Synthesis and Chromatography-Free Purification of PNA-PEO Conjugates for the Functionalisation of Gold Sensors

 ,
, 



Abstract
:
1. Introduction
2. Results and Discussion
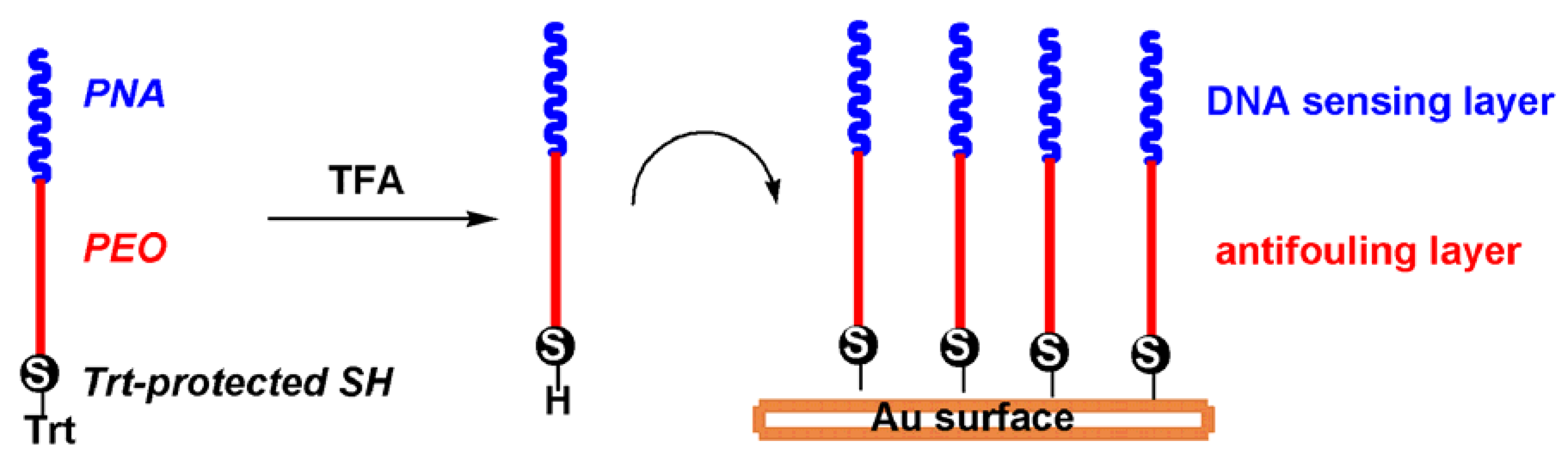
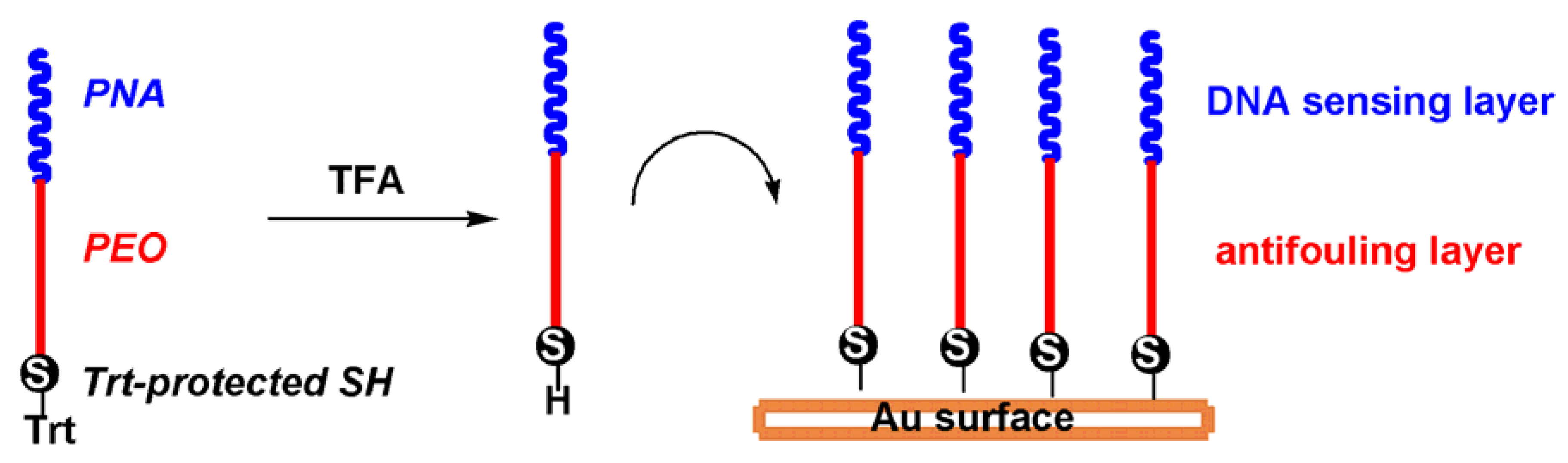
2.1. Probe Design
2.2. Selection of the PNA Sequence and Length
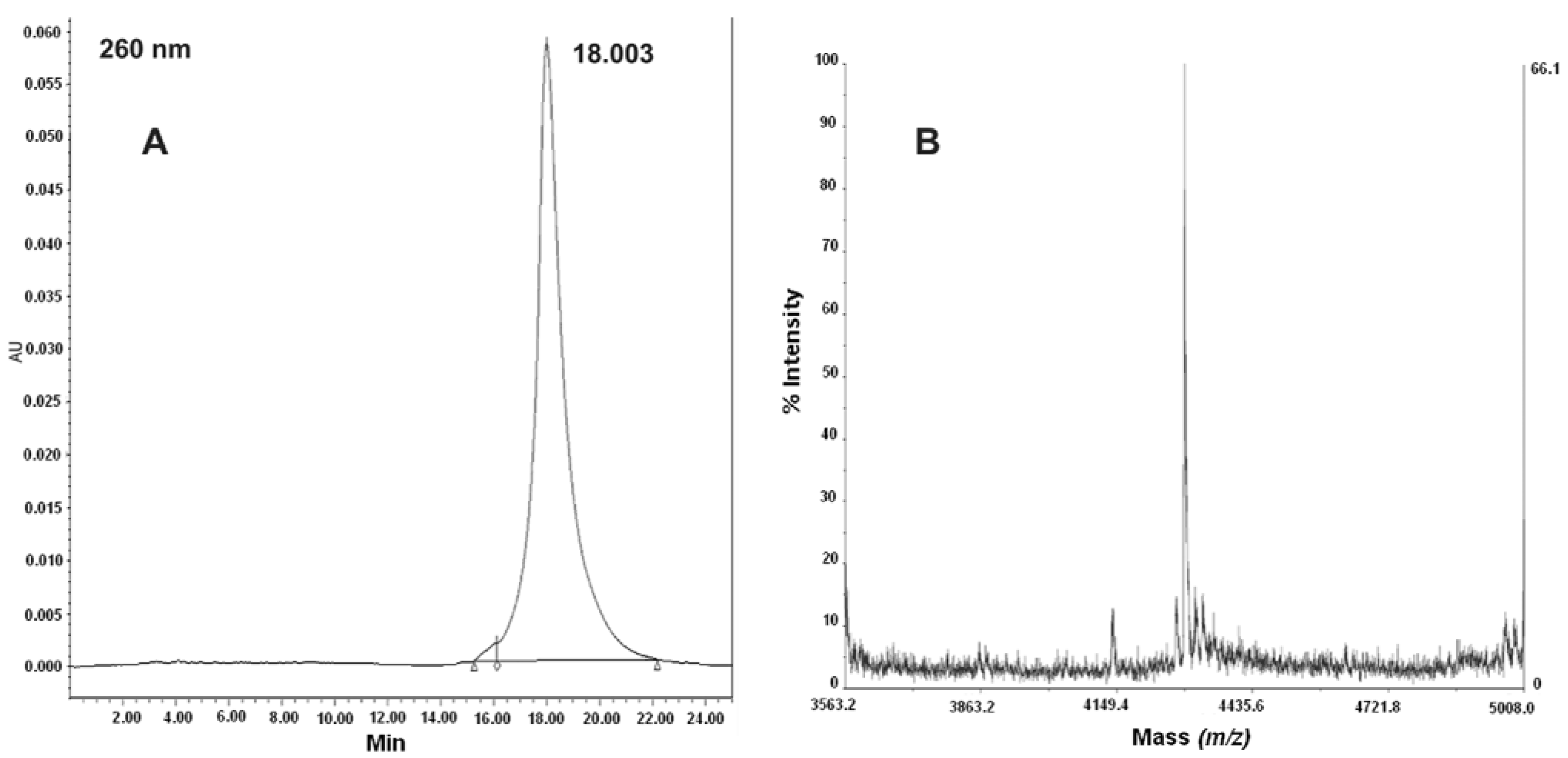
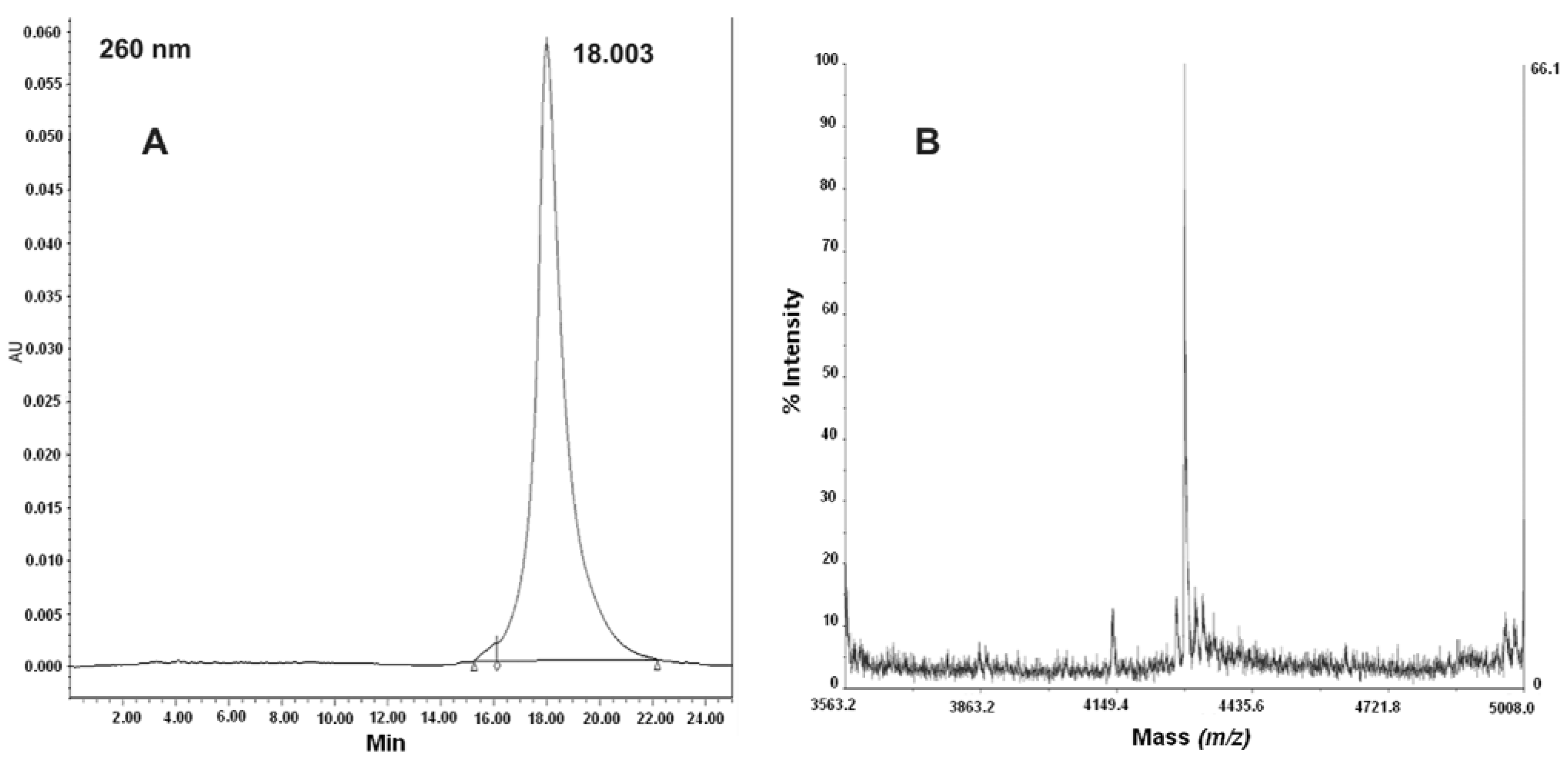
2.3. Synthesis and Characterization of PNA-RPOB15wt
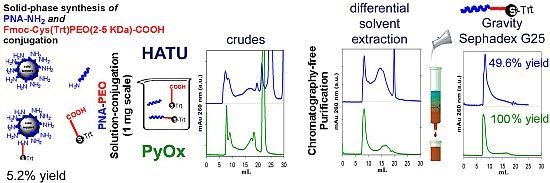
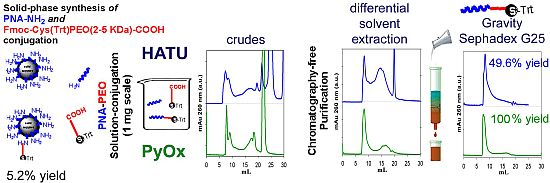
2.4. Solid-Phase Conjugation of Fmoc-Cys(Trt)-PEO5KDa-COOH to PNA
2.5. Conjugation of Fmoc-Cys(Trt)-PEO5KDa-COOH to PNA in Solution

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eq. ratio HATU/DMAP | % of amine modification | Eq. ratio PyOxim:TEA | % of amine modification |
|---|---|---|---|
| 1 | 0 | 10:2 | 0 |
| 10 | 44 | 10:10 | 33 |
| 30 | 90 | 10:20 | 52.1 |
| 36 | 100 | 20:20 | 72.3 |
| 40:30 | 100 |
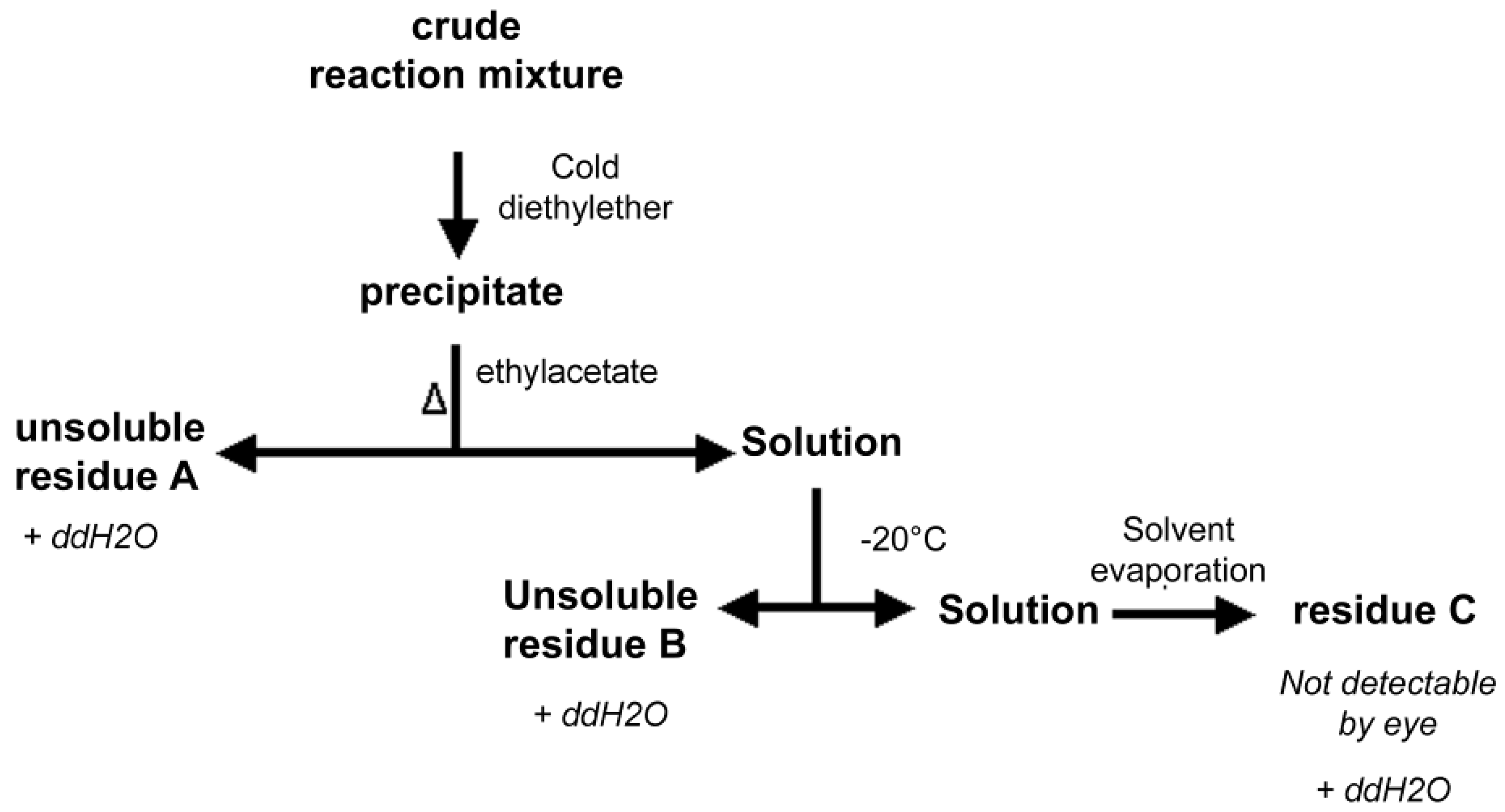
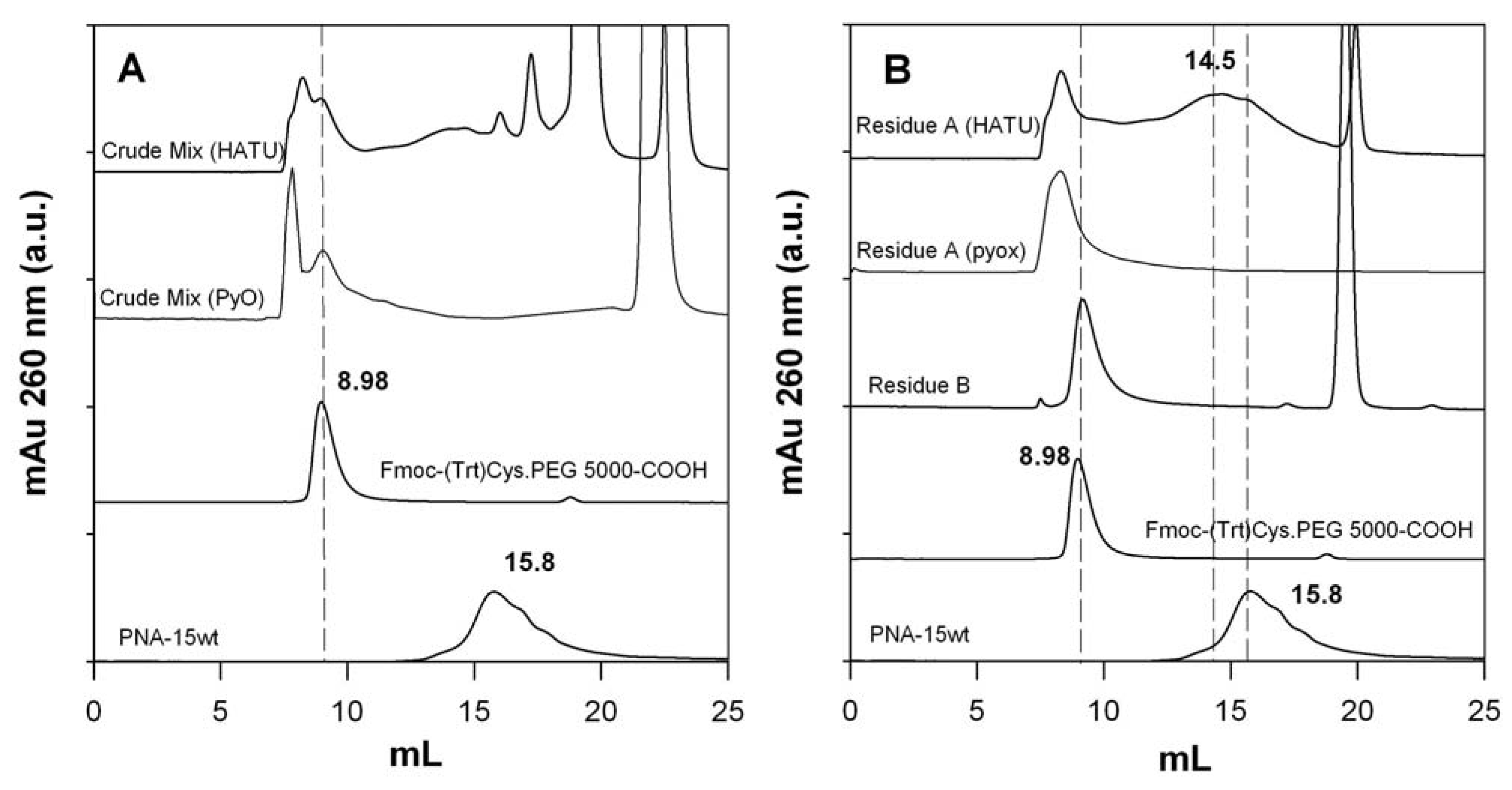
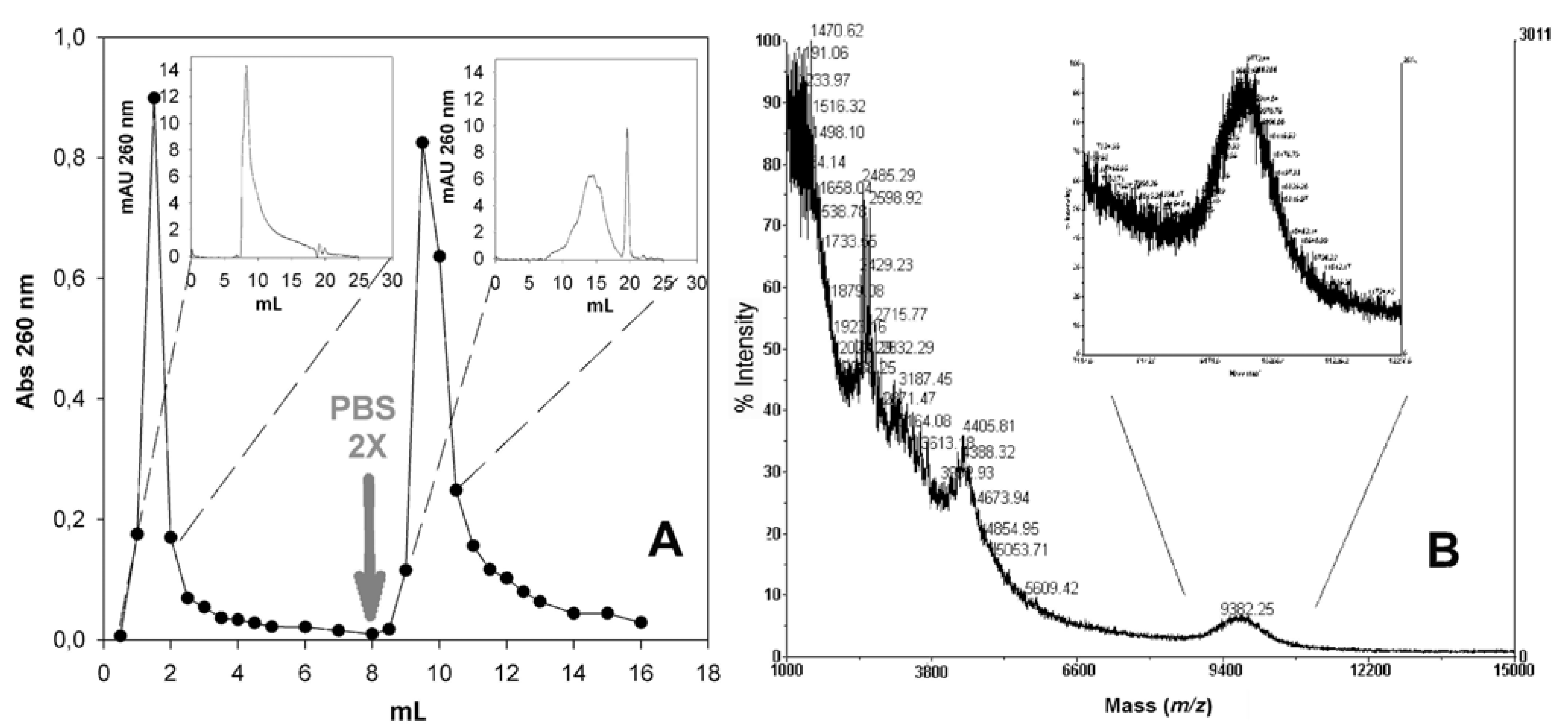
2.6. Isolation of PNA-PEO from Crude Reaction Mixture and Chromatographic Characterization
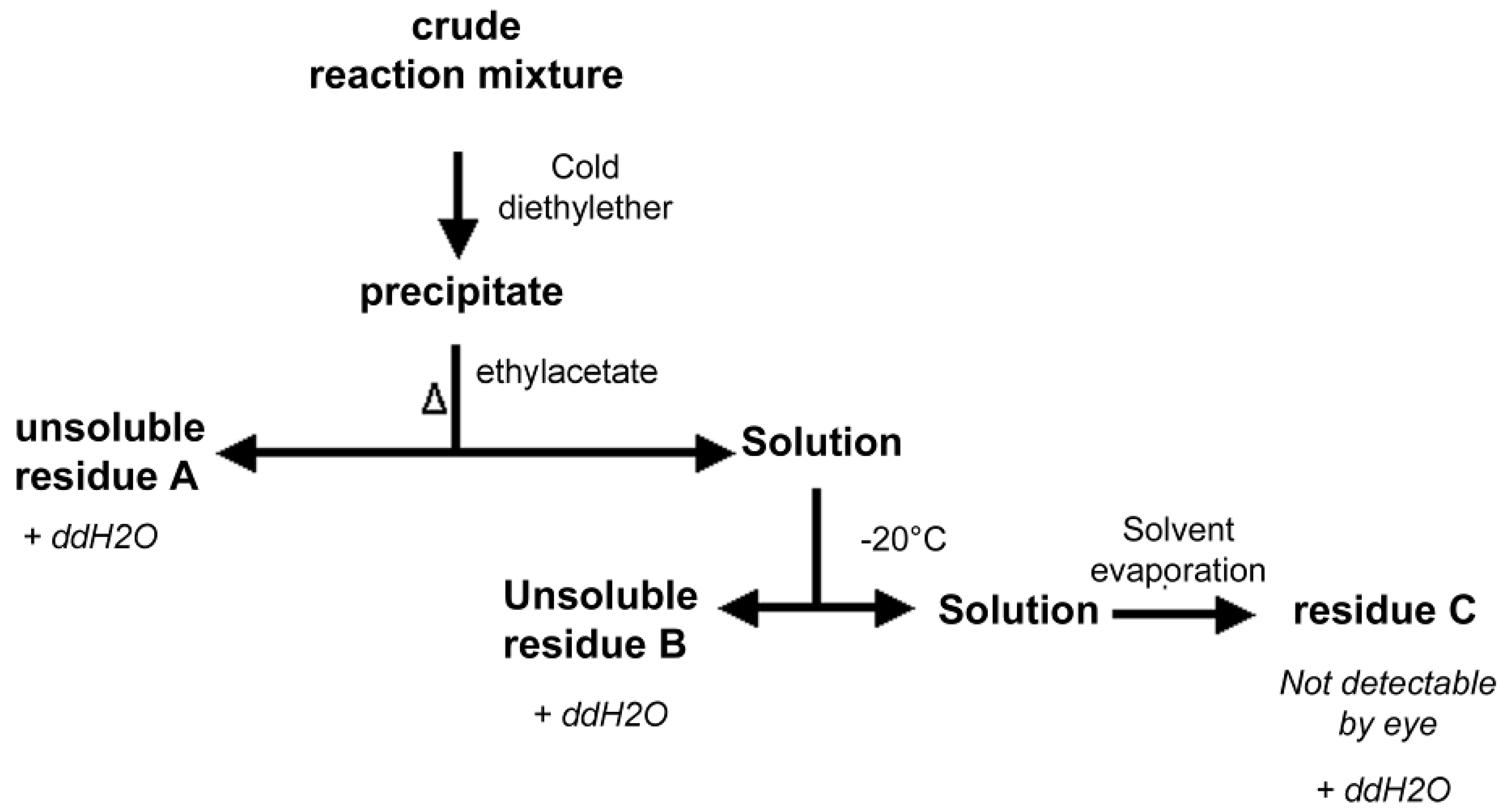
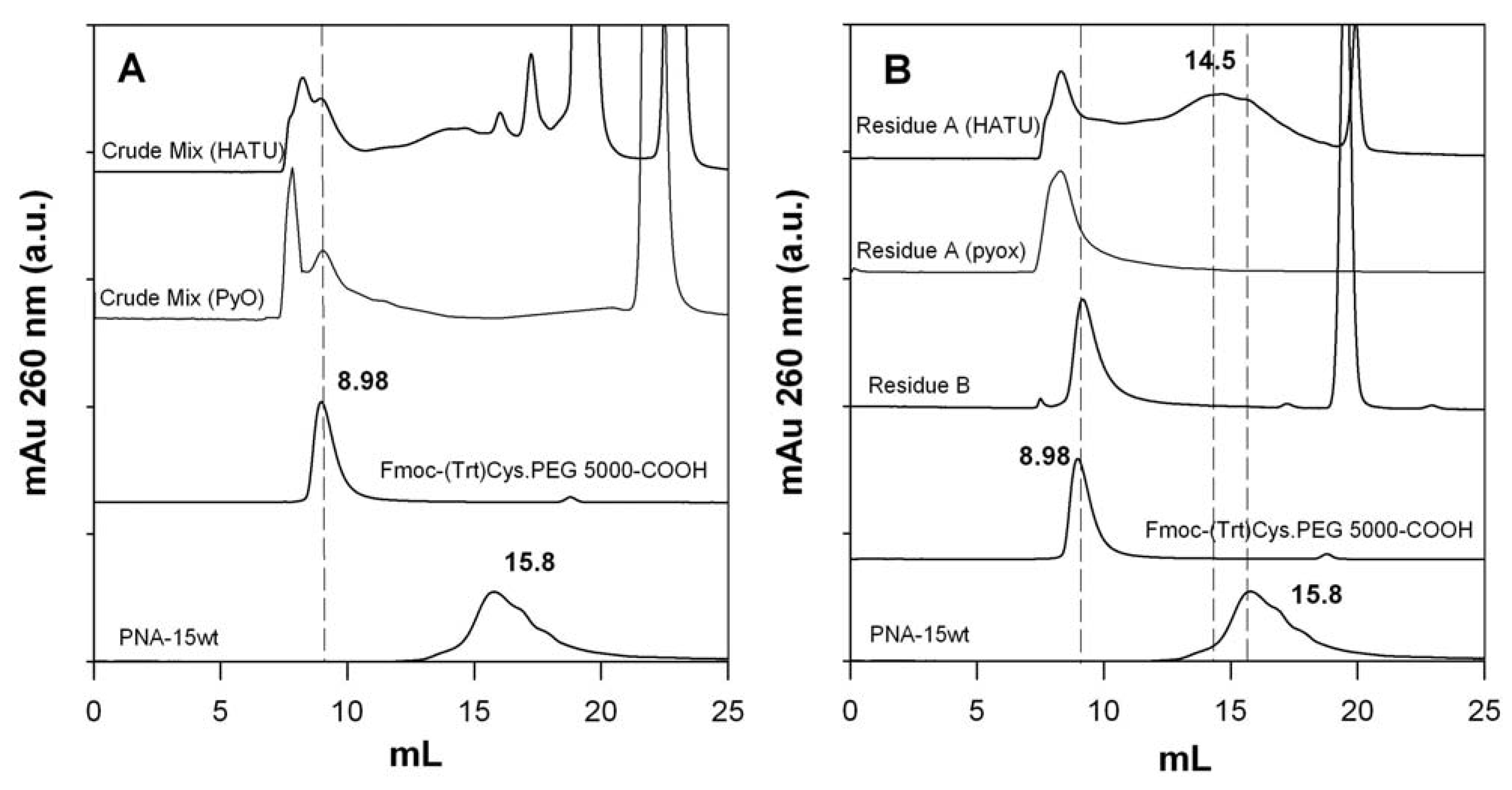
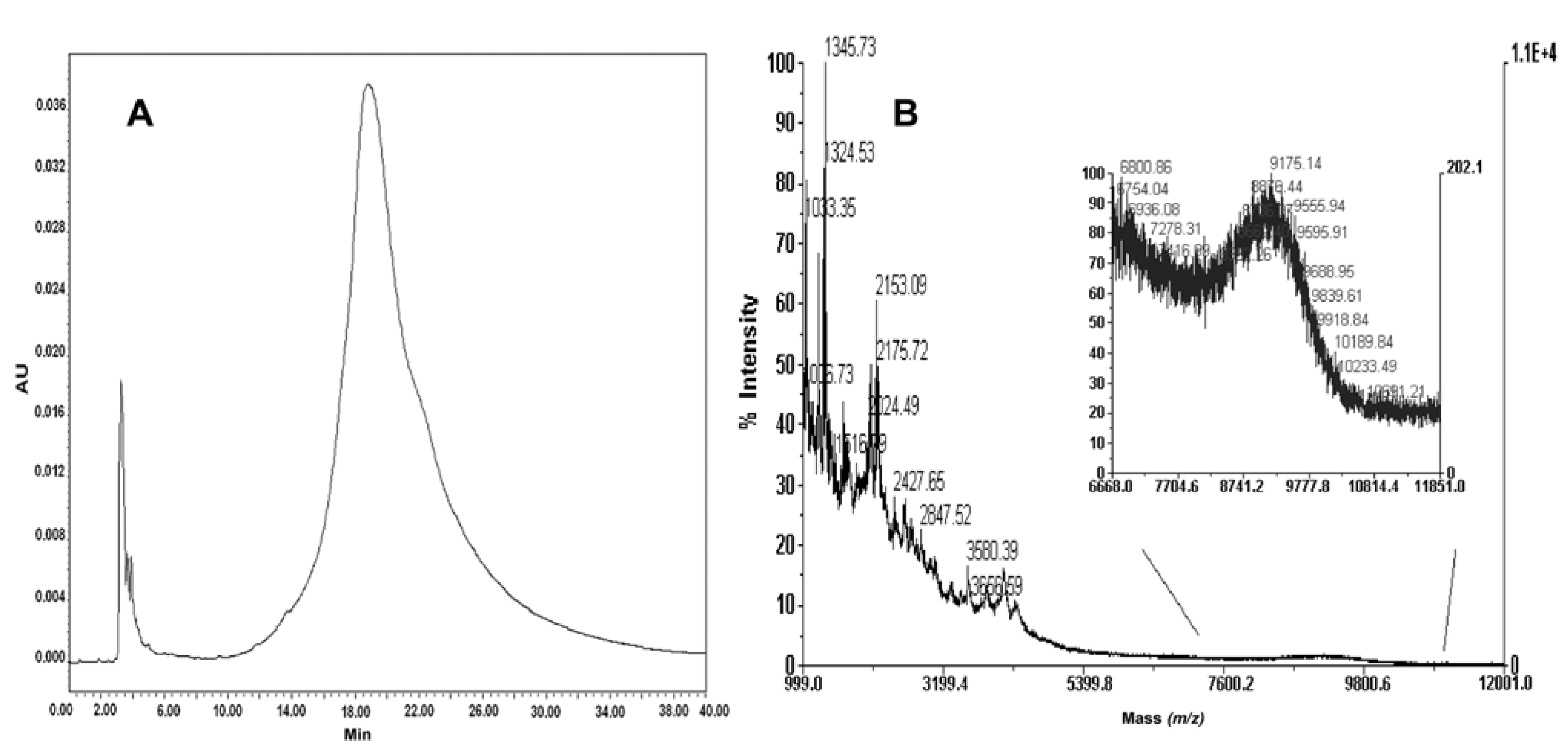
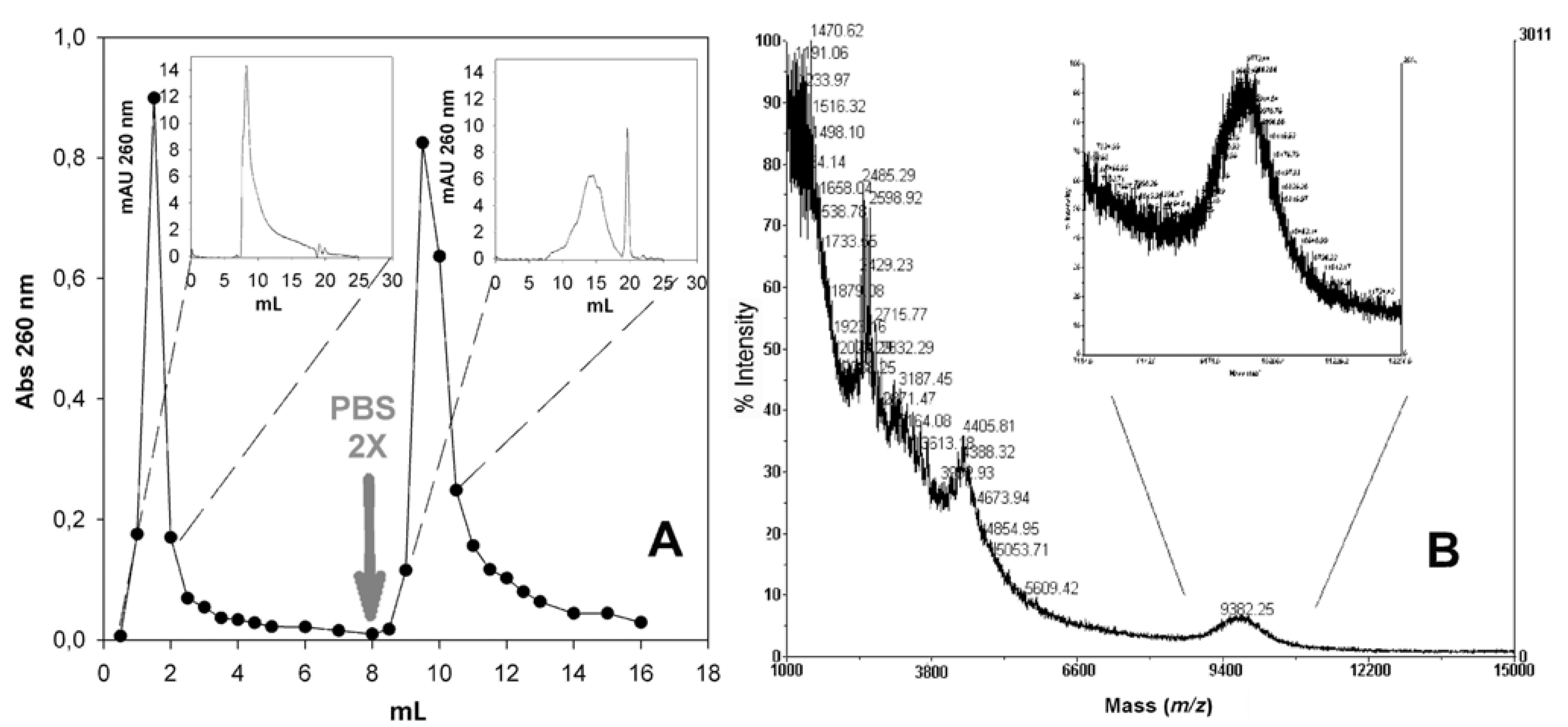
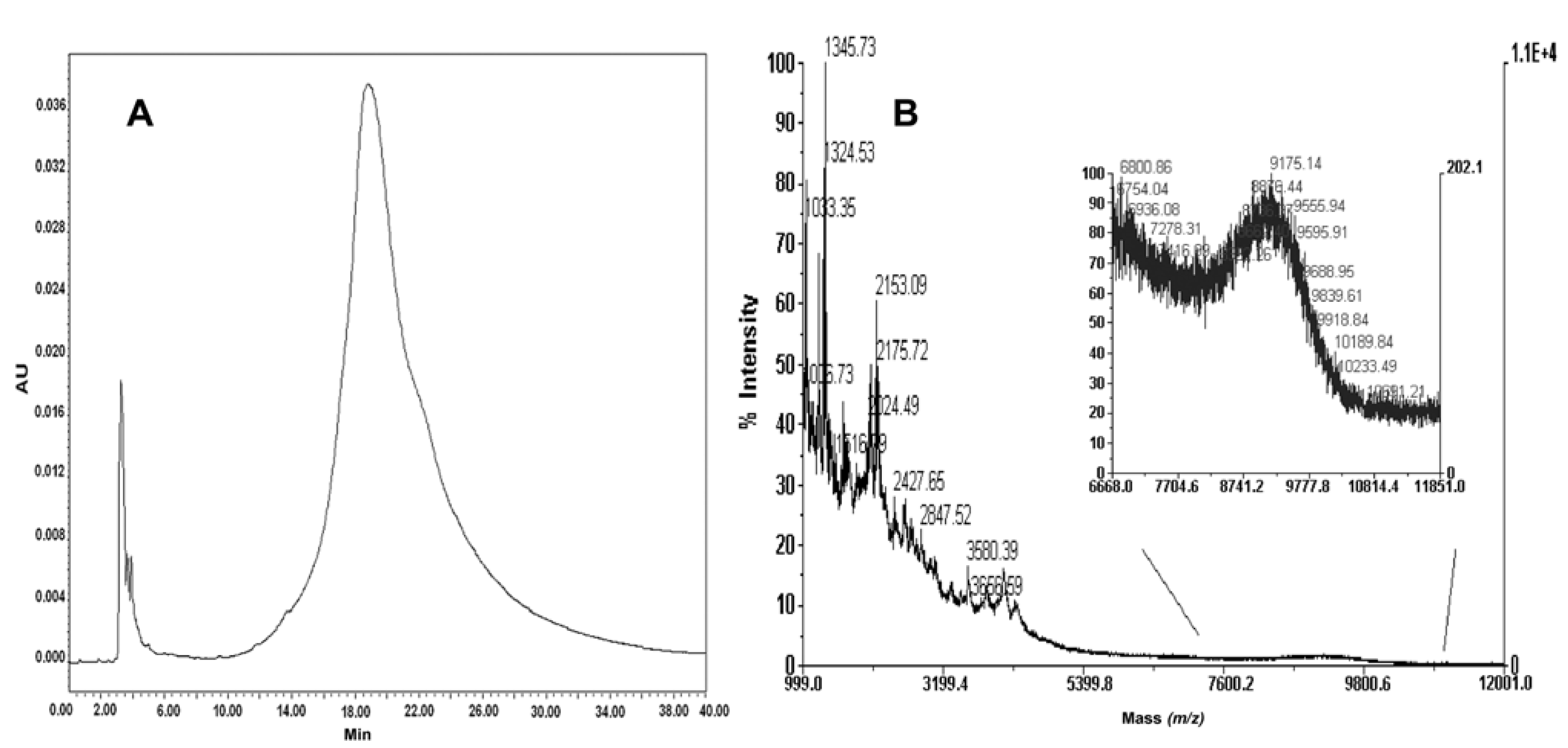
2.7. Conjugation of Fmoc-Cys(Trt)-PEO2KDa-COOH to PNA
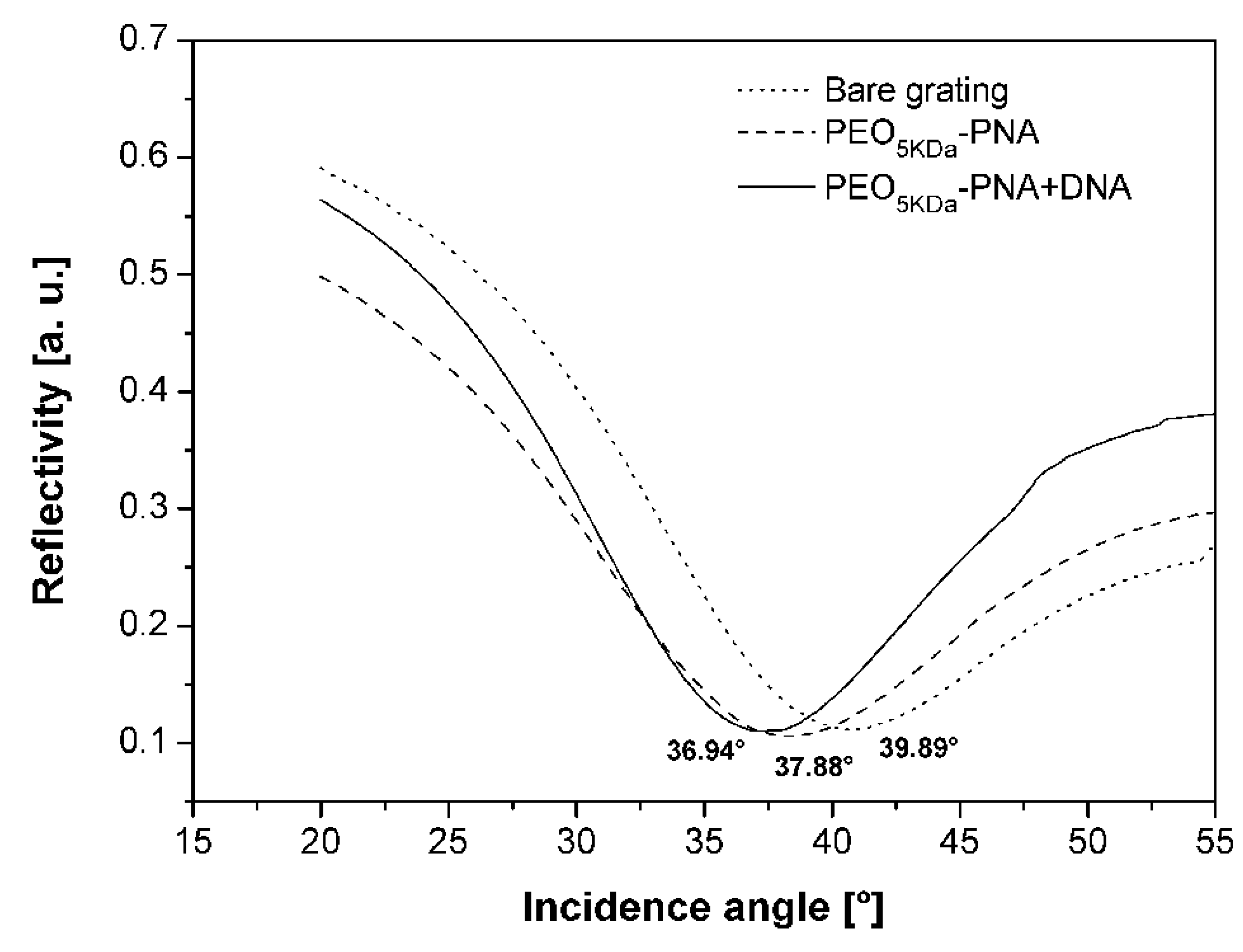
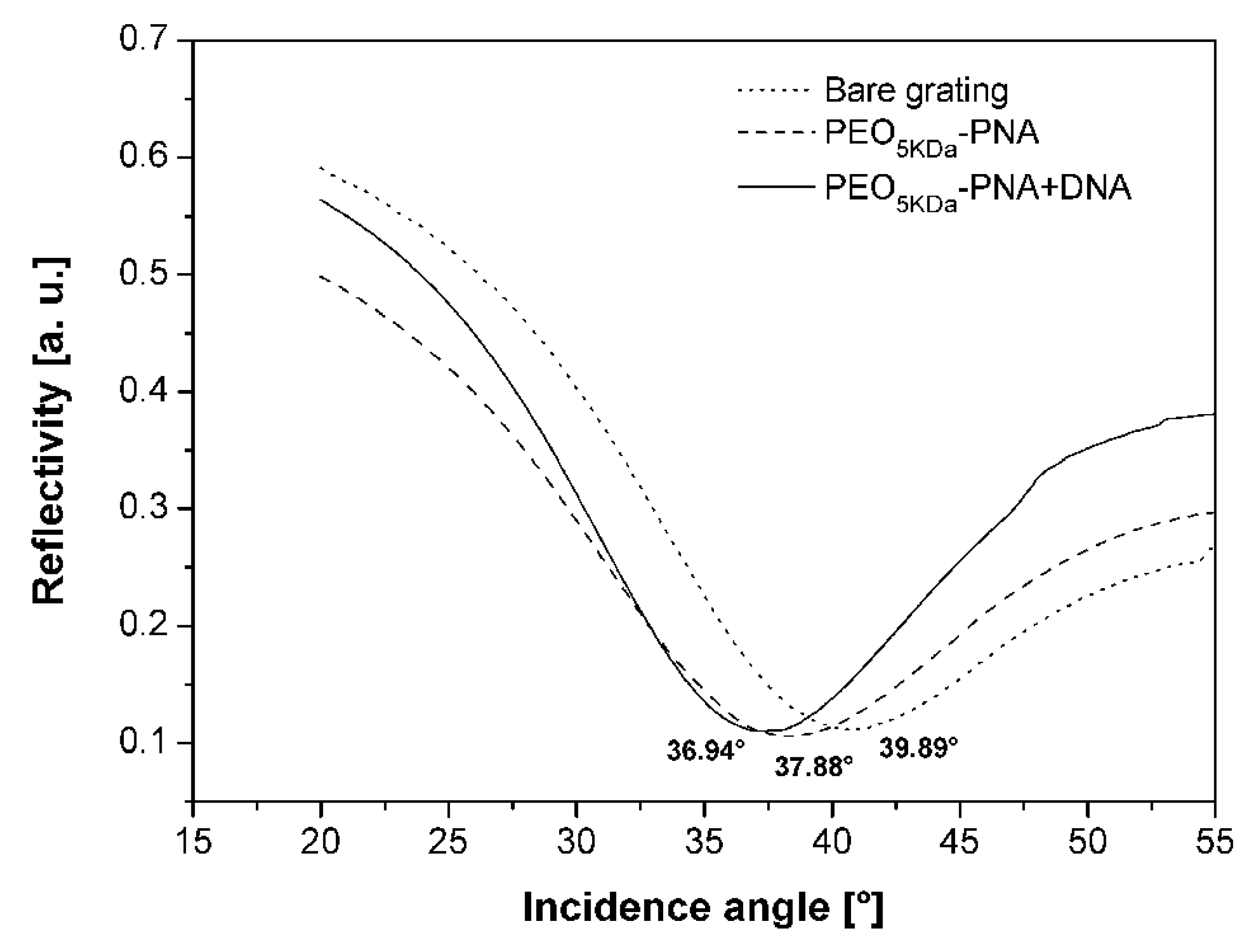
2.8. Application of the PNA-PEO Conjugate to SPR Detection
3. Experimental
3.1. Materials
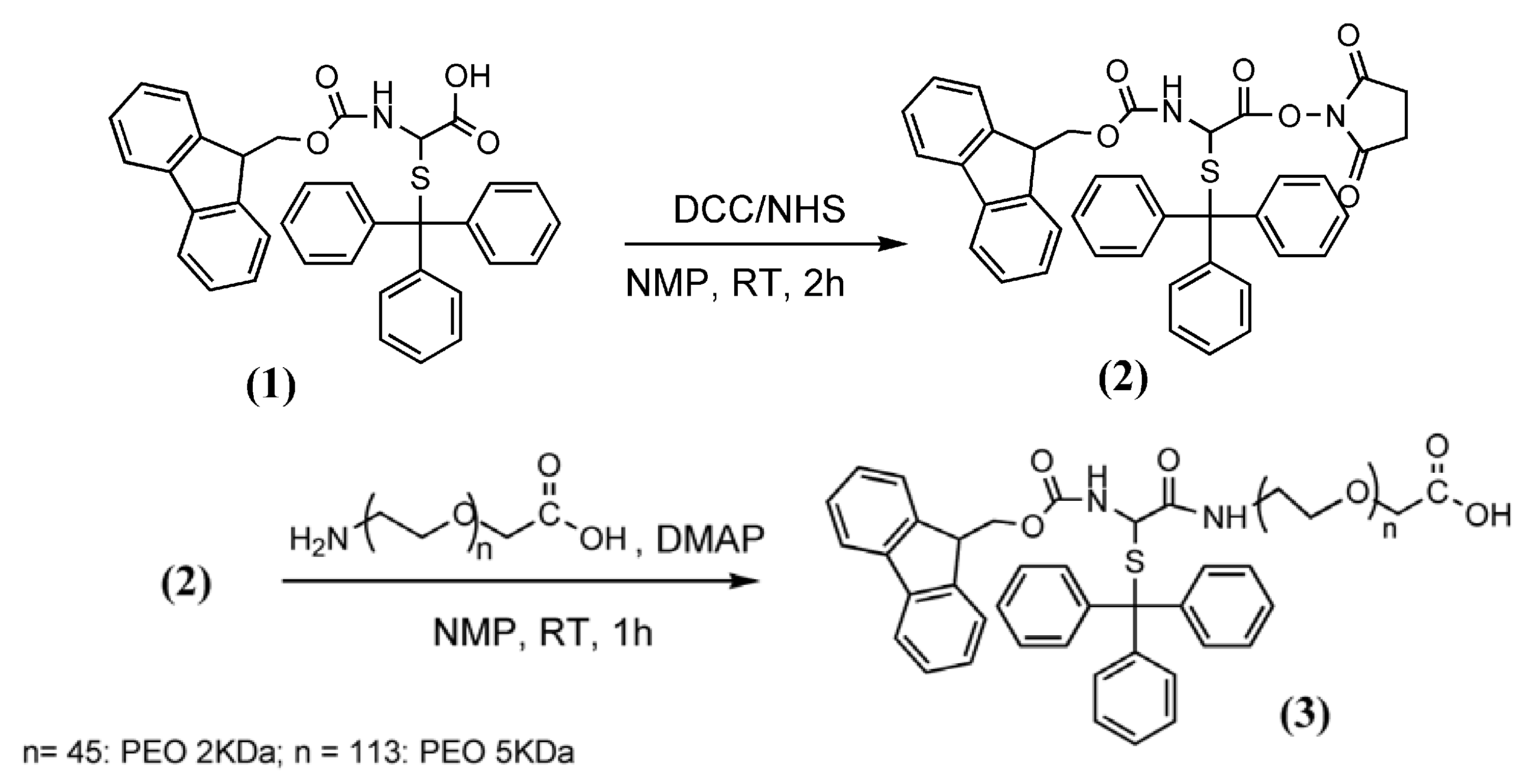
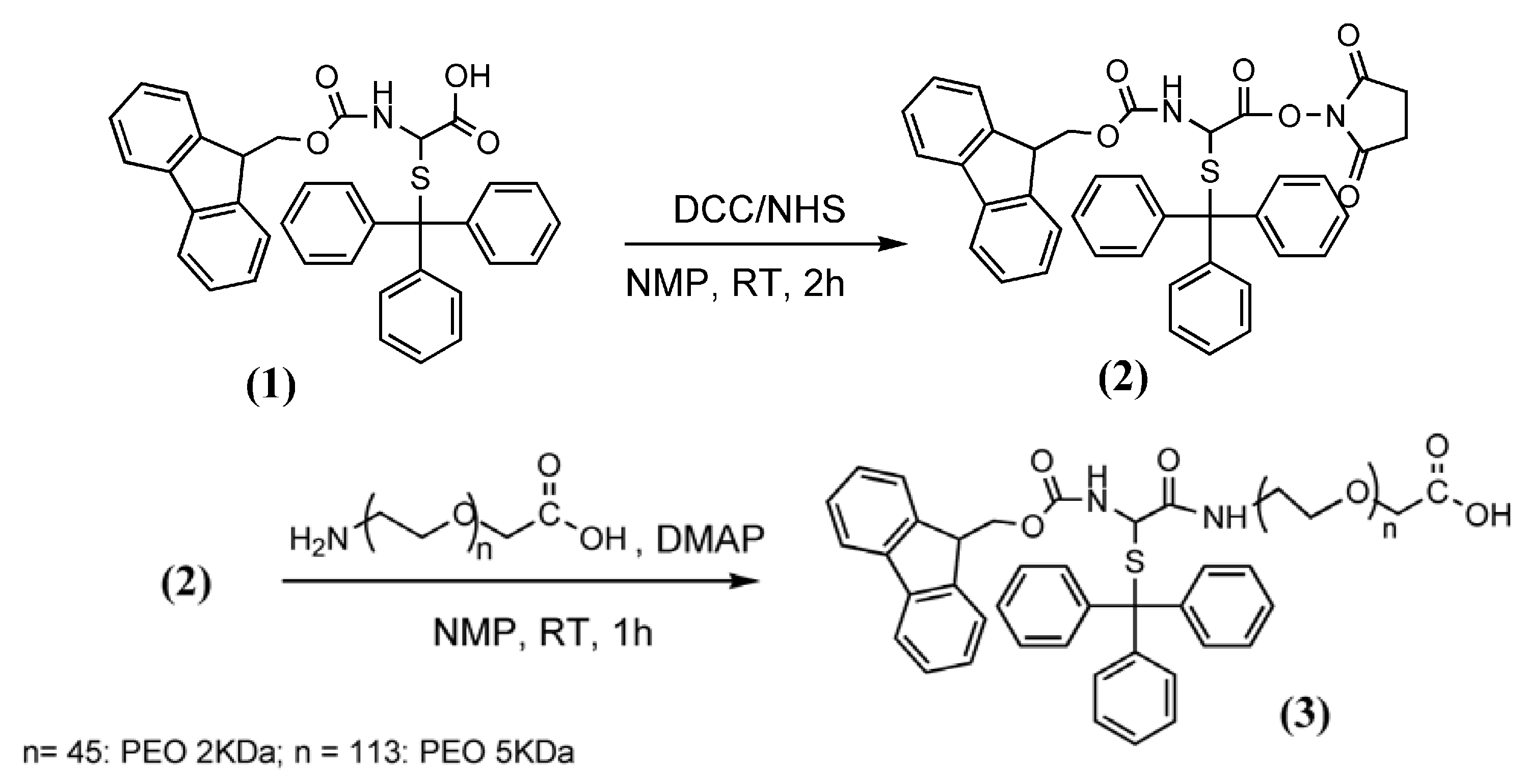
3.2. Synthesis of Fmoc-Cys(Trt)-PEO2KDa-COOH and Fmoc-Cys(Trt)-PEO5KDa-COOH
3.3. PNA Synthesis
3.3.1. Condensation of Amino Acids
3.3.2. Condensation of PNA Monomers
3.4. Solid-Phase Conjugation of Fmoc-Cys(Trt)-PEO5KDa-COOH to PNA
3.5. Solution Conjugation of Fmoc-Cys(Trt)-PEO-COOH to PNA
3.5.1. HATU/DMAP as Coupling Reagents
3.5.2. PyO/TEA as Coupling Reagents
3.6. PNA-PEO Purification from Crude Mixture
3.7. SPR Measurements
4. Conclusions
Supplementary Information
Acknowledgments
References
- Baldrich, E.; Laczka, O.; Del Campo, F.J.; Munoz, F.X. Gold immuno-functionalisation via self-assembled monolayers: Study of critical parameters and comparative performance for protein and bacteria detection. J. Immunol. Methods 2008, 336, 203–212. [Google Scholar] [CrossRef]
- Laib, S.; Krieg, A.; Rankl, M.; Seeger, S. Supercritical angle fluorescence biosensor for the detection of molecular interactions on cellulose-modified glass surfaces. Appl. Surf. Sci. 2006, 252, 7788–7793. [Google Scholar] [CrossRef]
- Johnsson, B.; Lofas, S.; Lindquist, G. Immobilization of Proteins to a Carboxymethyldextran-Modified Gold Surface for Biospecific Interaction Analysis in Surface-Plasmon Resonance Sensors. Anal. Biochem. 1991, 198, 268–277. [Google Scholar] [CrossRef]
- Wood, S.J. DNA-DNA Hybridization in Real-Time Using Biacore. Microchem. J. 1993, 47, 330–337. [Google Scholar] [CrossRef]
- Vikholm-Lundin, I.; Piskonen, R.; Albers, W.M. Hybridisation of surface-immobilised single-stranded oligonucleotides and polymer monitored by surface plasmon resonance. Biosens. Bioelectron. 2007, 22, 1323–1329. [Google Scholar] [CrossRef]
- Cattani-Scholz, A.; Pedone, D.; Blobner, F.; Abstreiter, G.; Schwartz, J.; Tornow, M.; Andruzzi, L. PNA-PEG modified silicon platforms as functional bio-interfaces for applications in DNA microarrays and biosensors. Biomacromolecules 2009, 10, 489–496. [Google Scholar] [CrossRef]
- Schlapak, R.; Pammer, P.; Armitage, D.; Zhu, R.; Hinterdorfer, P.; Vaupel, M.; Fruhwirth, T.; Howorka, S. Glass surfaces grafted with high-density poly(ethylene glycol) as substrates for DNA oligonucleotide microarrays. Langmuir 2006, 22, 277–285. [Google Scholar] [CrossRef]
- Chrisey, L.A.; Lee, G.U.; O’Ferrall, C.E. Covalent attachment of synthetic DNA to self-assembled monolayer films. Nucleic Acids Res. 1996, 24, 3031–3039. [Google Scholar] [CrossRef]
- Gong, P.; Lee, C.Y.; Gamble, L.J.; Castner, D.G.; Grainger, D.W. Hybridization behavior of mixed DNA/alkylthiol monolayers on gold: characterization by surface plasmon resonance and 32P radiometric assay. Anal. Chem. 2006, 78, 3326–3334. [Google Scholar] [CrossRef]
- Ito, M.; Nakamura, F.; Baba, A.; Tamada, K.; Ushijima, H.; Lau, K.H.A.; Manna, A.; Knoll, W. Enhancement of surface plasmon resonance signals by gold nanoparticles on high-density DNA microarrays. J. Phys. Chem. C Nanomater Interfaces 2007, 111, 11653–11662. [Google Scholar]
- Lao, A.I.K.; Su, X.D.; Aung, K.M.M. SPR study of DNA hybridization with DNA and PNA probes under stringent conditions. Biosens. Bioelectron. 2009, 24, 1717–1722. [Google Scholar] [CrossRef]
- Lee, C.Y.; Gong, P.; Harbers, G.M.; Grainger, D.W.; Castner, D.G.; Gamble, L.J. Surface coverage and structure of mixed DNA/alkylthiol monolayers on gold: characterization by XPS, NEXAFS, and fluorescence intensity measurements. Anal. Chem. 2006, 78, 3316–3325. [Google Scholar] [CrossRef]
- Liebermann, T.; Knoll, W.; Sluka, P.; Herrmann, R. Complement hybridization from solution to surface-attached probe-oligonucleotides observed by surface-plasmon-field-eahanced fluorescence spectroscopy. Colloid. Surface A 2000, 169, 337–350. [Google Scholar] [CrossRef]
- Oh, S.J.; Cho, S.J.; Kim, C.O.; Park, J.W. Characteristics of DNA microarrays fabricated on various aminosilane layers. Langmuir 2002, 18, 1764–1769. [Google Scholar] [CrossRef]
- Wang, Y.; Prokein, T.; Hinz, M.; Seliger, H.; Goedel, W.A. Immobilization and hybridization of oligonucleotides on maleimido-terminated self-assembled monolayers. Anal. Biochem. 2005, 344, 216–223. [Google Scholar]
- Metzger, S.W.; Natesan, M.; Yanavich, C.; Schneider, J.; Lee, G.U. Development and characterization of surface chemistries for microfabricated biosensors. J. Vac. Sci. Technol. A 1999, 17, 2623–2628. [Google Scholar] [CrossRef]
- Buxboim, A.; Bar-Dagan, M.; Frydman, V.; Zbaida, D.; Morpurgo, M.; Bar-Ziv, R. A single-step photolithographic interface for cell-free gene expression and active biochips. Small 2007, 3, 500–510. [Google Scholar] [CrossRef]
- Egholm, M.; Buchardt, O.; Nielsen, P.E.; Berg, R.H. Peptide NucleicAcids (PNA). Oligonucleotide Analogs with an Achiral Peptide Backbone. J. Am. Chem. Soc. 1992, 114, 1895–1897. [Google Scholar] [CrossRef]
- Nielsen, P.E. Peptide Nucleic Acids (PNA) in Chemical Biology and Drug Discovery. Chem. Biodivers. 2010, 7, 786–804. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-Selective Recognition of DNA by Strand Displacement with a Thymine-Substituted Polyamide. Science 1991, 254, 1497–1500. [Google Scholar]
- Ananthanawat, C.; Vilaivan, T.; Mekboonsonglarp, W.; Hoven, V.P. Thiolated pyrrolidinyl peptide nucleic acids for the detection of DNA hybridization using surface plasmon resonance. Biosens. Bioelectron. 2009, 24, 3544–3549. [Google Scholar] [CrossRef]
- Arlinghaus, H.F.; Schroder, M.; Feldner, J.C.; Brandt, O.; Hoheisel, J.D.; Lipinsky, D. Development of PNA microarrays for gene diagnostics with TOF-SIMS. Appl. Surf. Sci. 2004, 231, 392–396. [Google Scholar] [CrossRef]
- Hyrup, B.; Nielsen, P.E. Peptide nucleic acids (PNA): Synthesis, properties and potential applications. Bioorg. Med. Chem. 1996, 4, 5–23. [Google Scholar] [CrossRef]
- Kerman, K.; Matsubara, Y.; Morita, Y.; Takamura, Y.; Tamiya, E. Peptide nucleic acid modified magnetic beads for intercalator based electrochemical detection of DNA hybridization. Sci. Technol. Adv. Mat. 2004, 5, 351–357. [Google Scholar] [CrossRef]
- Jeon, S.I.; Andrade, J.D. Protein Surface Interactions in the Presence of Polyethylene Oxide. II. Effect of Protein Size. J. Colloid Interface Sci. 1991, 142, 159–166. [Google Scholar] [CrossRef]
- Jeon, S.I.; Lee, J.H.; Andrade, J.D.; Degennes, P.G. Protein Surface Interactions in the Presence of Polyethylene Oxide. I. Simplified Theory. J. Colloid Interface Sci. 1991, 142, 149–158. [Google Scholar] [CrossRef]
- Bonora, G.M.; Ivanova, E.; Zarytova, V.; Burcovich, B.; Veronese, F.M. Synthesis and characterization of high-molecular mass polyethylene glycol-conjugated oligonucleotides. Bioconjug. Chem. 1997, 8, 793–797. [Google Scholar] [CrossRef]
- Zacco, G.; Romanato, F.; Sonato, A.; Sammito, D.; Ruffato, G.; Morpurgo, M.; Silvestri, D.; Carli, M.; Schiavuta, P.; Brusatin, G. Sinusoidal plasmonic crystals for bio-detection sensors. Microelectron. Eng. 2011, 88, 1898–1901. [Google Scholar]
- Romanato, F.; Pilot, R.; Massari, M.; Ongarello, T.; Pirruccio, G.; Zilio, P.; Ruffato, G.; Carli, M.; Sammito, D.; Giorgis, V.; et al. Design, fabrication and characterization of plasmonic gratings for SERS. Microelectron. Eng. 2011, 88, 2717–2720. [Google Scholar]
- Romanato, F.; Lee, K.H.; Kang, H.K.; Ruffato, G.; Wong, C.C. Sensitivity enhancement in grating coupled surface plasmon resonance by azimuthal control. Opt. Express 2009, 17, 12145–12154. [Google Scholar]
- Prabhakar, N.; Arora, K.; Arya, S.K.; Solanki, P.R.; Iwamoto, M.; Singh, H.; Malhotra, B.D. Nucleic acid sensor for M-tuberculosis detection based on surface plasmon resonance. Analyst 2008, 133, 1587–1592. [Google Scholar] [CrossRef]
- McCammon, M.T.; Gillette, J.S.; Thomas, D.P.; Ramaswamy, S.V.; Graviss, E.A.; Kreiswirth, B.N.; Vijg, J.; Quitugua, T.N. Detection of rpoB mutations associated with rifampin resistance in Mycobacterium tuberculosis using denaturing gradient gel electrophoresis. Antimicrob. Agents Chemother. 2005, 49, 2200–2209. [Google Scholar] [CrossRef]
- Stuart, D.A.; Haes, A.J.; Yonzon, C.R.; Hicks, E.M.; va Duyne, R.P. Biological applications of localised surface plasmonic phenomenae. IEEE Proc. Nanobiotechnol. 2005, 152, 13–32. [Google Scholar] [CrossRef]
- Fiche, J.B.; Buhot, A.; Calemczuk, R.; Livache, T. Temperature effects on DNA chip experiments from surface plasmon resonance imaging: Isotherms and melting curves. Biophys. J. 2007, 92, 935–946. [Google Scholar] [CrossRef]
- Neidle, S. The structures of quadruplex nucleic acids and their drug complexes. Curr. Opin. Struct. Biol. 2009, 19, 239–250. [Google Scholar] [CrossRef]
- Subiros-Funosas, R.; El-Faham, A.; Albericio, F. PyOxP and PyOxB: The Oxyma-based novel family of phosphonium salts. Org. Biomol. Chem. 2010, 8, 3665–3673. [Google Scholar] [CrossRef]
- Udenfriend, S.; Stein, S.; Böhlen, P.; Dairman, W.; Leimgruber, W.; Weigele, M. Fluorescamine: A Reagent for Assay of Amino Acids, Peptides, Proteins, and Primary Amines in the Picomole Range. Science 1972, 178, 871–872. [Google Scholar]
- Golovchenko, N.P.; Kataeva, I.A.; Akimenko, V.K. Analysis of Ph-Dependent Protein Interactions with Gel-Filtration Medium. J. Chromatogr. 1992, 591, 121–128. [Google Scholar] [CrossRef]
- Ziegler, A.; Zaia, J. Size-exclusion chromatography of heparin oligosaccharides at high and low pressure. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2006, 837, 76–86. [Google Scholar] [CrossRef]
- Albericio, F.; Bofill, J.M.; El-Faham, A.; Kates, S.A. Use of onium salt-based coupling reagents in peptide synthesis. J. Org. Chem. 1998, 63, 9678–9683. [Google Scholar] [CrossRef]
- Harding, V.; Warneford, F. The ninhidrin reaction with amino-acids and amonium salts. J. Biol. Chem. 1916, 25, 319–335. [Google Scholar]
- Kayser, O.; Lemke, A.; Hernandez-Trejo, N. The impact of nanobiotechnology on the development of new drug delivery systems. Curr. Pharm. Biotechnol. 2005, 6, 3–5. [Google Scholar]
- Habeeb, A. Determination of free amino groups in proteins by trinitrobenzensulphonic acid. Anal. Biochem. 1966, 14, 328–336. [Google Scholar] [CrossRef]
- Morpurgo, M.; Veronese, F.M. Conjugates of peptides and proteins to polyethylene glycols. Methods Mol. Biol. 2004, 283, 45–70. [Google Scholar]
- Sims, G.E.; Snape, T.J. A method for the estimation of polyethylene glycol in plasma protein fractions. Anal. Biochem. 1980, 107, 60–63. [Google Scholar]
- Riddles, P.W.; Blakeley, R.L.; Zerner, B. Reassessment of Ellman’s reagent. Meth. Enzymol. 1983, 91, 49–60. [Google Scholar]
- Sample Availability: Samples of both 5 and 2 KDa PEO conjugates are available from the authors for experiments in collaboration.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dettin, M.; Silvestri, D.; Danesin, R.; Cretaio, E.; Picariello, G.; Casarin, E.; Sonato, A.; Romanato, F.; Morpurgo, M. Synthesis and Chromatography-Free Purification of PNA-PEO Conjugates for the Functionalisation of Gold Sensors. Molecules 2012, 17, 11026-11045. https://doi.org/10.3390/molecules170911026
Dettin M, Silvestri D, Danesin R, Cretaio E, Picariello G, Casarin E, Sonato A, Romanato F, Morpurgo M. Synthesis and Chromatography-Free Purification of PNA-PEO Conjugates for the Functionalisation of Gold Sensors. Molecules. 2012; 17(9):11026-11045. https://doi.org/10.3390/molecules170911026
Chicago/Turabian StyleDettin, Monica, Davide Silvestri, Roberta Danesin, Erica Cretaio, Gianluca Picariello, Elisabetta Casarin, Agnese Sonato, Filippo Romanato, and Margherita Morpurgo. 2012. "Synthesis and Chromatography-Free Purification of PNA-PEO Conjugates for the Functionalisation of Gold Sensors" Molecules 17, no. 9: 11026-11045. https://doi.org/10.3390/molecules170911026
APA StyleDettin, M., Silvestri, D., Danesin, R., Cretaio, E., Picariello, G., Casarin, E., Sonato, A., Romanato, F., & Morpurgo, M. (2012). Synthesis and Chromatography-Free Purification of PNA-PEO Conjugates for the Functionalisation of Gold Sensors. Molecules, 17(9), 11026-11045. https://doi.org/10.3390/molecules170911026