Benchmarking Ligand-Based Virtual High-Throughput Screening with the PubChem Database

Abstract
:
1. Introduction
1.1. Quantitative Structure Activity Relationships Relate Chemical Structure and Biological Activity
1.2. Molecular Descriptors Numerically Encode Chemical Structure
1.3. Machine Learning Techniques Have Viable Impact on the Generation of QSAR Models
1.4. Consensus of QSAR Models Has Potential to Improve Prediction Accuracy
1.5. Significance
2. Results and Discussion
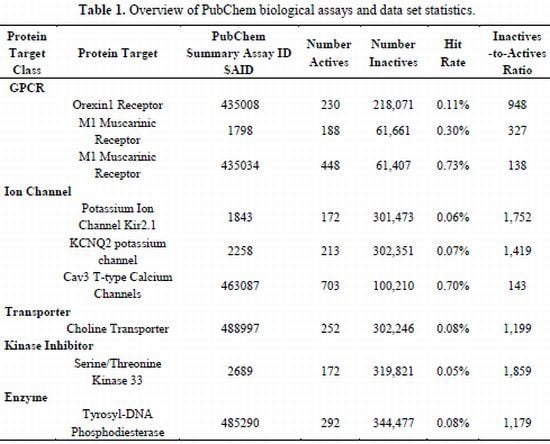
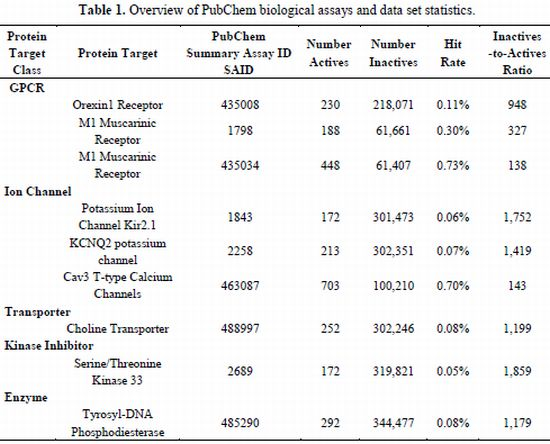
2.1. Compilation of Validated PubChem HTS Screens Provides Benchmark Data Sets for Training QSAR Models

{kind=link}
| Protein Target Class | Protein Target | PubChem Summary Assay ID SAID | Number Actives | NumberInactives | Hit Rate | Inactives -to-ActivesRatio | |
|---|---|---|---|---|---|---|---|
| GPCR | |||||||
| Orexin1 Receptor | 435008 | 230 | 218,071 | 0.11% | 948 | ||
| M1 Muscarinic Receptor | 1798 | 188 | 61,661 | 0.30% | 327 | ||
| M1 Muscarinic Receptor | 435034 | 448 | 61,407 | 0.73% | 138 | ||
| Ion Channel | |||||||
| Potassium Ion Channel Kir2.1 | 1843 | 172 | 301,473 | 0.06% | 1,752 | ||
| KCNQ2 potassium channel | 2258 | 213 | 302,351 | 0.07% | 1,419 | ||
| Cav3 T-type Calcium Channels | 463087 | 703 | 100,210 | 0.70% | 143 | ||
| Transporter | |||||||
| Choline Transporter | 488997 | 252 | 302,246 | 0.08% | 1,199 | ||
| Kinase Inhibitor | |||||||
| Serine/Threonine Kinase 33 | 2689 | 172 | 319,821 | 0.05% | 1,859 | ||
| Enzyme | |||||||
| Tyrosyl-DNA Phosphodiesterase | 485290 | 292 | 344,477 | 0.08% | 1,179 | ||
2.2. Machine Learning Algorithms Relate Chemical Structure to Biological Activity
2.3. Quality Measures Assess the Predictive Power of Machine Learning Algorithms
2.4. QSAR Model Quality Depends Critically on the Selection of Optimal Descriptor Set
| ANN | SVM | DT | KN | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PubChem | IG | FS | SF | IG | FS | SF | IG | FS | SF | IG | FS | SF | Mean |
| SAID | FS | FS | FS | FS | (Stdev) | ||||||||
| 435008 | 0.79 | 0.81 | 0.80 | 0.84 | 0.84 | 0.85 | 0.77 | 0.77 | 0.77 | 0.77 | 0.77 | 0.77 | 0.80 (0.03) |
| 1798 | 0.68 | 0.68 | 0.64 | 0.74 | 0.73 | 0.76 | 0.72 | 0.68 | 0.52 | 0.70 | 0.72 | 0.68 | 0.69 (0.06) |
| 435034 | 0.79 | 0.80 | 0.80 | 0.83 | 0.82 | 0.85 | 0.74 | 0.74 | 0.50 | 0.75 | 0.76 | 0.78 | 0.76 (0.09) |
| 2258 | 0.80 | 0.80 | 0.83 | 0.84 | 0.84 | 0.85 | 0.78 | 0.75 | 0.77 | 0.75 | 0.76 | 0.79 | 0.80 (0.04) |
| 1843 | 0.91 | 0.90 | 0.92 | 0.92 | 0.91 | 0.92 | 0.86 | 0.84 | 0.83 | 0.86 | 0.83 | 0.86 | 0.88 (0.04) |
| 463087 | 0.84 | 0.86 | 0.86 | 0.88 | 0.89 | 0.89 | 0.82 | 0.81 | 0.82 | 0.75 | 0.77 | 0.81 | 0.83 (0.05) |
| 488997 | 0.76 | 0.75 | 0.75 | 0.79 | 0.81 | 0.82 | 0.77 | 0.74 | 0.75 | 0.74 | 0.75 | 0.76 | 0.77 (0.03) |
| 2689 | 0.92 | 0.92 | 0.92 | 0.92 | 0.93 | 0.93 | 0.88 | 0.86 | 0.86 | 0.88 | 0.86 | 0.85 | 0.89 (0.03) |
| 485290 | 0.83 | 0.84 | 0.85 | 0.86 | 0.86 | 0.86 | 0.82 | 0.84 | 0.76 | 0.80 | 0.80 | 0.75 | 0.82 (0.04) |
| Mean (Stdev) | 0.81 (0.07) | 0.82 (0.07) | 0.82 (0.09) | 0.85 (0.06) | 0.85 (0.06) | 0.86 (0.05) | 0.79 (0.05) | 0.78 (0.06) | 0.73 (0.13) | 0.78 (0.06) | 0.78 (0.04 | 0.78 (0.05) | |
2.5. Consensus Prediction of Machine Learning Techniques Increases Prediction Accuracy
| SAID | FS | #r | INT | ENR | IG | #r | INT | ENR | SFFS | #r | INT | ENR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 435008 | ANN DT KN SVM | 1 | 0.249 | 27 | SVM | 1 | 0.245 | 24 | DT SVM | 1 | 0.247 | 40 |
| SVM | 2 | 0.245 | 26 | DT SVM | 2 | 0.245 | 22 | SVM | 2 | 0.246 | 39 | |
| DT SVM | 3 | 0.245 | 26 | KN SVM | 3 | 0.245 | 22 | ANN DT SVM | 3 | 0.249 | 27 | |
| Diff | - | - | 0.11% | Diff | - | - | 0.21% | Diff | - | - | 0.11% | |
| 1798 | ANN DT KN SVM | 1 | 0.240 | 10 | ANN DT KN SVM | 1 | 0.241 | 15 | ANN DT KN SVM | 1 | 0.243 | 7 |
| ANN DT SVM | 2 | 0.240 | 9 | ANN KN SVM | 2 | 0.241 | 10 | ANN KN SVM | 2 | 0.243 | 7 | |
| SVM | 6 | 0.240 | 8 | SVM | 8 | 0.242 | 7 | SVM | 8 | 0.244 | 5 | |
| Diff | - | - | 0.61% | Diff | - | - | 2.45% | Diff | - | - | 0.61% | |
| 435034 | ANN SVM | 1 | 0.246 | 18 | ANN SVM | 1 | 0.245 | 17 | ANN SVM | 1 | 0.246 | 18 |
| ANN DT SVM | 2 | 0.246 | 17 | ANN DT SVM | 2 | 0.245 | 16 | ANN DT SVM | 2 | 0.246 | 18 | |
| SVM | 7 | 0.245 | 14 | SVM | 4 | 0.246 | 16 | SVM | 3 | 0.246 | 17 | |
| Diff | - | - | 2.90% | Diff | - | - | 0.72% | Diff | - | - | 0.72% | |
| 1843 | ANN DT KN | 1 | 0.249 | 54 | ANN DT SVM | 1 | 0.250 | 68 | ANN DT KN SVM | 1 | 0.250 | 50 |
| DT KN SVM | 2 | 0.249 | 45 | ANN DT KN SVM | 2 | 0.250 | 67 | ANN DT SVM | 2 | 0.250 | 45 | |
| SVM | 11 | 0.249 | 32 | SVM | 10 | 0.250 | 45 | ANN | 11 | 0.250 | 30 | |
| Diff | - | - | 1.26% | Diff | - | - | 1.31% | Diff | - | - | 1.14% | |
| 2258 | ANN DT | 1 | 0.241 | 28 | ANN DT | 1 | 0.241 | 34 | ANN DT KN | 1 | 0.244 | 65 |
| ANN DT SVM | 2 | 0.246 | 26 | ANN DT KN SVM | 2 | 0.246 | 33 | DT KN SVM | 2 | 0.249 | 49 | |
| SVM | 8 | 0.246 | 18 | DT | 6 | 0.238 | 23 | SVM | 10 | 0.249 | 28 | |
| Diff | - | - | 0.70% | Diff | - | - | 0.78% | - | - | 2.61% | ||
| 463087 | ANN SVM | 1 | 0.250 | 23 | ANN DT KN SVM | 1 | 0.250 | 19 | ANN KN SVM | 1 | 0.250 | 29 |
| SVM | 2 | 0.250 | 22 | ANN DT SVM | 2 | 0.250 | 18 | ANN DT KN SVM | 2 | 0.250 | 28 | |
| DT SVM | 4 | 0.250 | 21 | SVM | 8 | 0.250 | 17 | SVM | 8 | 0.250 | 24 | |
| Diff | - | - | 0.70% | Diff | - | - | 1.40% | Diff | - | - | 3.50% | |
| 2689 | ANN DT KN SVM | 1 | 0.248 | 74 | ANN DT KN SVM | 1 | 0.248 | 58 | ANN DT SVM | 1 | 0.249 | 101 |
| ANN DT SVM | 2 | 0.248 | 63 | ANN DT SVM | 2 | 0.248 | 54 | ANN DT KN SVM | 2 | 0.249 | 91 | |
| ANN | 10 | 0.250 | 42 | SVM | 10 | 0.248 | 41 | ANN | 10 | 0.248 | 44 | |
| Diff | - | - | 2.67% | Diff | - | - | 1.42% | Diff | - | - | 4.75% | |
| 488997 | ANN DT SVM | 1 | 0.246 | 20 | DT KN SVM | 1 | 0.244 | 14 | ANN DT KN SVM | 1 | 0.243 | 49 |
| ANN DT KN SVM | 2 | 0.247 | 19 | ANN DT KN | 2 | 0.241 | 13 | ANN KN SVM | 2 | 0.243 | 44 | |
| SVM | 6 | 0.245 | 15 | DT | 7 | 0.242 | 12 | SVM | 11 | 0.244 | 31 | |
| Diff | - | - | 0.27% | Diff | - | - | 0.11% | Diff | - | - | 0.97% | |
| 485290 | ANN DT KN | 1 | 0.241 | 64 | DT KN SVM | 1 | 0.245 | 71 | ANN SVM | 1 | 0.244 | 30 |
| DT KN SVM | 2 | 0.245 | 58 | ANN DT KN SVM | 2 | 0.246 | 60 | ANN DT SVM | 2 | 0.244 | 28 | |
| SVM | 11 | 0.244 | 38 | SVM | 12 | 0.245 | 36 | SVM | 4 | 0.244 | 26 | |
| Diff | - | - | 2.22% | Diff | - | - | 2.96% | Diff | - | - | 0.28% |
3. Experimental
3.1. Determination of Confirmatory High-Throughput Screening Data Sets for Diverse Protein Targets
3.1.1. GPCR: Antagonist of the Orexin 1 Receptor (SAID 435008)
3.1.2. GPCR: Allosteric Modulators of M1 Muscarinic Receptor: Agonist (SAID 1798)
3.1.3. GPCR: Allosteric Modulators of M1 Muscarinic Receptor: Antagonist (SAID 435034)
3.1.4. Ion Channel: Potentiators of KCNQ2 Potassium Channel (SAID 2258)
3.1.5. Ion Channel: Identification of Compounds that Inhibit Inward-Rectifying Potassium Ion Channel Kir2.1 (SAID 1843)
3.1.6. Ion Channel: Inhibitors of the of Cav3 T-type Calcium Channels (SAID 463087)
3.1.7. Transporter: Inhibitors of the Choline Transporter (CHT, SAID 488997)
3.1.8. Kinase Inhibitor: Inhibitors of Serine/Threonine Kinase 33 (STK33, SAID 2689)
3.1.9. Enzyme: Inhibitors of Tyrosyl-DNA Phosphodiesterase 1 (TDP1, SAID 485290)
3.2. Numerical Representation of Biological Data Distinguishes Active from Inactive Compounds
3.3. Numerical Description of Molecules for QSAR Model Development
3.4. Monitoring Data Set is Used for Early Termination of Training Process
3.5. The Integral of the True-Negative-Rate–True-Positive-Rate Curve is a Viable Quality Measure for QSAR Models
3.6. Enrichment Measures Ratio of Fraction of Active Compounds Predicted Above Actives Rate
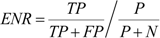
3.7. Orthogonal Supervised and Unsupervised Machine Learning Algorithms Seek Optimal Biological Activity Predictions
3.8. Cross-Validation Ascertains Robustness of QSAR Models
3.9. Selection of an Optimized Descriptor Set Guides QSAR Model Training
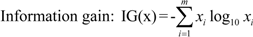
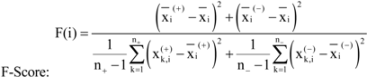
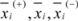 are the average of the i th feature of the whole, active, and inactive data sets, respectively;
are the average of the i th feature of the whole, active, and inactive data sets, respectively; 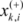 is the i th feature of the k th active instance, and
is the i th feature of the k th active instance, and 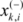 is the i th feature of the k th inactive instance.
is the i th feature of the k th inactive instance. 3.10. Consensus Predictions Seeks Improved Accuracies of Trained QSAR Models
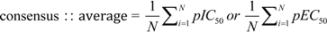
3.11. Implementation
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Geppert, H.; Vogt, M.; Bajorath, J.R. Current Trends in Ligand-Based Virtual Screening: Molecular Representations, Data Mining Methods, New Application Areas, and Performance Evaluation. J. Chem. Inf. Model. 2010, 50, 205–216. [Google Scholar] [CrossRef]
- Austin, C.P.; Brady, L.S.; Insel, T.R.; Collins, F.S. NIH Molecular Libraries Initiative. Science 2004, 306, 1138–1139. [Google Scholar] [CrossRef]
- Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002, 1, 882–894. [Google Scholar] [CrossRef]
- PubChem Home Page. Available online: http://pubchem.ncbi.nlm.nih.gov/ (accessed on 26 November 2012).
- Handen, J.S. The industrialization of drug discovery. Drug Discov. Today 2002, 7, 83–85. [Google Scholar] [CrossRef]
- Schneider, G.; Fechner, U. Computer-based de novo design of drug-like molecules. Nat. Rev.Drug Discov. 2005, 4, 649–663. [Google Scholar] [CrossRef]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef]
- Perkins, R.; Fang, H.; Tong, W.; Welsh, W.J. Quantitative structure-activity relationship methods: Perspectives on drug discovery and toxicology. Environ. Toxicol. Chem. 2003, 22, 1666–1679. [Google Scholar] [CrossRef]
- Dudek, A.Z.; Arodz, T.; Galvez, J. Computational methods in developing quantitative structure-activity relationships (QSAR): A review. Comb. Chem. High T. Scr. 2006, 9, 213–228. [Google Scholar]
- Du, Q.S.; Huang, R.B.; Chou, K.C. Recent advances in QSAR and their applications in predicting the activities of chemical molecules, peptides and proteins for drug design. Curr. Protein Pept. Sci. 2008, 9, 248–259. [Google Scholar] [CrossRef]
- Hansch, C. Use of quantitative structure-activity relationships (QSAR) in drug design (review). Pharm. Chem. J. 1980, 14, 678–691. [Google Scholar] [CrossRef]
- Hansch, C.; Maloney, P.P.; Fujita, T.; Muir, R.M. Correlation of Biological Activity of Phenoxyacetic Acids with Hammett Substituent Constants and Partition Coefficients. Nature 1962, 194, 178–180. [Google Scholar]
- Scior, T.; Medina-Franco, J.L.; Do, Q.T.; Martínez-Mayorga, K.; Rojas, Y.; Bernard, P. How to Recognize and Workaround Pitfalls in QSAR Studies: A Critical Review. Curr. Med. Chem. 2009, 16, 4297–4313. [Google Scholar] [CrossRef]
- Hillebrecht, A.; Klebe, G. Use of 3D QSAR models for database screening: A feasibility study. J. Chem. Inform. Model. 2008, 48, 384–396. [Google Scholar] [CrossRef]
- Fawcett, R.W. A radial distribution function analysis of an amorphous calcium phosphate with calcium to phosphorus molar ratio of 1.42. Calcif. Tissue Int. 1973, 13, 319–325. [Google Scholar] [CrossRef]
- Gonzalez, M.P.; Teran, C.; Teijeira, M.; Helguera, A.M. Radial distribution function descriptors: An alternative for predicting A2 A adenosine receptors agonists. Eur. J. Med. Chem. 2006, 41, 56–62. [Google Scholar] [CrossRef]
- Gonzalez, M.P.; Gandara, Z.; Fall, Y.; Gomez, G. Radial Distribution Function descriptors for predicting affinity for vitamin D receptor. Eur. J. Med. Chem. 2008, 43, 1360–1365. [Google Scholar] [CrossRef]
- Hollas, B. An Analysis of the Autocorrelation Descriptor for Molecules. J. Math. Chem. 2003, 33, 91–101. [Google Scholar] [CrossRef]
- Caballero, J.; Fernandez, M.; Gonzalez-Nilo, F.D. Structural requirements of pyrido[2,3-d]pyrimidin-7-one as CDK4/D inhibitors: 2D autocorrelation, CoMFA and CoMSIA analyses. Bioorg. Med. Chem. 2008, 16, 6103–6115. [Google Scholar] [CrossRef]
- Caballero, J.; Garriga, M.; Fernandez, M. 2D Autocorrelation modeling of the negative inotropic activity of calcium entry blockers using Bayesian-regularized genetic neural networks. Bioorg. Med. Chem. 2006, 14, 3330–3340. [Google Scholar] [CrossRef]
- Butkiewicz, M.; Mueller, R.; Selic, D.; Dawson, E.; Meiler, J. Application of Machine Learning Approaches on Quantitative Structure Activity Relationships. In Computational Intelligence in Bioinformatics and Computational Biology, 2009, Proceedings of the CIBCB '09. IEEE Symposium on, Nashville, TN, USA, 30 March–2 April 2009; pp. 255–262.
- King, R.; Hirst, J.; Sternberg, M. New approaches to QSAR: Neural networks and machine learning. Perspect. Drug Discov. 1993, 1, 279–290. [Google Scholar] [CrossRef]
- Bleckmann, A.; Meiler, J. Epothilones: Quantitative Structure Activity Relations Studied by Support Vector Machines and Artificial Neural Networks. QSAR Comb. Sci. 2003, 22, 719–721. [Google Scholar] [CrossRef]
- Mueller, R.; Rodriguez, A.L.; Dawson, E.S.; Butkiewicz, M.; Nguyen, T.T.; Oleszkiewicz, S.; Bleckmann, A.; Weaver, C.D.; Lindsley, C.W.; Conn, P.J.; et al. Identification of Metabotropic Glutamate Receptor Subtype 5 Potentiators Using Virtual High-Throughput Screening. ACS Chem. Neurosci. 2010, 1, 288–305. [Google Scholar] [CrossRef]
- Golla, S.; Neely, B.J.; Whitebay, E.; Madihally, S.; Robinson, R.L., Jr.; Gasem, K.A. Virtual design of chemical penetration enhancers for transdermal drug delivery. Chem. Biol. Drug Des. 2012, 79, 478–487. [Google Scholar] [CrossRef]
- Sun, H.; Shahane, S.; Xia, M.; Austin, C.P.; Huang, R. Structure Based Model for the Prediction of Phospholipidosis Induction Potential of Small Molecules. J. Chem. Inf. Model. 2012, 52, 1798–1805. [Google Scholar] [CrossRef]
- Shao, L.; Wu, L.; Fan, X.; Cheng, Y. Consensus Ranking Approach to Understanding the Underlying Mechanism With QSAR. J. Chem. Inf. Model. 2010, 50, 1941–1948. [Google Scholar] [CrossRef]
- Simmons, K.; Kinney, J.; Owens, A.; Kleier, D.A.; Bloch, K.; Argentar, D.; Walsh, A.; Vaidyanathan, G. Practical Outcomes of Applying Ensemble Machine Learning Classifiers to High-Throughput Screening (HTS) Data Analysis and Screening. J. Chem. Inf. Model 2008, 48, 2196–2206. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random Forest: A Classification and Regression Tool for Compound Classification and QSAR Modeling. J. Chem. Inform. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Hewitt, M.; Cronin, M.T.D.; Madden, J.C.; Rowe, P.H.; Johnson, C.; Obi, A.; Enoch, S.J. Consensus QSAR Models: Do the Benefits Outweigh the Complexity? J. Chem. Inf. Model. 2007, 47, 1460–1468. [Google Scholar] [CrossRef]
- Hecht-Nielsen, R. Counterpropagation networks. Appl. Optics 1987, 26, 4979–4983. [Google Scholar] [CrossRef]
- Winkler, D. Neural networks as robust tools in drug lead discovery and development. Mol. Biotechnol. 2004, 27, 139–167. [Google Scholar] [CrossRef]
- Patnaik, L.M.; Rajan, K. Target detection through image processing and resilient propagation algorithms. Neurocomputing 2000, 35, 123–135. [Google Scholar] [CrossRef]
- Smola, A.J.; Schoelkopf, B. A Tutorial on Support Vector Regression. Stat. Comput. 2004, 14, 199–222. [Google Scholar] [CrossRef]
- Hong, D.H.; Hwang, C. Support vector fuzzy regression machines. Fuzzy Set. Syst. 2003, 138, 271–281. [Google Scholar] [CrossRef]
- Drucker, H.; Burges, C.J.; Kaufman, L.; Smola, A.; Vapnik, V. Support vector regression machines. Adv. Neural Inf. Process. Syst. 1997, 155–161. [Google Scholar]
- Vapnik, V. The Nature of Statistical Learning Theory; Springer: New York, NY, USA, 1999. [Google Scholar]
- Hossain, M.M.; Hassan, Md.R.; Bailey, J. ROC-tree: A novel decision tree induction algorithm based on receiver operating characteristics to classify gene expression data. In Proceedings of SIAM International Conference on Data Mining, Atlanta, GA, USA, 2008; pp. 455–465.
- Quinlan, J.R. Induction of Decision Trees. Mach. Learn. 1986, 1, 81–106. [Google Scholar]
- White, A.P.; Liu, W.Z. Technical note: Bias in information-based measures in decision tree induction. Mach. Learn. 1994, 15, 321–329. [Google Scholar]
- Kohonen, T. The self-organizing map. Proc. IEEE 1990, 78, 1464–1480. [Google Scholar] [CrossRef]
- Kohonen, T. Self-organization and Associative Memory, 3rd ed; Springer-Verlag: Berlin, Germany.
- Kohonen, T. Self-organized formation of topologically correct feature maps. Biol. Cybern. 1982, 43, 59–69. [Google Scholar] [CrossRef]
- Bradley, A.P. The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern Recogn. 1997, 30, 1145–1159. [Google Scholar]
- Smith, R.J.; See, R.E.; Aston-Jones, G. Orexin/hypocretin signaling at the orexin 1 receptor regulates cue-elicited cocaine-seeking. Eur. J. Neurosci. 2009, 30, 493–503. [Google Scholar] [CrossRef]
- Winrow, C.J.; Tanis, K.Q.; Reiss, D.R.; Rigby, A.M.; Uslaner, J.M.; Uebele, V.N.; Doran, S.M.; Fox, S.V.; Garson, S.L.; Gotter, A.L.; et al. Orexin receptor antagonism prevents transcriptional and behavioral plasticity resulting from stimulant exposure. Neuropharmacology 2010, 58, 185–194. [Google Scholar] [CrossRef]
- Rodems, S.M.; Hamman, B.D.; Lin, C.; Zhao, J.; Shah, S.; Heidary, D.; Makings, L.; Stack, J.H.; Pollok, B.A. A FRET-based assay platform for ultra-high density drug screening of protein kinases and phosphatases. Assay Drug Dev. Technol. 2002, 1, 9–19. [Google Scholar]
- Bodick, N.C.; Offen, W.W.; Shannon, H.E.; Satterwhite, J.; Lucas, R.; van Lier, R.; Paul, S.M. The selective muscarinic agonist xanomeline improves both the cognitive deficits and behavioral symptoms of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1997, 11, S16–S22. [Google Scholar]
- Klett, C.P.; Bonner, T.I. Identification and characterization of the rat M1 muscarinic receptor promoter. J. Neurochem. 1999, 72, 900–909. [Google Scholar]
- Medina, A.; Bodick, N.; Goldberger, A.L.; Mac Mahon, M.; Lipsitz, L.A. Effects of central muscarinic-1 receptor stimulation on blood pressure regulation. Hypertension 1997, 29, 828–834. [Google Scholar] [CrossRef]
- Burford, N.T.; Nahorski, S.R. Muscarinic m1 receptor-stimulated adenylate cyclase activity in Chinese hamster ovary cells is mediated by Gs alpha and is not a consequence of phosphoinositidase C activation. Biochem. J. 1996, 315, 883–888. [Google Scholar]
- Arumugam, B.; McBrien, N.A. Muscarinic antagonist control of myopia: evidence for m4 and m1 receptor-based pathways in the inhibition of experimentally-induced axial myopia in the tree shrew. Invest. Ophthalmol. Vis. Sci. 2012, 53, 5827–5837. [Google Scholar] [CrossRef]
- Wu, W.; Saunders, R.C.; Mishkin, M.; Turchi, J. Differential effects of m1 and m2 receptor antagonists in perirhinal cortex on visual recognition memory in monkeys. Neurobiol. Learn. Mem. 2012, 98, 41–46. [Google Scholar] [CrossRef]
- Charlier, C.; Singh, N.A.; Ryan, S.G.; Lewis, T.B.; Reus, B.E.; Leach, R.J.; Leppert, M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat. Genet. 1998, 18, 53–55. [Google Scholar] [CrossRef]
- Gutman, G.A.; Chandy, K.G.; Adelman, J.P.; Aiyar, J.; Bayliss, D.A.; Clapham, D.E.; Covarriubias, M.; Desir, G.V.; Furuichi, K.; Ganetzky, B.; et al. International Union of Pharmacology. XLI. Compendium of voltage-gated ion channels: potassium channels. Pharmacol. Rev. 2003, 55, 583–586. [Google Scholar] [CrossRef]
- Dhamoon, A.S.; Pandit, S.V.; Sarmast, F.; Parisian, K.R.; Guha, P.; Li, Y.; Bagwe, S.; Taffet, S.M.; Anumonwo, J.M. Unique Kir2.x properties determine regional and species differences in the cardiac inward rectifier K+ current. Circ. Res. 2004, 94, 1332–1339. [Google Scholar] [CrossRef]
- Kaczorowski, G.J.; McManus, O.B.; Priest, B.T.; Garcia, M.L. Ion channels as drug targets: The next GPCRs. J. Gen. Physiol. 2008, 131, 399–405. [Google Scholar] [CrossRef]
- Sun, H.; Liu, X.; Xiong, Q.; Shikano, S.; Li, M. Chronic inhibition of cardiac Kir2.1 and HERG potassium channels by celastrol with dual effects on both ion conductivity and protein trafficking. J. Biol. Chem. 2006, 281, 5877–5884. [Google Scholar]
- Jensen, B.F.; Vind, C.; Padkjaer, S.B.; Brockhoff, P.B.; Refsgaard, H.H.F. In silico prediction of cytochrome P450 2D6 and 3A4 inhibition using Gaussian kernel weighted k-nearest neighbor and extended connectivity fingerprints, including structural fragment analysis of inhibitors versus noninhibitors. J. Med. Chem. 2007, 50, 501–511. [Google Scholar] [CrossRef]
- Nelson, M.T.; Joksovic, P.M.; Perez-Reyes, E.; Todorovic, S.M. The endogenous redox agent L-cysteine induces T-type Ca2+ channel-dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J. Neurosci. 2005, 25, 8766–8775. [Google Scholar] [CrossRef]
- Nelson, M.T.; Todorovic, S.M.; Perez-Reyes, E. The role of T-type calcium channels in epilepsy and pain. Curr. Pharm. Des. 2006, 12, 2189–2197. [Google Scholar] [CrossRef]
- Perez-Reyes, E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol. Rev. 2003, 83, 117–161. [Google Scholar]
- Ferguson, S.M.; Blakely, R.D. The choline transporter resurfaces: New roles for synaptic vesicles? Mol. Interv. 2004, 4, 22–37. [Google Scholar] [CrossRef]
- Iwamoto, H.; Blakely, R.D.; De Felice, L.J. Na+, Cl−, and pH dependence of the human choline transporter (hCHT) in Xenopus oocytes: The proton inactivation hypothesis of hCHT in synaptic vesicles. J. Neurosci. 2006, 26, 9851–9859. [Google Scholar] [CrossRef]
- Liao, Z.; Thibaut, L.; Jobson, A.; Pommier, Y. Inhibition of human tyrosyl-DNA phosphodiesterase by aminoglycoside antibiotics and ribosome inhibitors. Mol. Pharmacol. 2006, 70, 366–372. [Google Scholar]
- Dexheimer, T.S.; Antony, S.; Marchand, C.; Pommier, Y. Tyrosyl-DNA phosphodiesterase as a target for anticancer therapy. Anti-Cancer Agents Med. Chem. 2008, 8, 381–389. [Google Scholar]
- Antony, S.; Marchand, C.; Stephen, A.G.; Thibaut, L.; Agama, K.K.; Fisher, R.J.; Pommier, Y. Novel high-throughput electrochemiluminescent assay for identification of human tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors and characterization of furamidine (NSC 305831) as an inhibitor of Tdp1. Nucleic Acids Res. 2007, 35, 4474–4484. [Google Scholar]
- Marchand, C.; Lea, W.A.; Jadhav, A.; Dexheimer, T.S.; Austin, C.P.; Inglese, J.; Pommier, Y.; Simeonov, A. Identification of phosphotyrosine mimetic inhibitors of human tyrosyl-DNA phosphodiesterase I by a novel AlphaScreen high-throughput assay. Mol. Cancer Ther. 2009, 8, 240–248. [Google Scholar] [CrossRef]
- Gilson, M.K.; Gilson, H.S.R.; Potter, M.J. Fast Assignment of Accurate Partial Atomic Charges: An Electronegativity Equalization Method that Accounts for Alternate Resonance Forms. J. Chem. Inf. Comput. Sci. 2003, 43, 1982–1997. [Google Scholar]
- Gasteiger, J.; Rudolph, C.; Sadowski, J. Automatic generation of 3D atomic coordinates for organic molecules. Tetrahedron Comput. Methodol. 1990, 3, 537–547. [Google Scholar] [CrossRef]
- Meiler, J.; Will, M. Automated Structure Elucidation of Organic Molecules from 13C-NMR Spectra using Genetic Algorithms and Neural Networks. J. Chem. Inform. Comput. Sci. 2001, 41, 1535–1546. [Google Scholar]
- Polanski, J.; Gasteiger, J.; Wagener, M.; Sadowski, J. The comparison of molecular surfaces by neural networks and its applications to quantitative structure activity studies. Quant. Struct.-Act. Relat. 1998, 17, 27–36. [Google Scholar] [CrossRef]
- Meiler, J.; Müller, M.; Zeidler, A.; Schmäschke, F. Generation and Evaluation of Dimension Reduced Amino Acid Parameter Representations by Artificial Neural Networks. J. Mol. Model. 2001, 7, 360–369. [Google Scholar] [CrossRef]
- Hecht, D.; Fogel, G. High-throughput ligand screening via preclustering and evolved neural networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2007, 4, 476–484. [Google Scholar] [CrossRef]
- Jorissen, R.N.; Gilson, M.K. Virtual screening of molecular databases using a support vector machine. J. Chem. Inf. Model. 2005, 45, 549–561. [Google Scholar] [CrossRef]
- Yap, C.W.; Cai, C.Z.; Xue, Y.; Chen, Y.Z. Prediction of torsade-causing potential of drugs by support vector machine approach. Toxicol. Sci. 2004, 79, 170–177. [Google Scholar] [CrossRef]
- Zernov, V.V.; Balakin, K.V.; Ivaschenko, A.A.; Savchuk, N.P.; Pletnev, I.V. Drug discovery using support vector machines. The case studies of drug-likeness, agrochemical-likeness, and enzyme inhibition predictions. J. Chem. Inf. Comput. Sci. 2003, 43, 2048–2056. [Google Scholar]
- Schoelkopf, B.; Smola, A.J. Learning with Kernels; The MIT Press: Cambridge, MA, USA, 2002. [Google Scholar]
- Sadras, V.; Bongiovanni, R. Use of Lorenz curves and Gini coefficients to assess yield inequality within paddocks. Field Crops Res. 2004, 90, 303–310. [Google Scholar] [CrossRef]
- Kent, J.T. Information gain and a general measure of correlation. Biometrika 1983, 70, 163–173. [Google Scholar] [CrossRef]
- Chen, Y.-W.; Lin, C.-J. Combining SVMs with Various Feature Selection Strategies. In Feature Extraction; Guyon, I., Nikravesh, M., Gunn, S., Zadeh, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 207, pp. 315–324. [Google Scholar]
- Mao, K.Z. Orthogonal forward selection and backward elimination algorithms for feature subset selection. IEEE Trans. Syst. Man Cybern. 2004, 34, 629–634. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Butkiewicz, M.; Lowe, E.W., Jr.; Mueller, R.; Mendenhall, J.L.; Teixeira, P.L.; Weaver, C.D.; Meiler, J. Benchmarking Ligand-Based Virtual High-Throughput Screening with the PubChem Database. Molecules 2013, 18, 735-756. https://doi.org/10.3390/molecules18010735
Butkiewicz M, Lowe EW Jr., Mueller R, Mendenhall JL, Teixeira PL, Weaver CD, Meiler J. Benchmarking Ligand-Based Virtual High-Throughput Screening with the PubChem Database. Molecules. 2013; 18(1):735-756. https://doi.org/10.3390/molecules18010735
Chicago/Turabian StyleButkiewicz, Mariusz, Edward W. Lowe, Jr., Ralf Mueller, Jeffrey L. Mendenhall, Pedro L. Teixeira, C. David Weaver, and Jens Meiler. 2013. "Benchmarking Ligand-Based Virtual High-Throughput Screening with the PubChem Database" Molecules 18, no. 1: 735-756. https://doi.org/10.3390/molecules18010735
APA StyleButkiewicz, M., Lowe, E. W., Jr., Mueller, R., Mendenhall, J. L., Teixeira, P. L., Weaver, C. D., & Meiler, J. (2013). Benchmarking Ligand-Based Virtual High-Throughput Screening with the PubChem Database. Molecules, 18(1), 735-756. https://doi.org/10.3390/molecules18010735