Diastereoselective Synthesis of 5-Hydroxy-8-methoxy-1-oxaspiro[5,5]undeca-7,10-diene-9-one

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
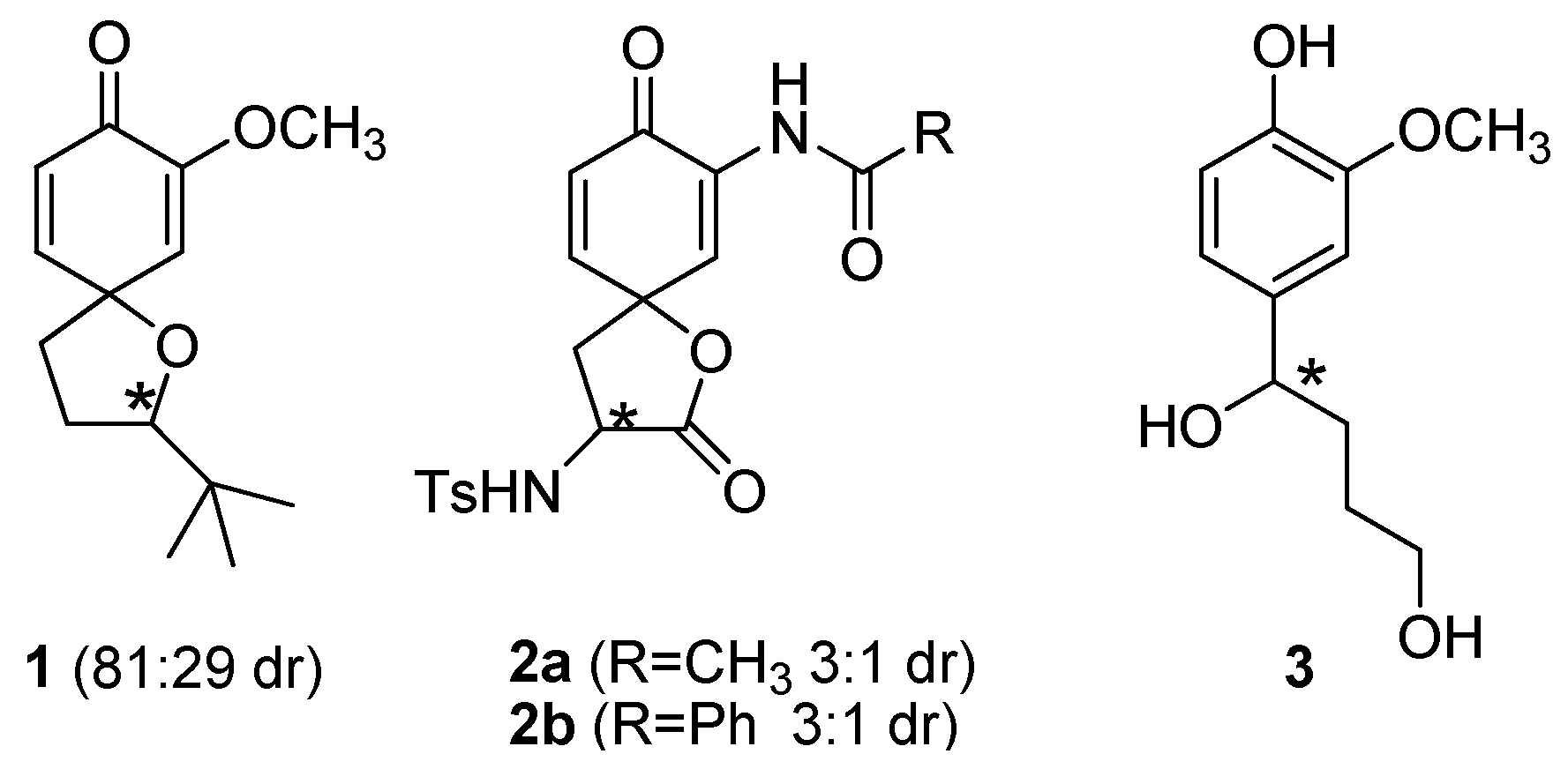
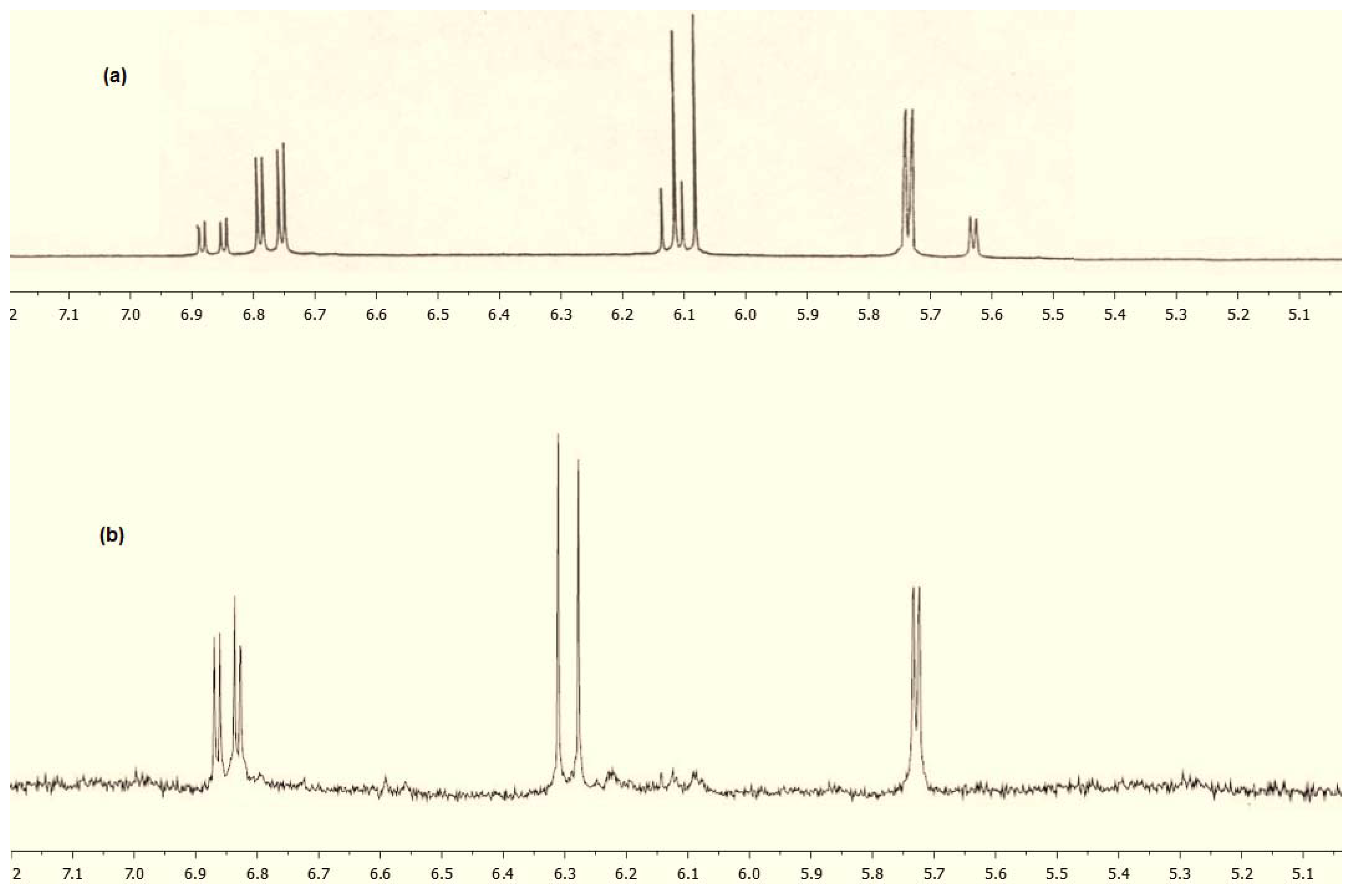
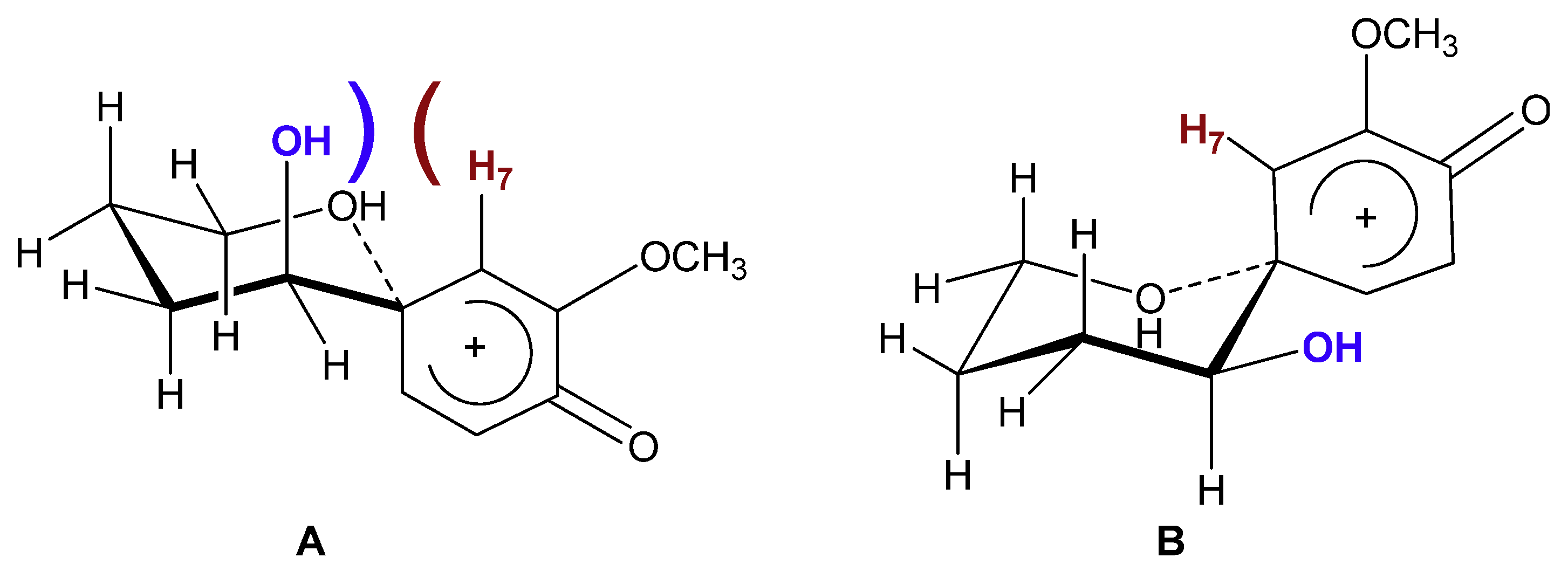
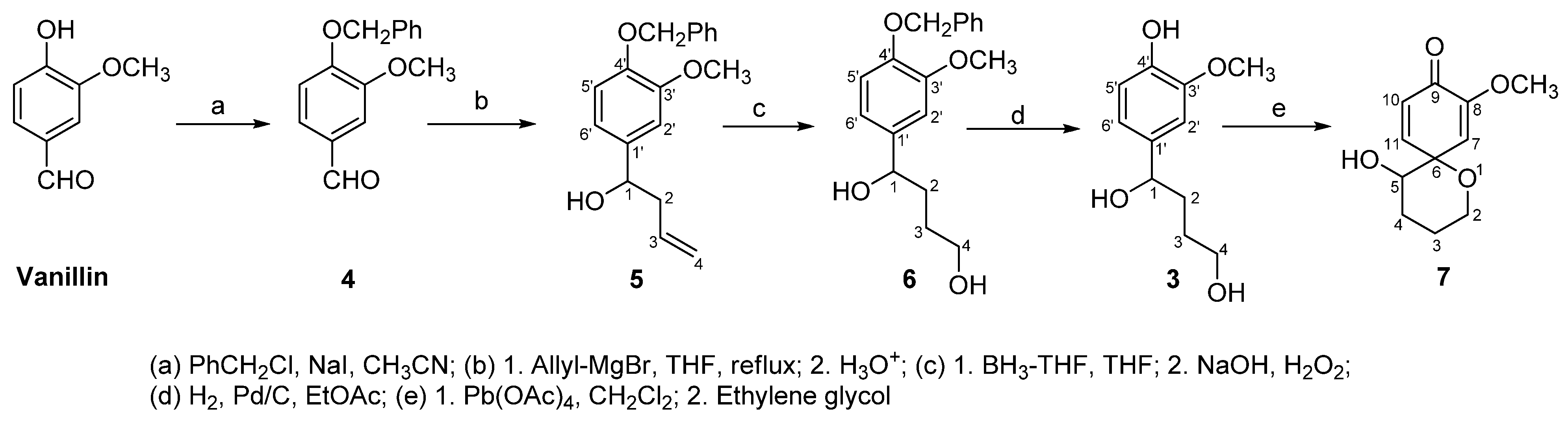
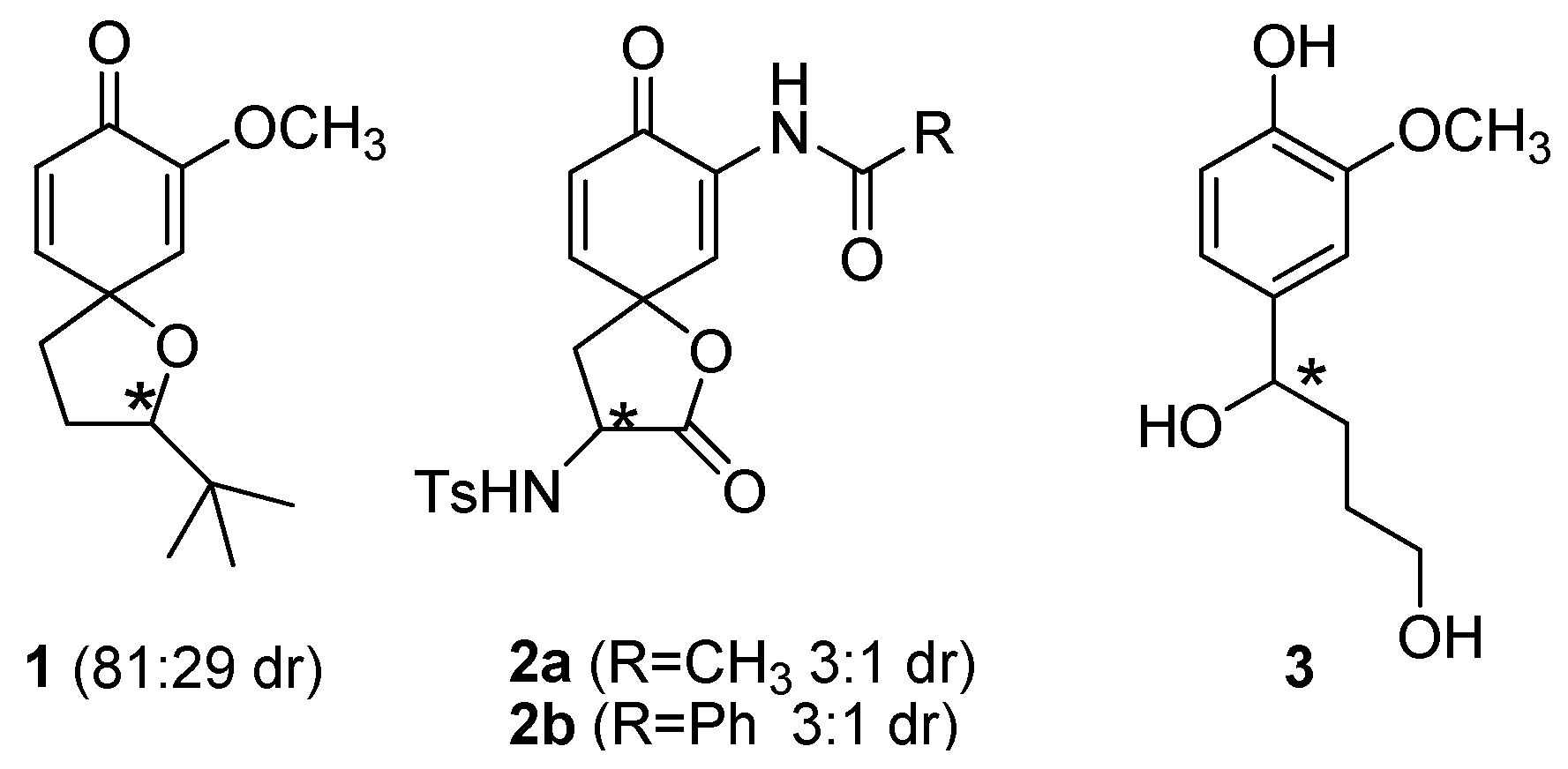
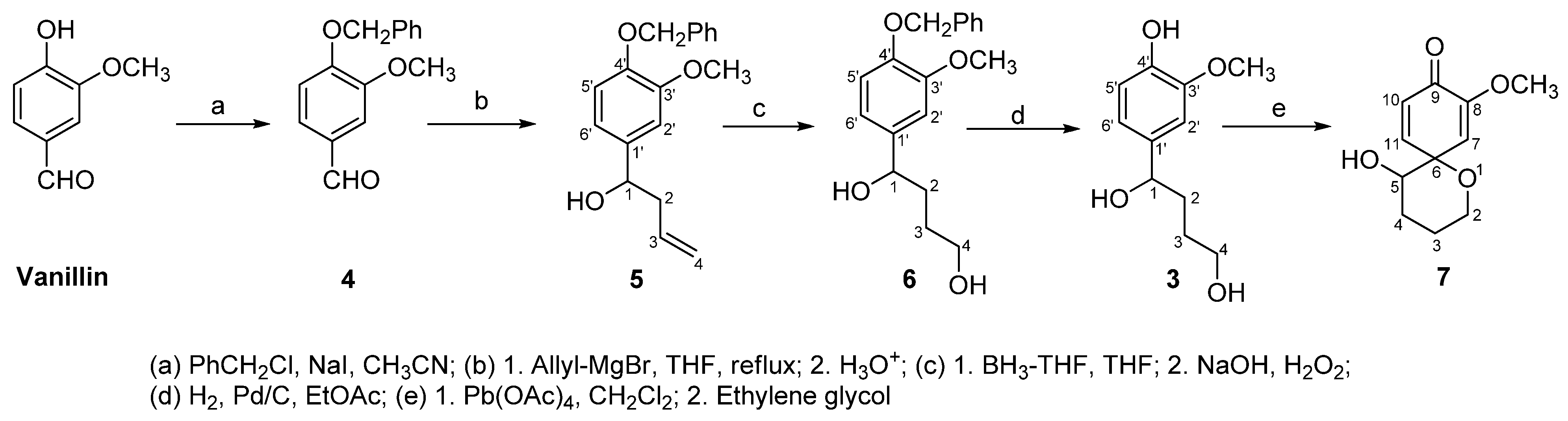
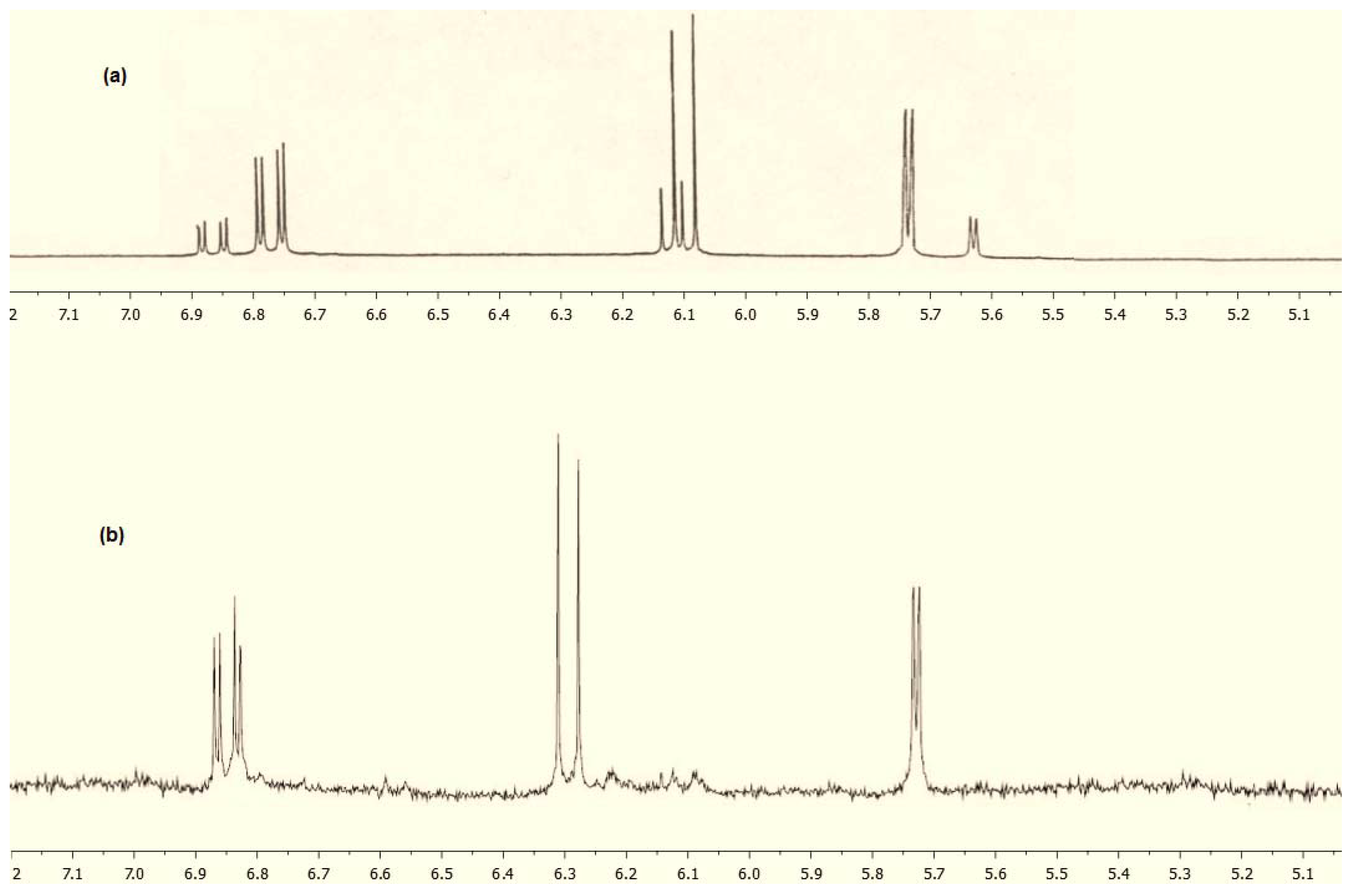
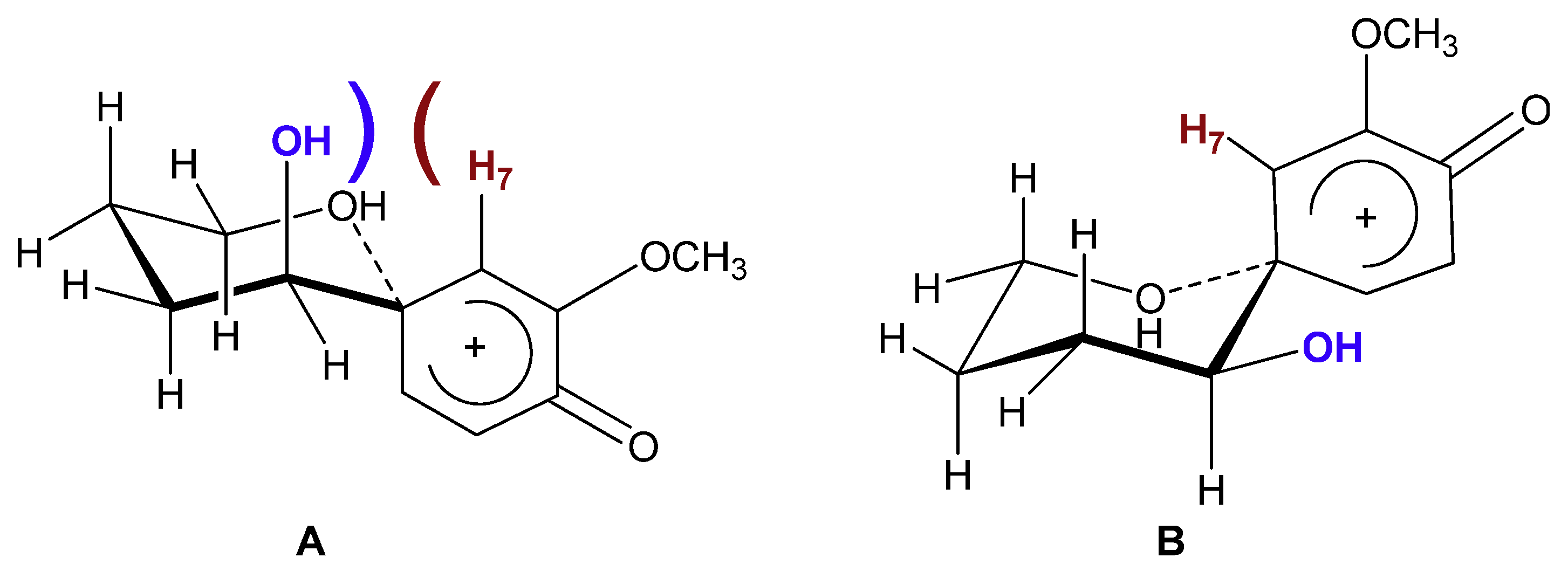
2. Results and Discussion
3. Experimental
General
4. Conclusions
References
- Plourde, G.L. Studies towards the diastereoselective spiroannulation of phenolic derivatives. Tetrahedron Lett. 2002, 43, 3597–3599. [Google Scholar] [CrossRef]
- Plourde, G.L.; Spaetzel, R.R.; Kwasnitza, J.S.; Scully, T.W. Diastereoselective spiroannulation of phenolic substrates: Advances towards the asymmetric formation of the manumycin m-C7N core skeleton. Molecules 2007, 12, 2215–2222. [Google Scholar] [CrossRef] [PubMed]
- Plourde, G.L.; Susag, L.M.; Dick, D.G. Determination of the absolute configurations of (+)-N-((3S)-3-{[(4-methylphenyl)sulfonyl]amino}-1-oxaspiro[4.5]deca-6,9-dien-2,8-dion-7-yl) acetamide and benzamide. Molbank 2008, M579. [Google Scholar] [CrossRef]
- Plourde, G.L.; English, N.J. Diastereoselective spiroannulation of phenolic substrates: Synthesis of (+/−)-2-tbutyl-6-methoxy-1-oxaspiro[4,5]deca-6,9-diene-8-one. Molecules 2005, 10, 1335–1339. [Google Scholar] [CrossRef] [PubMed]
- Plourde, G.L.; Susag, L.M. Diastereoselective spiroannulation of phenolic derivatives: Effect of the O-alkoxy substituent on the diastereoselectivity. Int. J. Chem. 2011, 3, 3–9. [Google Scholar] [CrossRef]
- Mendelson, W.L.; Holmes, M.; Dougherty, J. The regioselective 4-benzylation of 2,4-dihydroxybenzaldehyde. Synth. Commun. 1996, 26, 593–601. [Google Scholar] [CrossRef]
- Bukas, A.; Dufour, C. N-Vanilylpelargonamide. Ann. Pharm. Fr. 1959, 17, 453–455. [Google Scholar]
- Samaresh, J.; Chandrani, G.; Subhas, C.R. Mild and efficient allylation of aldehydes mediated by titanium (III) chloride. Tetrahedron Lett. 2004, 45, 6575–6577. [Google Scholar]
- Plourde, G.L.; Fisher, B.B. Synthesis of 6-methoxy-1-oxaspiro[4,5]-6,9-diene-8-one. Molecules 2002, 7, 315–319. [Google Scholar] [CrossRef]
- Plourde, G.L.; Speatzel, R.R. Synthesis of N-(2,8-Dioxo-1-oxaspiro[4,5]deca-6,9-dien-7-yl) acetamide and benzamide. Molbank 2009, M599. [Google Scholar] [CrossRef]
- Plourde, G.L. (+/−)-7-Methoxy-2-methyl-1-oxaspiro[4,5]deca-6,9-diene-8-one. Molbank 2003, M316. [Google Scholar] [CrossRef]
- Plourde, G.L. (+/−)-7-Methoxy-2-isopropyl-1-oxaspiro[4,5]deca-6,9-diene-8-one. Molbank 2003, M319. [Google Scholar] [CrossRef]
- Plourde, G.L. (+/−)-2-tButyl-7-methoxy-1-oxaspiro[4,5]deca-6,9-diene-8-one. Molbank 2003, M322. [Google Scholar] [CrossRef]
- Plourde, G.L.; Fairchild, M.D.; Sarohia, G.S. Synthesis of a new spirolactone: 7,10-Dimethoxy-1-oxaspiro[4,5]deca-6,9-diene-2,8-dione. Int. J. Chem. 2012, 4, 2–6. [Google Scholar] [CrossRef]
- Quideau, S.; Looney, M.A.; Pouységu, L. Oxidized arenol intermediates in intermolecular carbon-carbon bond formation. Naphthoid 2,4-cyclohexadienones via oxidative nucleophilic substitution. Org. Lett. 1999, 1, 1651–1654. [Google Scholar] [CrossRef]
- Dohi, T.; Uchiyama, T.; Yamashita, D.; Washimi, N.; Kita, Y. Efficient phenolic oxidations using u-oxo-bridged phenyliodine trifluoroacetate. Tetrahedron Lett. 2011, 52, 2212–2215. [Google Scholar] [CrossRef]
- Minamitsuji, Y.; Kato, D.; Fujoka, H.; Dohi, T.; Kita, Y. Organo-catalyzed oxidative spirocyclization of phenols using peracetic acid as a green and economic terminal oxidant. Aust. J. Chem. 2009, 62, 648–652. [Google Scholar] [CrossRef]
- Taylor, E.C.; Andrade, J.G.; Rall, G.J.H.; Turchi, I.J.; Steliou, K.; Jagdmann, G.E., Jr.; McKillop, A. Thallium in organic synthesis. 61. Intramolecular capture of radical cations from thallium (III) trifluoroacetate oxidation of arylalkanoic acids and arylalkanols. New routes to oxygen heterocycles. J. Am. Chem. Soc. 1981, 103, 6856–6863. [Google Scholar] [CrossRef]
- El-Mobayed, M.; Ismail, N.; Abo El-Enein, G.; Abd El-Haleem, E. Anodic oxidation of α-naphthol derivative. A facile route to ring closure products. J. Chem. Soc. Pakistan 1986, 8, 305–310. [Google Scholar]
- Fujioka, H.; Komatsu, H.; Nakamura, T.; Miyoshi, A.; Hata, K.; Ganesh, J.; Murai, K.; Kita, Y. Organic synthesis using a hypervalent iodine reagent: Unexpected and novel domino reaction to spiro cyclohexadienone lactones. Chem. Commun. 2010, 46, 4133–4135. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Plourde, G.L.; Scully, T.W. Diastereoselective Synthesis of 5-Hydroxy-8-methoxy-1-oxaspiro[5,5]undeca-7,10-diene-9-one. Molecules 2013, 18, 1174-1180. https://doi.org/10.3390/molecules18011174
Plourde GL, Scully TW. Diastereoselective Synthesis of 5-Hydroxy-8-methoxy-1-oxaspiro[5,5]undeca-7,10-diene-9-one. Molecules. 2013; 18(1):1174-1180. https://doi.org/10.3390/molecules18011174
Chicago/Turabian StylePlourde, Guy L., and Thomas W. Scully. 2013. "Diastereoselective Synthesis of 5-Hydroxy-8-methoxy-1-oxaspiro[5,5]undeca-7,10-diene-9-one" Molecules 18, no. 1: 1174-1180. https://doi.org/10.3390/molecules18011174
APA StylePlourde, G. L., & Scully, T. W. (2013). Diastereoselective Synthesis of 5-Hydroxy-8-methoxy-1-oxaspiro[5,5]undeca-7,10-diene-9-one. Molecules, 18(1), 1174-1180. https://doi.org/10.3390/molecules18011174