Abstract
In the presence of trifluoromethanesulfonic acid (TfOH) or bis(trifluoromethane-sulfonyl)imide (Tf2NH), iodosobenzene (PhI=O) efficiently promoted the reactions of dicarbonyl compounds as well as monocarbonyl compounds with nitriles to give 2,4-disubstituted and 2,4,5-trisubstituted oxazole in a single step under the mild conditions.
1. Introduction
The synthesis of oxazole compounds has attracted a great deal of attention due to the widespread application of oxazole derivatives in biologically active compounds as well as versatile building blocks in organic synthesis [1,2,3]. For the purpose of synthesizing highly substituted oxazole compounds, an intramolecular reaction, the so-called Robinson-Gabriel cyclocondensation of α-acylamino ketones in the presence of dehydrating reagents [H2SO4, POCl3, (CF3SO2)2O, and so on] has been commonly employed [4,5,6]. As for the intermolecular approaches to highly substituted oxazoles, α-diazo ketones [7,8,9], α-halo ketones [10,11], α-sulfonyloxy ketones [12], and iodonium ylides of ketones [13] have been used as a reactive synthetic intermediate. The preparation of α-acylamino ketones or these intermediates, however, requires a multi-step synthesis and/or the harsh reaction conditions. Although the direct synthesis of oxazoles from simple ketones and nitriles with oxidants based on Tl(III) [14], Hg(II) [15], Fe(III) [16], or Cu(II) [17] have been developed as a convenient procedure, these procedures have met with the drawbacks including the limitation of the substrates and/or the use of toxic oxidants.
Hypervalent iodine(III) reagents have gained increasing popularity in organic syntheses due to their low toxicity, mild reactivity, high stability, easy handling, and so on [18]. Among them, [hydroxyl-(tosyloxy)iodo]benzene (Koser’s reagent) and related reagents have been reported to work well for the α-sulfoxylations of ketones [19,20]. [Hydroxy(2,4-dinitrobenzenesulfonyloxy)iodo]benzene (HDNIB) has been used in the stepwise and one-pot synthesis of oxazoles via the formation of α-sulfonyloxy ketones as intermediate from simple ketones [21]. In addition, phenyliodine(III) diacetate (PIDA) with trifluoromethanesulfonic acid (TfOH) [22] or the iodoarene-oxone-TfOH system [23] efficiently promoted the direct synthesis of oxazoles from monocarbonyl compounds, such as alkyl aryl ketones with nitriles. To the best of our knowledge, however, there is no report about the direct synthesis of oxazoles from simple dicarbonyl compounds and nitriles [21,24,25]. As a part of our study on the iodine(III)-mediated synthesis of heterocycles [26,27], we carried out research on iodine(III) reagents for the direct synthesis of oxazoles from monocarbonyl or dicarbonyl compounds. In this article, we describe a single-step synthesis of highly substituted oxazoles by the reaction of ketones with nitriles in the presence of iodosobenzene (PhI=O) and a Brønsted acid.
2. Results and Discussion
2.1. Evaluation of Oxidants for the Direct Synthesis of Oxazole
At the outset, we focused on the investigation of reactive oxidants for the reaction of monocarbonyl compounds with nitriles as shown in Table 1. In the presence of PIDA (1.2 equiv.) with TfOH (4.5 equiv.), the reaction of acetophenone (1a) in acetonitrile (MeCN) afforded the oxazole 2a in 94% yield at 80 °C for 2 h (entry 1) [23], albeit low yield (22%) at ambient temperature for 3 h (entry 2). Exploring milder conditions, we examined the oxidants which were prepared from miscellaneous Brønsted acids (1.5–3.0 equiv.) and PhI=O (1.5 equiv.) [28,29], in the reaction of 1a in MeCN (entries 3–8). Although the use of p-toluenesulfonic aicd (TsOH) gave α-tosyloxy ketone 3 as a main product (entry 3), the other acids led to the desired formation of oxazole compound 2a (entries 4–8). Thus, 3.0 equiv. TfOH showed the similar results to entry 1 (entry 7) and 3.0 equiv. bis(trifluoromethane-sulfonyl)imide (Tf2NH) produced a good yield of 2a (86%) after 3 h at ambient temperature (entry 9). It should be mentioned that the use of 10 equiv. of MeCN in 1,2-dichloroethane instead of MeCN solvent decreased the yield of 2a, even under the similar conditions mediated by PhI=O with TfOH or Tf2NH.
For the formation of oxazole 5a from β-keto ester 4a in MeCN, PhI=O/Tf2NH system turned out to display superior activity to iodine(III) reagents/TfOH (Table 2). Thus, PhI=O (1.5 equiv.) with Tf2NH (3.0 equiv.) improved the yield of 5a to 51% at 80 °C for 16 h (entry 4), compared to the use of 1.5 equiv. PIDA or 1.5 equiv. PhI=O in the presence of TfOH (3.0–4.5 equiv.), in which 4a were converted to 5a in only 4–7% yields even at 80 °C for 24–25 h (entries 1 and 2). In the case of the extension of the reaction time (72 h) or the increase in the amount of Tf2NH (6.0 equiv.), 5a was obtained in 79–81% yields (entries 5 and 6).

Table 1.
Evaluation of oxidants for the reaction of acetophenone (1a) in MeCN.
| Entry | Oxidant (equiv) | Acid (equiv.) | Temp. (°C) | Time (h) | 2a (%) [a] | |
|---|---|---|---|---|---|---|
| 1 [b] | PIDA (1.2) | TfOH (4.5) | 80 | 2 | 94 | |
| 2 | PIDA (1.2) | TfOH (4.5) | rt | 3 | 22 | |
| 3 | PhI=O (1.5) | TsOH (1.5) | 80 | 18 | - | ( 3 69) |
| 4 | PhI=O (1.5) | HBF4/Et2O (1.5) | 80 | 18 | 54 | |
| 5 | PhI=O (1.5) | TfOH (1.5) | 80 | 18 | 69 | |
| 6 | PhI=O (1.5) | Tf2O (1.5) | 80 | 18 | 21 [c] | |
| 7 | PhI=O (1.5) | TfOH (3.0) | 80 | 3 | 94 | |
| 8 | PhI=O (1.5) | Tf2NH (1.5) | 80 | 3 | 40 | ( 1a 16) |
| 9 | PhI=O (1.5) | Tf2NH (3.0) | rt | 3 | 86 |  |
[a] Yields were determined by 1H-NMR analysis; [b] Ref. [21]; [c] 2,4-Dimethyl-6-phenylpyrimidine was obtained in 38% yield.
Table 2.
Evaluation of oxidants for the reaction of benzoylacetate 4a in MeCN.
| Entry | Oxidant | Acid | Time (h) | 5a (%) [a] | |
|---|---|---|---|---|---|
| 1 | PIDA | TfOH [b] | 24 | 4 | |
| 2 | PhI=O | TfOH | 25 | 7 | |
| 3 | PhI=O | TfOH | 115 | 41 | |
| 4 | PhI=O | Tf2NH | 16 | 51 | |
| 5 | PhI=O | Tf2NH | 72 | 79 | |
| 6 | PhI=O | Tf2NH [c] | 3 | 81  |
[a] Yields were determined by 1H-NMR analysis; [b] TfOH: 4.5 equiv.; [c] TfOH: 6.0 equiv.
2.2. Scope of the Direct Synthesis of Oxazoles Using PhI=O with TfOH or Tf2NH
The scope of monocarbonyl compounds 1 and dicarbonyl compounds 4 by means of the PhI=O (1.5 equiv.)-mediated procedure A (acid: 3.0 equiv. TfOH), B (acid: 3.0 equiv. Tf2NH), or C (acid: 6.0 equiv. Tf2NH) was shown in Table 3 and Table 4 and Scheme 1. Procedure A could be applied to the reactions of monocarbonyl compounds 1a–g in MeCN, and the corresponding oxazoles 2a–g were obtained in 53–94% yields at 80 °C (Table 3, entries 1, 3–6, 8, and 9). Furthermore, procedure B brought about the formation of 2a or 2e at ambient temperature (entries 2 and 7). In the case of benzoylacetonitrile (1g), an increase in the amount of Tf2NH (procedure C) improved the yield of 5f up to 69% at ambient temperature for 24 h (entry 11). Although the dicarbonyl compounds 4a,b required the long time (72–139 h) to give good yields of products through procedure B (entries 12 and 14), procedure C reduced the reaction times (3 h) giving rise to 5a–c in 67–83% yields (entries 13, 15 and 17). Bicyclic oxazole 5d could be formed by procedure C, albeit in only 40% yield after 167 h (entry 18). Unfortunately, p-methoxyacetophenone and 4-phenyl-2-butanone did not give the desired products with any of the procedures. By procedure A, the reaction of acetophenone (1a) in propionitrile (EtCN) or benzonitrile (PhCN) instead of MeCN smoothly proceeded at 80 °C for 4 h to yield the corresponding oxazole 6 or 7 in good yields (Scheme 1). The procedure C could be applied to the reaction of dicarbonyl compounds 4a–c in EtCN or PhCN (Table 4).
Table 3.
The reactions of 1 or 4 in MeCN by the means of procedures A, B, or C [a].
| Entry | 1 or 4 | R1 | R2 | Procedure | (°C) | (h) | 2 or 5 | Yield (%) [b] |
|---|---|---|---|---|---|---|---|---|
| 1 | 1a | Ph | H | A | 80 | 3 | 2a | 88 |
| 2 | 1a | B | rt | 3 | 2a | 86 | ||
| 3 | 1b | m-Me-C6H4 | H | A | 80 | 3 | 2b | 75 |
| 4 | 1c | p-Cl-C6H4 | H | A | 80 | 3 | 2c | 86 |
| 5 | 1d | p-NO2-C6H4 | H | A | 80 | 3 | 2d | 73 |
| 6 | 1e | Ph | Me | A | 80 | 3 | 2e | 94 |
| 7 | 1e | B | rt | 2 | 2e | 91 | ||
| 8 | 1f | Ph | Cl | A | 80 | 49 | 2f | 68 |
| 9 | 1g | Ph | CN | A | 80 | 20 | 2g | 53 |
| 10 | 1g | B | 80 | 20 | 2g | 38 | ||
| 11 | 1g | C | rt | 24 | 2g | 69 | ||
| 12 | 4a | Ph | OEt | B | 80 | 72 | 5a | 78 |
| 13 | 4a | C | 80 | 3 | 5a | 81 | ||
| 14 | 4b | Ph | Ph | B | 80 | 139 | 5b | 89 |
| 15 | 4b | C | 80 | 3 | 5b | 83 | ||
| 16 | 4c | Me | Me | B | 80 | 120 | 5c | 35 |
| 17 | 4c | C | 80 | 3 | 5c | 67 | ||
| 18 | 4d | -(CH2)4- | C | 80 | 167 | 5d | 40 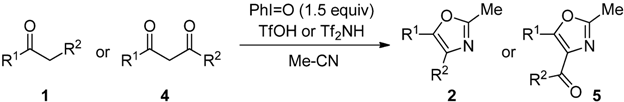 | |
[a] Procedure A: 3 equiv. TfOH was used as a Brønsted acid. Procedure B: 3 equiv. Tf2NH was used as a Brønsted acid. Procedure C: 6 equiv. Tf2NH was used as a Brønsted acid; [b] Isolated yields.
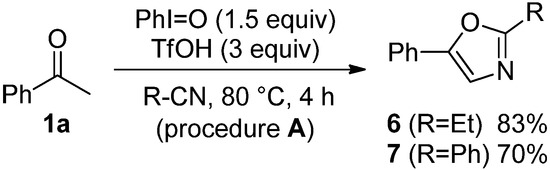
Scheme 1.
The reactions of 1a in EtCN or PhCN by the means of procedure A.
Table 4.
The reactions of 4a–c in EtCN or PhCN by the means of procedure C.
| Entry | 4 | R1 | R2 | R3 | (h) | 2 or 5 | Yield (%) [a] |
|---|---|---|---|---|---|---|---|
| 1 | 4a | Ph | OEt | Et | 2 | 8a | 83 |
| 2 | 4a | Ph | 3 | 9a | 72 | ||
| 3 | 4b | Ph | Ph | Et | 3 | 8b | 89 |
| 4 | 4b | Ph | 3 | 9b | 67 | ||
| 5 | 4c | Me | Me | Et | 20 | 8c | 56 |
| 6 | 4c | Ph | 20 | 9c | 60 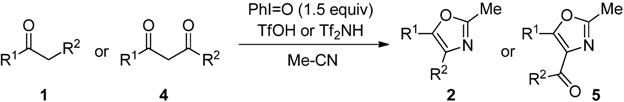 |
[a] Isolated yields.
2.3. Mechanistic Considerations
Since it has been known that PhI=O reacts with two equiv. of TfOH in CH2Cl2 to produce to the oxidant 10 [30], to better understand the present oxazole formation, we examined the reaction of acetophenone (1a) with 10 (Scheme 2). Thus, under conditions similar to those of entry 7 in Table 1, 1a was treated with 10 (0.75 equiv.) instead of PhI=O (1.5 equiv.) and TfOH (3 equiv.) in MeCN to give 2a in only 11% yield at 80 °C for 3 h. Therefore, 10 would not be considered to take part in the present oxazole formation.
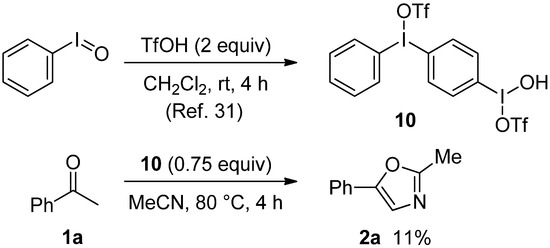
Scheme 2.
Preparation of 10 and the reaction of 1a with 10.
On the basis of abovementioned observations and the previous reports about the iodine(III)-mediated synthesis of oxazoles [21,22,23], the mechanism for the present oxazole formation from ketones 1 or 4 with nitriles as shown in Scheme 3 is proposed. That is, α-iodanyl ketone Int-A, which is generated from 1 or 4 with PhI=O and H-X (TfOH or Tf2NH), would be converted to Int-B by the Ritter-type reaction with R3CN. Int-C generated by the reductive elimination of ArI might also be a possible intermediate for the formation of Int-B, and the subsequent cyclization of Int-B gives oxazoles 2 or 5. The formation of Int-A and/or Int-C is supported by the formation of α-tosyloxy ketone 3 (69%) in the case of TsOH as an acid (Table 1, entry 3) [19,23].
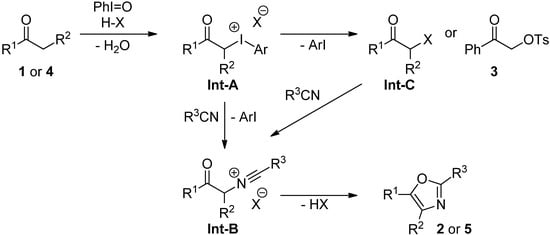
Scheme 3.
Proposed reaction mechanism of direct synthesis of oxazoles.
3. Experimental
3.1. General
All starting materials and reagents were commercially available. Dried organic solvents were purchased and used without further drying. Unless otherwise stated, all reactions were conducted under an argon atmosphere. Melting points were measured on a Yanaco SP-M1 melting point apparatus (Yanagimoto Co.) and were uncorrected. IR spectra were recorded on a HORIBA FT-710 FT-IR spectrometer. 1H and 13C-NMR spectra were measured in CDCl3 with a Bruker AV300M FT NMR spectrometer at 300 and 75 MHz, and the chemical shifts are given in ppm using CHCl3 (7.26 ppm) for 1H-NMR and CDCl3 (77.0 ppm) for 13C-NMR as an internal standard, respectively. Mass spectra and HRMS were recorded by FAB method on a JMS-HX110 Mass spectrometer. Elemental analysis was measured on a Perkin-Elmer 240B or Elemental Vavio EL. For the TLC analysis, Merck precoated TLC plates (silica gel 60 F254) were used. Column chromatography was performed on Silica gel 60N (63–200 μm, Kanto Kagaku Co., Ltd.).
3.2. General Procedure for the Iodine(III)-Mediated Synthesis of Oxazoles
Ketone 1 or 4 (0.4 mmol) was added to a solution of iodosobenzene (132 mg, 0.6 mmol) and trifluoromethanesulfonic acid (106 μL, 1.2 mmol) or bis(trifluoromethanesulfonyl)imide (337 or 675 mg, 1.2 or 2.4 mmol), which were premixed in acetonitrile, propionitrile, or benzonitrile (2 mL) at 0 °C for 5 min, and the reaction mixture was stirred at ambient temperature or 80 °C until the consumption of substrate by TLC analysis. The mixture was diluted with ether and filtered through a short alumina column. After concentration of the filtrate to dryness, the subsequent purification gave the corresponding oxazole 2 or 5. 2a [21], 2c [21], 2d [31], 2e [21], 6 [21], 7 [32], and 9a–c [33] were identified by the comparison with 1H-NMR spectra reported in the appropriate literature.
2-Methyl-5-(3′-methylphenyl)oxazole (2b). Colorless oil. IR (neat) ν cm−1; 3054, 2925, 2863, 1610, 1560, 1519, 784, 748. 1H-NMR (CDCl3) δ 2.38 (s, 3H), 2.51 (s, 3H), 7.02 (d, J = 7.6 Hz, 1H), 7.18 (s, 1H), 7.28 (t, J = 7.6 Hz, 1H), 7.40 (d, J = 9.2 Hz, 2H). 13C-NMR (CDCl3) δ 14.0, 21.4, 121.0, 121.6, 124.5, 128.0, 128.7, 128.9, 138.5, 151.2. FAB-LM m/z: 174.2 (M++H). FAB-HM Calcd for C11H12NO: 174.0919, Found: 174.0906.
4-Chloro-2-methyl-5-phenyloxazole (2e). White solid. Mp 56 °C. IR (KBr) ν cm−1; 3050, 3002, 2923, 2856, 1438, 1378. 1H-NMR (CDCl3) δ 2.50 (s, 3H), 7.30–7.36 (m, 1H), 7.41–7.46 (m, 2H), 7.83–7.84 (m, 2H). 13C-NMR (CDCl3) δ 14.2, 124.8, 127.0, 128.7, 143.8, 159.4. FAB-LM m/z: 194.1 (M++H). FAB-HM Calcd for C10H9ClNO: 194.0373, Found: 194.0383. Anal. Calcd for C10H8ClNO: C, 62.03; H, 4.06; N, 7.23. Found: C, 62.21; H, 4.47; N, 7.23.
4-Cyano-2-methyl-5-phenyloxazole (2f). White solid. Mp 49 °C. IR (KBr) ν cm−1; 3062, 2927, 2854, 2225. 1H-NMR (CDCl3) δ 2.56 (s, 3H), 7.48–7.50 (m, 3H), 7.89–7.92 (m, 2H). 13C-NMR (CDCl3) δ 13.9, 108.0, 113.8, 125.2, 125.5, 129.3, 130.9, 157.8, 161.1. FAB-LM m/z: 185.2 (M++H). Anal. Calcd for C11H8N2O: C, 71.73; H, 4.38; N, 15.21. Found: C, 71.91; H, 4.39; N, 15.02.
4-Ethoxycarbonyl-2-methyl-5-phenyloxazole (5a). White solid. Mp 64 °C. IR (KBr) ν cm−1; 3054, 3006, 2977, 2927, 2857, 1598, 1560, 1488. 1H-NMR (CDCl3) δ 1.38 (t, J = 7.1 Hz, 3H), 2.54 (s, 3H), 4.40 (q, J = 7.1 Hz, 2H), 7.43–7.45 (m, 3H), 8.02–8.05 (m, 2H). 13C-NMR (CDCl3) δ; 13.9, 14.3, 61.3, 106.9, 127.1, 128.3, 128.3, 130.1, 155.3, 159.9, 162.2. FAB-LM m/z: 232.2 (M++H). FAB-HM Calcd for C13H14NO3: 232.0974, Found: 232.0977. Anal. Calcd for C13H13NO3: C, 67.52; H, 5.67; N, 6.06. Found: C, 67.80; H, 5.81; N, 5.89.
4-Benzoyl-2-methyl-5-phenyloxazole (5b). Colorless oil. IR (neat) ν cm−1; 3031, 2965, 2927, 2927, 2856, 1710. 1H-NMR (CDCl3) δ 2.58 (s, 3H), 7.40–7.47 (m, 5H), 7.47–7.55 (m, 1H), 7.95–7.98 (m, 2H), 8.05–8.08 (m, 2H). 13C-NMR (CDCl3) δ; 13.8, 127.3, 127.6, 128.1, 128.4, 130.0, 130.2, 132.9, 133.7, 137.5, 154.7, 159.0. FAB-LM m/z: 264.2 (M++H). FAB-HM Calcd for C18H14NO2: 264.1025, Found: 264.1014. Anal. Calcd for C17H13NO2: C, 77.55; H, 4.98; N, 5.32. Found: C, 77.38; H, 5.16; N, 5.40.
4-Acetyl-2,5-dimethyloxazole (5c). White solid. Mp 39 °C. IR (KBr) ν cm−1; 1681. 1H-NMR (CDCl3) δ 2.73 (s, 3H), 2.43 (s, 3H), 2.50 (s, 3H). 13C-NMR (CDCl3) δ 12.0, 13.6, 27.8, 134.5, 154.2, 158.4, 194.7. EI-LM m/z: 139.1 (M+). Anal. Calcd for C7H9NO2: C, 60.42; H, 6.52; N, 10.07. Found: C, 60.02; H, 6.16; N, 9.82.
6,7-Dihydro-2-methylbenzo[d]oxazole-4(5H)-one (5d). Colorless oil. IR (neat) ν cm−1; 2952, 2927, 2854, 1682. 1H-NMR (CDCl3) δ 2.12–2.20 (m, 2H), 2.52 (s, 3H), 2.57 (t, J = 5.9 Hz, 2H), 2.81 (t, J = 6.1 Hz, 2H). 13C-NMR (CDCl3) δ 14.4, 23.2, 23.8, 38.0, 144.2, 156.1, 165.9, 185.8. FAB-HM Calcd for C8H10NO2: 152.0712, Found: 152.0697.
4-Ethoxycarbonyl-2-ethyl-5-phenyloxazole (8a). White solid. Mp 37 °C. IR (KBr) ν cm−1; 3066, 2983, 2933, 2875, 1714, 1240, 1189. 1H-NMR (CDCl3) δ 1.38 (t, J = 7.1 Hz, 3H), 1.39 (t, J = 7.6 Hz, 3H), 2.88 (q, J = 7.6 Hz, 2H), 4.40 (q, J = 7.1 Hz, 2H), 7.42–7.47 (m, 3H), 8.01–8.04 (m, 2H). 13C-NMR (CDCl3) δ 11.2, 14.3, 21.6, 61.3, 126.7, 127.1, 128.3, 130.0, 155.0, 162.2, 164.1. FAB-LM m/z: 264.2 (M++H). FAB-HM Calcd for C14H16NO3: 246.1130, Found: 246.1126. Anal. Calcd for C14H15NO3: C, 68.56; H, 6.16; N, 5.71. Found: C, 68.79; H, 6.06; N, 5.61.
4-Benzoyl-2-ethyl-5-phenyloxazole (8b). Colorless oil. IR (neat) ν cm−1; 3064, 2981, 2937, 1656. 1H-NMR (CDCl3) δ 1.43 (t, J = 7.6 Hz, 3H), 2.92 (q, J = 7.6 Hz, 2H), 7.40–7.45 (m, 5H), 7.53–7.58 (m, 1H), 7.94–7.97 (m, 2H), 8.06–8.10 (m, 2H). 13C-NMR (CDCl3) δ 11.3, 21.7, 127.5, 127.7, 128.2, 128.5, 130.0, 130.4, 133.0, 133.7, 137.5, 154.5, 163.4. FAB-LM m/z: 278.2 (M++H). FAB-HM Calcd for C18H16NO2: 278.1181, Found: 278.1177. Anal. Calcd for C18H15NO2: C, 77.96; H, 5.45; N, 5.05. Found: C, 77.95; H,5.42; N,5.10.
4-Acetyl-2-ethyl-5-menyloxazole (8c). Colorless oil. IR (neat) ν cm−1; 1685. 1H-NMR (CDCl3) δ 1.25 (t, J = 7.6 Hz, 3H), 2.42 (s, 3H), 2.50 (s, 3H), 2.68 (q, J = 7.6 Hz, 2H). 13C-NMR (CDCl3) δ; 11.1, 12.1, 21.4, 27.8, 134.4, 154.0, 162.7, 194.9. FAB-LM m/z: 154.1 (M++H). FAB-HM Calcd for C8H12NO2: 154.0868, Found: 154.0873.
3.3. Formation of α-Tosyloxy Ketone 3 under the Iodine(III)-Mediated Conditions
Ketone 1a (47 μL, 0.4 mmol) was added to a solution of iodosobenzene (132 mg, 0.6 mmol) and p-toluenesulfonic acid monohydrate (114 mg, 0.6 mmol), which were premixed in MeCN (2 mL) at 0 °C for 5min, and the reaction mixture was stirred at 80 °C for 18 h. The mixture was diluted with ether and filtered through a short alumina column. After concentration of the filtrate to dryness, the subsequent purification gave 3 (73.2 mg, 0.25 mmol, 63%). Compound 3 was identified by the comparison with the 1H-NMR spectrum reported in the literature [20].
4. Conclusions
We have demonstrated the single-step synthesis of highly substituted oxazoles from ketones and nitriles by the use of iodosobenzene with trifluoromethanesulfonic acid or bis(trifluoromethane-sulfonyl)imide. The present procedure could be applied not only to monocarbonyl compounds, but also to dicarbonyl ones. In particular, we believe that the reactivity of iodosobenzene with bis(trifluoro-methanesulfonyl)imide sheds light on a new possibility for the use of hypervalent iodine compounds in organic synthesis.
Acknowledgments
This work was supported by Grant-in-Aid for Young Scientists (B), MEXT Japan (No 24790024).
References
- Wipf, P. Synthetic Studies of Biologically Active Marine Cyclopeptides. Chem. Rev. 1995, 95, 2115–2134. [Google Scholar] [CrossRef]
- Palmer, D.C.; Venkatraman, S.; Lowe, D.; Levin, J.L.; Laakso, L.M.; Gribble, G.W. The Chemistry of Heterocyclic Compounds, Oxazoles: Synthesis, Reactions, and Spectroscopy, Part A; Palmer, D.C., Ed.; Wiley & Sons: Hoboken, NJ, USA, 2003; Volume 60. [Google Scholar]
- Kunieda, T.; Matsunaga, H.; Palmer, D.C.; Cativiela, C.; Diaz-de-Villegas, M.D.; Wong, G.S.K.; Wu, W.; Ghosh, A.; Bilcer, G.; Fidanze, S. The Chemistry of Heterocyclic Compounds, Oxazoles: Synthesis, Reactions, and Spectroscopy, Part B; Palmer, D.C., Ed.; Wiley & Sons: Hoboken, NJ, USA, 2004; Volume 60. [Google Scholar]
- Robinson, R. A new synthesis of oxazole derivatives. J. Chem. Soc. 1909, 95, 2167–2174. [Google Scholar] [CrossRef]
- Gabriel, S. Synthese von Oxazolen und Thiazolen II. Ber. Dtsch. Chem. Soc. 1910, 43, 1283–1287. [Google Scholar] [CrossRef]
- Thalhammer, A.; Mecinović, J.; Schofield, C.J. Triflic anhydride-mediated synthesis of oxazoles. Tetrahedron Lett. 2009, 50, 1045–1047 and references cited therein. [Google Scholar]
- Huisgen, R. 1,3-Dipolar Cycloadditions. Past and Future. Angew. Chem. Int. Ed. 1963, 2, 565–598. [Google Scholar]
- Ibata, T.; Fukushima, K. Formation and reaction of acyl-substituted nitrile ylide through the rhodium complex Rh2(OAc)4-catalyzed reaction of α-diazocarbonyl compounds with benzonitrile. Chem. Lett. 1992, 2197–2200. [Google Scholar]
- Austeri, M.; Rix, D.; Zeghida, W.; Lacour, J. CpRu-Catalyzed O-H Insertion and Condensation Reactions of α-Diazocarbonyl Compounds. Org. Lett. 2011, 13, 1394–1397. [Google Scholar]
- Lora-Tamayo, M.; Madroñero, R.; Leipprand, H. Die Anwendung Der Nitriliumsalze bei der Synthese heterocyclischer Verbindungen, V. Derivate des 4.5-Diphenyl-oxazols. Chem. Ber. 1964, 97, 2230–2233. [Google Scholar]
- Schmittel, M.; Roeck, M. Enol cation radicals in solution. 3. Reaction of enol cation radicals in the presence of nucleophiles. Chem. Ber. 1992, 125, 1611–1620. [Google Scholar] [CrossRef]
- Lai, P.-S.; Taylor, M.S. Preparation of substituted oxazoles by Ritter reactions of α-oxo tosylates. Synthesis 2010, 1449–1452. [Google Scholar]
- Gogonas, E.P.; Hadjiarapoglou, L.P. [3+2]-Cycloaddition reactions of 2-phenyliodonio-5,5-dimethyl-1,3-dioxacyclohexanemethylide. Tetrahedron Lett. 2000, 41, 9299–9303. [Google Scholar] [CrossRef]
- Lee, J.C.; Hong, T. A novel and direct synthesis of 2-alkyl-5-aryl disubstituted oxazoles. Tetrahedron Lett. 1997, 38, 8959–8960. [Google Scholar] [CrossRef]
- Lee, J.C.; Song, I.-G. Mercury(II) p-toluenesulfonate mediated synthesis of oxazoles under microwave irradiation. Tetrahedron Lett. 2000, 41, 5891–5894. [Google Scholar]
- Kotani, E.; Kobayashi, S.; Adachi, M.; Tsujioka, T.; Nakamura, K.; Tobinaga, S. Synthesis of oxazole by the reaction of ketones with Iron(III) solvates of nitriles. Chem. Pharm. Bull. 1989, 37, 606–609. [Google Scholar]
- Nagayoshi, K.; Sato, T. One-step Synthesis of oxazole from ketones and nitriles using copper(II) trifluoromethanesulfonate as a key reagent. Chem. Lett. 1983, 1355–1356. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Stang, P.J. Chemistry of polyvalent iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef]
- Koser, G.F. Hydroxy(tosyloxy)iodo]benzene and closely related iodanes: The second stage of development. Aldrich. Acta 2001, 34, 89–102. [Google Scholar]
- Moroda, A.; Togo, H. Biphenyl- and terphenyl-based recyclable organic trivalent iodine reagents. Tetrahedron 2006, 62, 12408–12414. [Google Scholar] [CrossRef]
- Lee, J.C.; Choi, H.J.; Lee, Y.C. Efficient synthesis of multi-substituted oxazoles under solvent-free microwave irradiation. Tetrahedron Lett. 2003, 44, 123–125. [Google Scholar] [CrossRef]
- Varma, R.S.; Kumar, D. A facile one-pot synthesis of 2,5-disubstituted oxazoles using iodobenzene diacetate. J. Heterocyclic Chem. 1998, 35, 1533–1534. [Google Scholar] [CrossRef]
- Ishiwata, Y.; Togo, H. Iodoarene-mediated one-pot preparation of 2,5-disubstituted and 2,4,5-trisubstituted oxazoles from alkyl aryl ketones with oxone in nitriles. Tetrahedron 2009, 65, 10720–10724. [Google Scholar] [CrossRef]
- Lee, J.E.; Koh, H.Y.; Seo, S.H.; Baek, Y.Y.; Rhim, H.; Cho, Y.S.; Choo, H.; Pae, A.N. Synthesis and biological evaluation of oxazole derivatives as T-type calcium channel blockers. Bioorg. Med. Chem. Lett. 2010, 20, 4219–4222. [Google Scholar] [CrossRef]
- Very recently, an elegant method using tetra-n-butylammonium iodide and tert-butyl hydroperoxide has been reported for the single-step synthesis of oxazole from dicarbonyl compounds and benzylamine derivatives. See, Xie, J.; Jiang, H.; Cheng, Y.; Zhu, C. Metal-free, organocatalytic cascade formation of C-N and C-O bonds through dual sp3 C-H activation: oxidative synthesis of oxazole derivatives. Chem. Commun. 2012, 48, 979–981. [Google Scholar] [CrossRef]
- Saito, A.; Matsumoto, A.; Hanzawa, Y. PIDA-mediated synthesis of oxazoles through oxidative cycloisomerization of propargylamides. Tetrahedron Lett. 2010, 51, 2247–2250. [Google Scholar]
- Saito, A.; Anzai, T.; Matsumoto, A.; Hanzawa, Y. PIFA-mediated oxidative cycloisomerization of 2-propargyl-1,3-dicarbonyl compounds: divergent synthesis of furfuryl alcohols and furfurals. Tetrahedron Lett. 2011, 52, 4658–4661. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Tykwinski, R.; Caple, R.; Berglund, B.; Koz'min, A.S.; Zefirov, N.S. Reaction of PhIO·HBF4/silyl enol ether adduct with olefins as general approach to carbon-carbon bond formation in AdE reactions using hypervalent iodine reagents. Tetrahedron Lett. 1988, 29, 3703–3704. [Google Scholar]
- Wenderski, T.A.; Hoarau, C.; Mejorado, L.; Pettus, T.R.R. Dearomatization applications of I(III) reagents and some unusual reactivity amongst resorcinol derived cyclohexadienones. Tetrahedron Lett. 2010, 66, 5873–5883. [Google Scholar]
- Kitamura, T.; Nagata, K.; Nakamura, T.; Furuki, R. Self-condensation of iodosylbenzene. Formation of a (p-phenylene) type of bisiodine(III) reagents. Tetrahedron 1995, 51, 6229–6236. [Google Scholar]
- Kuwano, Y.; Togo, H. Iodoarene-catalyzed one-pot preparation of 2,4,5-trisubstituted oxazoles from alkyl aryl ketones with mCPBA in nitriles. Tetrahedron 2006, 62, 6251–6256. [Google Scholar]
- Schuh (nee Müller), K.; Glorius, F. A domino copper-catalyzed C-N-/C-O-coupling process for the synthesis of oxazoles. Synthesis 2007, 2297–2306. [Google Scholar]
- Wan, C.; Zhang, J.; Wang, S.; Fan, J.; Wang, Z. Facile Synthesis of polysubstituted oxazoles via acopper-catalyzed tandem oxidative cyclization. Org. Lett. 2010, 12, 2338–2341. [Google Scholar]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).