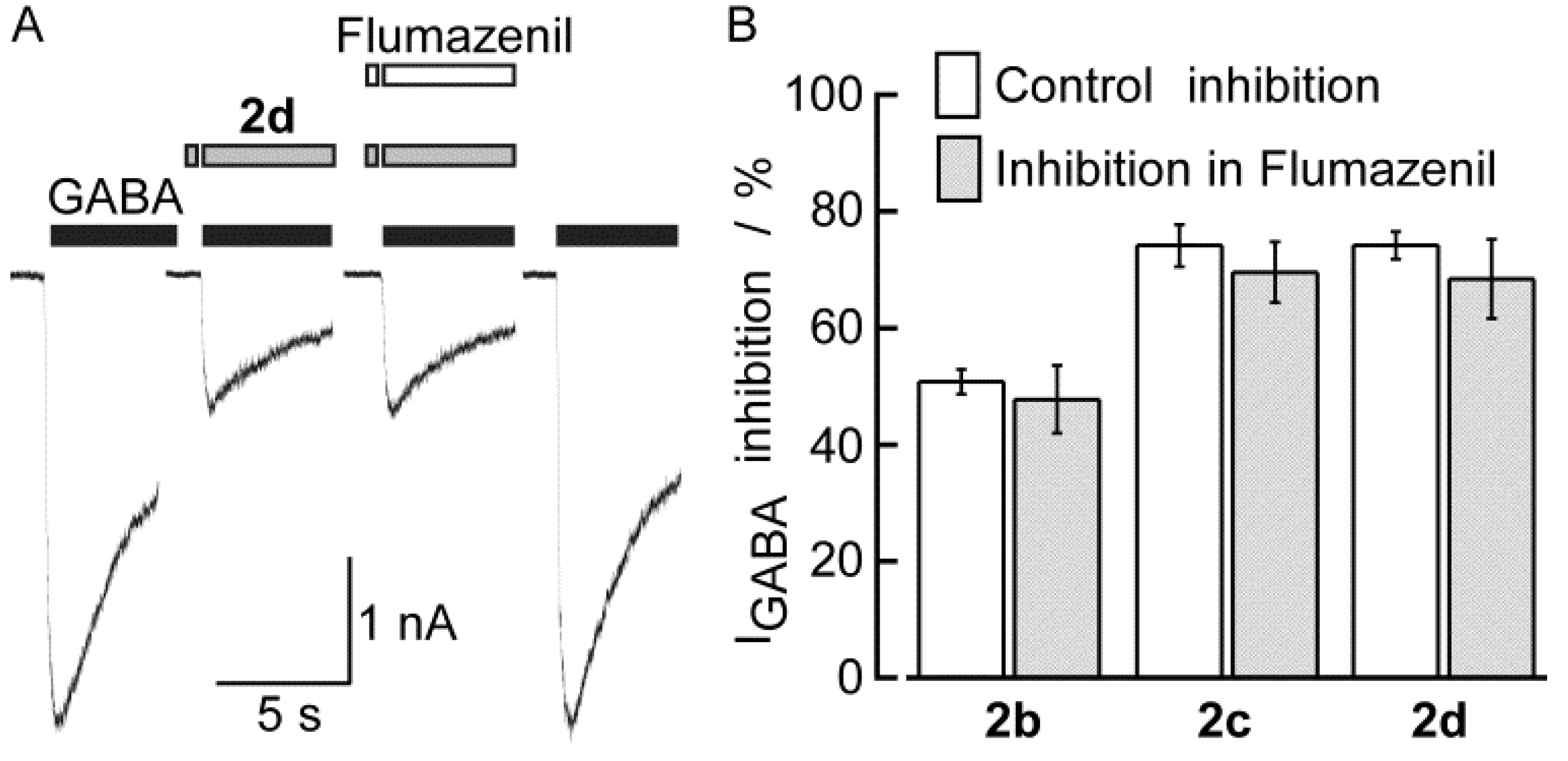
3. Experimental
3.1. Chemistry
3.1.1. General
3.1.2. General Procedures for Synthesizing N-(4-(R)-Phenyl)-2-nitrobenzenesulfonamides 5a–f
3.1.3. General Procedures for the Synthesis of 2-Amino-N-(4-(R) phenyl)benzenesulfonamides 6a-f
3.1.4. General Procedures for Synthesizing 2-Azido-N-(4-(R)phenyl)benzenesulfonamides 4a-f
3.1.5. General Procedures for Synthesizing 9-(R)-6,11-Dihydrodibenzo[c,f][1,2,5]thiadiazepine-5,5-dioxides 2a–f
3.1.6. Procedure for Synthesizing 6,11-Dihydrodibenzo[c,f][1,2,5]thiadiazepine-9-carboxylic acid 5,5-dioxide (2g)
3.2. Biological Methods
3.2.1. Primary Cultures of the Myenteric Neurons
3.2.2. Whole-Cell Recordings of the Membrane Currents Induced by GABA
3.2.3. Solutions and Reagents
3.2.4. Data Analysis
3.2.5. Theoretical Calculations
4. Conclusions
Acknowledgments
Conflicts of Interest
- Sample Availability: Contact the corresponding authors.
References
- Weber, A. 5,5-dioxodibenzo[1,2,5]-thiadiazepines and intermediates therefor. U.S. Patent 3268557, 1966. [Google Scholar]
- Weber, A.; Frossard, J. Research on tricyclic psychotropic drugs. Dibenzothiadizepines. Ann. Pharm. Fr. 1966, 24, 445–450. [Google Scholar]
- Weber, A. 5,5-dioxodibenzo[1,2,5]thiadiazepines derivatives and method of use. U.S. Patent 3274058, 1966. [Google Scholar]
- Giannotti, D.; Viti, G.; Sbraci, P.; Pestellini, V.; Volterra, G.; Borsini, F.; Lecci, A.; Meli, A.; Dapporto, P.; Paoli, P. New dibenzothiadiazepine derivatives with antidepressant activities. J. Med. Chem. 1991, 34, 1356–1362. [Google Scholar] [CrossRef]
- Bellarosa, D.; Antonelli, G.; Bambacioni, F.; Giannotti, D.; Viti, G.; Nannicini, R.; Giachetti, A.; Dianzani, F.; Witvrouw, M.; Pauwels, R.; et al. New arylpyrido-diazepine and -thiodiazepine derivatives are potent and highly selective HIV-1 inhibitors targeted at the reverse transcriptase. Antiviral. Res. 1996, 30, 109–124. [Google Scholar] [CrossRef]
- Silvestri, R.; Marfe, G.; Artico, M.; La Regina, G.; Lavecchia, A.; Novellino, E.; Morgante, E.; Di Stefano, C.; Catalano, G.; Filomeni, G.; et al. Pyrrolo[1,2-b][1,2,5]benzothiadiazepines (PBTDs): A new class of agents with high apoptotic activity in chronic myelogenous leukemia K562 cells and in cells from patients at onset and who were imatinib-resistant. J. Med. Chem. 2006, 49, 5840–5844. [Google Scholar] [CrossRef]
- Goldberg, I. Ueber Phenylirungen bei Gegenwart von Kupfer als Katalysator. Ber. Dtsch. Chem. Ges. 1906, 39, 1691–1692. [Google Scholar] [CrossRef]
- Altamura, M.; Fedi, V.; Giannotti, D.; Paoli, P.; Rossi, P. Privileged structures: Synthesis and structural investigations on tricyclic sulfonamides. New J. Chem. 2009, 33, 2219–2231. [Google Scholar] [CrossRef]
- Jian, H.H.; Tour, J.M. En route to surface-bound electric field-driven molecular motors. J. Org. Chem. 2003, 68, 5091–5103. [Google Scholar] [CrossRef]
- Tsao, M.L.; Gritsan, N.; James, T.R.; Platz, M.S.; Hrovat, D.A.; Borden, W.T. Study of the chemistry of ortho- and para-biphenylnitrenes by laser flash photolysis and time-resolved IR experiments and by B3LYP and CASPT2 calculations. J. Am. Chem. Soc. 2003, 125, 9343–9358. [Google Scholar]
- Shou, W.G.; Li, J.A.; Guo, T.X.; Lin, Z.Y.; Jia, G.C. Ruthenium-Catalyzed Intramolecular Amination Reactions of Aryl- and Vinylazides. Organometallics 2009, 28, 6847–6854. [Google Scholar] [CrossRef]
- Stokes, B.J.; Jovanovic, B.; Dong, H.J.; Richert, K.J.; Riell, R.D.; Driver, T.G. Rh-2(II)-Catalyzed Synthesis of Carbazoles from Biaryl Azides. J. Org. Chem. 2009, 74, 3225–3228. [Google Scholar]
- Cherney, R.J.; Duan, J.J.W.; Voss, M.E.; Chen, L.H.; Wang, L.; Meyer, D.T.; Wasserman, Z.R.; Hardman, K.D.; Liu, R.Q.; Covington, M.B.; et al. Design, synthesis, and evaluation of benzothiadiazepine hydroxamates as selective tumor necrosis factor-alpha converting enzyme inhibitors. J. Med. Chem. 2003, 46, 1811–1823. [Google Scholar]
- Giannotti, D.; Viti, G.; Nannicini, R.; Pestellini, V.; Bellarosa, D. Bioorg. Med. Chem. Lett. 1995, 5, 1461–1466. [CrossRef]
- Enna, S.J. The GABA Receptors. In The Receptors; Enna, S.J., Mohler, H., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2012. [Google Scholar]
- Krystal, J.H.; Sanacora, G.; Blumberg, H.; Anand, A.; Charney, D.S.; Marek, G.; Epperson, C.N.; Goddard, A.; Mason, G.F. Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol. Psychiatr. 2002, 7, S71–S80. [Google Scholar] [CrossRef]
- Brickley, S.G.; Mody, I. Extrasynaptic GABA(A) receptors: Their function in the CNS and implications for disease. Neuron 2012, 73, 23–34. [Google Scholar]
- Zhou, X.; Galligan, J.J. GABA(A) receptors on calbindin-immunoreactive myenteric neurons of guinea pig intestine. J. Auton. Nerv. Syst. 2000, 78, 122–135. [Google Scholar] [CrossRef]
- Karanjia, R.; Garcia-Hernandez, L.M.; Miranda-Morales, M.; Somani, N.; Espinosa-Luna, R.; Montano, L.M.; Barajas-Lopez, C. Cross-inhibitory interactions between GABAA and P2X channels in myenteric neurones. Eur. J. Neurosci. 2006, 23, 3259–3268. [Google Scholar] [CrossRef]
- Miranda-Morales, M.; Garcia-Hernandez, L.M.; Ochoa-Cortes, F.; Espinosa-Luna, R.; Naranjo-Rodriguez, E.B.; Barajas-Lopez, C. Cross-talking between 5-HT3 and GABAA receptors in cultured myenteric neurons. Synapse 2007, 61, 732–740. [Google Scholar] [CrossRef]
- Krehan, D.; Storustovu, S.I.; Liljefors, T.; Ebert, B.; Nielsen, B.; Krogsgaard-Larsen, P.; Frolund, B. Potent 4-arylalkyl-substituted 3-isothiazolol GABA(A) competitive/noncompetitive antagonists: synthesis and pharmacology. J. Med. Chem. 2006, 49, 1388–1396. [Google Scholar]
- Kenakin, R. Pharmacologic Analysis of Drug–Receptor Interaction, 2nd ed; Raven Press Ltd.: New York, NY, USA, 1993. [Google Scholar]
- Olsen, R.W. Picrotoxin-like channel blockers of GABAA receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 6081–6082. [Google Scholar] [CrossRef]
- Sieghart, W.; Ramerstorfer, J.; Sarto-Jackson, I.; Varagic, Z.; Ernst, M. A novel GABAA receptor pharmacology: Drugs interacting with the a(+) b(-) interface. Br. J. Pharmacol. 2012, 166, 476–485. [Google Scholar] [CrossRef]
- Saeed, A.; Rama, N.H. Synthesis of some new 2-Aryl-2,3-dihydro-1,2,3-benzothiadiazol-1,1-dioxides. J. Chem. Soc. Pak. 1997, 19, 236–239. [Google Scholar]
- Barajas-Lopez, C.; Peres, A.L.; Espinosa-Luna, R. Cellular mechanisms underlying adenosine actions on cholinergic transmission in enteric neurons. Am. J. Physiol. 1996, 271, C264–C275. [Google Scholar]
- Wellendorph, P.; Brauner-Osborne, H. Molecular basis for amino acid sensing by family C G-protein-coupled receptors. Br. J. Pharmacol. 2009, 156, 869–884. [Google Scholar] [CrossRef]
- Cherubini, E.; North, R.A. Actions of gamma-aminobutyric acid on neurones of guinea-pig myenteric plexus. Br. J. Pharmacol. 1984, 82, 93–100. [Google Scholar] [CrossRef]
- Krantis, A. GABA in the Mammalian Enteric Nervous System. News Physiol. Sci. 2000, 15, 284–290. [Google Scholar]
- Frisch, G.W.T.M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, .T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; et al. Gaussian v03; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ramírez-Martínez, J.F.; González-Chávez, R.; Guerrero-Alba, R.; Reyes-Gutiérrez, P.E.; Martínez, R.; Miranda-Morales, M.; Espinosa-Luna, R.; González-Chávez, M.M.; Barajas-López, C. Dibenzo[1,2,5]thiadiazepines Are Non-Competitive GABAA Receptor Antagonists. Molecules 2013, 18, 894-913. https://doi.org/10.3390/molecules18010894
Ramírez-Martínez JF, González-Chávez R, Guerrero-Alba R, Reyes-Gutiérrez PE, Martínez R, Miranda-Morales M, Espinosa-Luna R, González-Chávez MM, Barajas-López C. Dibenzo[1,2,5]thiadiazepines Are Non-Competitive GABAA Receptor Antagonists. Molecules. 2013; 18(1):894-913. https://doi.org/10.3390/molecules18010894
Chicago/Turabian StyleRamírez-Martínez, Juan F., Rodolfo González-Chávez, Raquel Guerrero-Alba, Paul E. Reyes-Gutiérrez, Roberto Martínez, Marcela Miranda-Morales, Rosa Espinosa-Luna, Marco M. González-Chávez, and Carlos Barajas-López. 2013. "Dibenzo[1,2,5]thiadiazepines Are Non-Competitive GABAA Receptor Antagonists" Molecules 18, no. 1: 894-913. https://doi.org/10.3390/molecules18010894
APA StyleRamírez-Martínez, J. F., González-Chávez, R., Guerrero-Alba, R., Reyes-Gutiérrez, P. E., Martínez, R., Miranda-Morales, M., Espinosa-Luna, R., González-Chávez, M. M., & Barajas-López, C. (2013). Dibenzo[1,2,5]thiadiazepines Are Non-Competitive GABAA Receptor Antagonists. Molecules, 18(1), 894-913. https://doi.org/10.3390/molecules18010894