Synthesis and Bioactivity of Secondary Metabolites from Marine Sponges Containing Dibrominated Indolic Systems

Abstract
:1. Introduction
1.1. Metabolites from Hyrtios sp.
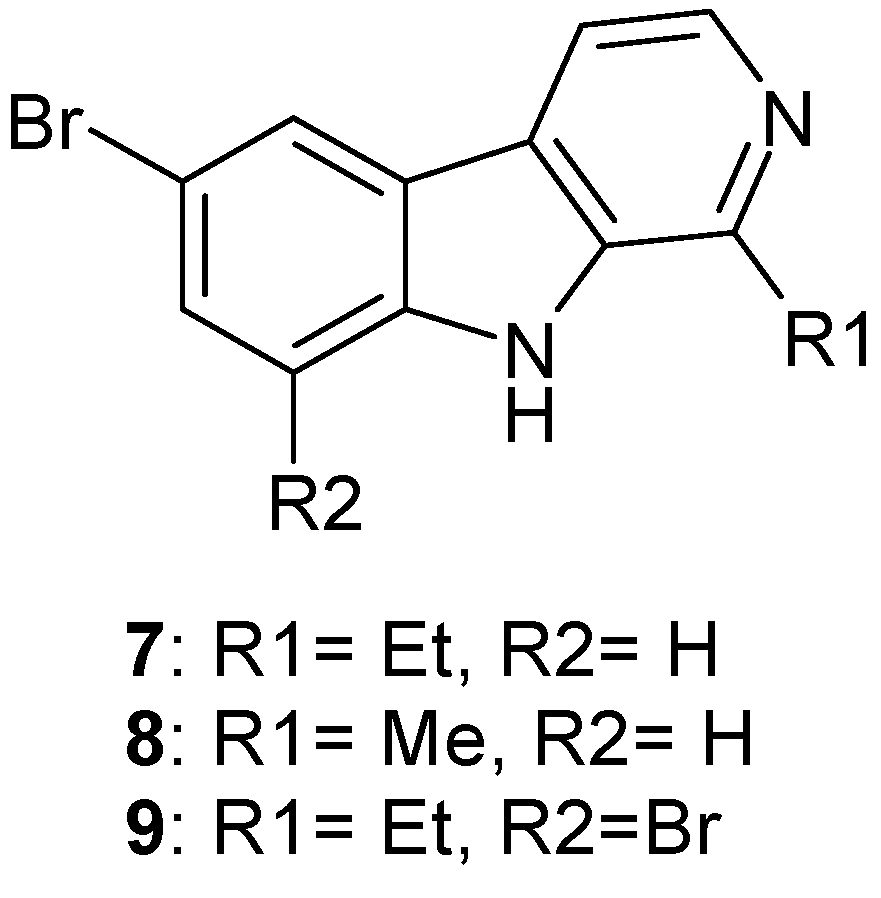
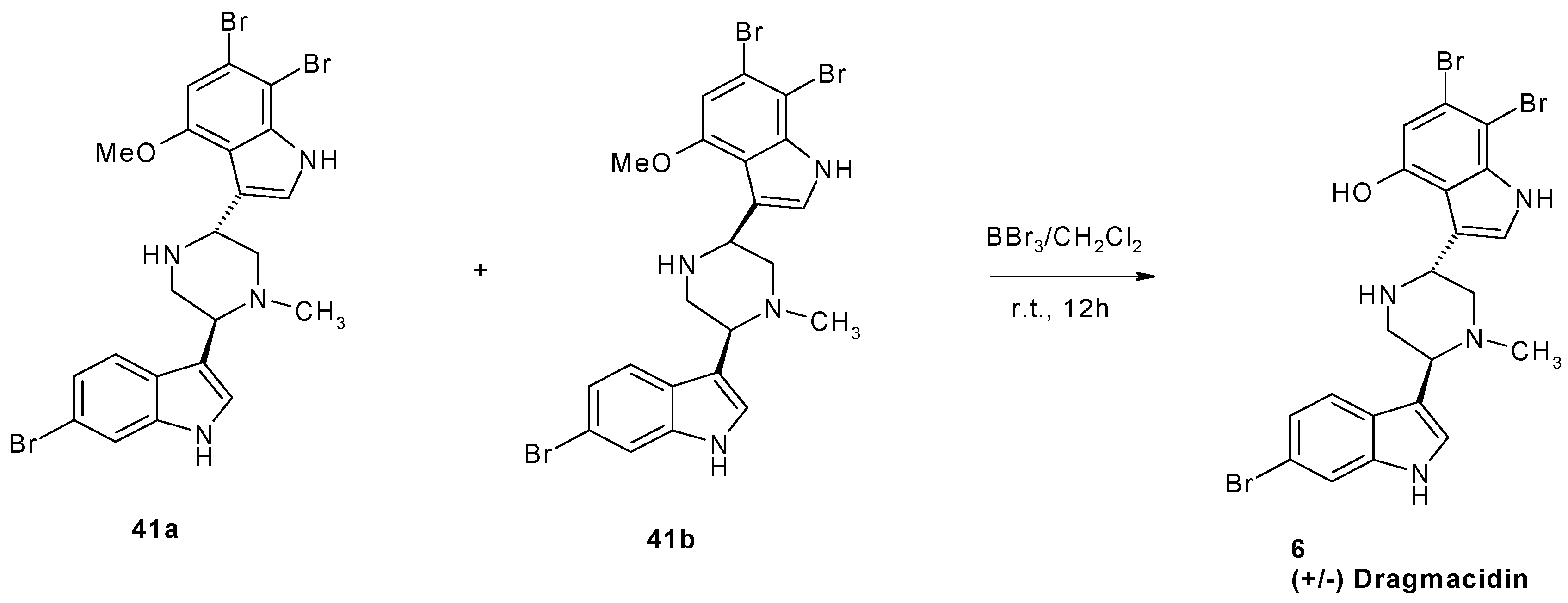
1.2. Metabolites from Dragmacidin sp.
1.3. Metabolites from Aglophenia pleuma
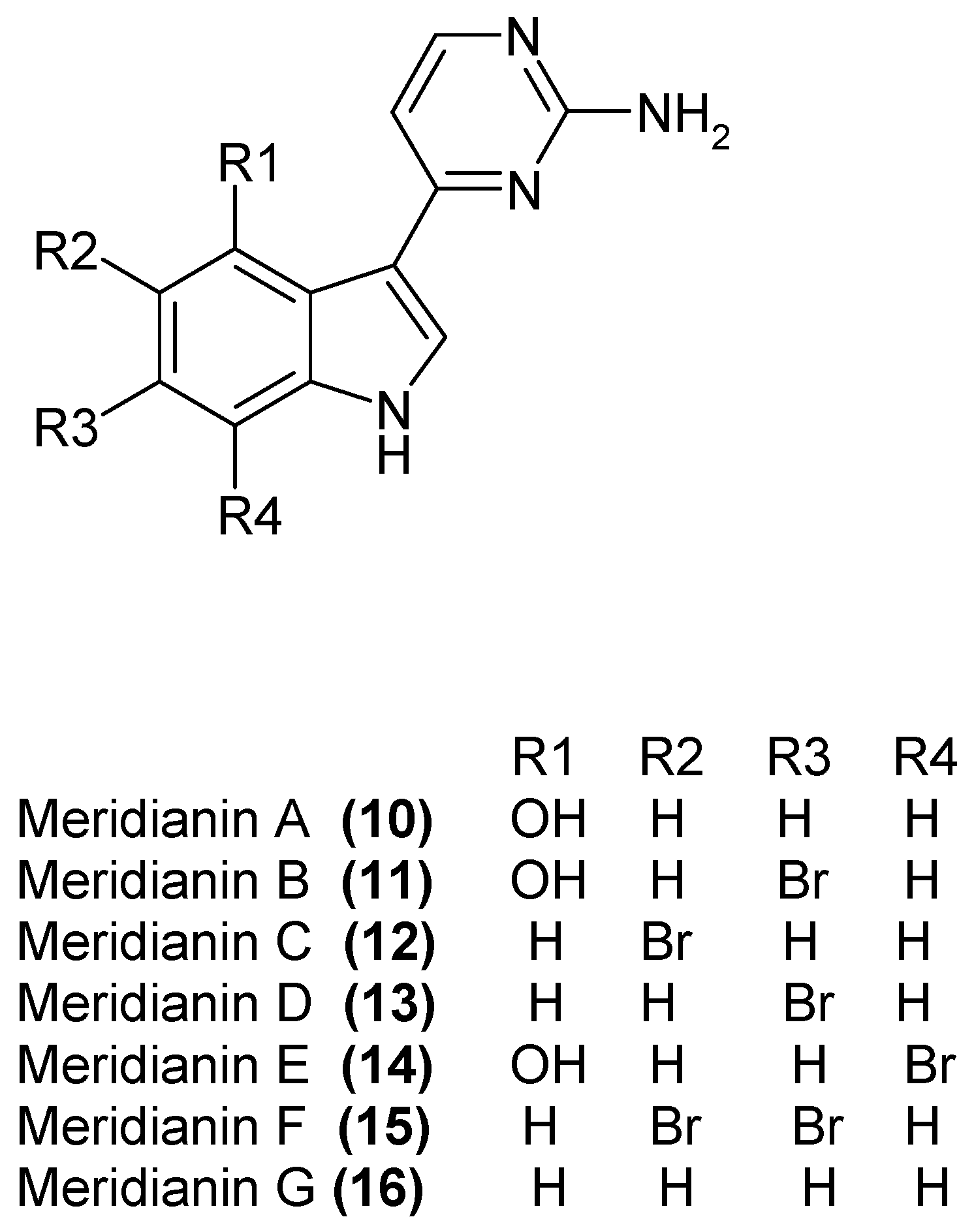
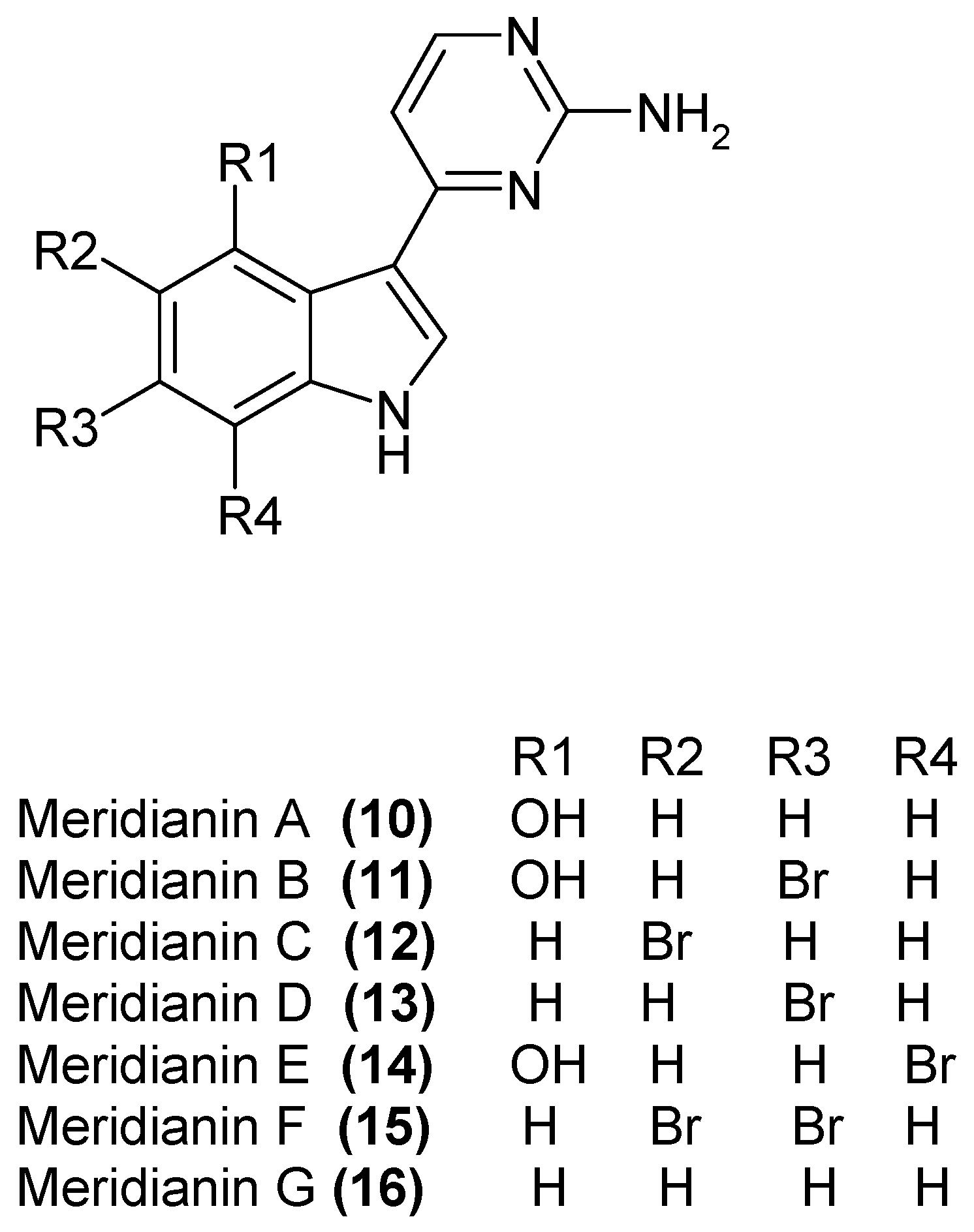
1.4. Metabolites from Aplidium meridianum
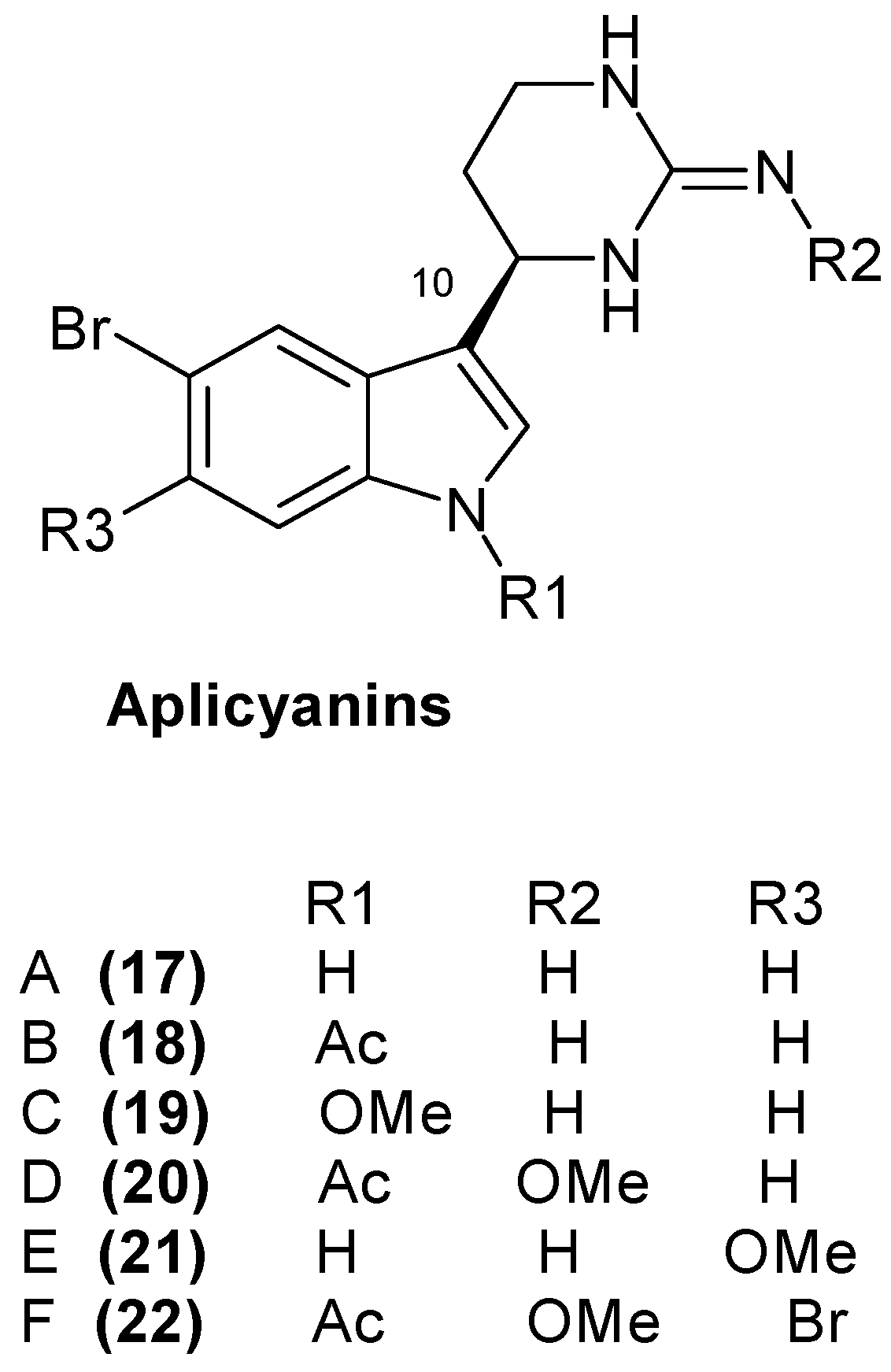
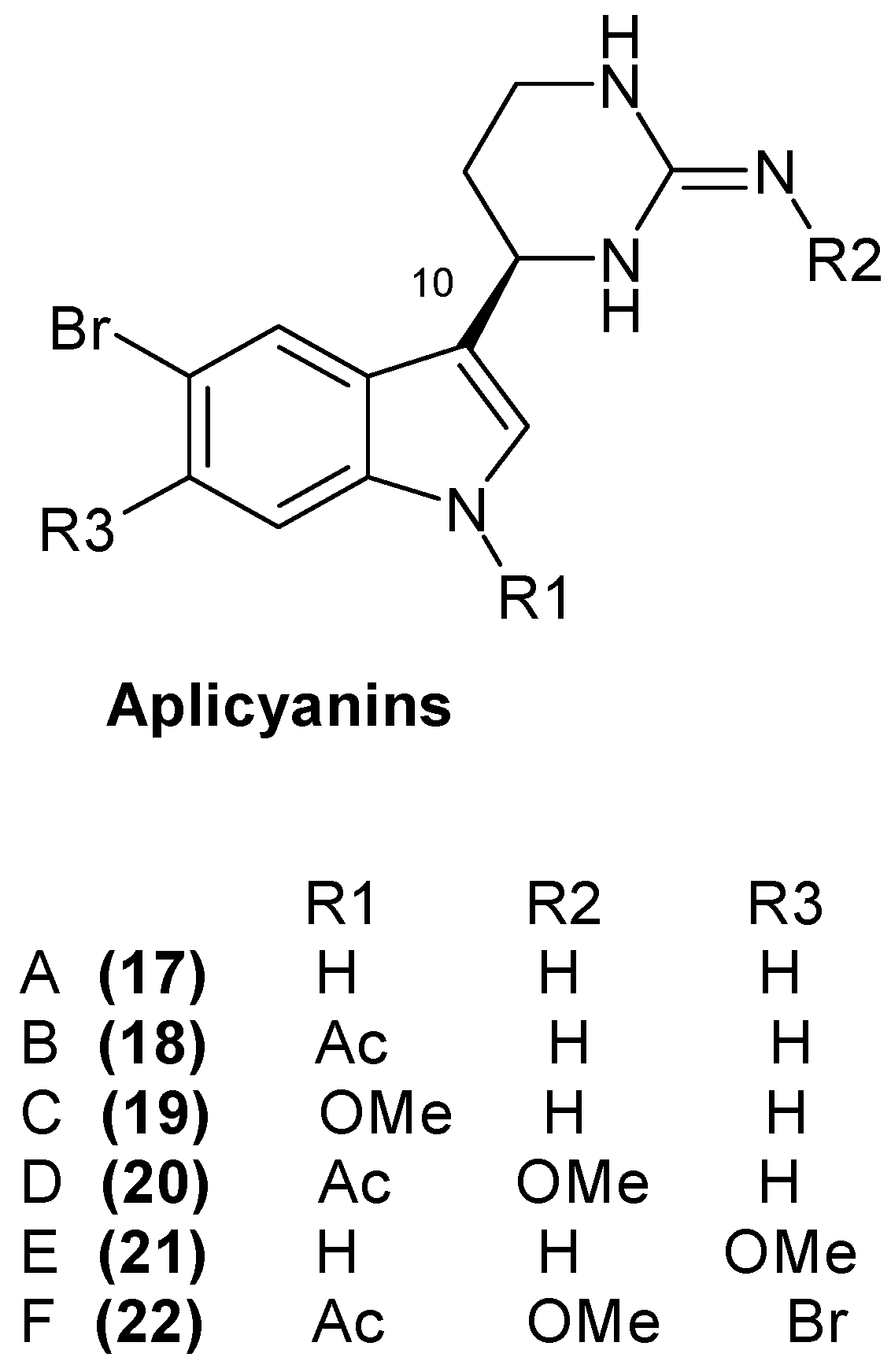
1.5. Metabolites from Aplidium cyaneum
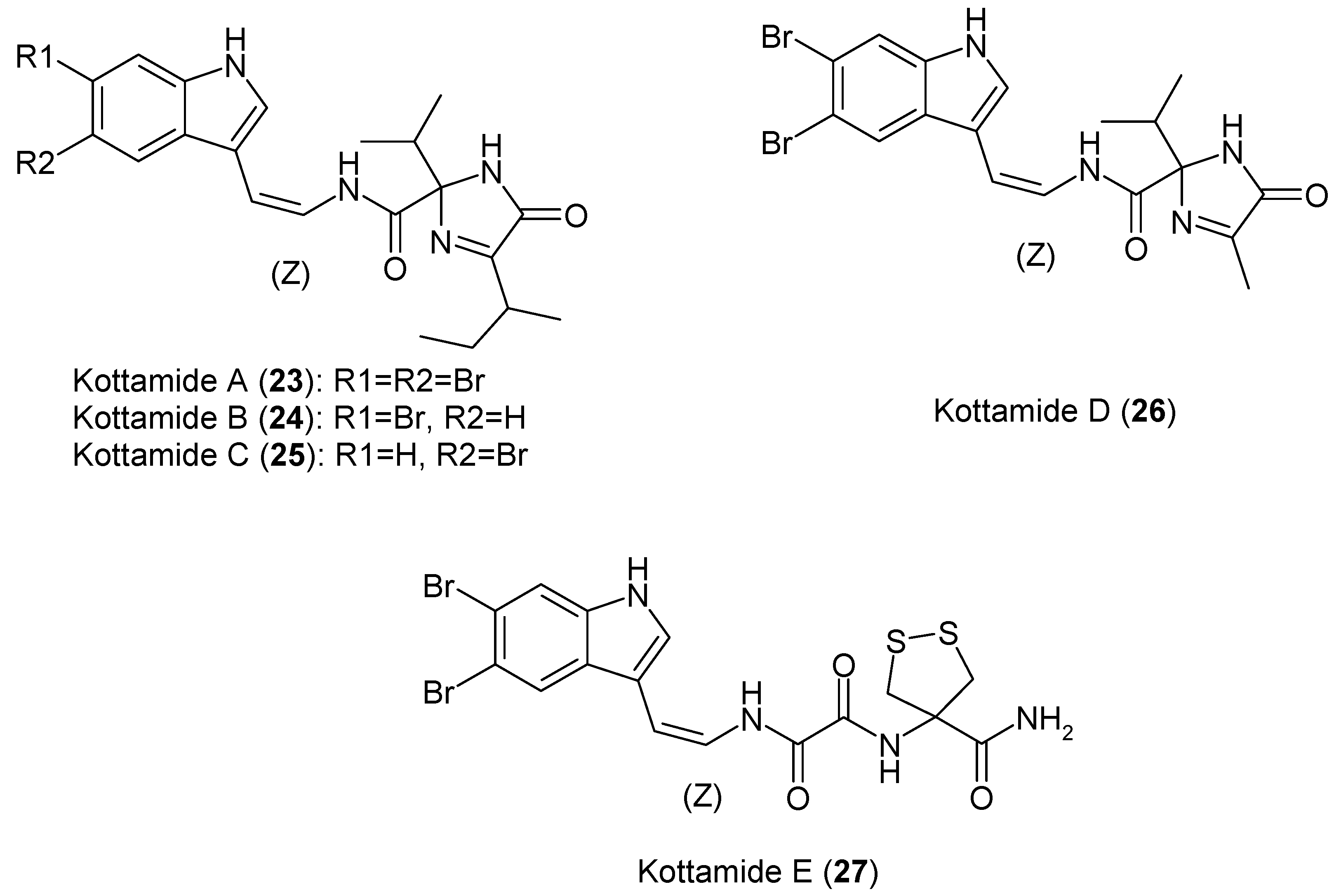
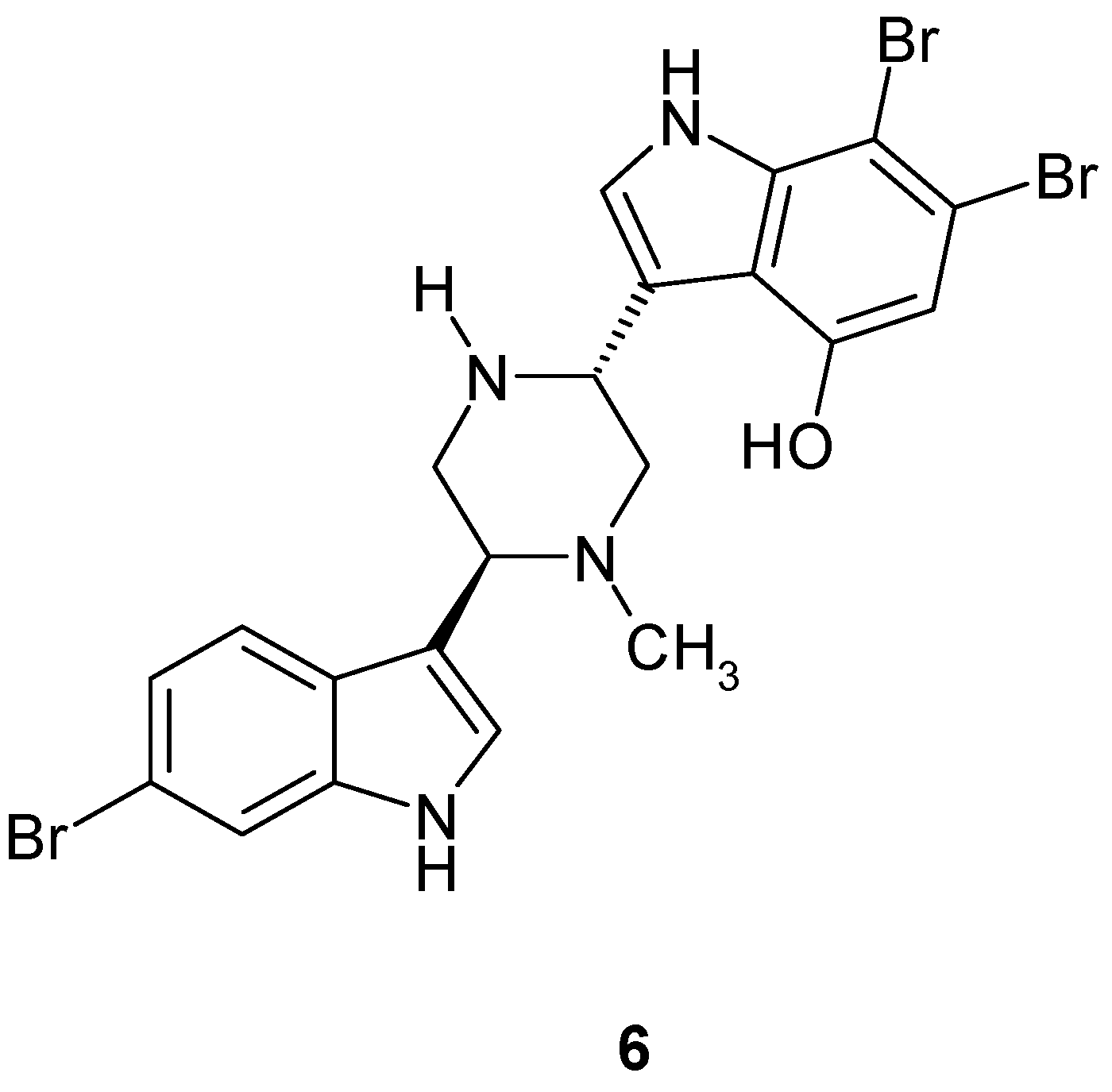
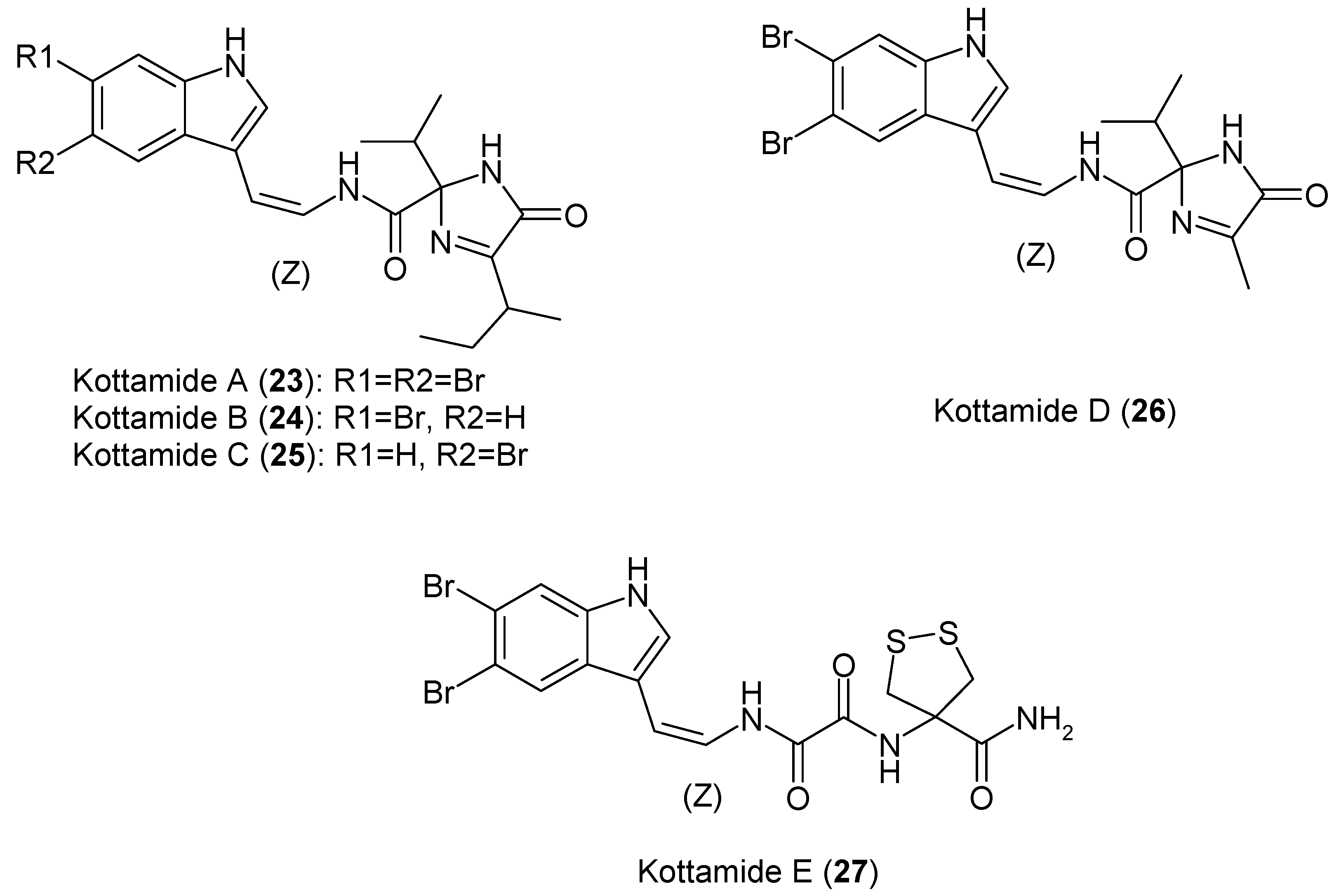
1.6. Metabolites from Pycnoclavella kottae
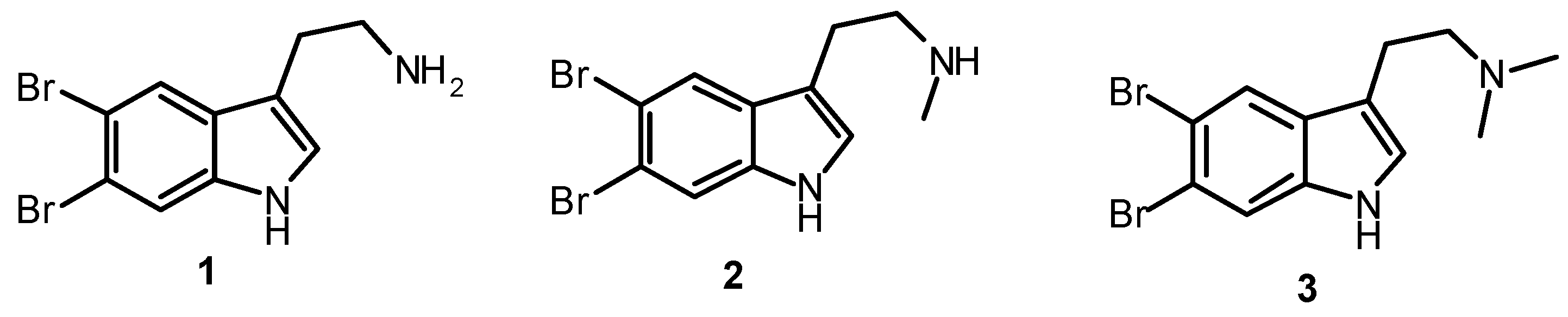
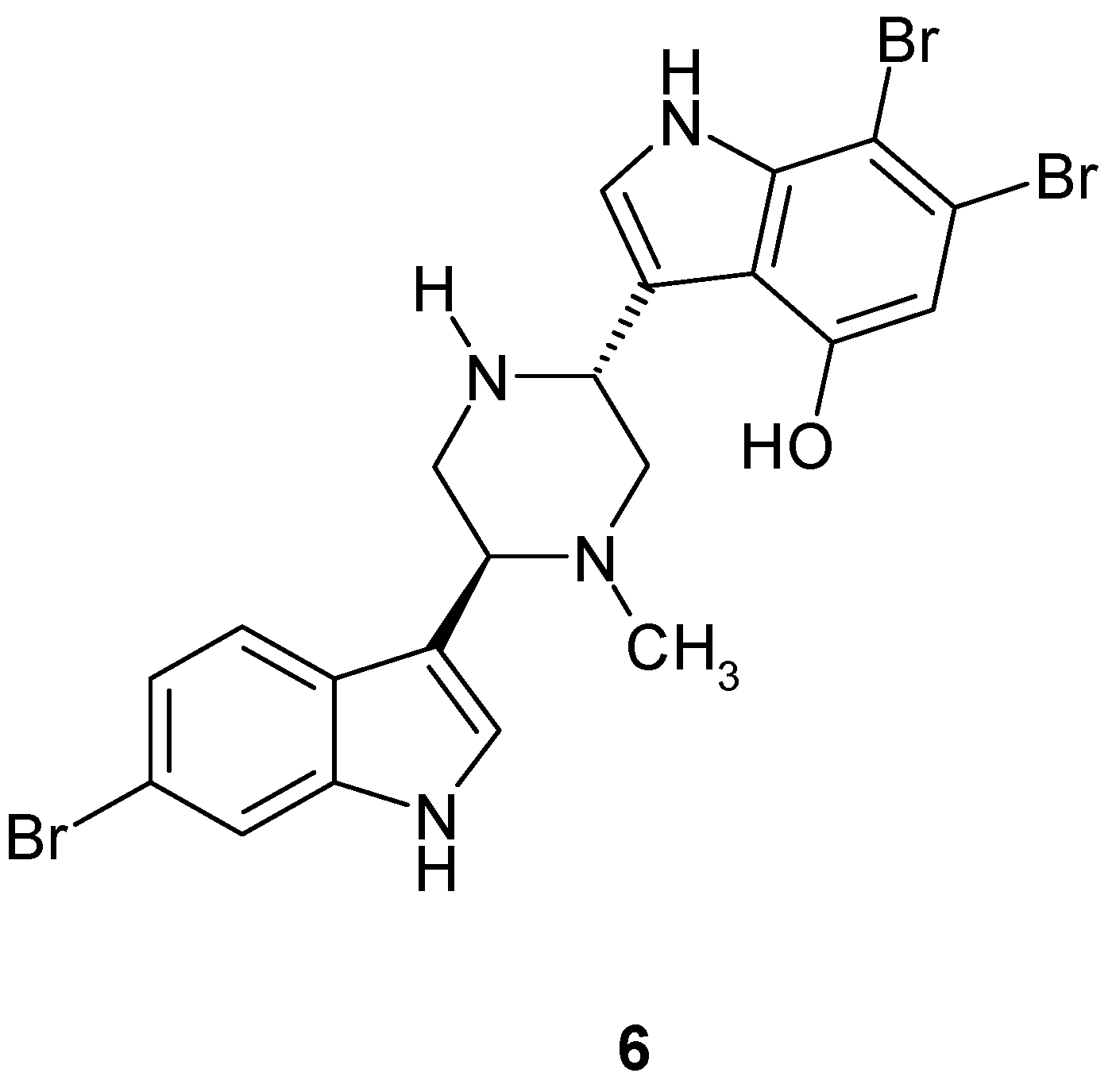
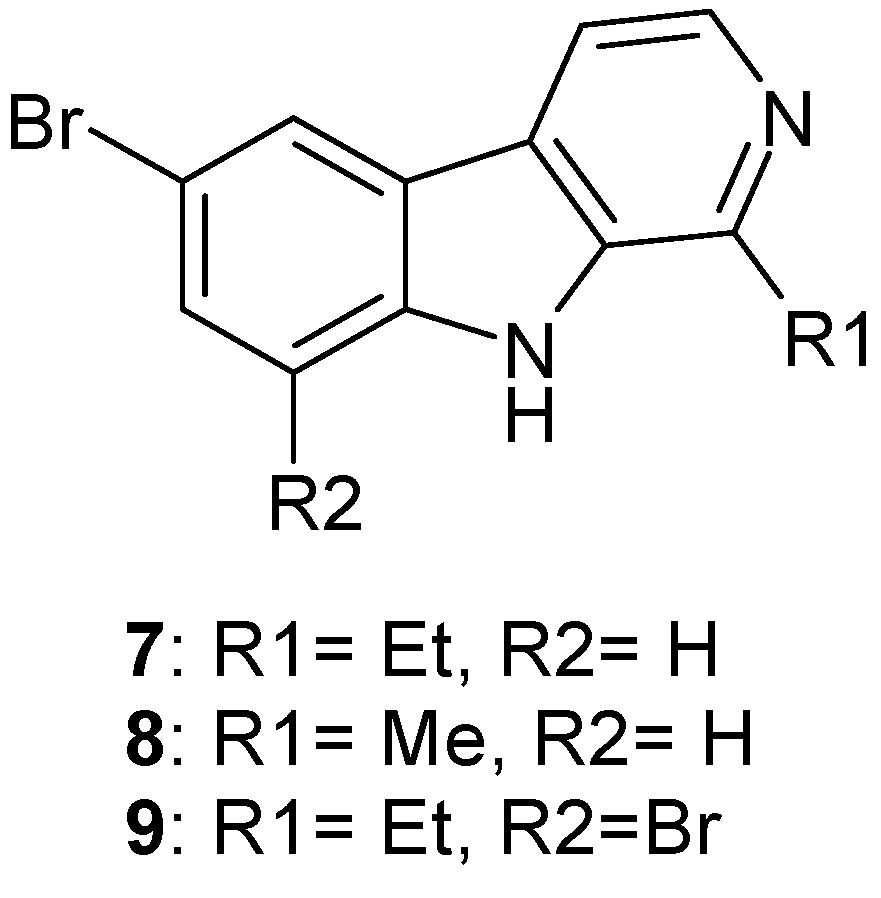
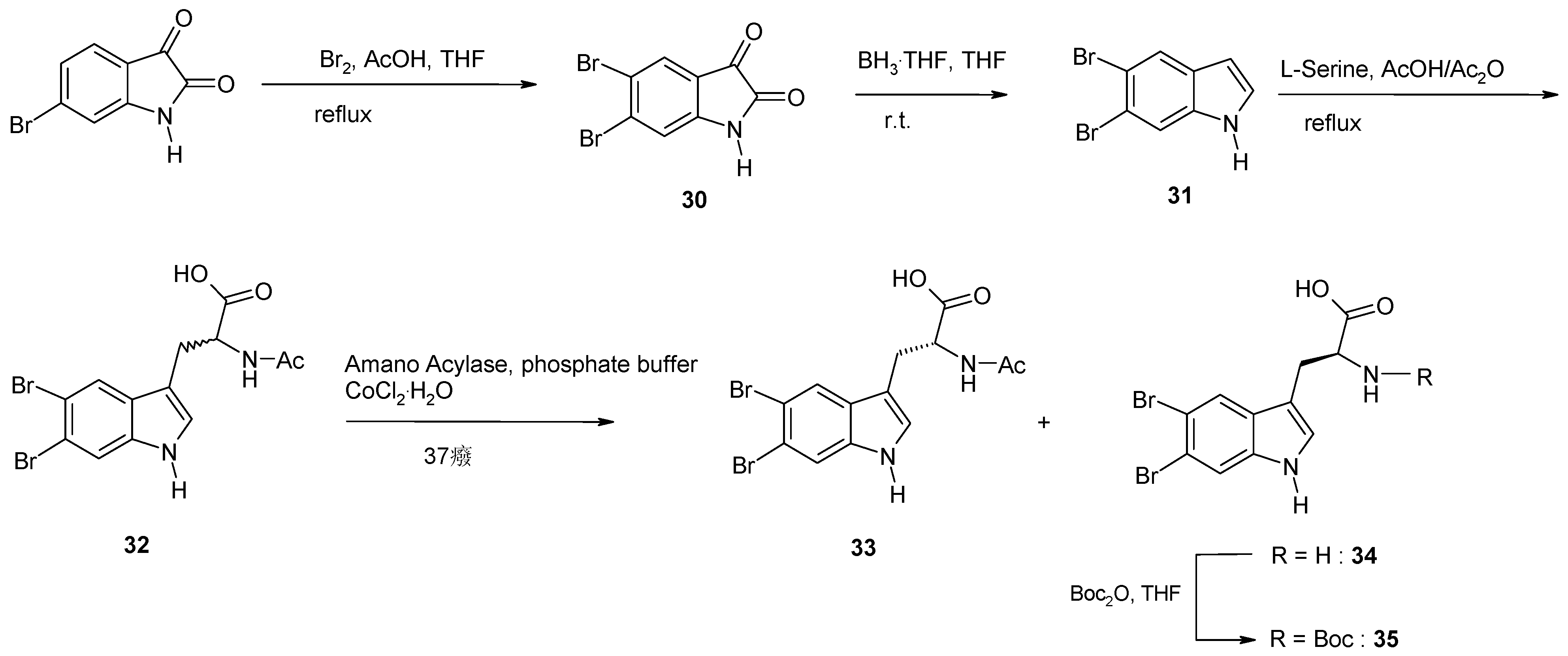
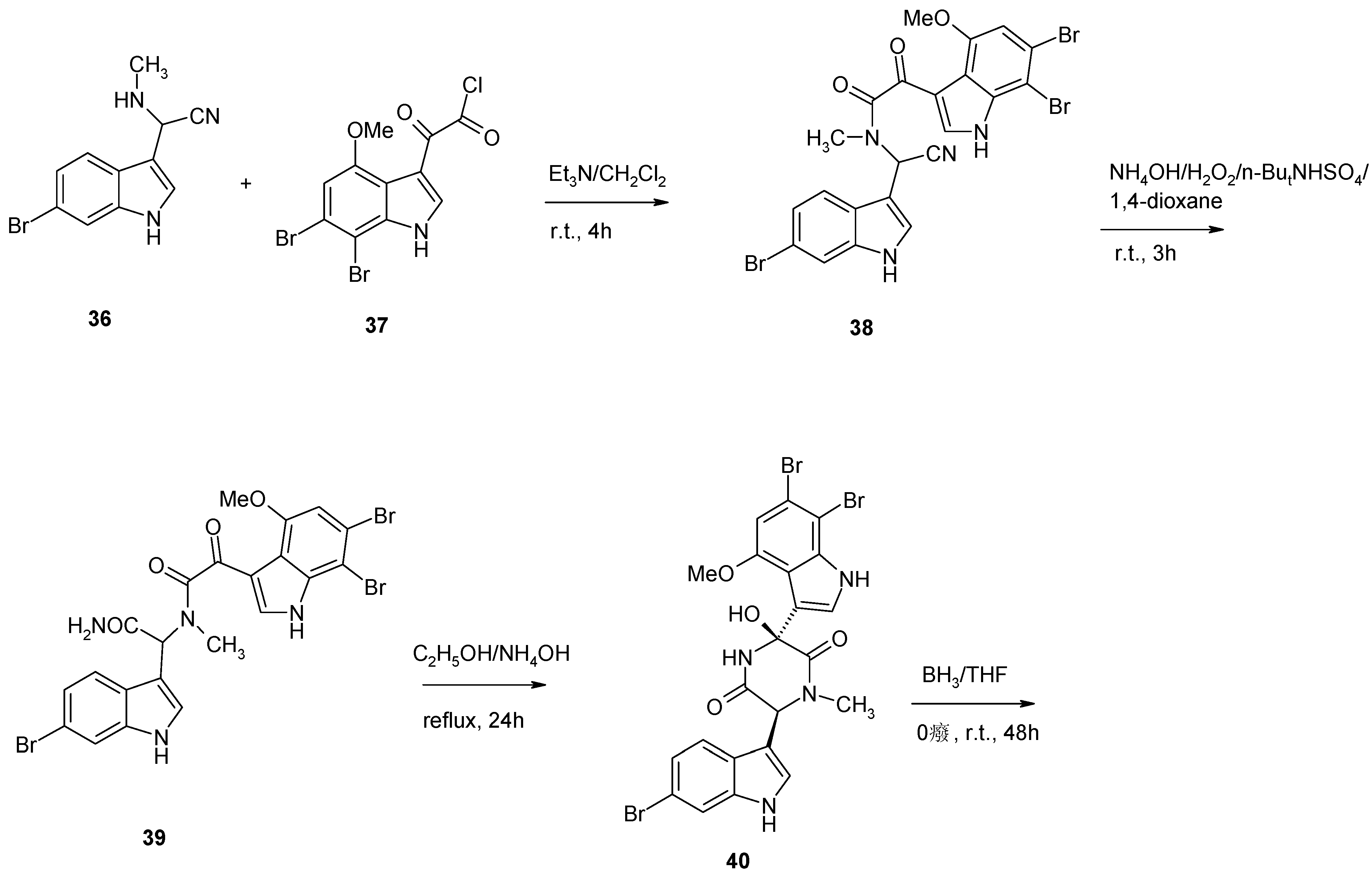
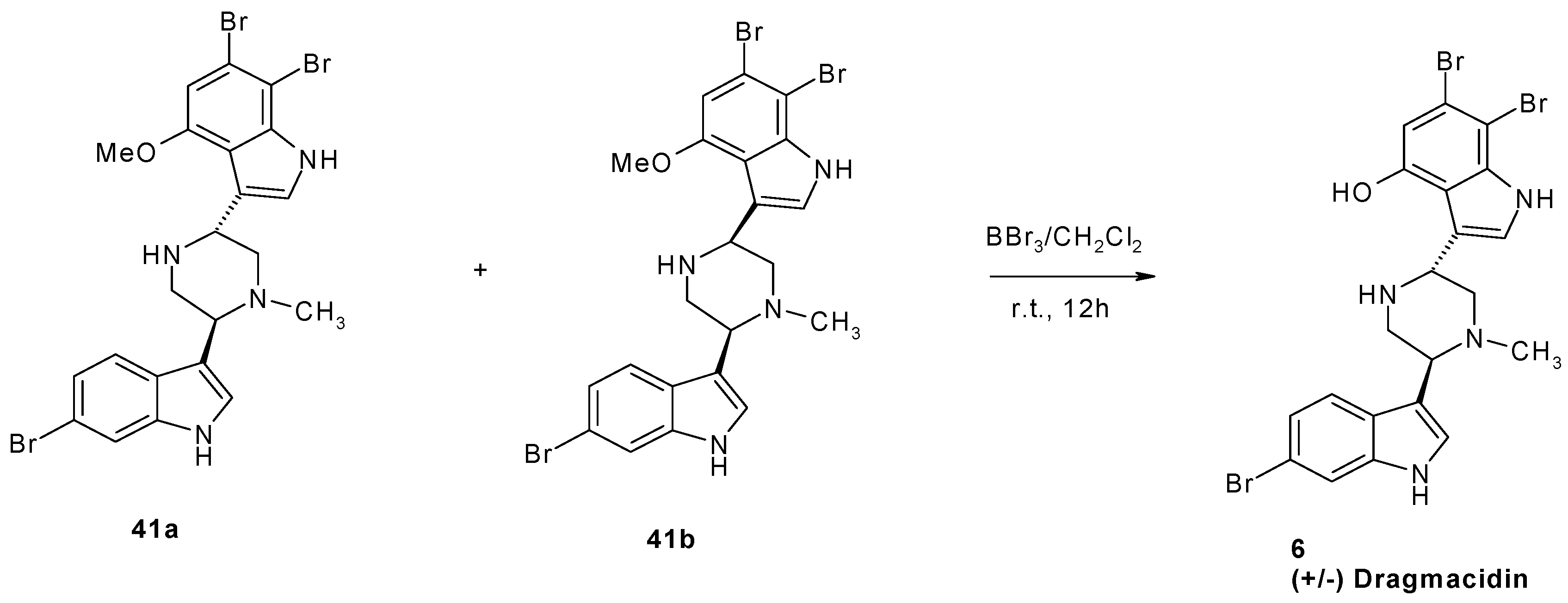
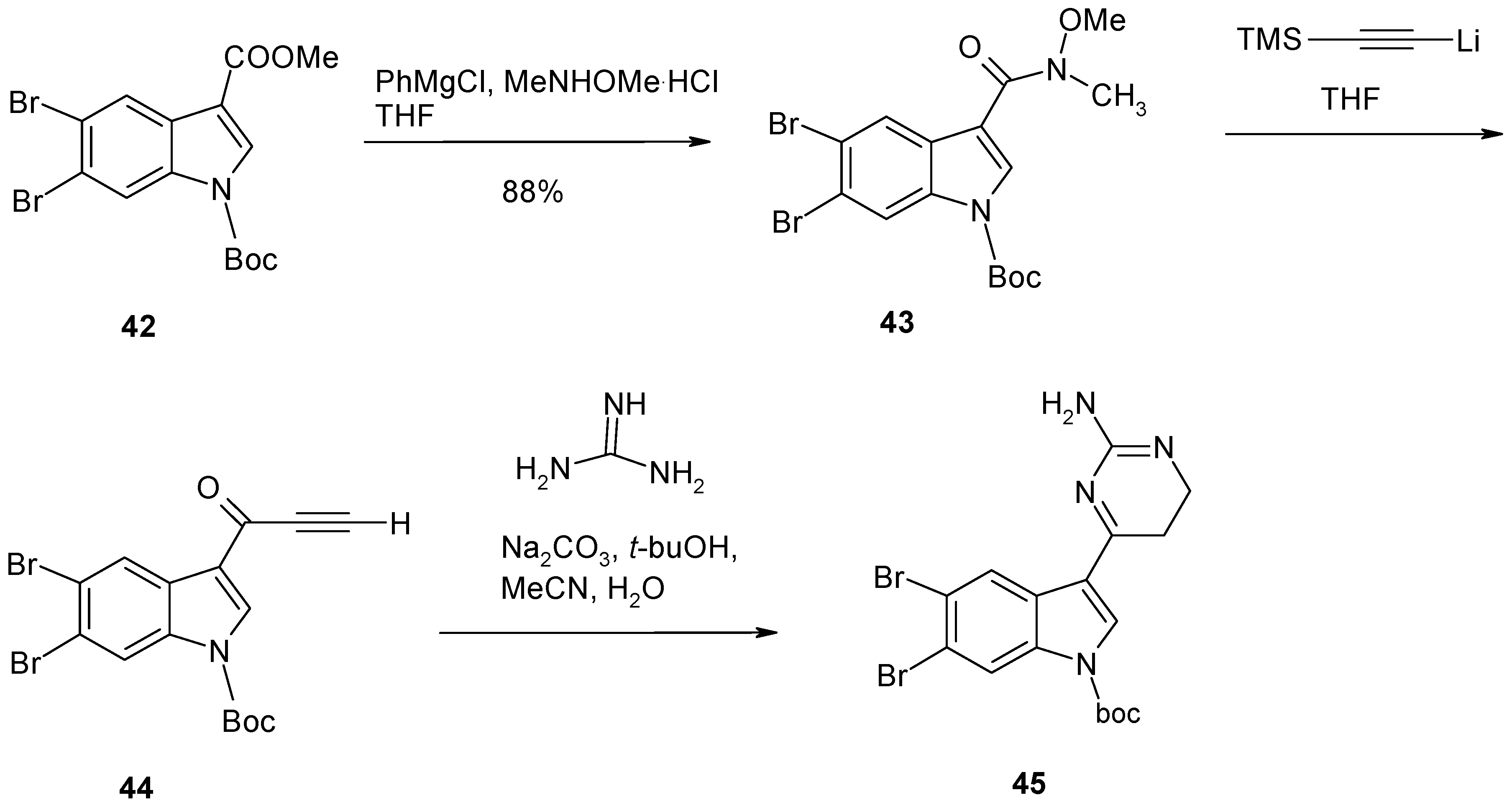
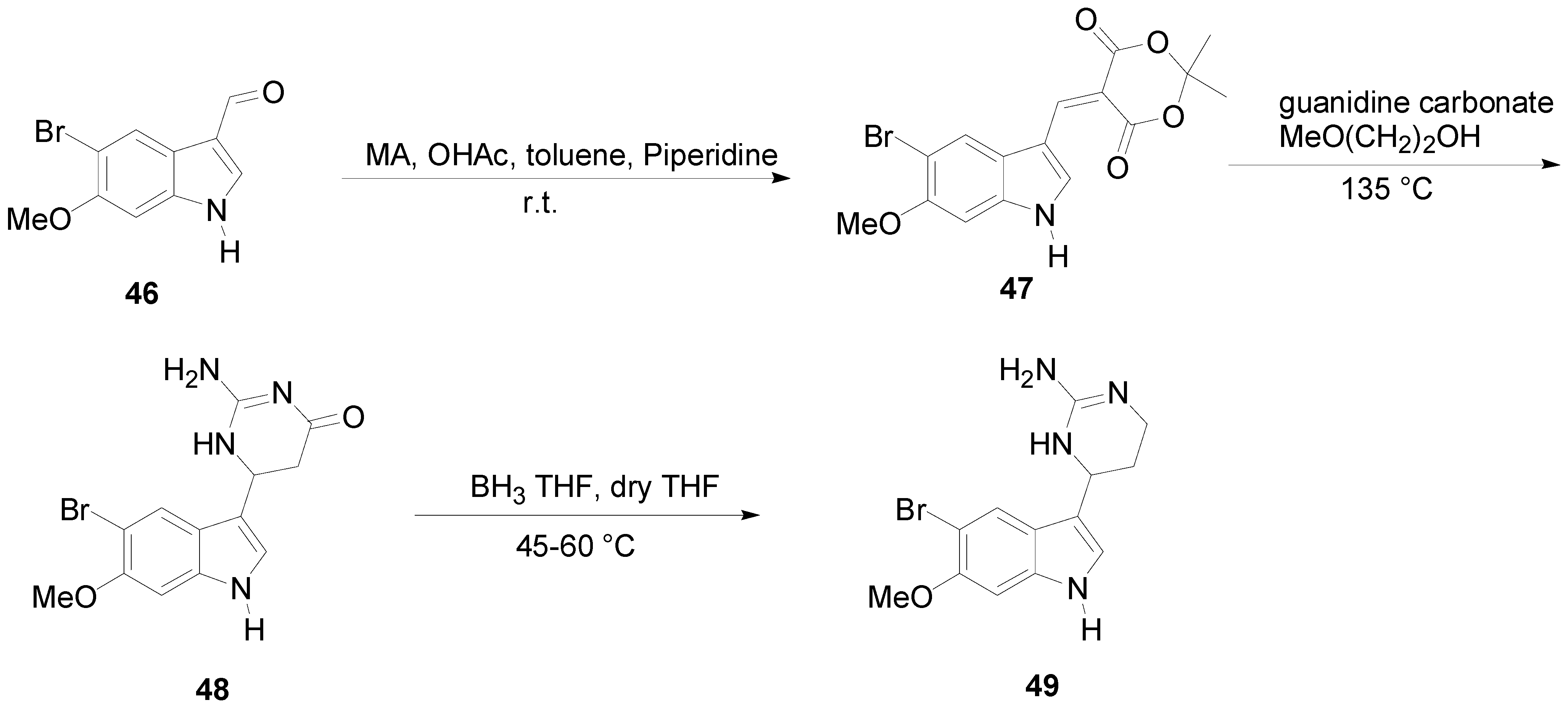
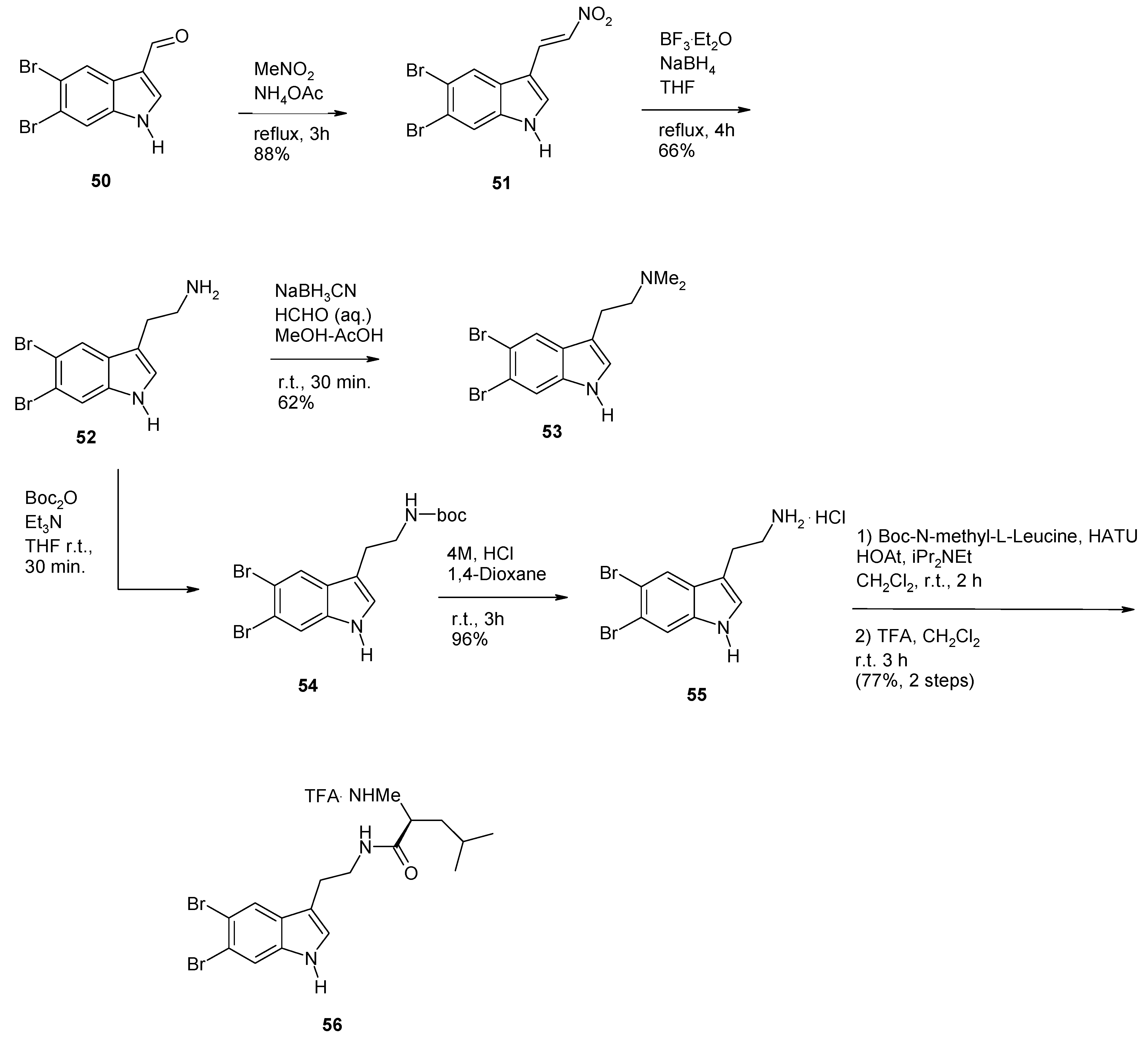
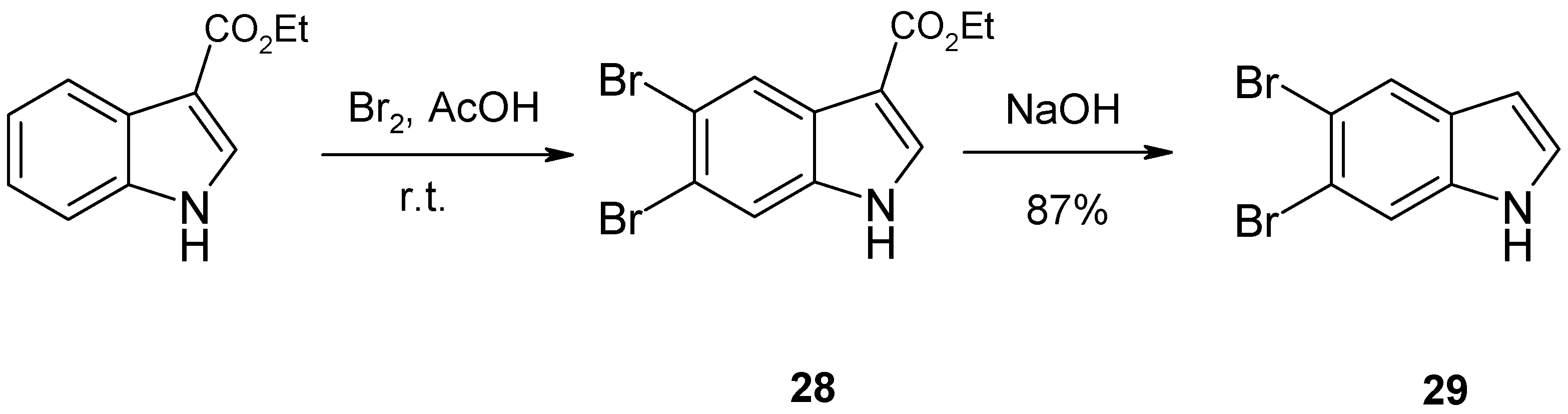
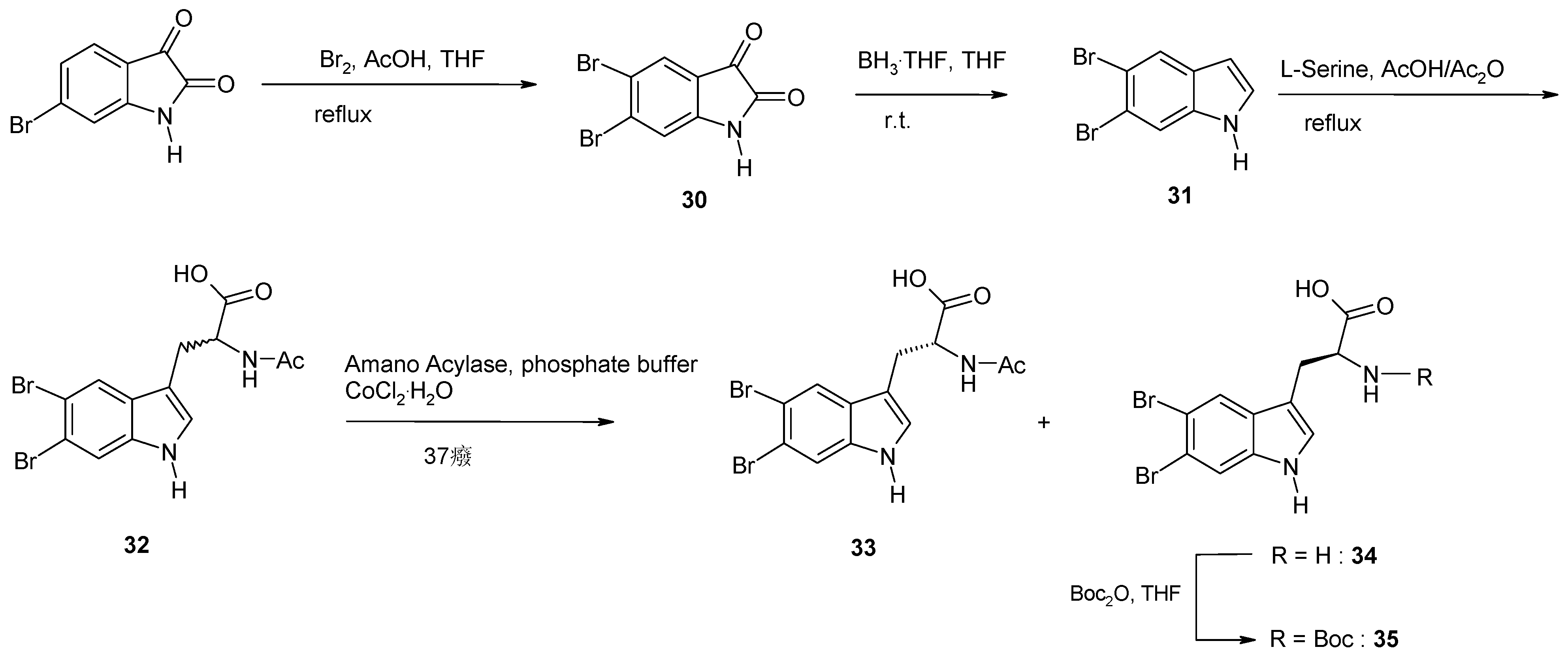
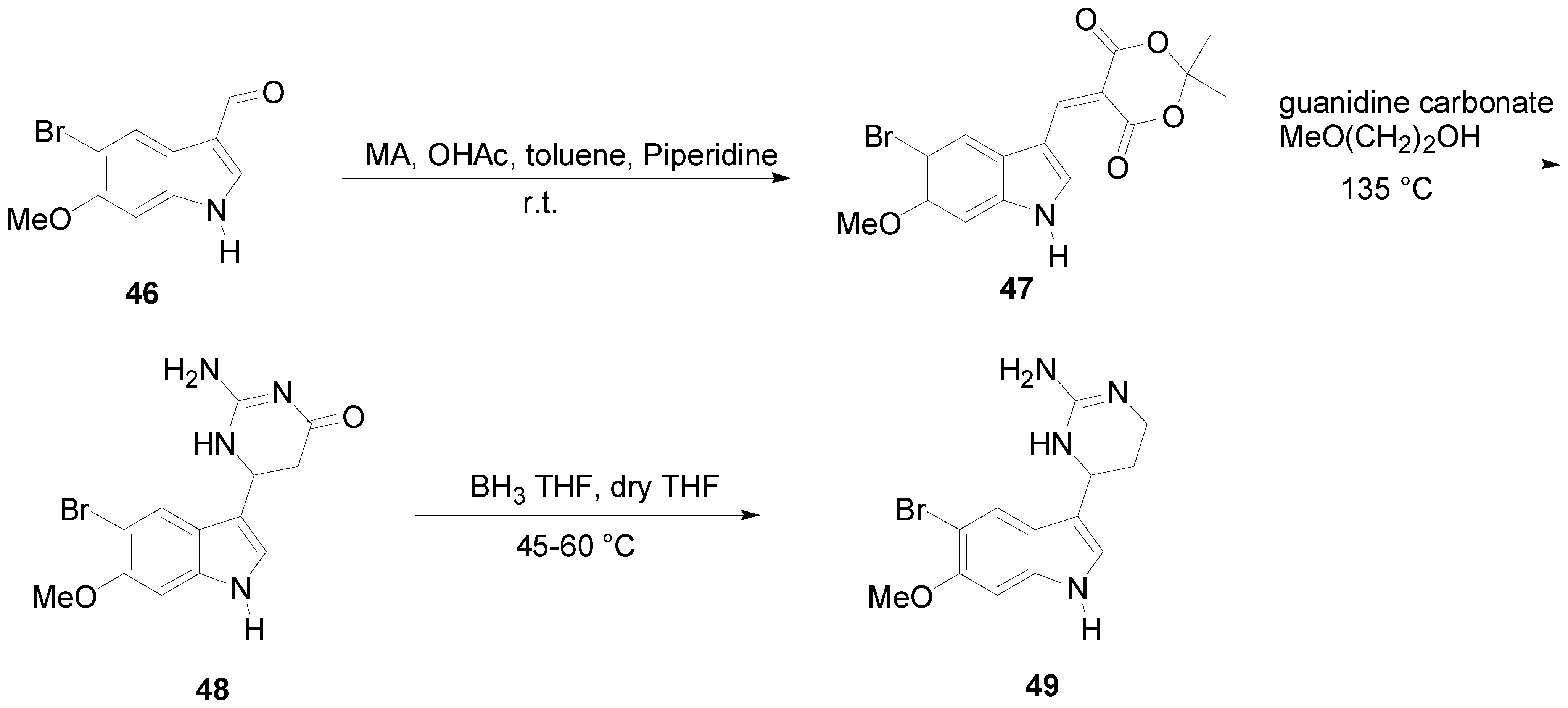
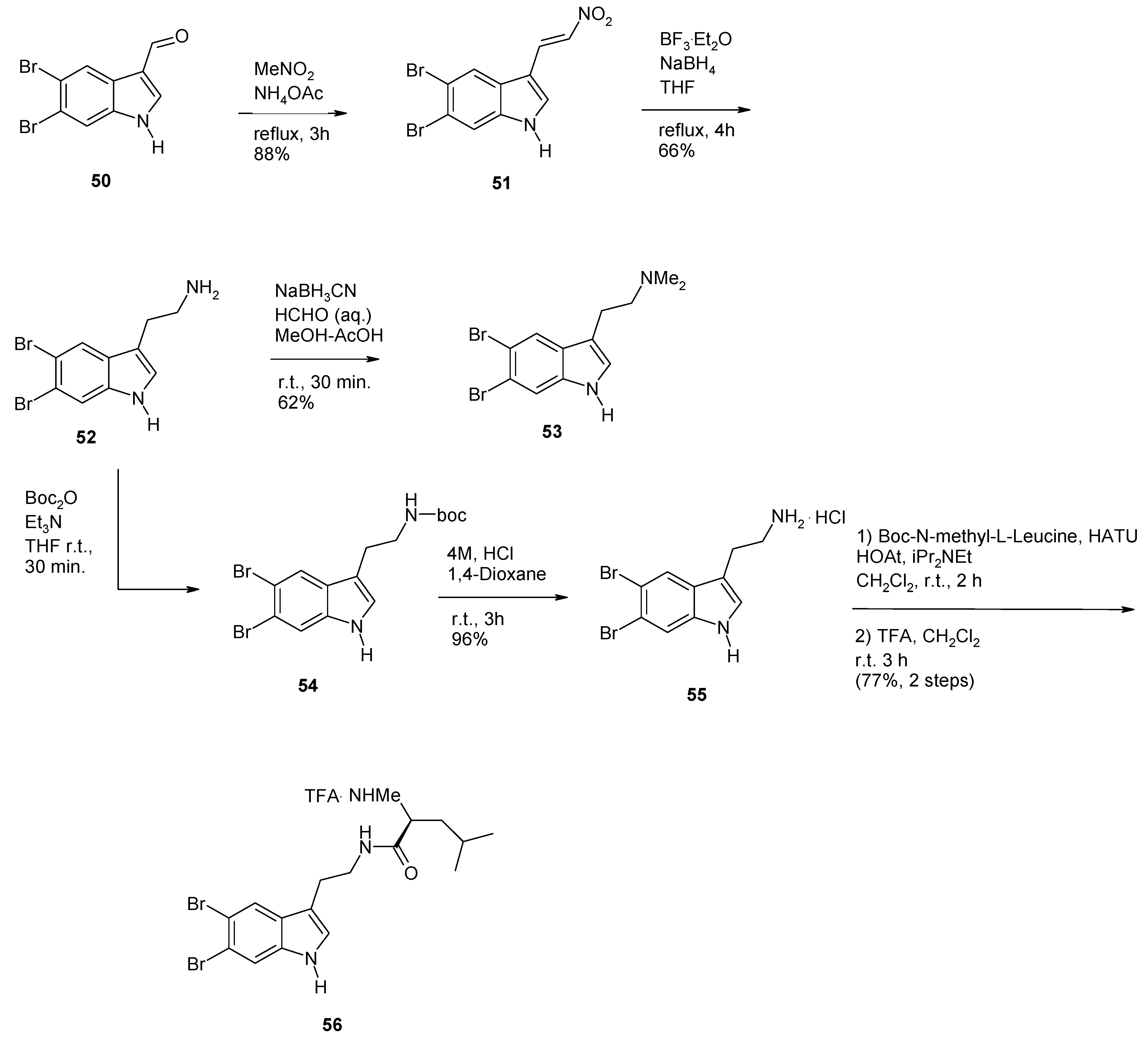
2. Synthesis of Dibrominated Indole-Containing Derivatives
3. Conclusions
References and Notes
- Locatelli, M.; Governatori, L.; Carlucci, G.; Genovese, S.; Mollica, A.; Epifano, F. Recent Application of Analytical Methods to Phase I and Phase II Drugs Development: A Review. Biomed. Chromatogr. 2012, 26, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Laport, M.S.; Santos, O.C.S.; Muricy, G. Marine sponges: Potential sources of new antimicrobial drugs. Curr. Pharm. Biotechnol. 2009, 10, 86–105. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2011, 28, 196–268. [Google Scholar] [CrossRef] [PubMed]
- Ebada, S.S.; Edrada, R.A.; Lin, W.; Proksch, P. Methods for isolation, purification and structural elucidation of bioactive secondary metabolites from marine invertebrates. Nat. Protoc. 2008, 3, 1820–1831. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Melucci, D.; Carlucci, G.; Locatelli, C. Recent HPLC strategies to improve sensitivity and selectivity for the analsis of complex matrices. Instrum. Sci. Technol. 2012, 40, 112–137. [Google Scholar] [CrossRef]
- Longeon, A.; Copp, B.R.; Quévrain, E.; Roué, M.; Kientz, B.; Cresteil, T.; Petek, S.; Debitus, C.; Bourguet-Kondracki, M.L. Bioactive Indole Derivatives from the South Pacific Marine Sponges Rhopaloeides odorabile and Hyrtios sp. Mar. Drugs 2011, 9, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, D.; Bugni, T.S.; Mangalindan, G.C.; Concepción, G.P.; Harper, M.K.; Ireland, C.M. New bromotryptophan derivatives from the Philippine marine sponge Smenospongia sp. Zeitschrift für Naturforschung Sec. C 2002, 57, 914–923. [Google Scholar] [CrossRef]
- Fattorusso, E.; Lanzotti, V.; Magno, S.; Novellino, E. Tryptophan Derivatives from a Mediterranean Anthozoan, Astroides calycularis. J. Nat. Prod. 1985, 48, 924–927. [Google Scholar] [CrossRef]
- Kochanowska, A.J.; Rao, K.V.; Childress, S.; Alfy, A.E.; Matsumoto, R.R.; Kelly, M.; Stewart, G.S.; Sufka, K.J.; Hamann, M.T. Secondary Metabolites from Three Florida Sponges with Antidepressant Activity. J. Nat. Prod. 2008, 71, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Smallheer, J.M.; Ly, C.A.; Wuonola, M.A. Total Synthesis of (+/–)-Dragmacidin: A Cytotoxic Bis(indole)alkaloid of Marine Origin. J. Org. Chem. 1994, 59, 6823–6827. [Google Scholar] [CrossRef]
- Bittner, S.; Scherzer, R.; Harlev, E. The five bromotryptophans. Amino Acids 2007, 33, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.R.; Carter, E.J.; Huff, B.C.; Morris, J.C. Variolins and Related Alkaloids. Chem. Rev. 2009, 109, 3080–3098. [Google Scholar] [CrossRef] [PubMed]
- Hernàndez Franco, L.; Bal de Kier Joffè, E.; Puricelli, L.; Tatian, M.; Seldes, A.M.; Palermo, J.A. Indole Alkaloids from the Tunicate Aplidium meridianum. J. Nat. Prod. 1998, 61, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Seldes, A.M.; Brasco, M.F.R.; Hernandez Franco, L.; Palermo, J.A. Identification of two meridianins from the crude extract of the tunicate Aplidium meridianum by tandem mass spectrometry. Nat. Prod. Res. 2007, 21, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Reyes, F.; Fernandez, R.; Rodriguez, A.; Francesch, A.; Taboada, S.; Avila, C.; Cuevas, C. Aplicyanins A–F, new cytotoxic bromoindole derivatives from the marine tunicate Aplidium cyaneum. Tetrahedron 2008, 64, 5119–5123. [Google Scholar] [CrossRef]
- Appleton, D.R.; Page, M.J.; Lambert, G.; Berridge, M.V.; Copp, B.R. Kottamides A-D: Novel Bioactive Imidazolone-Containing Alkaloids from the New Zealand Ascidian Pycnoclavella kottae. J. Org. Chem. 2002, 67, 5402–5404. [Google Scholar] [CrossRef] [PubMed]
- Aschi, M.; Lucente, G.; Mazza, F.; Mollica, A.; Morera, E.; Nalli, F.; Paglialunga Paradisi, M. Peptide backbone folding induced by the Cα-tetrasubstituted cyclic α-amino acids 4-amino-1,2-dithiolane-4-carboxylic acid (Adt) and 1-aminocyclopentane-1-carboxylic acid (Ac5c). A joint computational and experimental study. Org. Biomol. Chem. 2003, 1, 1980–1988. [Google Scholar] [CrossRef] [PubMed]
- Morera, E.; Nalli, M.; Mollica, A.; Paglialunga Paradisi, M.; Aschi, M.; Gavuzzo, E.; Mazza, F.; Lucente, G. Peptides containing 4-amino-1,2-dithiolane-4-carboxylic acid (adt): Conformation of Boc-Adt-Adt-NHMe and NH...S interactions. J. Pept. Sci. 2005, 11, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Tan, A.S. High-capacity redox control at the plasma membrane of mammalian cells: Transmembrane, Cell surface, and Serum NADH-oxidases. Antiox Redox Signal. Protoplasma 1998, 205, 74–82. [Google Scholar] [CrossRef]
- Tan, A.S.; Berridge, M.V. Superoxide produced by activated neutrophils efficiently reduces the tetrazolium salt, WST-1 to produce a soluble formazan: A simple colorimetric assay for measuring respiratory burst activation and for screening anti-inflammatory agents. J. Immunol. Methods 2000, 238, 59–68. [Google Scholar] [CrossRef]
- Majima, R.; Kotake, M. Synthetische Versuche in der Indol gruppe. VII. Uber Nitrieren und Bromieren des β-indol-carbonsaure-Esters und eine neue Synthese des Farbstoffs des antiken Purpurs. Ber. Dtsch. Chem. Ges. 1930, 63, 2237–2245. [Google Scholar] [CrossRef]
- Mollica, A.; Stefanucci, A.; Feliciani, F.; Lucente, G.; Pinnen, F. Synthesis of (S)-5,6-dibromo-tryptophan derivatives as building blocks for peptide chemistry. Tetrahedron Lett. 2011, 52, 2583–2585. [Google Scholar] [CrossRef]
- Vine, K.L.; Locke, J.M.; Ranson, M.; Pyne, S.G.; Bremner, J.B. In vitro cytotoxicity evaluation of some substituted isatin derivatives. Bioorg. Med. Chem. 2007, 15, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Tsintsadze, T.G.; Khoshtariya, T.E.; Kurkovskaya, L.N.; Mirziashvili, N.T.; Sikharulidze, M.I. Novel route for obtaining isomeric benzo[b]thiophenoindoles. Chem. Heterocycl. Compd. 2002, 38, 472–476. [Google Scholar] [CrossRef]
- Angelini, E.; Balsamini, C.; Bartoccini, F.; Lucarini, S.; Piersanti, G. Switchable Reactivity of Acylated α,β-Dehydroamino Ester in the Friedel-Crafts Alkylation of Indoles by Changing the Lewis Acid. J. Org. Chem. 2008, 73, 5654–5657. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.Y.; Bae, S.K.; Lee, W. Comparative studies between covalently immobilized and coated chiral stationary phases based on polysaccharide derivatives for enantiomer separation of N-protected α-amino acids and their ester derivatives. Chirality 2009, 21, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Gompel, M.; Leost, M.; Bal de Kier Joffè, E.; Puricelli, L.; Franco, L.H.; Palermo, J.; Meijer, L. Meridianins, a new family of protein kinase inhibitors isolated from the Ascidian Aplidium meridianum. Bioorg. Med. Chem. Lett. 2004, 14, 1703–1707. [Google Scholar] [CrossRef] [PubMed]
- Síša, M.; Pla, D.; Altuna, M.; Francesch, A.; Cuevas, C.; Albericio, F.; Alvarez, M. Total Synthesis and Antiproliferative Activity Screening of (+/−)-Aplicyanins A, B and E and Related Analogues. J. Med. Chem. 2009, 52, 6217–6223. [Google Scholar] [CrossRef] [PubMed]
- Ostras, K.S.; Gorobets, N.Y.; Desenko, S.M.; Musatov, V.I. An easy access to 2-amino-5,6-dihydro-3H-pyrimidin-4-one building blocks: The reaction under conventional and microwave conditions. Mol. Divers. 2006, 10, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Benzies, D.W.M.; Martínez-Fresneda, P.; Jones, R.A.; McNab, H. Flash-vacuum pyrolysis of 5-(indol-2-and -3-ylmethylene)-2,2-dimethyl-1,3-dioxane-4,6-diones. J. Chem. Soc. Perkin Trans. 1 1986, 1, 1651–1654. [Google Scholar] [CrossRef]
- Agami, C.; Dechoux, L.; Melaimi, M. Chemoselective reduction of pyrimidines. An access to enantiopure tetrahydropyrimidinones. Tetrahedron Lett. 2001, 42, 8629–8631. [Google Scholar] [CrossRef]
- Sperry, J. Concise syntheses of 5,6-dibromotryptamine and 5,6-dibromo-N,N-dimethyltryptamine en route to the antibiotic alternatamide D. Tetrahedron Lett. 2011, 52, 4042–4044. [Google Scholar] [CrossRef]
- Bernauer, K.; Mahboobi, S. Synthesis of Esters of 3-(2-Aminoethyl)-1H-indole-2-acetic Acid and-(2-Aminoethyl)-1H-indole-2-malonic Acid (2-(3-(2-Aminoethyl)1H-indol-2-l)Propanedioic acid). Helv. Chim. Acta 1988, 71, 2034–2041. [Google Scholar]
- Varma, R.S.; Kabalka, G.W. A Simple Route to Alkylaminea. Via The Reduction of Nitroalkenes. Synth. Commun. 1985, 15, 843–847. [Google Scholar] [CrossRef]
- Djura, P.; Stierle, D.B.; Sullivan, B.; Faulkner, D.J.; Arnold, E.; Clardy, J. Some Metabolites of the Marine Sponges Smenospongia aurea and Smenospongia (Polyfibrospongia) echina. J. Org. Chem. 1980, 45, 1435–1441. [Google Scholar] [CrossRef]
- Fink, B.E.; Chen, L.; Chen, P.; Dodd, D.S.; Gaval, A.V.; Kim, S.H.; Vaccaro, W.; Zhang, L.H. US Patent 20100323994, 2009.
- Han, S.Y.; Kim, Y.A. Recent development of peptide coupling reagents in organic synthesis. Tetrahedron 2004, 60, 2447–2467. [Google Scholar] [CrossRef]
- Gordaliza, M. Natural products as leads to anticancer drugs. Clin. Transl. Oncol. 2007, 9, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.K.; Basu, S.; Sarkat, N.; Gosh, A.C. Advances in cancer therapy with plant based natural products. Curr. Med. Chem. 2001, 8, 1467–1486. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H. Novel antitumor agents from higher plants. Med. Res. Rev. 1999, 310, 569–596. [Google Scholar] [CrossRef]
- Torino, D.; Mollica, A.; Pinnen, F.; Lucente, G.; Feliciani, F.; Davis, P.; Lai, J.; Ma, S.W.; Porreca, F.; Hruby, J.V. Synthesis and evaluation of new endomorphin analogues modified at the Pro2 residue. Bioorg. Med. Chem. Lett. 2009, 19, 4115–4118. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Davis, P.; Ma, S.-W.; Lai, J.; Porreca, F.; Hruby, V.J. Synthesis and biological evaluation of new biphalin analogues with non-hydrazine linkers. Bioorg. Med. Chem. Lett. 2005, 15, 2471–2475. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Stefanucci, A.; Feliciani, F.; Cacciatore, I.; Cornacchia, C.; Pinnen, F. Delivery methods of Camptothecin and its hydrosoluble analogue Irinotecan for treatment of colorectal cancer. Curr. Drug Del. 2012, 9, 122–131. [Google Scholar] [CrossRef]
- Mollica, A.; Paglialunga Paradisi, M.; Varani, K.; Spisani, S.; Lucente, G. Chemotactic peptides: fMLF-OMe analogues incorporating proline-methionine chimeras as N-terminal residue. Bioorg. Med. Chem. 2006, 14, 2253–2265. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Paglialunga Paradisi, M.; Torino, D.; Spisani, S.; Lucente, G. Hybrid α/β-peptides: For-Met-Pleu-Phe-OMe analogues containing geminally disubstituted β2,2- and β3,3-amino acids at the central position. Amino Acids 2006, 30, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Davis, P.; Ma, S.W.; Porreca, F.; Lai, J.; Hruby, V.J. Synthesis and biological activity of the first cyclic biphalin analogues. Bioorg. Med. Chem. Lett. 2006, 16, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Torino, D.; Mollica, A.; Pinnen, F.; Feliciani, F.; Spisani, S.; Lucente, G. Novel chemotactic For-Met-Leu-Phe-OMe (fMLF-OMe) analogues based on Met residue replacement by 4-amino-proline scaffold: Synthesis and bioactivity. Bioorg. Med. Chem. 2009, 17, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Lucente, G.; Mollica, A.; Nalli, M.; Zecchini Pagani, G.; Paglialunga Paradisi, M.; Gavuzzo, E.; Mazza, F.; Spisani, S. Hybrid α/β3-peptides with proteinogenic side chains. Monosubstituted analogues of the chemotactic tripeptide For-Met-Leu-Phe-OME. J. Pept. Sci. 2004, 10, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef] [PubMed]
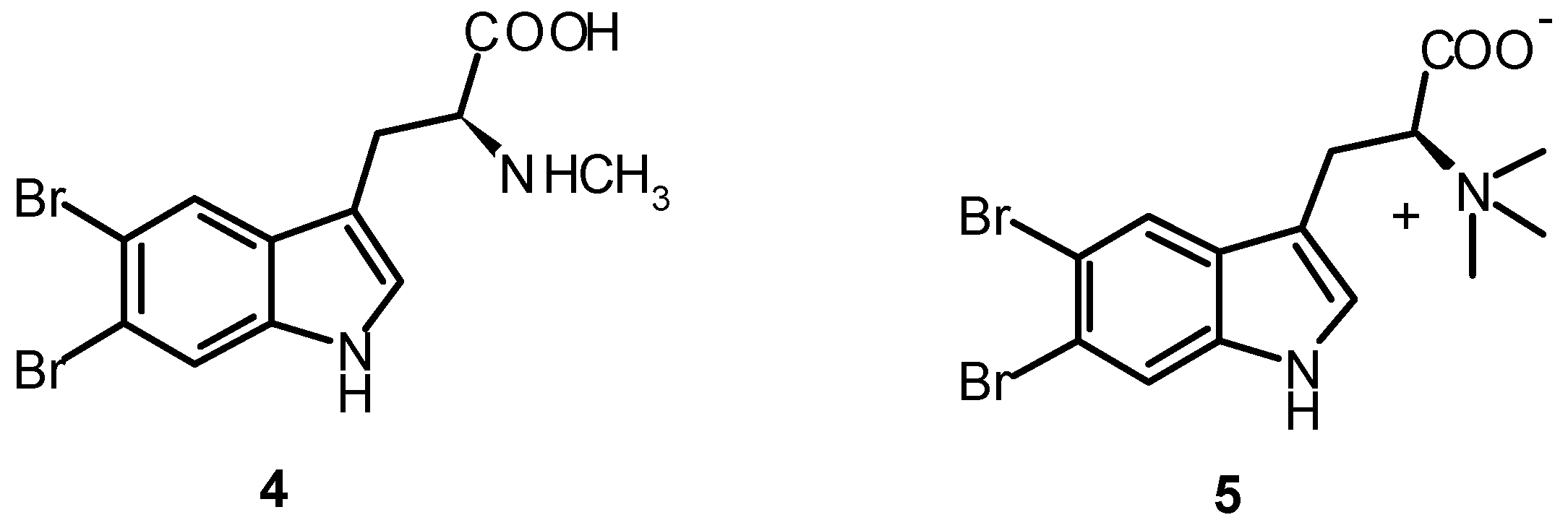


{kind=link}
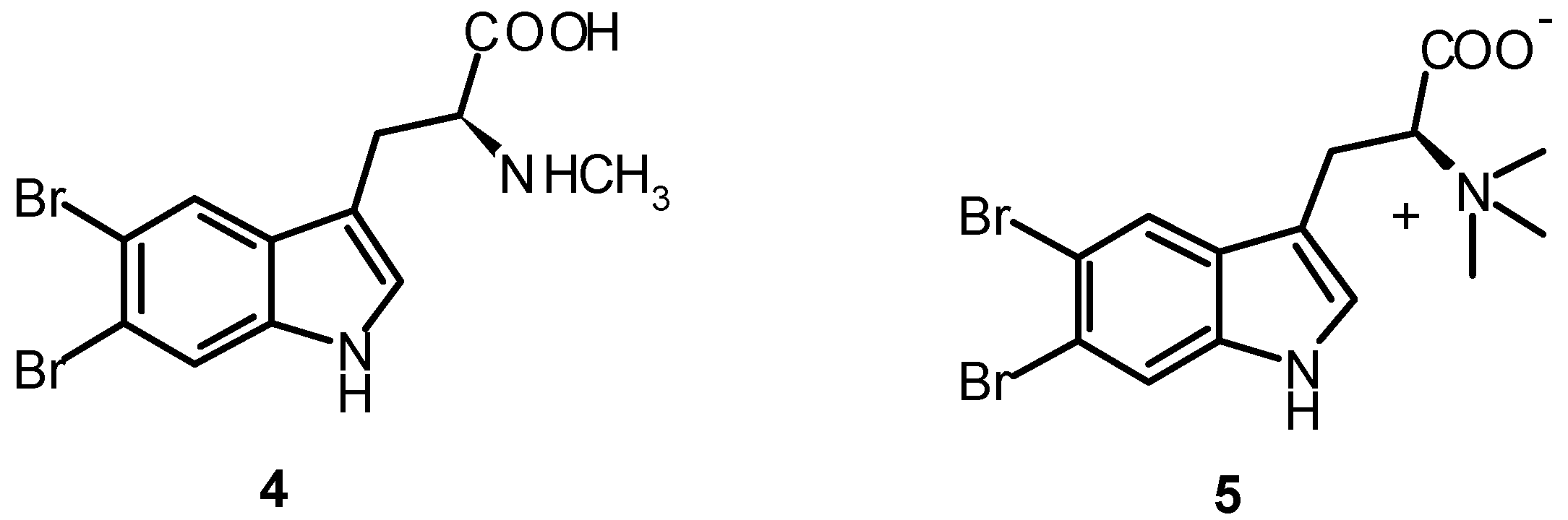{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | PLA2 a | ORAC FL b |
|---|---|---|
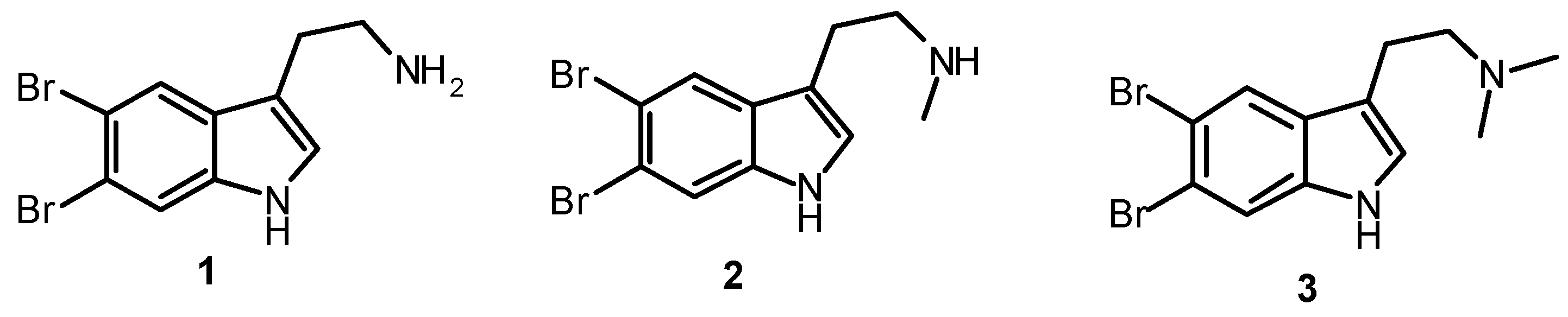
| 5,6-Dibromotryptamine (1) | 0.62 ± 0.01 | nt |
| N-Methyl-5,6-dibromotryptamine (2) | 0.33 ± 0.03 | nt |
| N,N-Dimethyl-5,6-dibromotryptamine (3) | 0.77 ± 0.05 | 0.06 ± 0.01 |
| 5,6-Dibromoabrine (4) | 0.30 ± 0.01 | 0.07 ± 0.01 |
| 5,6-Dibromo-L-hypaphorine (5) | 0.20 ± 0.01 | 0.22 ± 0.04 |
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mollica, A.; Locatelli, M.; Stefanucci, A.; Pinnen, F. Synthesis and Bioactivity of Secondary Metabolites from Marine Sponges Containing Dibrominated Indolic Systems. Molecules 2012, 17, 6083-6099. https://doi.org/10.3390/molecules17056083
Mollica A, Locatelli M, Stefanucci A, Pinnen F. Synthesis and Bioactivity of Secondary Metabolites from Marine Sponges Containing Dibrominated Indolic Systems. Molecules. 2012; 17(5):6083-6099. https://doi.org/10.3390/molecules17056083
Chicago/Turabian StyleMollica, Adriano, Marcello Locatelli, Azzurra Stefanucci, and Francesco Pinnen. 2012. "Synthesis and Bioactivity of Secondary Metabolites from Marine Sponges Containing Dibrominated Indolic Systems" Molecules 17, no. 5: 6083-6099. https://doi.org/10.3390/molecules17056083
APA StyleMollica, A., Locatelli, M., Stefanucci, A., & Pinnen, F. (2012). Synthesis and Bioactivity of Secondary Metabolites from Marine Sponges Containing Dibrominated Indolic Systems. Molecules, 17(5), 6083-6099. https://doi.org/10.3390/molecules17056083