Frontiers and Approaches to Chemical Synthesis of Oligodeoxyribonucleotides


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
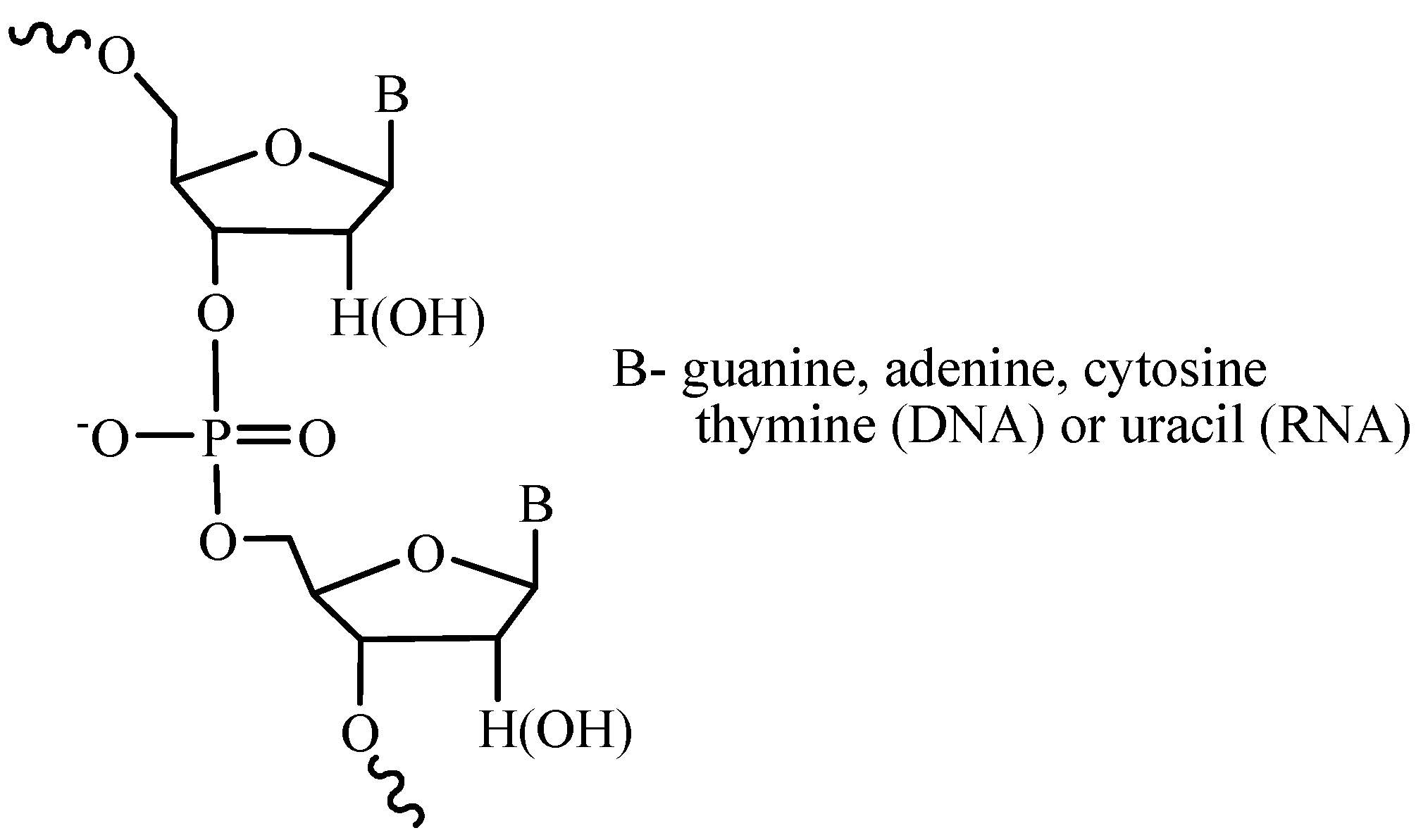
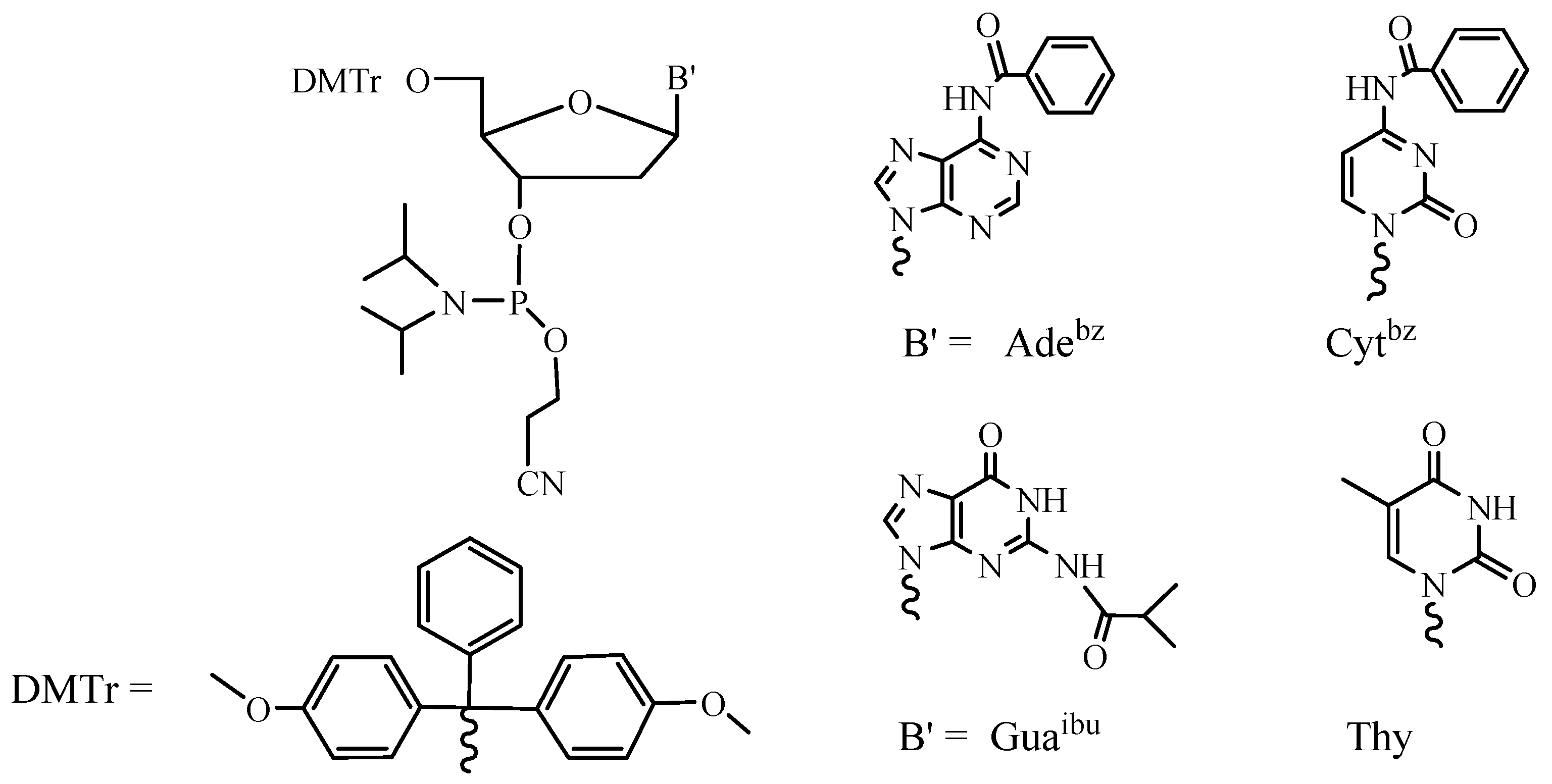
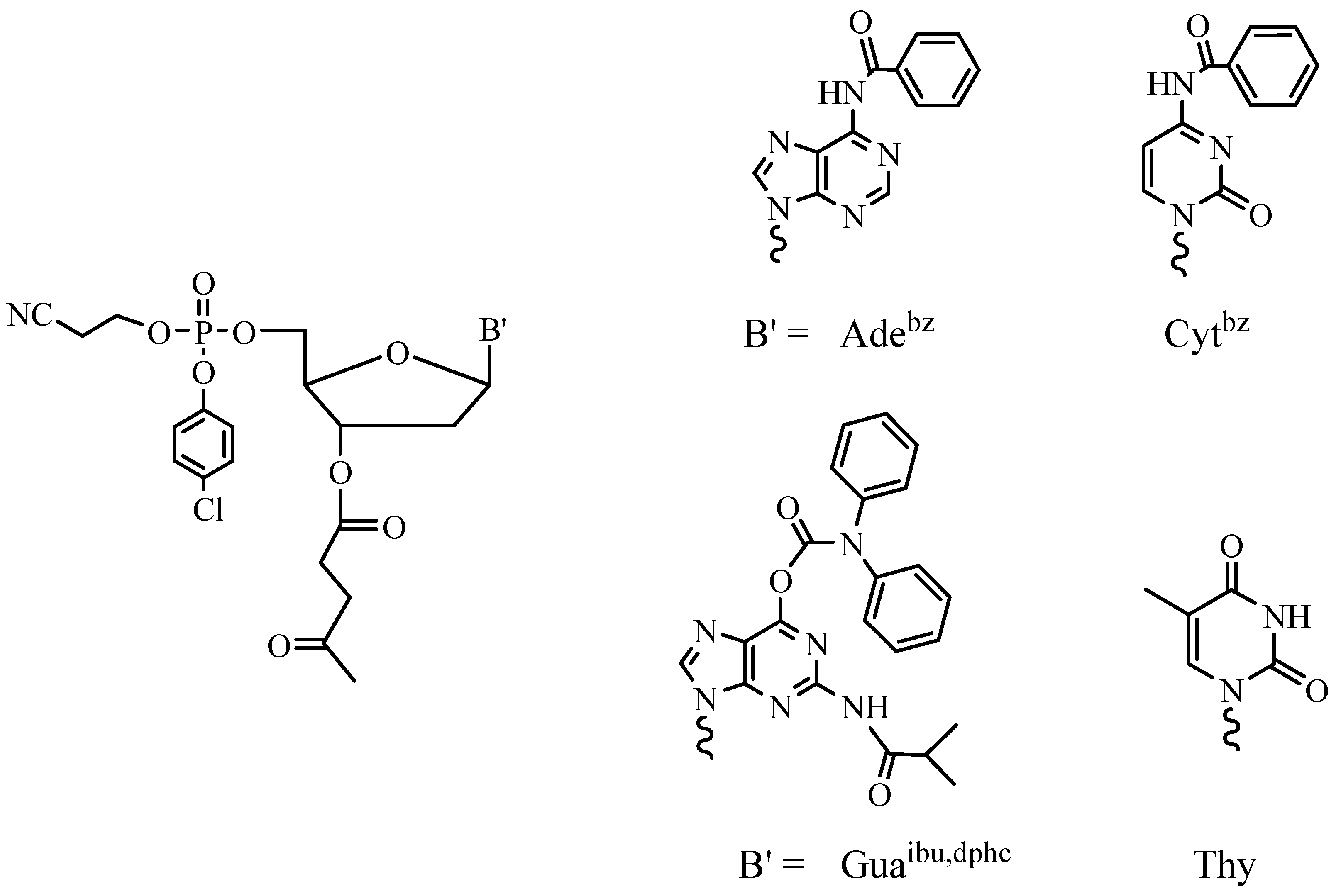
2. Main Steps of Internucleotide Bond Formation
3. Problems of Modern ODN Synthesis
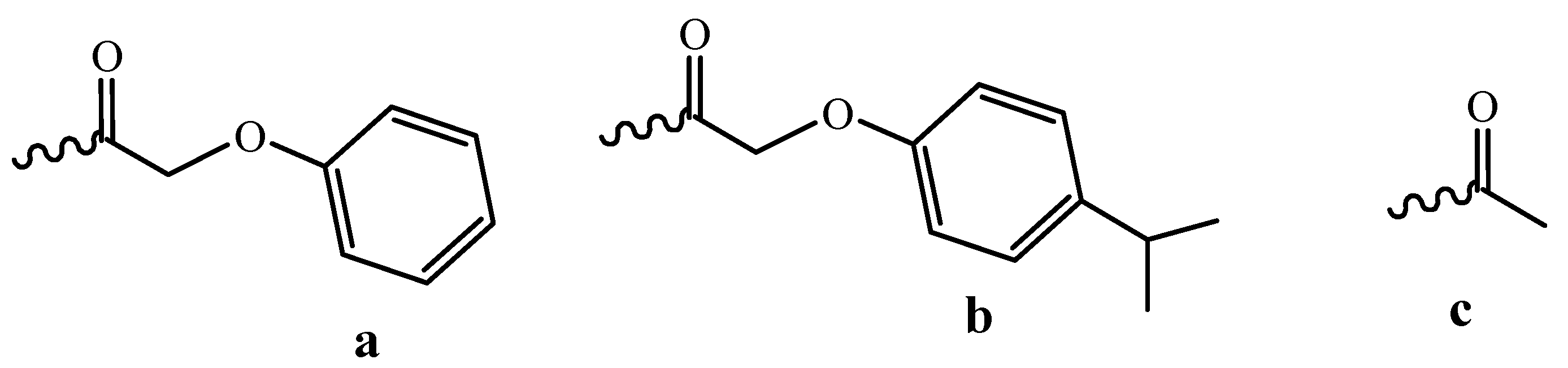
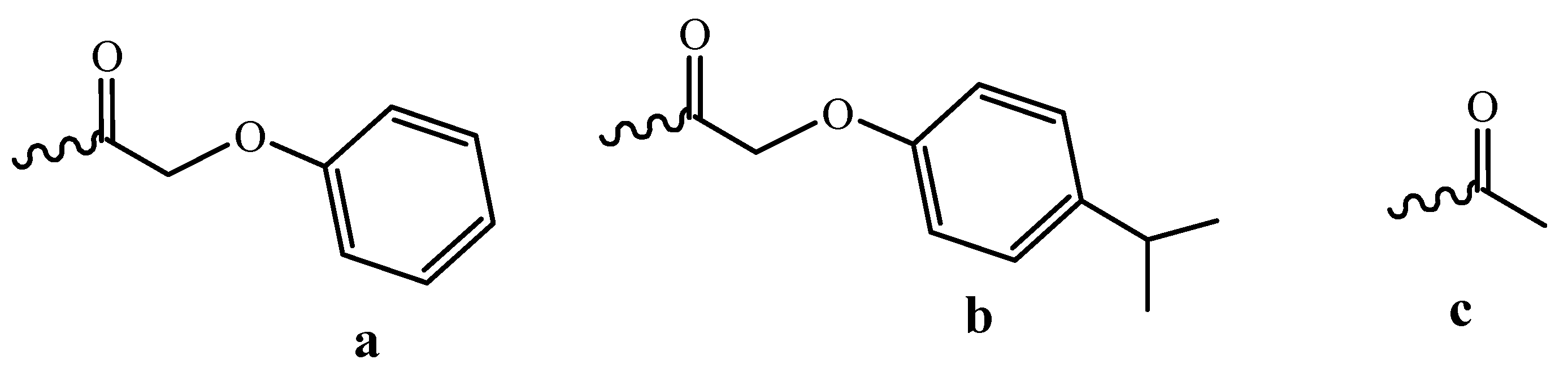
3.1. Fast Oligomer Deprotection
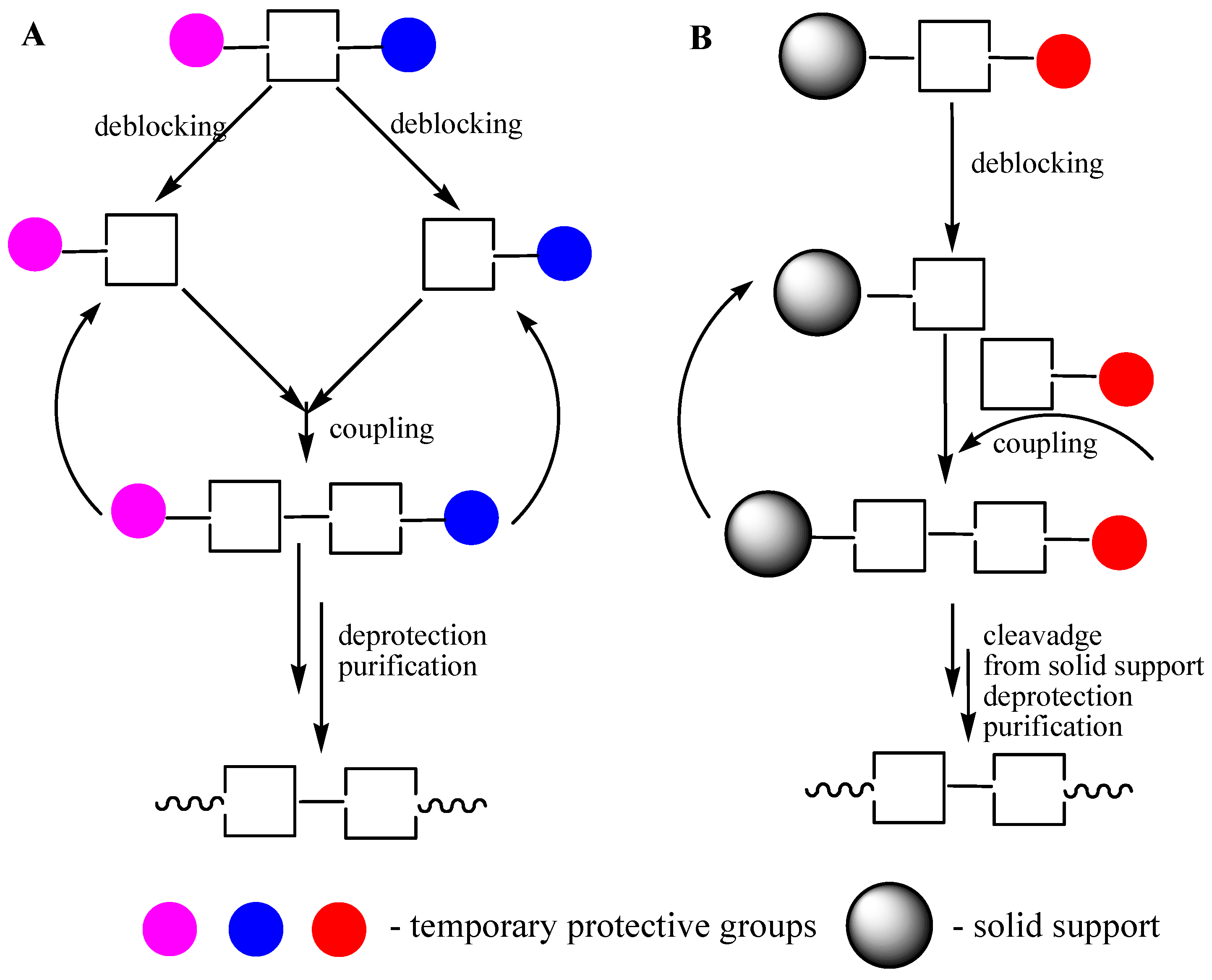
3.2. High-Throughput Parallel Synthesis of ODNs
3.3. Preparative ODN Synthesis
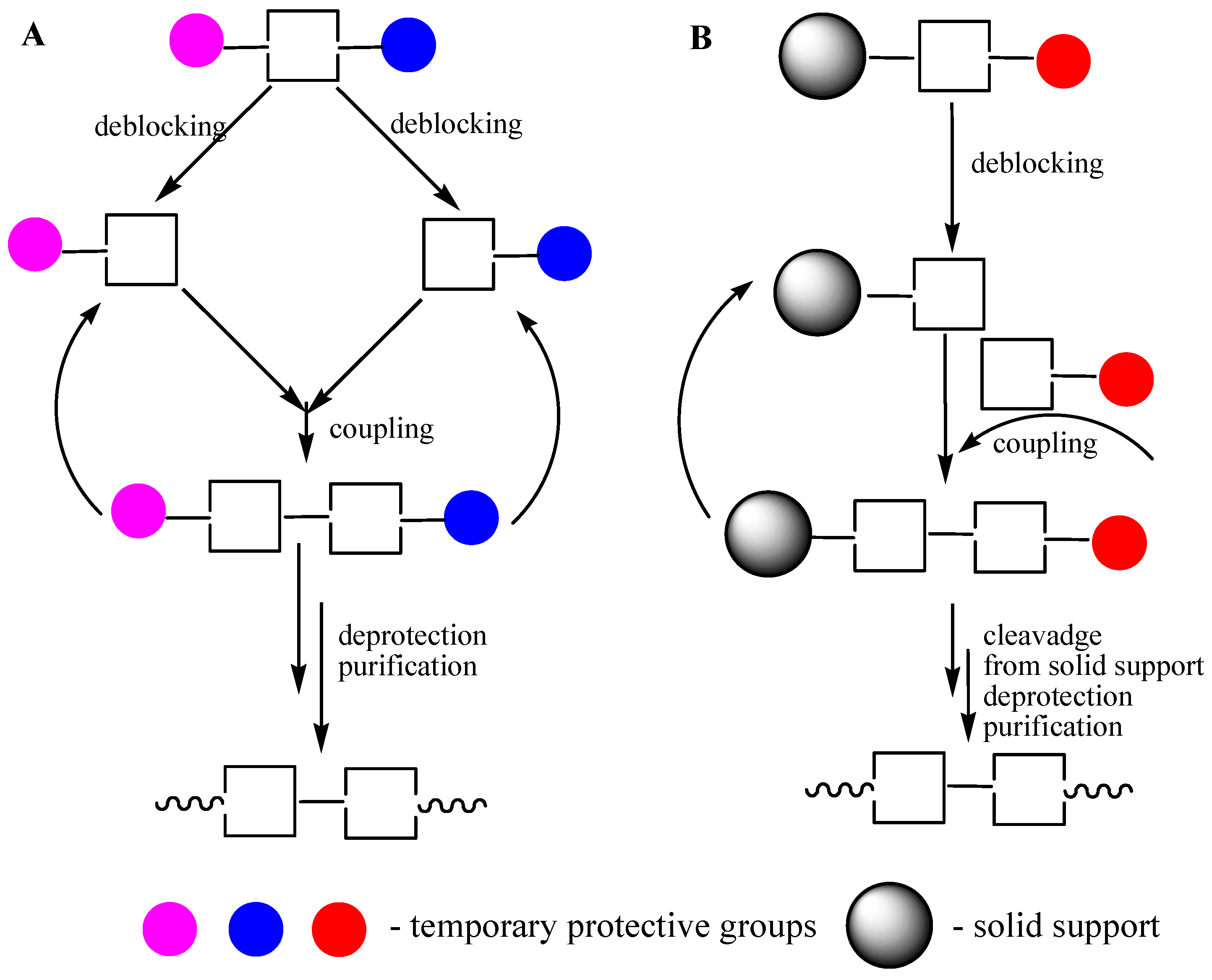
3.4. ODN Synthesis in Liquid Phase
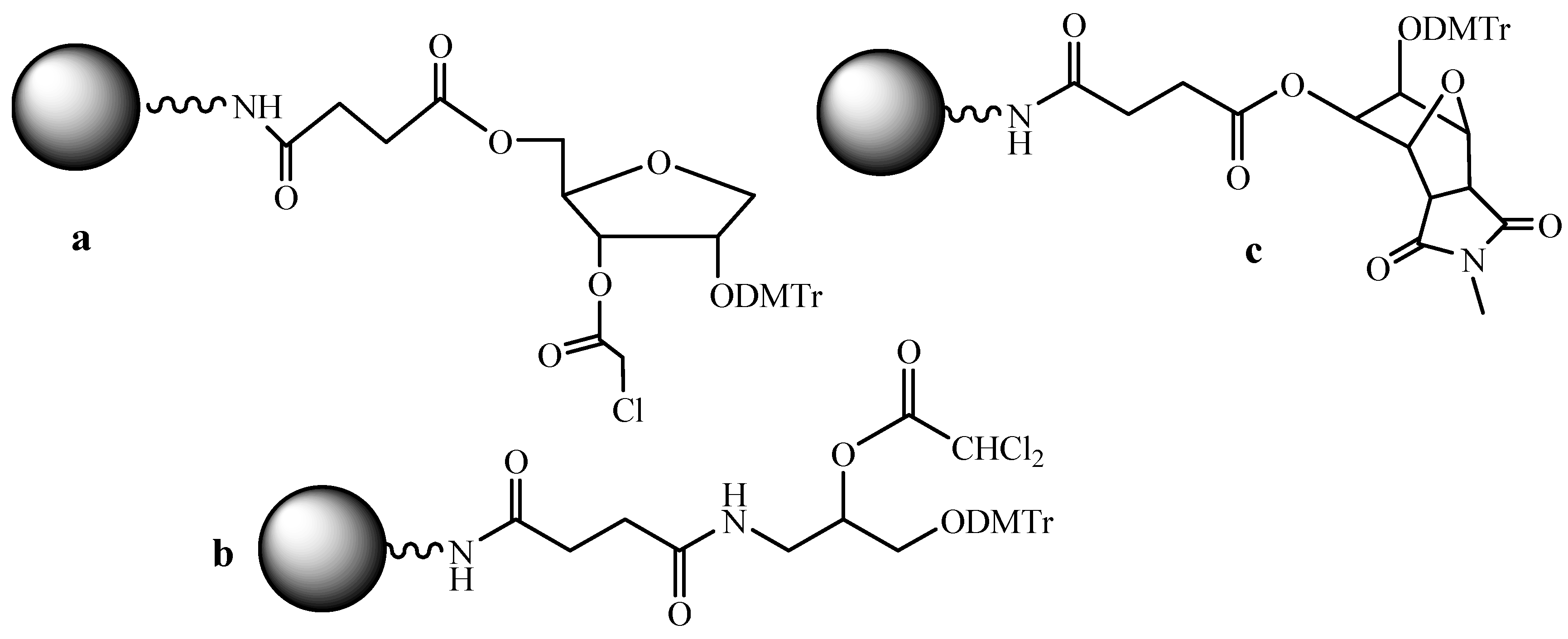
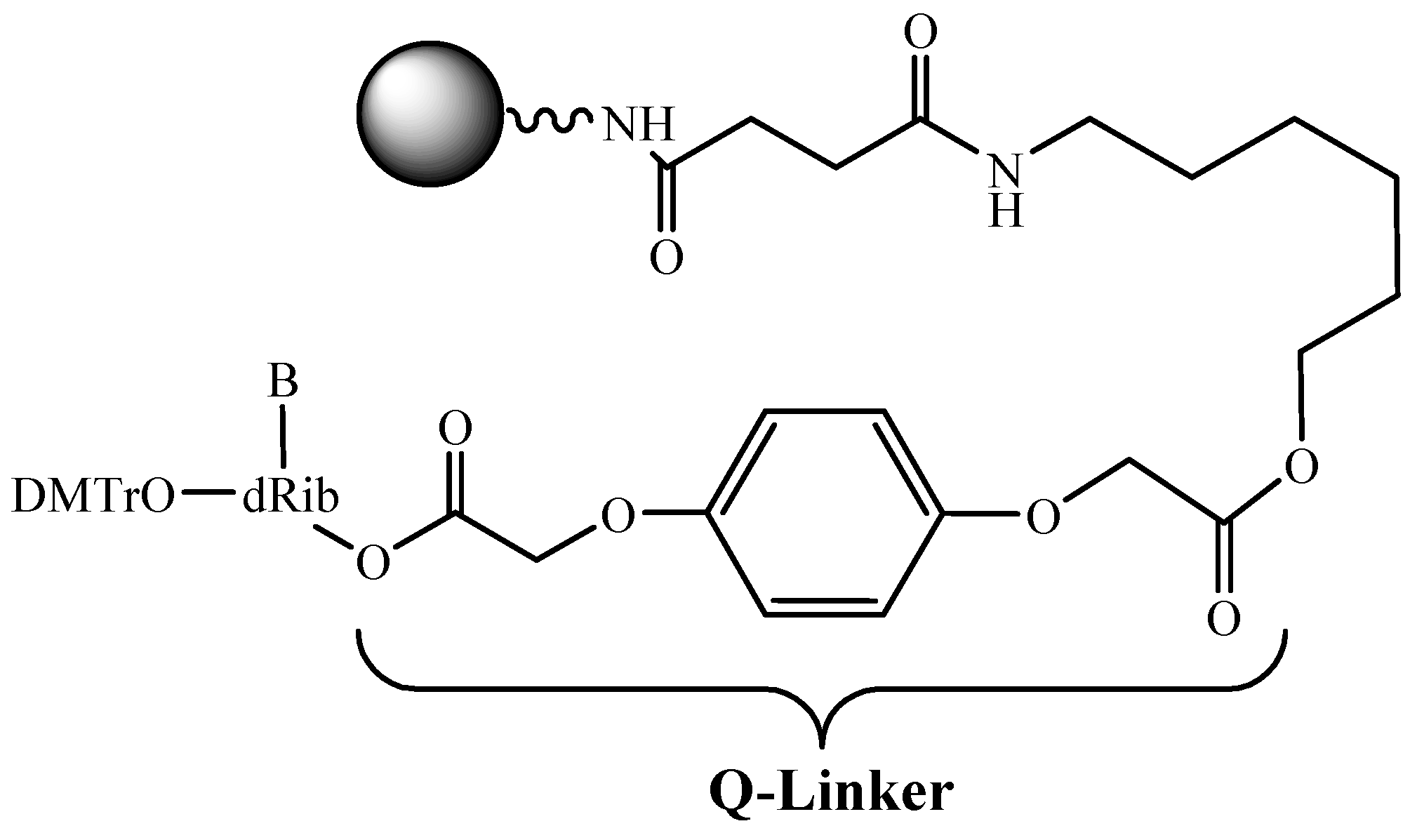
3.5. Soluble Supports in ODN Synthesis
4. Conclusions
Acknowledgments
References
- Reese, C.B. Oligo- and poly-nucleotides: 50 years of chemical synthesis. Org. Biomol. Chem. 2005, 3, 3851–3858. [Google Scholar] [CrossRef] [PubMed]
- Abramova, T.V.; Silnikov, V.N. Synthesis and properties of carbohydrate-phosphate backbone-modified oligonucleotide analogues and nucleic acid mimetics. Russ. Chem. Rev. 2011, 80, 429–452. [Google Scholar] [CrossRef]
- Crooke, S.T. Antisense Drug Technology: Principles, Strategies and Applications, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Watts, J.K.; Corey, D.R. Silencing disease genes in the laboratory and the clinic. J. Pathol. 2012, 226, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Deleavay, G.F.; Damha, M.J. Designing chemically modified oligonucleotides for targeted gene silencing. Chem. Biol. 2012, 19, 937–954. [Google Scholar] [CrossRef] [PubMed]
- Lönnberg, H. Solid-phase synthesis of oligonucleotide conjugates useful for delivery and targeting of potential nucleic acid therapeutics. Bioconjug. Chem. 2009, 20, 1065–1094. [Google Scholar] [CrossRef] [PubMed]
- Gissot, A.; Campo, M.; Grinstaff, M.W.; Barthélémy, P. Nucleoside, nucleotide and oligonucleotide based amphiphiles: A successful marriage of nucleic acids with lipids. Org. Biomol. Chem. 2008, 6, 1324–1333. [Google Scholar] [CrossRef] [PubMed]
- Michelson, A.M.; Todd, A.R. Nucleotides part XXXII. Synthesis of a dithymidine dinucleotide containing a 3′:5′-internucleotidic linkage. J. Chem. Soc. 1955, 2632–2638. [Google Scholar] [CrossRef]
- Khorana, H.G.; Razzel, W.E.; Gilham, P.T.; Tener, G.M.; Pol, E.H. Synthesis of dideoxyribonucleotides. J. Am. Chem. Soc. 1957, 79, 1002–1003. [Google Scholar] [CrossRef]
- Khorana, H.G. Nucleic acid synthesis. Pure Appl. Chem. 1968, 17, 349–381. [Google Scholar] [CrossRef]
- Agarwal, K.L.; Büchi, H.; Caruthers, M.H.; Gupta, N.; Khorana, H.G.; Kleppe, K.; Kumar, A.; Ohtsuka, E.; Rajbhangary, U.L.; van de Sande, J.H.; et al. Total synthesis of the gene for an alanine transfer ribonucleic acid from yeast. Nature 1970, 227, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Khorana, H.G.; Agarwal, K.L.; Besmer, P.; Büchi, H.; Caruthers, M.H.; Cashion, P.J.; Fridkin, M.; Jay, E.; Kleppe, K.; Kleppe, R.; et al. Total synthesis of the structural gene for the precursor of a tyrosine suppressor transfer RNA from Esherichia coli. J. Biol. Chem. 1976, 251, 565–570. [Google Scholar] [PubMed]
- Reese, C.B.; Saffhill, R. Oligonucleotide synthesis via phosphotriester intermediates: The phenyl-protecting group. Chem. Commun. 1968, 1968, 767–768. [Google Scholar] [CrossRef]
- Letsinger, R.L.; Mahadevan, V. Oligonucleotide synthesis on a polymer support. J. Am. Chem Soc. 1965, 87, 3526–3527. [Google Scholar] [CrossRef] [PubMed]
- Berlin, Y.A.; Chakhmakhcheva, O.G.; Efimov, V.A.; Kolosov, M.N.; Korobko, V.G. Arenesulfonyl imidazolides, new reagents for polynucleotide synthesis. Tetrahedron Lett. 1973, 14, 1353–1354. [Google Scholar] [CrossRef]
- Itakura, K.; Bahl, P.; Katagiri, N.; Michniewicrz, J.J.; Wightman, H.; Narang, S.A. A modified triester method for the synthesis of deoxyribopolynucleotides. Can. J. Chem. 1973, 51, 3649–3651. [Google Scholar] [CrossRef]
- Brown, T.; Brown, D.J.S. Modern machine-aided methods of oligodeoxyribonucleotide synthesis. In Oligonucleotides and Analogs. A Practical Approach; Eckstein, F., Ed.; IRL Press: Oxford, UK, 1991; pp. 1–24. [Google Scholar]
- Beaucage, S.L.; Caruthers, M.H. Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett. 1981, 22, 1859–1862. [Google Scholar] [CrossRef]
- Garegg, P.J.; Lindh, I.; Redberg, T.; Stawinski, J.; Strömberg, R.; Henrichson, C. Nucleoside H-phosphonates. III. Chemical synthesis of oligodeoxyribonucleotides by hydrogenphosphonate approach. Tetrahedron Lett. 1986, 27, 4051–4054. [Google Scholar] [CrossRef]
- Zon, G.; Stec, W. Phosphorothioate oligonucleotides. In Oligonucleotides and Analogs. A Practical Approach; Eckstein, F., Ed.; IRL Press: Oxford, UK, 1991; pp. 87–108. [Google Scholar]
- Sanghvi, Y.S. Large-scale oligonucleotide synthesis. Org. Process Res. Dev. 2000, 4, 168–169. [Google Scholar]
- Deprotection—volume 1—deprotect to completion. Available online: http://www.glenresearch.com/GlenReports/GR20-24.html/ (accessed on 9 January 2013).
- Deprotection—volume 3—dye-containing oligonucleotides. Available online: http://www.glenresearch.com/GlenReports/GR21-28.html/ (accessed on 9 January 2013).
- Polushin, N.N.; Morocho, A.M.; Chen, B.-C.; Cohen, J.S. On the rapid deprotection of synthetic oligonucleotides and analogs. Nucleic Acids Res. 1994, 22, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.P.; Hanna, N.B.; Farooqui, F. Fast cleavage and deprotection of oligonucleotides. Tetrahedron Letts. 1994, 35, 4311–4314. [Google Scholar] [CrossRef]
- Chmielewski, M.K.; Tykarska, E.; Markiewicz, W.T.; Rypniewski, W. Engineering N-(2-pyridyl)aminoethyl alcohols as potential precursors of thermolabile protecting groups. New J. Chem. 2012, 36, 603–612. [Google Scholar] [CrossRef]
- Chmielewski, M.K. Novel thermolabile protecting groups with higher stability at ambient temperature. Tetrahedron Lett. 2012, 53, 666–669. [Google Scholar] [CrossRef]
- Chmielewski, M.K. Protecting of a thermolabile protecting group: “Click-clack” approach. Org. Lett. 2009, 11, 3742–3745. [Google Scholar] [CrossRef] [PubMed]
- Lashkari, D.A.; Hunicke-Smith, S.P.; Norge, R.M.; Davis, R.W.; Brennan, T. An automated multiplex oligonucleotide synthesizer: Development of high-throughput, low-cost DNA synthesis. Proc. Natl. Acad. Sci. USA 1995, 92, 7912–7915. [Google Scholar] [CrossRef] [PubMed]
- Sindelar, L.E.; Jaklevic, J.M. High-throughput DNA synthesis in a multichannel forma. Nucleic Acids Res. 1995, 23, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Rainer, S.; Brignac, S.; Bumeister, R.; Belosludsev, Y.; Ward, T.; Grant, O.; O’Brien, K.; Evans, G.A.; Garner, H.R. MerMade: An oligodeoxyribonucleotide synthesizer for high throughput oligonucleotide production in dual 96-well plates. Genome Res. 1998, 8, 741–747. [Google Scholar] [CrossRef]
- Cheng, J.-Y.; Chen, H.-H.; Kao, Y.-S.; Peck, K. High throughput parallel synthesis of oligonucleotides with 1536 channel synthesizer. Nucleic Acids Res. 2002, 30, e93. [Google Scholar] [CrossRef] [PubMed]
- Scheuer-Larsen, C.; Rosenbohm, C.; Jørgensen, T.J.D.; Wengel, J. Introduction of universal solid support for oligonucleotide synthesis. Nucleos. Nucleot. 1997, 16, 67–80. [Google Scholar] [CrossRef]
- Azhaev, A.V.; Antopolsky, M.L. Amide group assisted 3′-dephosphorilation of oligonucleotides synthesizes on universal A-supports. Tetrahedron 2001, 57, 4977–4986. [Google Scholar] [CrossRef]
- Azhaev, A.V.; Antopolsky, M.L.; Tennilä, T.M.L.; Randolph, J.B. A comparative study of commercially available universal supports for oligonucleotide synthesis. The Glen Res. 2004, 17, 1–8. [Google Scholar]
- Guzaev, A.P.; Manokharan, M. A conformationally preorganized universal solid support for efficient oligonucleotide synthesis. J. Am. Chem. Soc. 2003, 125, 2380–2381. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.G.; Anderson, N.L.; Taylor, J.; Goodman, J. Large-scale oligonucleotide synthesizers. Appl. Biochem. Biotech. 1995, 54, 19–42. [Google Scholar] [CrossRef]
- Pon, R.T.; Yu, S.; Guo, Z.; Sanghvi, Y.S. Multiple oligoribonucleotide synthesis on a reusable solid-phase CPG support via the hydroquinone-O,O′-diacetic acid (Q-linker) linker arm. Nucleic Acids Res. 1999, 27, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Sanghvi, Y.S.; Guo, Z.; Pfundheller, H.M.; Converso, A. Improved process for the preparation of nucleosidic phosphoramidites using a safer and cheaper activator. Org. Process Res. Dev. 2000, 4, 175–181. [Google Scholar] [CrossRef]
- Eleuteri, A.; Capaldi, D.C.; Krotz, A.H.; Cole, D.L.; Ravikumar, V.T. Pyridinium trifluoroacetate/N-methylimidazole as an efficient activator for oligonucleotide synthesis via the phosphoramidite method. Org. Process Res Dev. 2000, 4, 182–189. [Google Scholar] [CrossRef]
- Krotz, A.H.; Carty, R.L.; Scozzari, A.N.; Cole, D.L.; Ravicumar, V.T. Large-scale synthesis of antisense oligonucleotides without chlorinated solvents. Org. Process Res. Dev. 2000, 4, 190–193. [Google Scholar] [CrossRef]
- Cheruvallah, Z.S.; Carty, R.L.; Moore, M.N.; Capaldi, D.C.; Krotz, A.H.; Wheeler, P.D.; Turney, B.J.; Craig, S.R.; Gaus, H.J.; Scozzari, A.N.; et al. Synthesis of antisense oligonucleotides: Replacement of 3H-1,2-bezodithiol-3-one 1,1-dioxide (Beaucage reagent) with phenylacetyl disulfide (PADS) as efficient sulfurization reagent: From bench to bulk manufacture of active pharmaceutical ingredient. Org. Process Res. Dev. 2000, 4, 199–204. [Google Scholar] [CrossRef]
- Deshmukh, R.R.; Miller, J.E.; de Leon, P.; Leitch, W.E. II.; Cole, D.L.; Sanghvi, Y.S. Process development for purification of theurapeutic antisense oligonucleotides by anion-exchange chromatography. Org. Process Res. Dev. 2000, 4, 205–213. [Google Scholar] [CrossRef]
- Shanagar, J. Purification of a synthetic oligonucleotides by anion exchange chromatography: Method optimization and scale-up. J. Biochem. Biophys. Methods 2005, 64, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Josten, A.; Bauer, G.; Götz, P. Large-scale purification of oligonucleotides by extraction and precipitation with butanol. Biotechnol. Bioeng. 2005, 89, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Efimov, V.A.; Buryakova, A.A.; Reverdatto, S.V.; Chakhmakhcheva, O.G.; Ovchinnikov, Y.A. Rapid synthesis of long-chain deoxyribonucleotides by the N-methylimidazole phosphotriester method. Nucleic Acids Res. 1983, 11, 8369–8387. [Google Scholar] [CrossRef] [PubMed]
- Zarytova, V.F.; Ivanova, E.M.; Romanenko, V.P. Synthesis of oligonucleotides by the phosphotriester method in chloroform. Bioorg. Khim. (Russ.) 1983, 9, 516–521. [Google Scholar]
- Abramova, T.V.; Komarova, N.I.; Mundus, D.A.; Pereboeva, O.S. Effective synthesis of preparative quantities of 5′-phosphorylated oligodeoxyribonucleotides. Izv. Sib. Otd. Akad. Nauk. Khim. (Russ.) 1990, 5, 45–51. [Google Scholar]
- Mazzei, M.; Balbi, A.; Grandi, T.; Sottofattori, E.; Garzoglio, R.; Abramova, T.; Ivanova, E. Synthesis in solution of oligonucleotides and some their 5′- and 3′-linked derivatives. Farmaco 1993, 48, 1649–1661. [Google Scholar] [PubMed]
- Boutorine, A.S.; Sun, J.S. Postsynthetic functionalization of triple helix-forming oligonucleotides. In Methods in Molecular Biology; Walker, J.M., Ed.; Humana Press: Clifton, NJ, USA, 2005; Volume 288, pp. 251–260. [Google Scholar]
- Mukaiyama, T. Die Oxidations-reductions-kondensation. Angew. Chem. Int. Ed. Engl. 1976, 88, 111–120. [Google Scholar] [CrossRef]
- Abramova, T.V. Methodology for obtaining modified oligodeoxyrobonucleotides for creation effective tools of molecular biology investigations. D.Sc. Thesis, Institute of Chemical Biology and Fundamental Medicine, SB of RAN, Novosibirsk, Russia, 2012. [Google Scholar]
- Froehler, B.C.; Matteucci, M.D. 1-Methyl-2-(2-hydroxyphenyl)imidazole: A catalytic phosphate protecting group in oligodeoxynucleotide synthesis. J. Am. Chem. Soc. 1985, 107, 278–279. [Google Scholar] [CrossRef]
- Efimov, V.A.; Molchanova, N.S.; Chakhmakhcheva, O.G. Approach to the synthesis of natural and modified oligonucleotides by the phosphotriester method using O-nucleophilic intramolecular catalysis. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Rejman, D.; Masojíková, M.; Rosenberg, I. Nucleosidyl-O-Methylphosphonates: A pool of monomers for modified oligonucleotides. Nucleosides Nucleotides Nucleic Acids 2004, 23, 1683–1705. [Google Scholar] [CrossRef] [PubMed]
- Aralov, A.V.; Klykov, V.N.; Chakhmakhcheva, O.G.; Efimov, V.A. Monomers containing 2′-O-alkoxymethyl groups as synthons for the oligonucleotide synthesis by the phosphotriester method. Russ. J. Bioorg. Chem. 2011, 37, 586–592. [Google Scholar] [CrossRef]
- Efimov, V.A.; Aralov, A.V.; Fedunin, S.V.; Klykov, V.N.; Chakhmakhcheva, O.G. An azidomethyl protective group in the synthesis of oligoribonucleotides by the phosphotriester method. Russ. J. Bioorg. Chem. 2009, 35, 250–253. [Google Scholar] [CrossRef]
- Reese, C.B.; Yan, H. Solution phase synthesis of ISIS 2922 (Vitravene) by modified H-phosphonate approach. J. Chem. Soc. Perkin Trans. I 2002, 1, 2619–2633. [Google Scholar] [CrossRef]
- De Koning, M.C.; Ghisaidoobe, A.B.T.; Duynstee, H.I.; Ten Kortenaar, P.B.W.; Filippov, D.V.; van der Marel, G.A. Simple and efficient solution-phase synthesis of oligonucleotides using extractive work-up. Org. Process Res. Dev. 2006, 10, 1238–1245. [Google Scholar] [CrossRef]
- Arunachalam, T.S.; Wichert, C.; Appel, B.; Müller, S. Mixed oligonucleotides for random mutagenesis: Best way of making them. Org. Biomol. Chem. 2012, 10, 4641–4650. [Google Scholar] [CrossRef] [PubMed]
- Bonora, G.M.; Scremin, C.L.; Colonna, F.P.; Garbesi, A. HELP (High Efficiency Liquid Phase) new oligonucleotide synthesis on soluble polymeric support. Nucleic Acids Res. 1990, 18, 3155–3159. [Google Scholar] [CrossRef] [PubMed]
- Bonora, G.M.; Biancotto, G.; Maffini, M.; Scremin, C.L. Large scale, liquid phase synthesis of olgonucleotides by the phosphoramidite approach. Nucleic Acids Res. 1993, 21, 1213–1217. [Google Scholar] [CrossRef] [PubMed]
- Bonora, G.M. Large-scale preparation of conjugated oligonucleoside phosphorothioates by the high-efficiency liquid-phase (HELP) method. Curr. Protoc. Nucleic Acid Chem. 2005. [Google Scholar] [CrossRef]
- Molina, A.G.; Kungurtsev, V.; Virta, P.; Lönnberg, H. Acetylated and methylated β-cyclodextrins as viable soluble supports for the synthesis of short 2′-oligodeoxyribonucleotides in solution. Molecules 2012, 17, 12102–12120. [Google Scholar] [CrossRef] [PubMed]
- Dueymes, C.; Schönberger, A.; Adamo, I.; Navarro, A.E.; Meyer, A.; Lange, M.; Imbach, J.L.; Link, F.; Morvan, F.; Vasseur, J.J. High-yield solution-phase synthesis of di- and trinucleotideblocks assisted by polymer-supported reagents. Org. Lett. 2005, 7, 3485–3488. [Google Scholar] [CrossRef] [PubMed]
- Adamo, I.; Dueymes, C.; Schönberger, A.; Navarro, A.E.; Meyer, A.; Lange, M.; Imbach, J.L.; Link, F.; Morvan, F.; Vasseur, J.J. Solution-phase synthesis of phosphorothioate oligonucleotides using a solid-supported acyl chloride with H-Phosphonate Chemistry. Eur. J. Org. Chem. 2006, 436–448. [Google Scholar] [CrossRef]
- Gao, X.L.; Gulari, E.; Zhou, X.C. In situ synthesis of oligonucleotide microarrays. Biopolymers 2004, 73, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Coleman, J.R.; Wimmer, E. Putting synthesis into biology: A viral view of genetic engineering through de novo gene and genome synthesis. Chem. Biol. 2009, 16, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.S.; Carr, P.A.; Chen, L.; Zhang, S.G.; Jacobson, J.M. Parallel gene synthesis in microfluidic device. Nucleic Acids Res. 2007, 35, e61. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Castrataro, P.; Lee, C.-C.; Quaqe, S.P. Solvent resistant microfluidic synthesizer. Lab Chip 2007, 7, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.C.; Ye, H.; Kuan, Y.K.; Li, M.H.; Ying, J.Y. Integrated two-step gene synthesis in microfluidic device. Lab Chip 2009, 9, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-C.; Snyder, T.M.; Quaqe, S.R. A microfluidic oligonucleotide synthesizer. Nucleic Acids Res. 2010, 38, 2514–2531. [Google Scholar] [CrossRef] [PubMed]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Abramova, T. Frontiers and Approaches to Chemical Synthesis of Oligodeoxyribonucleotides. Molecules 2013, 18, 1063-1075. https://doi.org/10.3390/molecules18011063
Abramova T. Frontiers and Approaches to Chemical Synthesis of Oligodeoxyribonucleotides. Molecules. 2013; 18(1):1063-1075. https://doi.org/10.3390/molecules18011063
Chicago/Turabian StyleAbramova, Tatyana. 2013. "Frontiers and Approaches to Chemical Synthesis of Oligodeoxyribonucleotides" Molecules 18, no. 1: 1063-1075. https://doi.org/10.3390/molecules18011063
APA StyleAbramova, T. (2013). Frontiers and Approaches to Chemical Synthesis of Oligodeoxyribonucleotides. Molecules, 18(1), 1063-1075. https://doi.org/10.3390/molecules18011063