From BACE1 Inhibitor to Multifunctionality of Tryptoline and Tryptamine Triazole Derivatives for Alzheimer’s Disease

Abstract
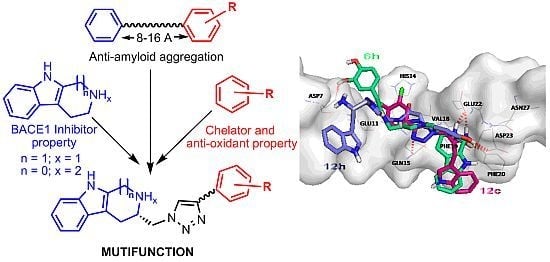
:
1. Introduction
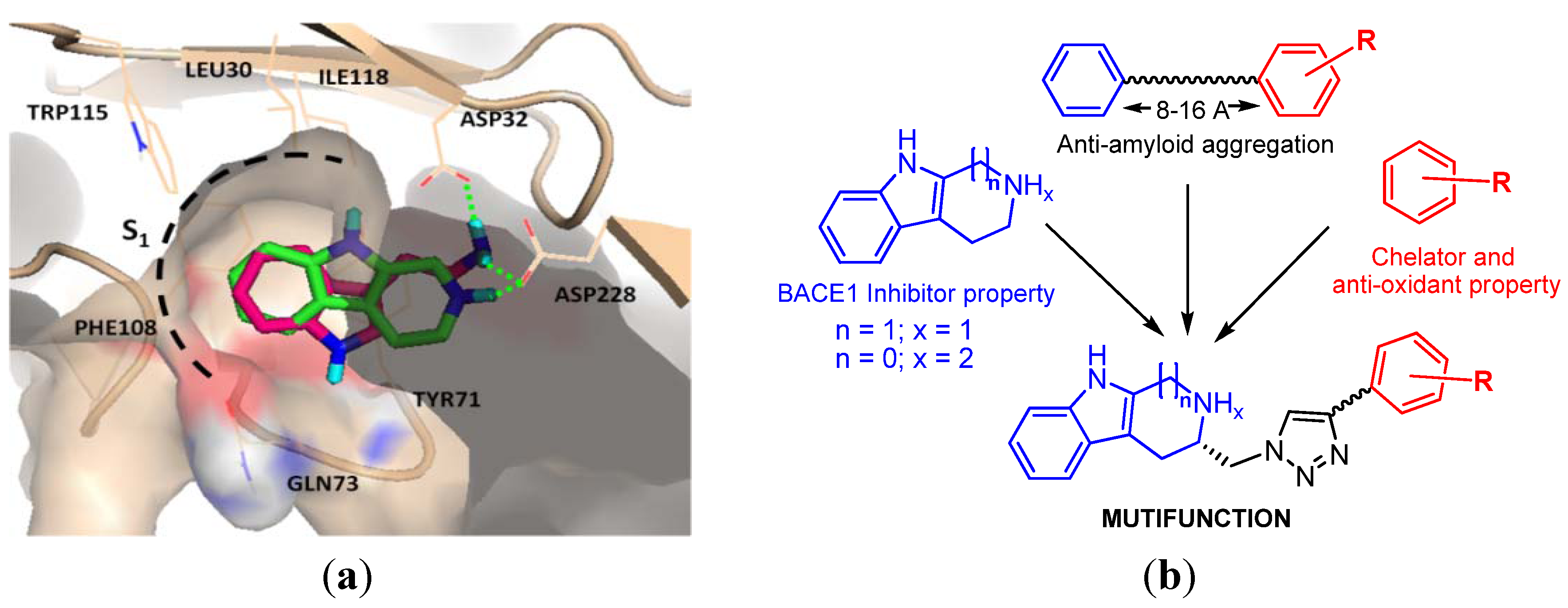
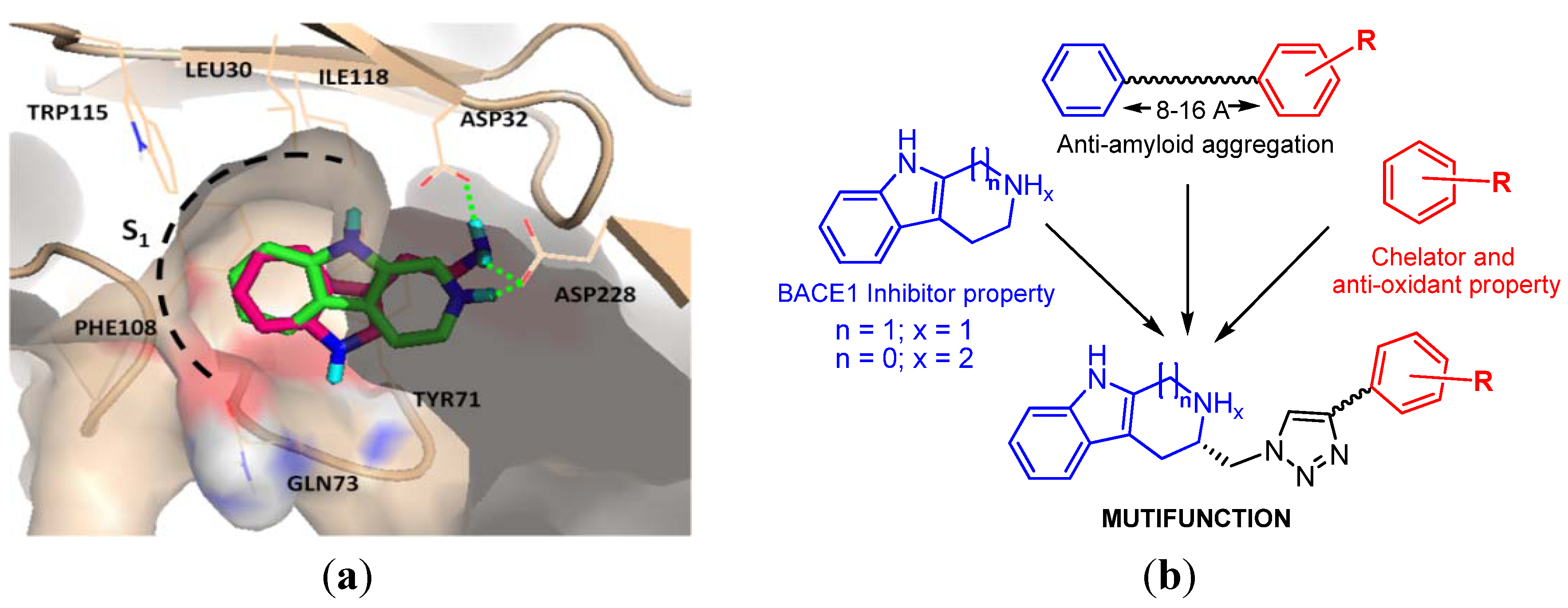
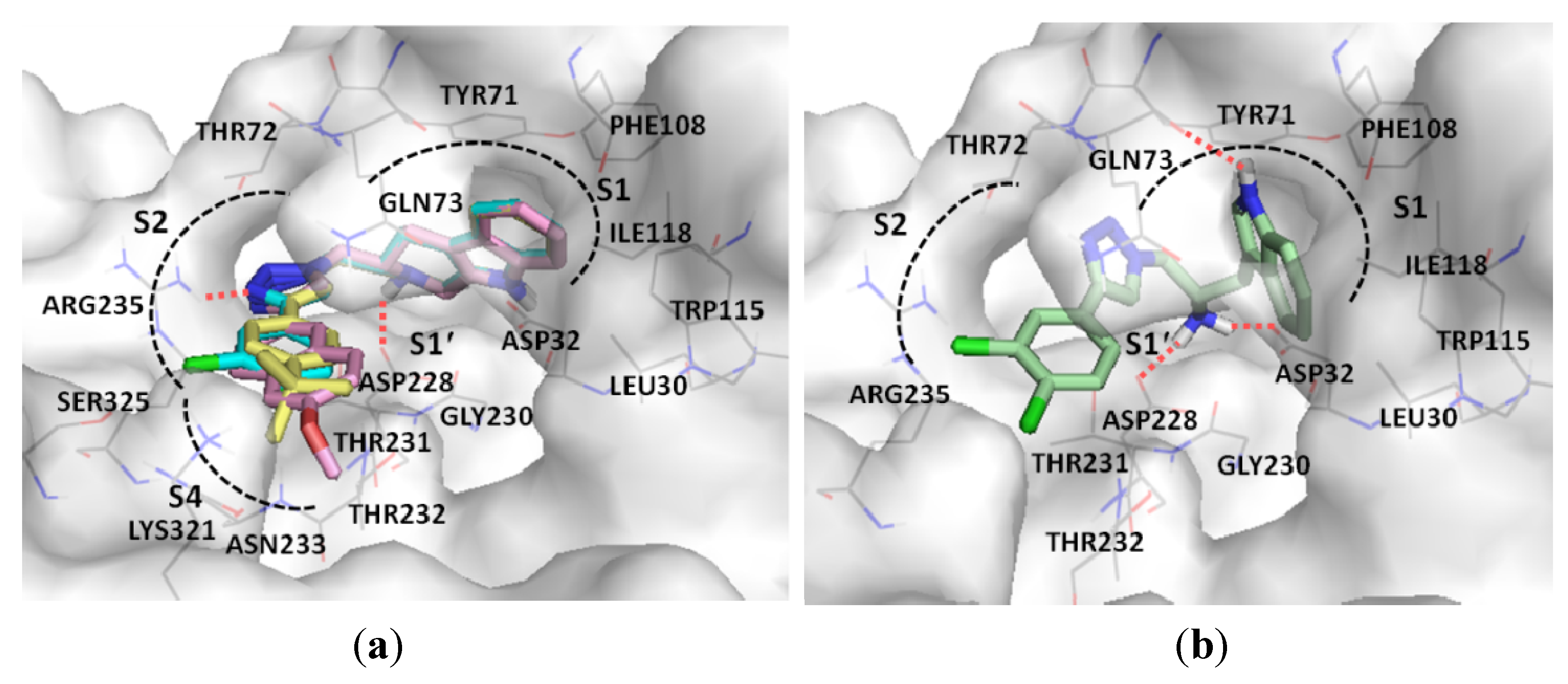
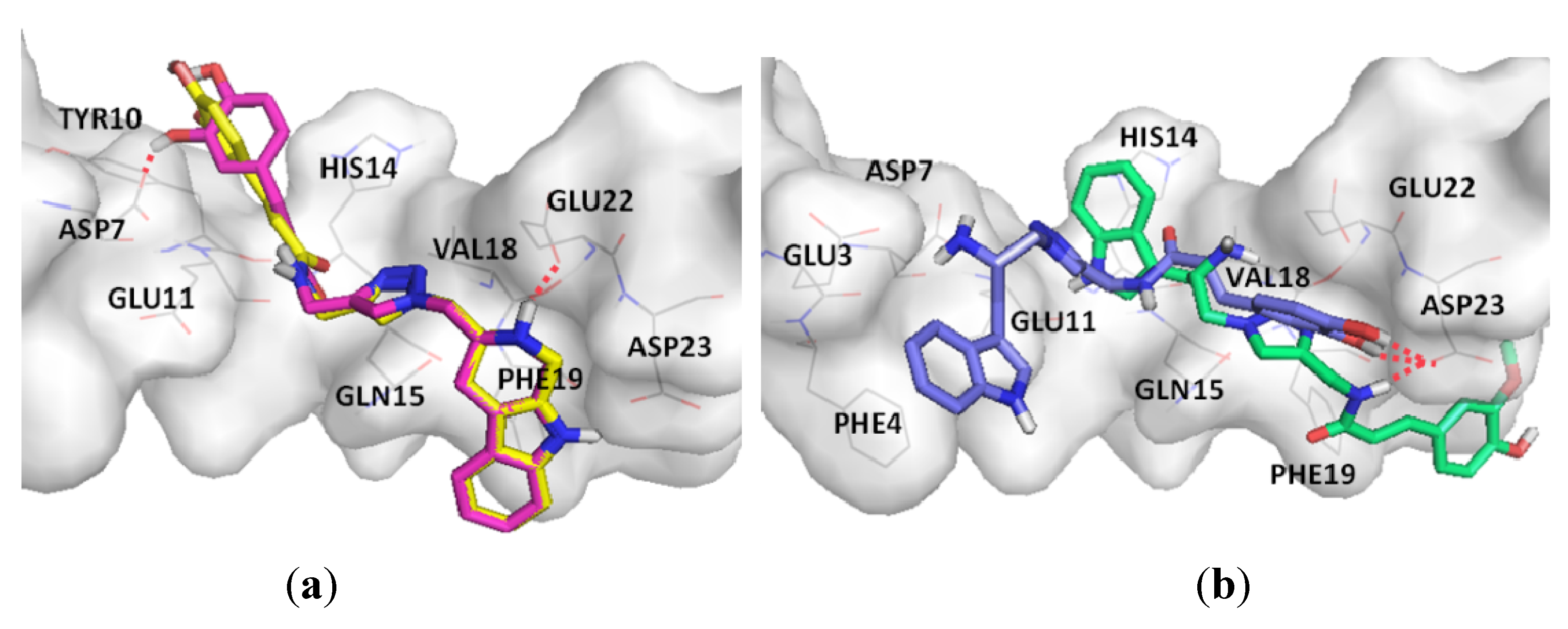
2. Results and Discussion
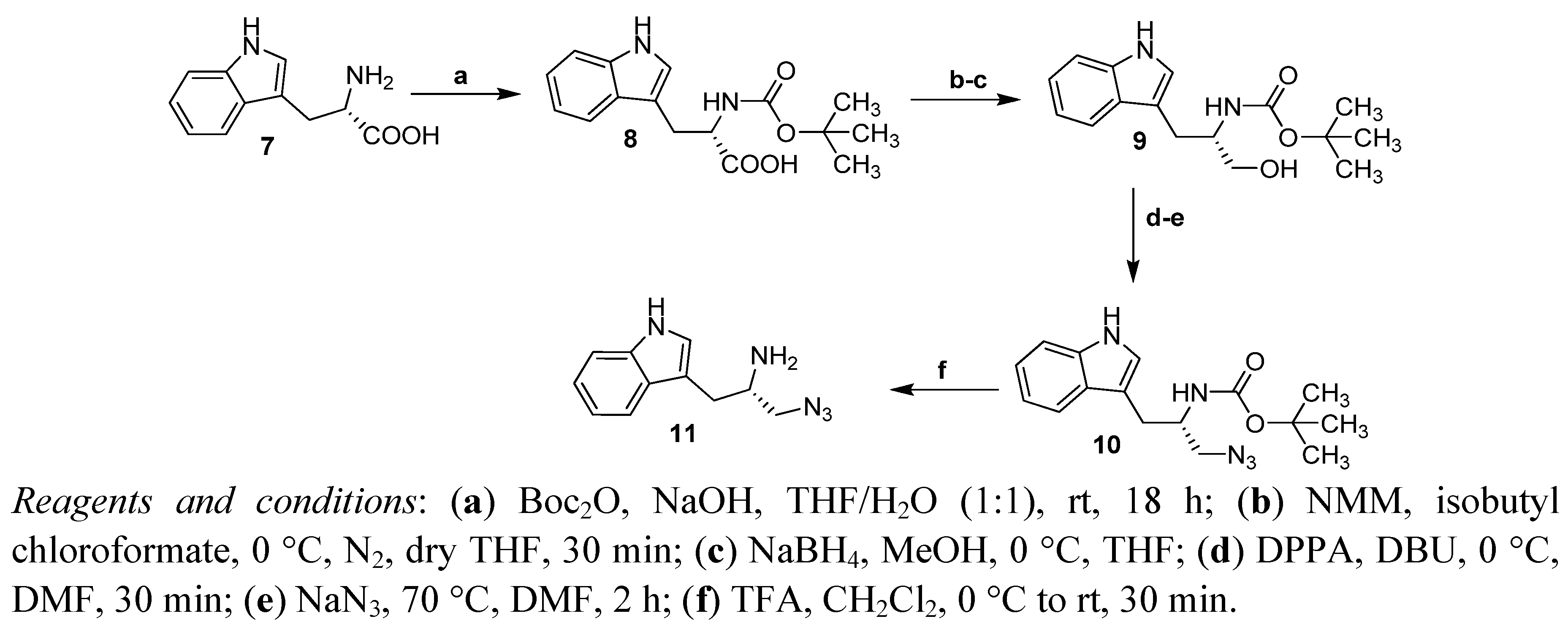
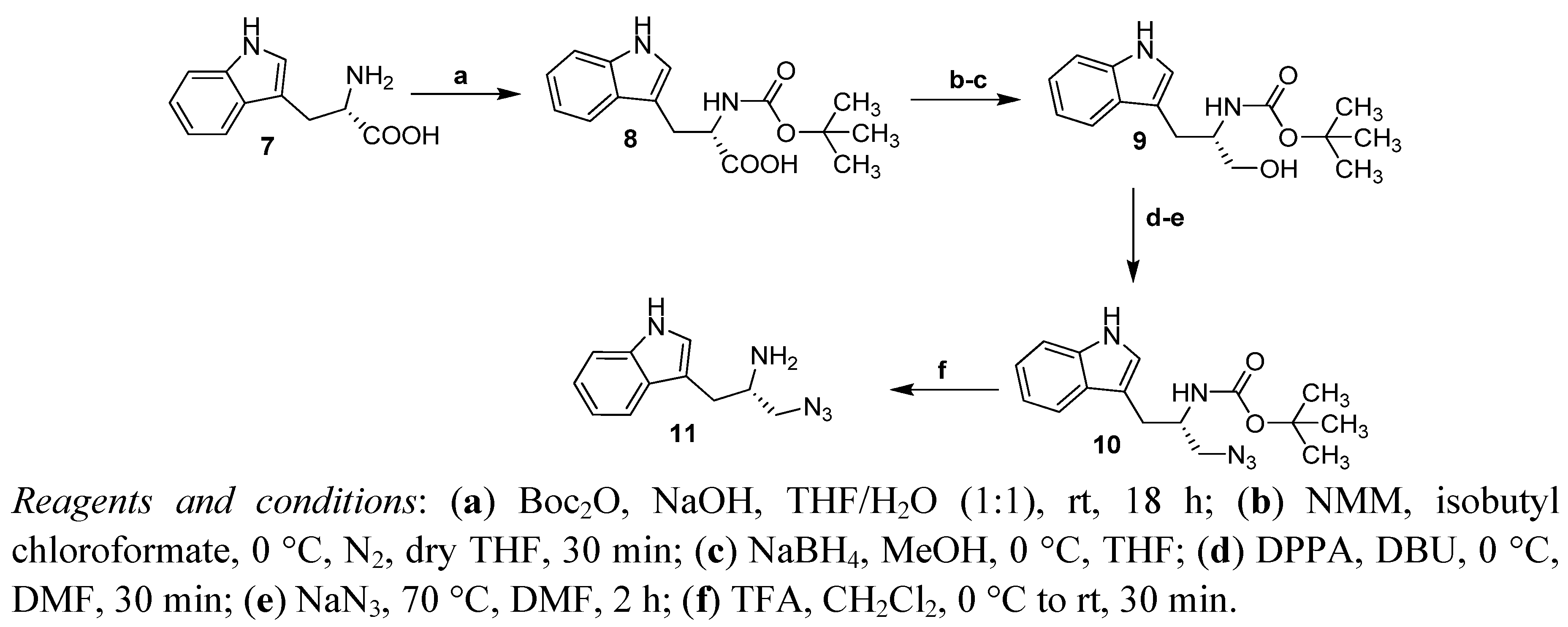
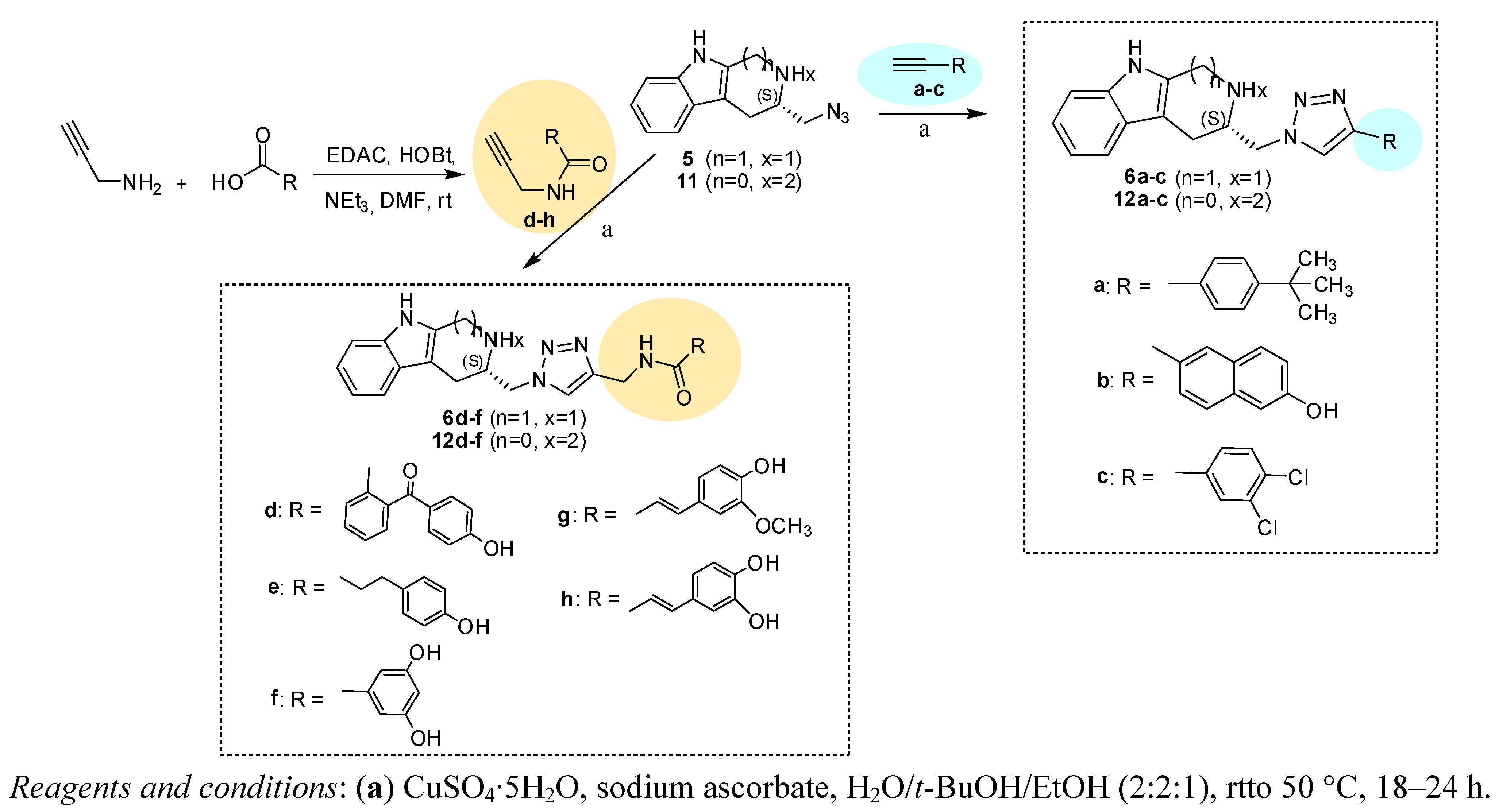
2.1. Synthesis

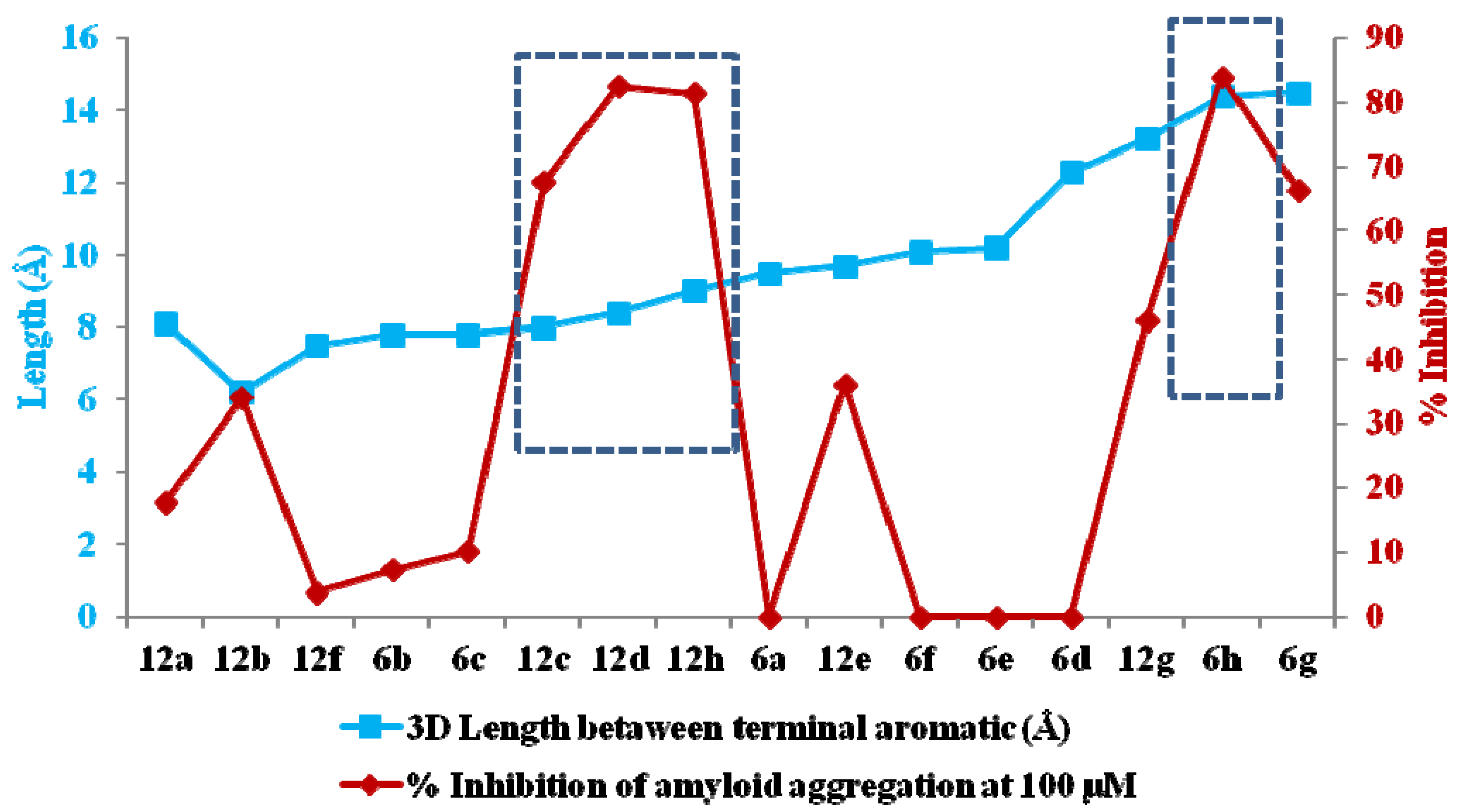
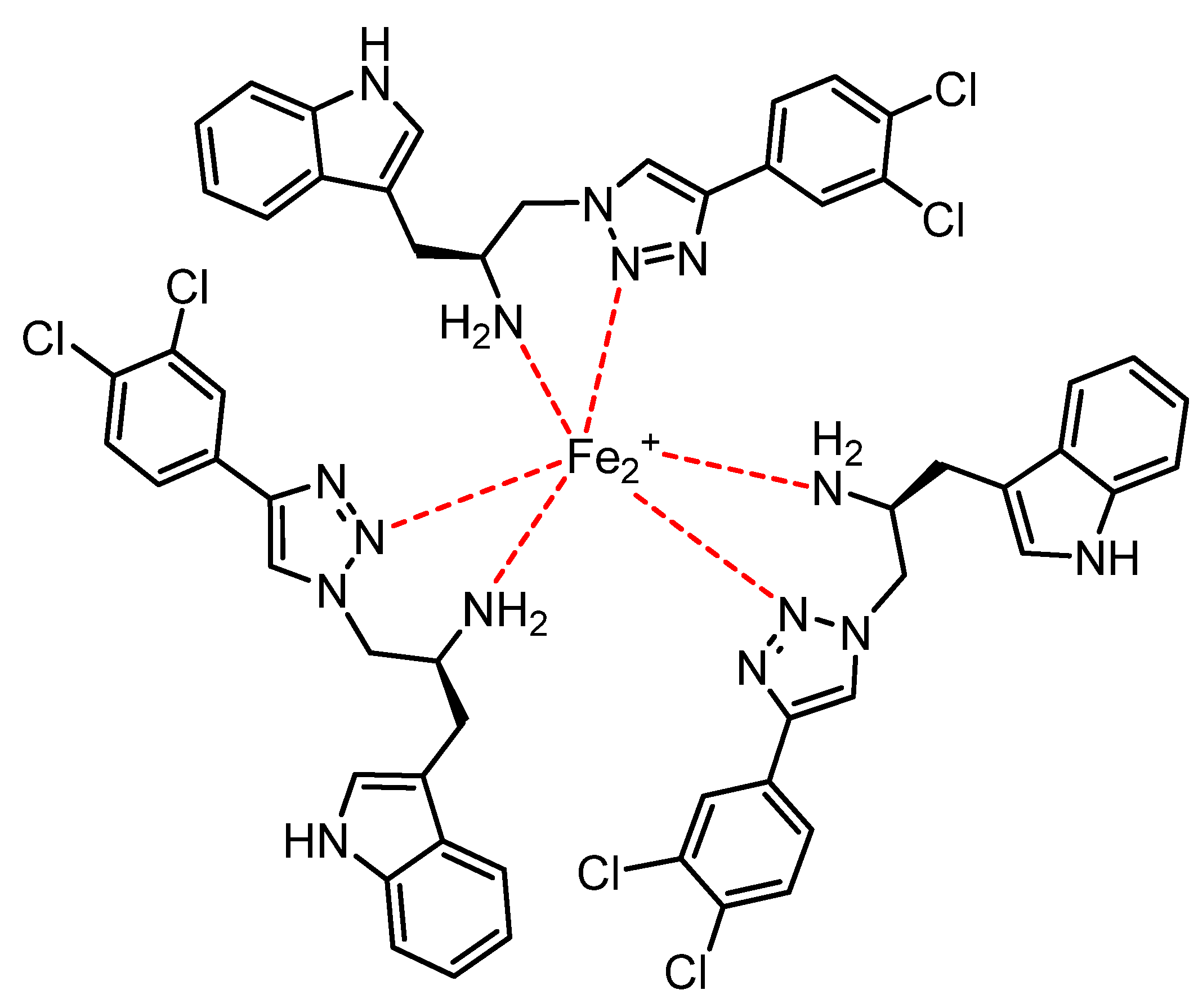
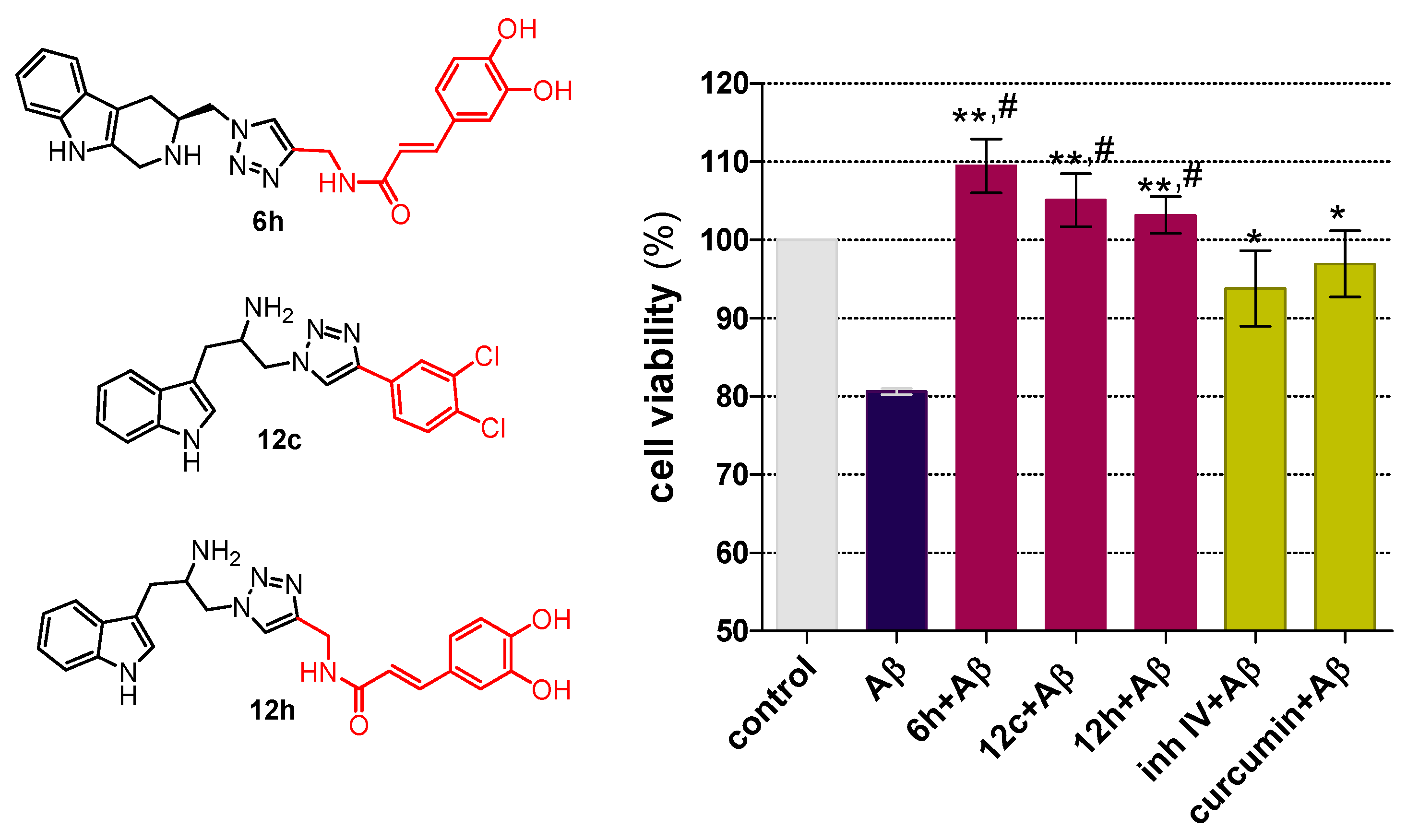
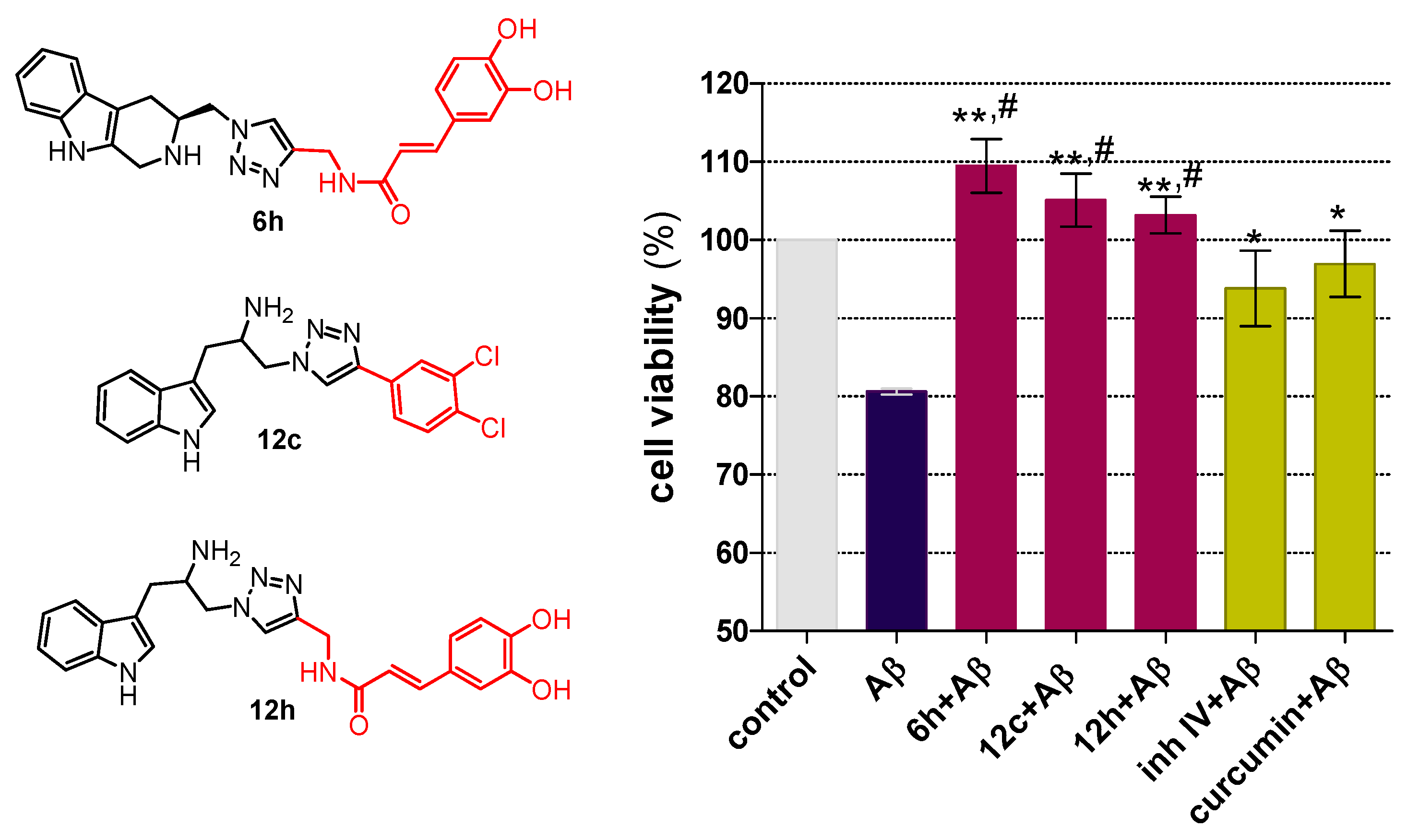
2.2. Chemical and Biological Assays

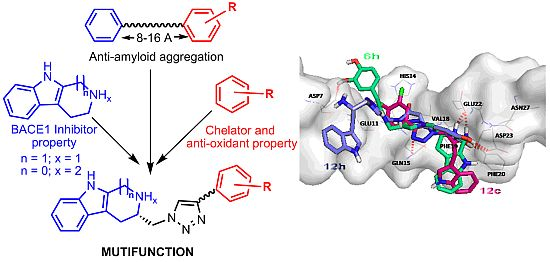{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | Log P | BACE1 | Anti-Aβ aggregation | Fe chelation | DPPH | ||||
|---|---|---|---|---|---|---|---|---|---|
| % Inhibition (at 25 µM) (±SEM) | IC50 (µM) | % Inhibition (at 100 µM) (±SEM) | IC50 (µM) | % Capacity (at 100 µM) (±SEM) | Stoichio-metric ratio (Fe:cpd) | % Inhibition (at 100 µM) (±SEM) | IC50 (µM) | ||
| 6a | 4.69 | 67.95 (±0.35) | 19.82 | NA | - | 9.67 (±0.32) | - | NA | - |
| 6b a | 3.86 | 73.63 (±1.47) | 18.86 | 7.19 (±1.19) | - | 12.35 (±0.68) | - | NA | - |
| 6c | 4.10 | 78.91 (±2.83) | 18.03 | 10.12 (±0.86) | - | 15.88 (±0.21) | - | NA | - |
| 6d | 3.01 | 14.10 (±1.79) | - | NA | - | 32.86 (±0.19) | - | 2.81 (±0.37) | - |
| 6e | 2.16 | 12.57 (±1.10) | - | NA | - | 20.63 (±0.35) | - | 5.45 (±0.33) | - |
| 6f | 1.41 | 21.28 (±1.92) | - | NA | - | 5.80 (±0.04) | - | NA | - |
| 6g | 2.01 | 21.14 (±1.13) | - | 66.36 (±2.02) | 82.90 | 8.88 (±0.25) | - | 47.03 (±0.12) | 106.41 |
| 6h | 1.75 | 21.72 (±0.33) | - | 84.13 (±2.49) | 29.86 | 42.74 (±0.30) | 1:3 | 92.29 (±0.02) | 42.91 |
| 12a | 4.42 | 18.91 (±0.40) | - | 17.81 (±0.86) | - | 42.65 (±1.07) | - | NA | - |
| 12b | 3.58 | 30.42 (±3.54) | - | 34.02 (±10.13) | - | 10.54 (±0.22) | - | NA | - |
| 12c | 3.83 | 61.46 (±1.83) | 20.75 | 67.56 (±0.72) | 83.23 | 60.90 (±0.51) | 1:3 | NA | - |
| 12d | 2.73 | 32.97 (±0.37) | - | 82.52 (±1.26) | 47.51 | 13.87 (±0.59) | - | 1.56 (±0.51) | - |
| 12e | 1.88 | 7.53 (±1.79) | - | 36.03 (±1.92) | - | 41.42 (±1.13) | - | NA | - |
| 12f | 1.13 | 16.57 (±2.91) | - | 3.80 (±1.56) | - | 13.06 (±0.16) | - | 1.32 (±0.78) | - |
| 12g | 1.74 | 16.23 (±0.45) | - | 46.06 (±1.63) | 109.83 | 77.70 (±0.67) | 1:3 | 40.79 (±0.31) | 130.44 |
| 12h | 1.47 | 40.03 (±0.95) | - | 81.48 (±2.54) | 56.39 | 66.45 (±0.37) | 1:3 | 50.58 (±0.17) | 92.70 |
| Inh IV (Merck®) | 1.23 | 96.51 (±1.33) | 0.015 b | - | - | - | - | - | - |
| Curcumin | 2.56 | - | - | 82.90 (±0.82) | 0.63c | - | - | - | - |
| EDTA | −2.69 | - | - | - | - | 98.00 (±0.34) | - | - | |
| Ascorbic acid | −3.36 | - | - | - | - | - | - | 53.64 (±0.11) | 94.92 |
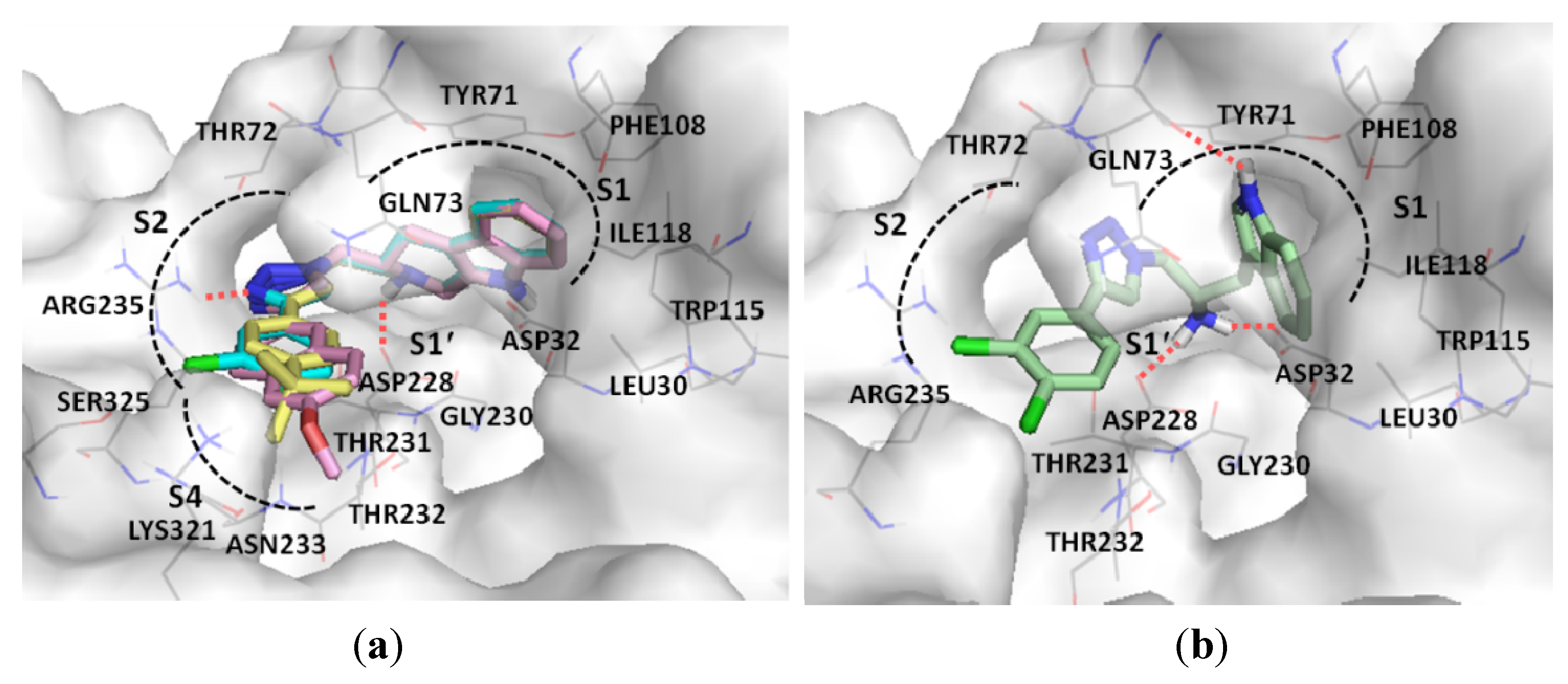
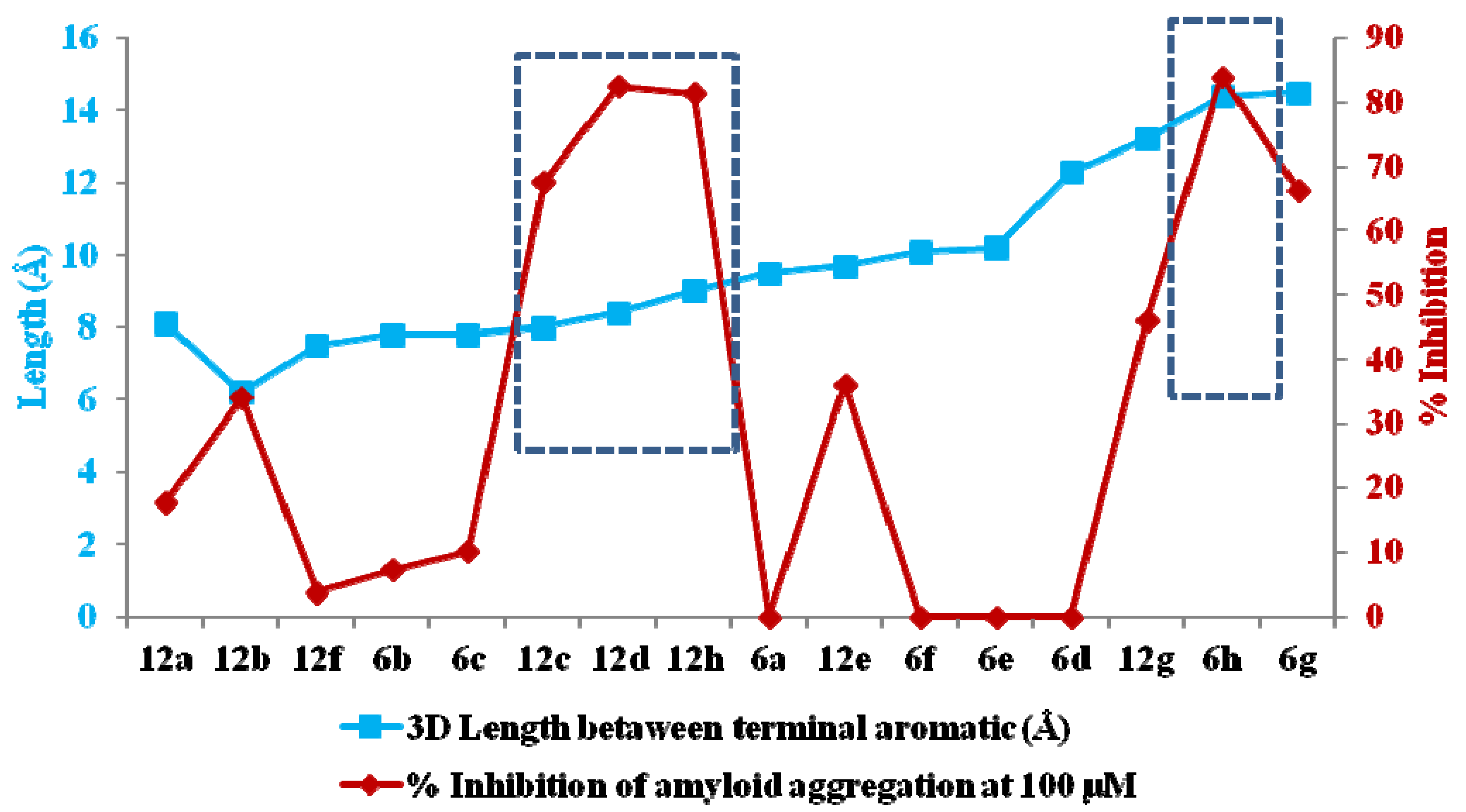
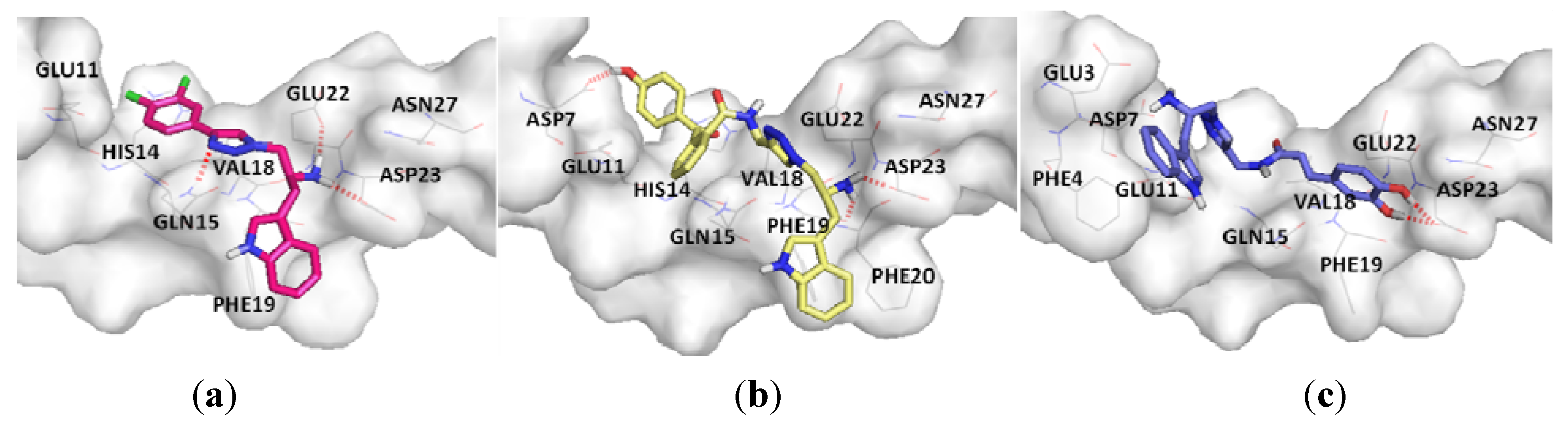

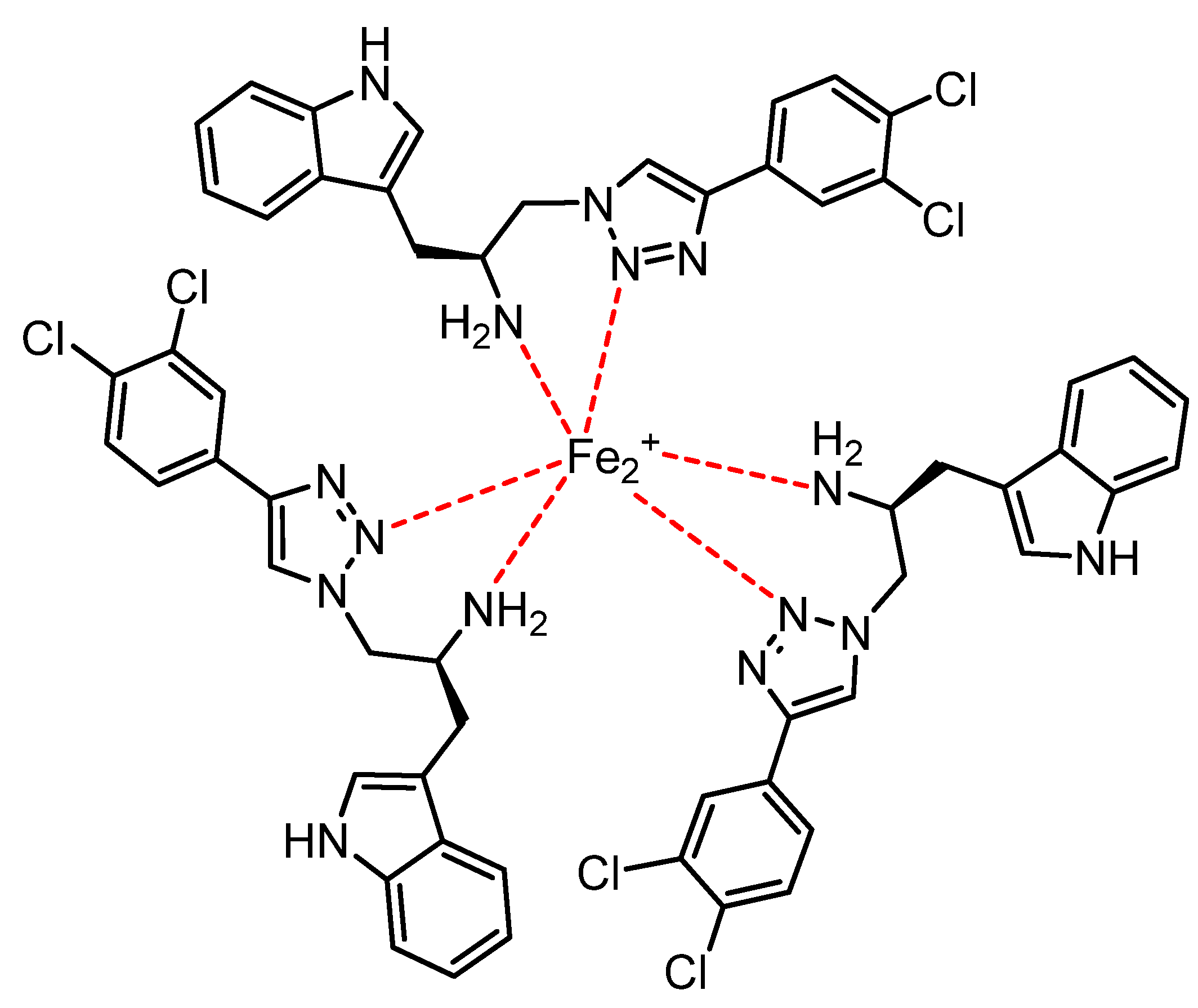
3. Experimental
3.1. General
3.2. Docking Study of β-Secretase (BACE1)
3.3. Docking Study of Amyloid β (Aβ)
3.4. Preparation of Azidomethyl Tryptamine Intermediates
3.5. Preparation of Alkynes d–h
3.6. Preparation of Triazolylmethyl Tryptolines 6a–i and Triazolylmethyl Tryptamines 12a–h
3.7. β-Secretase inhibition assay
3.8. Cathepsin D assay
3.9. Amyloid β Preparation and Aggregation Assay
3.10. Fe (II) Chelation Capcity Assay
3.11. Free Radical Scavenging Assay
3.12. Cell Culture and Cell Viability Assay by MTT method
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Jakob-Roetne, R.; Jacobsen, H. Alzheimer’s disease: From pathology to therapeutic approaches. Angew. Chem. Int. Ed. Engl. 2009, 48, 3030–3059. [Google Scholar] [CrossRef]
- Mandel, S.; Amit, T.; Bar-Am, O.; Youdim, M.B. Iron dysregulation in alzheimer’s disease: Multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Prog. Neurobiol. 2007, 82, 348–360. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Galimberti, D.; Scarpini, E. Progress in Alzheimer’s disease. J. Neurol. 2012, 259, 201–211. [Google Scholar] [CrossRef]
- Budimir, A. Metal ions, Alzheimer’s disease and chelation therapy. Acta Pharm. 2011, 61, 1–14. [Google Scholar] [CrossRef]
- Tabner, B.J.; Mayes, J.; Allsop, D. Hypothesis: Soluble Aβ oligomers in association with redox-active metal ions are the optimal generators of reactive oxygen species in Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2011, 2011. [Google Scholar]
- Bolognesi, M.L.; Cavalli, A.; Melchiorre, C. Memoquin: A Multi-target-directed ligand as an innovative therapeutic opportunity for Alzhemer’s disease. Neurotherapeutics 2009, 6, 152–162. [Google Scholar] [CrossRef]
- Jiaranaikulwanitch, J.; Boonyarat, C.; Fokin, V.V.; Vajragupta, O. Triazolyltryptoline derivatives as β-secretase inhibitors. Bioorg. Med. Chem. Lett. 2010, 10, 6572–6576. [Google Scholar]
- Reinke, A.A.; Gestwicki, J.E. Structure-activity relationships of amyloid beta-aggregation inhibitiors based on curcumin: Influence of linker length and flexibility. Chem. Biol. Drug. Des. 2007, 70, 206–215. [Google Scholar] [CrossRef]
- Zheng, W.; Wang, S.Y. Antioxidant activity and phenolic compounds in selected herbs. J. Agric. Food. Chem. 2001, 49, 5165–5170. [Google Scholar] [CrossRef]
- Ebrahimzadeh, M.A.; Pourmorad, F.; Bekhradnia, A.R. Iron chelating activity, phenol and flavonoid content of some medicinal plants from Iran. Afr. J. Biotechnol. 2008, 7, 3188–3192. [Google Scholar]
- Xiao, J.; Xu, J.; Cui, S.; Liu, H.; Wang, S.; Li, Y. Supramolecular helix of an amphiphilic pyrene derivative induced by chiral tryptophan through electrostatic interactions. Org. Lett. 2008, 10, 645–648. [Google Scholar] [CrossRef]
- Page, D.; Naismith, A.; Schmidt, R.; Coupal, M.; Labarre, M.; Gosselin, M.; Bellemare, D.; Payza, K.; Brown, W. Novel C-terminus modifications of the Dmt-Tic Motif: A new class of dipeptide analogues showing altered pharmacological profiles toward the opioid receptors. J. Med. Chem. 2001, 44, 2387–2390. [Google Scholar]
- Galli, U.; Ercolano, E.; Carraro, L.; Blasi Roman, C.R.; Sorba, G.; Canonico, P.L.; Genazzani, A.A.; Tron, G.C.; Billington, R.A. Synthesis and biological evaluation of isosteric analogues of FK866, an inhibitor of NAD salvage. ChemMedChem 2008, 3, 771–779. [Google Scholar]
- Xie, J.; Seto, C.T. A two stage click-based library of protein tyrosine phosphatase inhibitors. Bioorg. Med. Chem. 2007, 15, 458–473. [Google Scholar]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper (I)-catalyzed regioselective ligation of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Kumagai, N.; Muncipinto, G.; Schreiber, S.L. Short synthesis of skeletally and stereochemically diverse small molecules by coupling petasis condensation reactions to cyclization reactions. Angew. Chem. Int. Ed. Engl. 2006, 45, 3635–3638. [Google Scholar]
- Brady, S.F.; Singh, S.; Crouthamel, M.C.; Holloway, M.K.; Coburn, C.A.; Garsky, V.M.; Boqusky, M.; Pennington, M.W.; Vacca, J.P.; Hazuda, D.; et al. Rational design and synthesis of selective BACE-1 inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 601–604. [Google Scholar]
- Stachel, S.J.; Coburn, C.A.; Steele, T.G.; Jones, K.G.; Loutzenhiser, E.F.; Gregro, A.R. Structure-based design of potent and selective cell-permeable inhibitors of human β-secretase (BACE1). J. Med. Chem. 2004, 47, 6447–6450. [Google Scholar] [CrossRef]
- Ono, K.; Haseqawa, K.; Naiki, H.; Yamada, M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s beta-amyloid fibrils in vitro. J. Neurosci. Res. 2004, 75, 742–750. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Mouton-Liger, F.; Paquet, C.; Dumurgier, J.; Bouras, C.; Pradier, L.; Gray, F.; Hugon, J. Oxidative stress increase BACE1 protein levels through activation of the PKR-eIF2α pathway. Biochim. Biophys. Acta 2012, 1822, 885–896. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Aragno, M.; Borghi, R.; Autelli, R.; Giliberto, L.; Muraca, G.; Danni, O.; Zhu, X.; Smith, M.A. Oxidative stress activates a positive feedback between the gamma- and betasecretase cleavages of the beta-amyloid precursor protein. J. Neurochem. 2008, 104, 683–695. [Google Scholar]
- Quiroz-Baez, R.; Rojas, E.; Arias, C. Oxidative stress promotes JNK-dependent amyloidogenic processing of normally expressed human APP by differential modification of alpha-, beta- and gamma-secretase expression. Neurochem. Int. 2009, 55, 662–670. [Google Scholar] [CrossRef]
- Kwak, Y.D.; Wang, R.; Li, J.J.; Zhang, Y.W.; Xu, H.; Liao, F.F. Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol. Neurodegener. 2011, 6. [Google Scholar]
- Rajapakse, H.A.; Nantermet, P.G.; Selnick, H.G.; Munshi, S.; McGaughey, G.B.; Lindsley, S.R.; Young, M.B.; Lai, M.T.; Espeseth, A.S.; Shi, X.P.; et al. Discovery of oxadiazoyl tertiary carbinamine inhibitors of β-secretase (BACE-1). J. Med. Chem. 2006, 49, 7270–7273. [Google Scholar] [CrossRef]
- Hong, L.; Koelsch, G.; Lin, X.; Wu, S.; Terzyan, S.; Ghosh, A.K.; Zhang, X.C.; Tang, J. Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science 2000, 290, 150–153. [Google Scholar]
- Tomaselli, S.; Esposito, V.; Vangone, P.; van Nuland, N.A.; Bonvin, A.M.; Guerrini, R.; Tancredi, T.; Temussi, P.A.; Picone, D. The alpha-to-beta conformational transition of Alzheimer’s Abeta-(1-42) peptide in aqueous media is reversible: A step by step conformational analysis suggests the location of beta conformation seeding. ChemBioChem 2006, 2, 257–267. [Google Scholar]
- Ermolieff, J.; Loy, J.A.; Koelsch, G.; Tang, J. Proteolytic activation of recombinant pro-memapsin 2 (pro-β-secretase) studied with new fluorogenic substrates. Biochemistry 2000, 39, 12450–12456. [Google Scholar] [CrossRef]
- Levine, H., III. Thioflavine T interaction with synthetic Alzheimer’s disease β-amyloid peptides: Detection of amyloid aggregation in solution. Prot. Sci. 1993, 2, 404–410. [Google Scholar] [CrossRef]
- Stookey, L.L. Ferrozine-a new spectrophotometric reagent for Iron. Anal. Chem. 1970, 42, 779–781. [Google Scholar] [CrossRef]
- Karamac, M. Chelation of Cu(II), Zn(II) and Fe(II) by tannin constituents of selected edible nuts. Int. J. Mol. Sci. 2009, 10, 5485–5497. [Google Scholar] [CrossRef]
- Sharma, O.P.; Bhat, T.K. DPPH antioxidant assay revisited. Food Chem. 2009, 113, 1202–1205. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jiaranaikulwanitch, J.; Govitrapong, P.; Fokin, V.V.; Vajragupta, O. From BACE1 Inhibitor to Multifunctionality of Tryptoline and Tryptamine Triazole Derivatives for Alzheimer’s Disease. Molecules 2012, 17, 8312-8333. https://doi.org/10.3390/molecules17078312
Jiaranaikulwanitch J, Govitrapong P, Fokin VV, Vajragupta O. From BACE1 Inhibitor to Multifunctionality of Tryptoline and Tryptamine Triazole Derivatives for Alzheimer’s Disease. Molecules. 2012; 17(7):8312-8333. https://doi.org/10.3390/molecules17078312
Chicago/Turabian StyleJiaranaikulwanitch, Jutamas, Piyarat Govitrapong, Valery V. Fokin, and Opa Vajragupta. 2012. "From BACE1 Inhibitor to Multifunctionality of Tryptoline and Tryptamine Triazole Derivatives for Alzheimer’s Disease" Molecules 17, no. 7: 8312-8333. https://doi.org/10.3390/molecules17078312
APA StyleJiaranaikulwanitch, J., Govitrapong, P., Fokin, V. V., & Vajragupta, O. (2012). From BACE1 Inhibitor to Multifunctionality of Tryptoline and Tryptamine Triazole Derivatives for Alzheimer’s Disease. Molecules, 17(7), 8312-8333. https://doi.org/10.3390/molecules17078312