Synthesis of a New Group of Aliphatic Hydrazide Derivatives and the Correlations between Their Molecular Structure and Biological Activity

Abstract
:1. Introduction
2. Results and Discussion
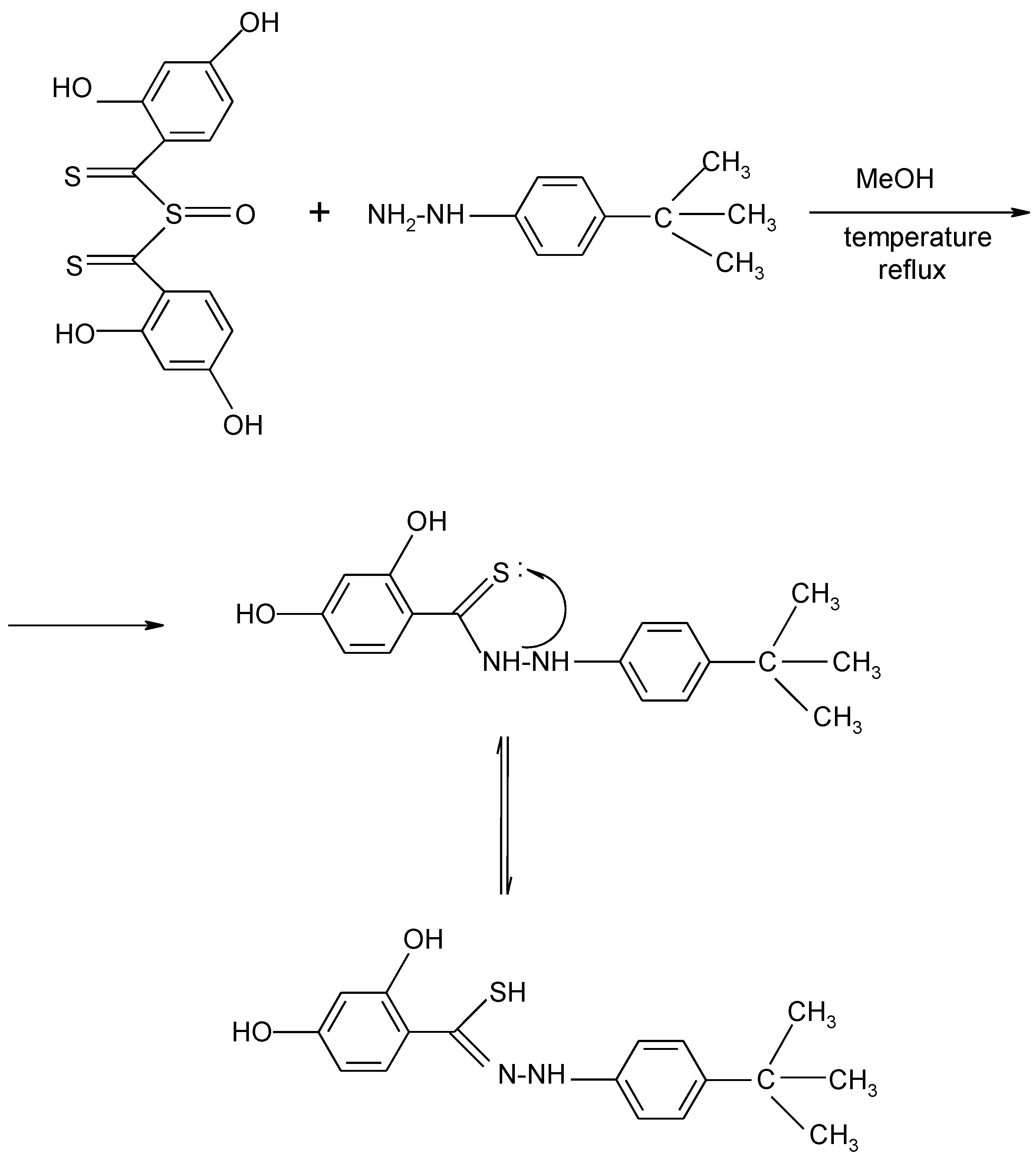

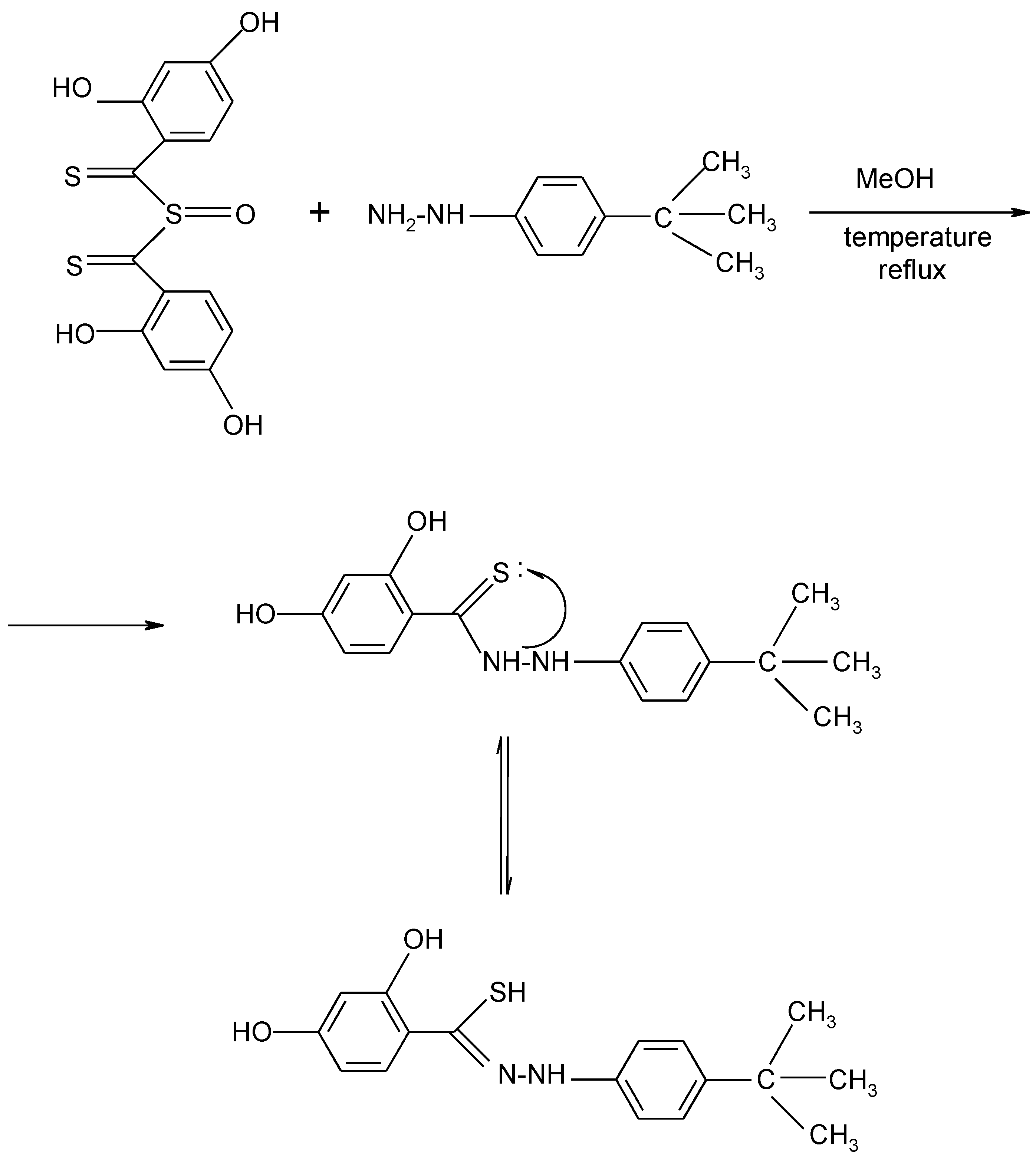
{kind=link}
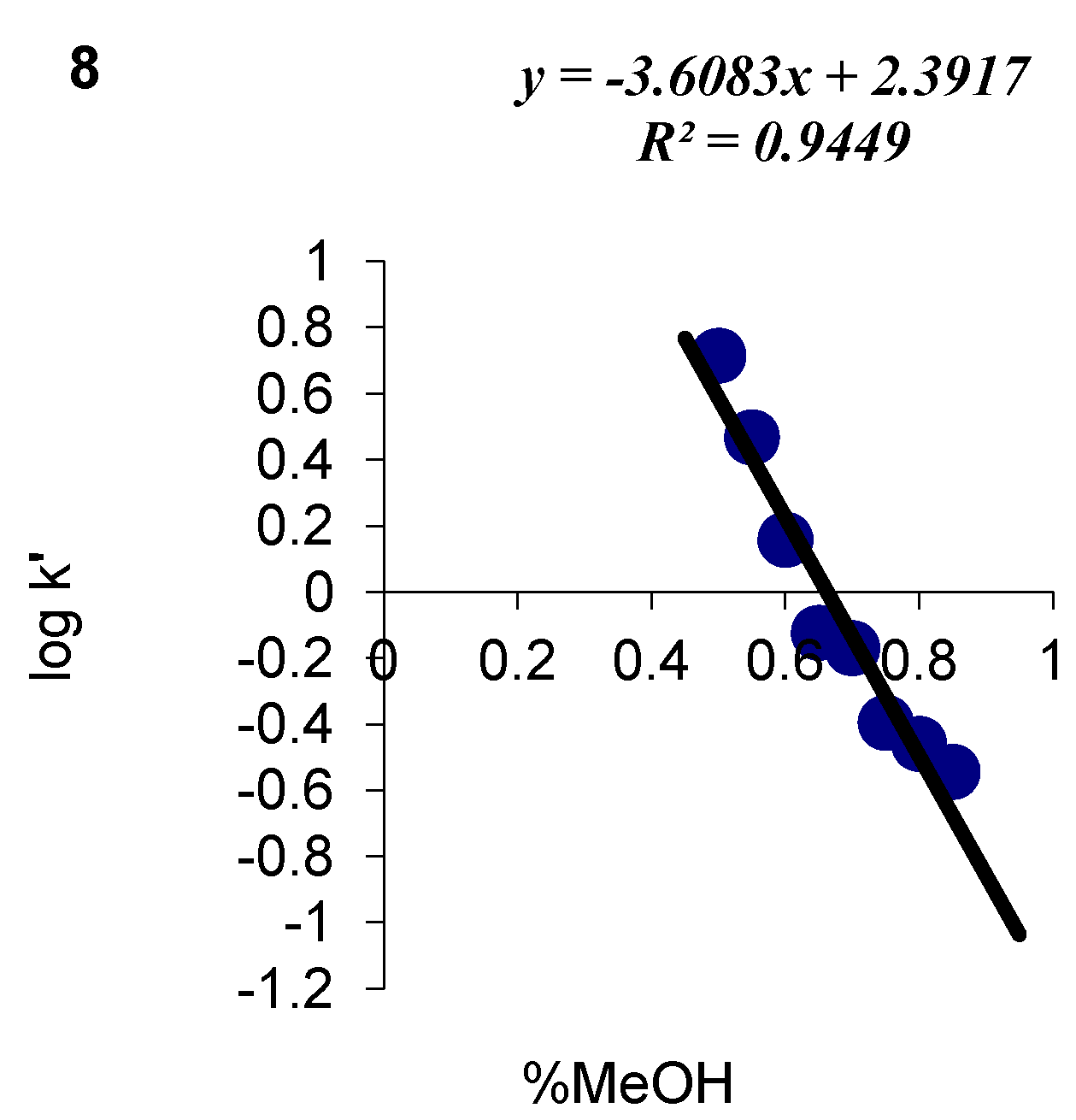{kind=link}
{kind=link}
| No | Molecular Formula | Yield [%] | Composition [%] calculated | Composition [%] found | Formula Weight | Molar Volume | m.p. °C |
|---|---|---|---|---|---|---|---|
| 1 | C9H9F3N2O2S | 86 | C (40.60) H (3.41) N (10.52) | C (40.5) H (3.42) | 266.24 | 176.2 ± 3.0 cm3 | 115–117 |
| 2 | C9H12N2O2S | 73 | C (50.92) H (5.70) N (13.20) | N (13.15) | 212.26 | 159.8 ± 3.0 cm3 | 171–173 |
| 3 | C14H11N3O2S | 97 | C (58.93) H (3.89) N (14.73) | -- | 285.32 | 191.1 ± 5.0 cm3 | 175 |
| 4 | C13H11N3O4S | 84 | C (51.14) H (3.63) N (13.76) | C (51.1) H (3.61) | 305.30 | 193.6 ± 3.0 cm3 | 166–167 |
| 5 | C13H10F2N2O2S | 79 | C (52.70) H (3.40) N (9.45) | N (9.48) | 296.29 | 190.2 ± 3.0 cm3 | 187–189 |
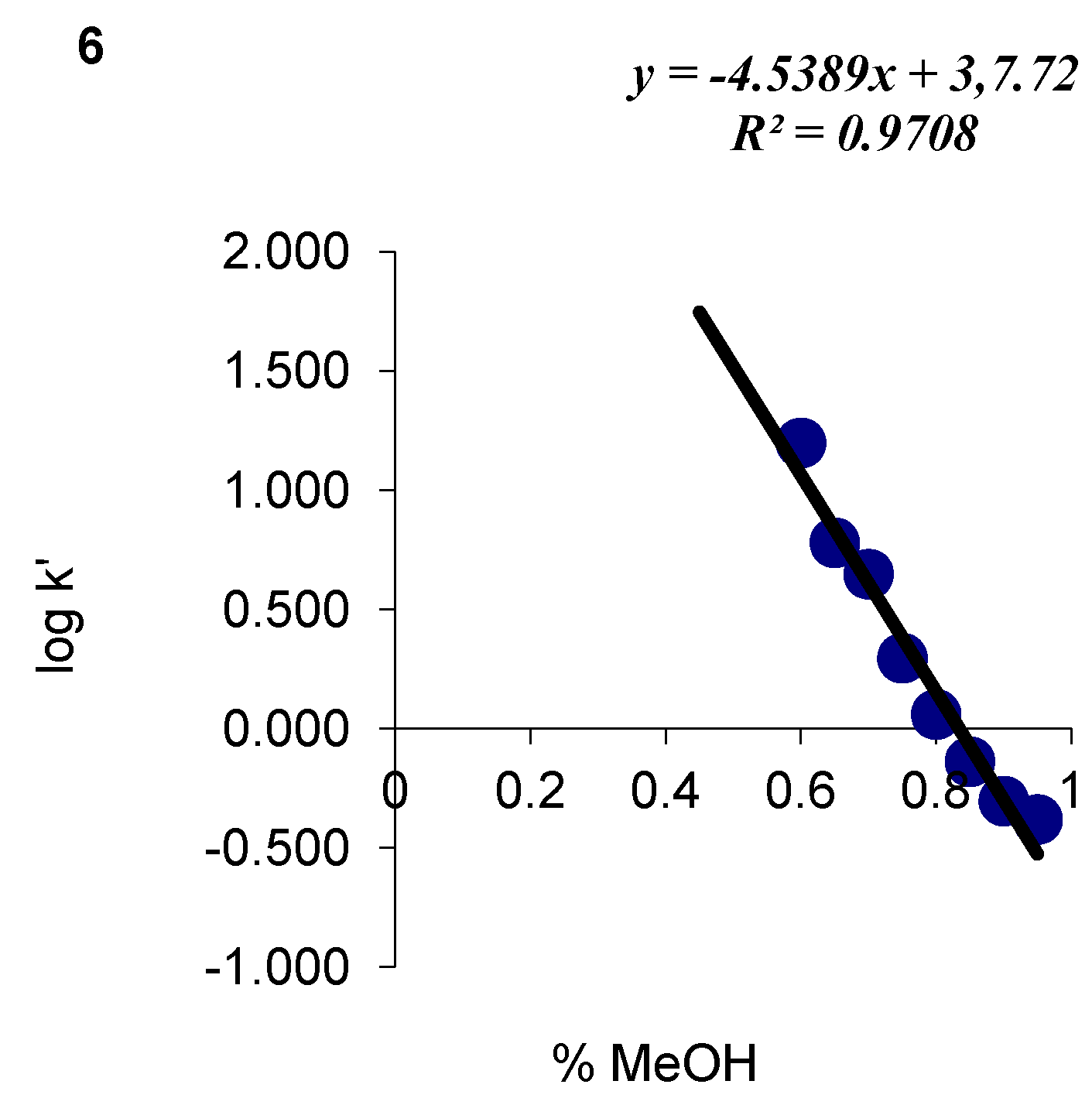
| 6 | C17H20N2O2S | 77 | C (64.53) H (6.37) N (8.85) | C (64.45) H (6.32) N(10.03) | 316.41 | 248.5 ± 3.0 cm3 | 104–107 |
| 7 | C15H16N2O2S | 75 | C (62.48) H (5.59) N (9.71) | N (9.68) | 288.36 | 214.3 ± 3.0 cm3 | 121–124 |
| 8 | C14H13ClN2O2S | 84 | C (54.46) H (4.24) N (9.07) | -- | 308.78 | 210.0 ± 3.0 cm3 | 110–112 |
| 9 | C13H12N2O2S | 98 | C (59.98) H (4.65) N (10.76) | -- | 260.31 | 181.8 ± 3.0 cm3 | 137–139 |
| 10 | C13H11FN2O2S | 82 | C (56.10) H (3.98) N (10.07) | C (56.15) H (4.0) N (10.03) | 278.30 | 186.0 ± 3.0 cm3 | 141–142 |
| No | Structure | Log kw | S |
|---|---|---|---|
| 1 |  | 3.05 | 3.17 |
| 2 | 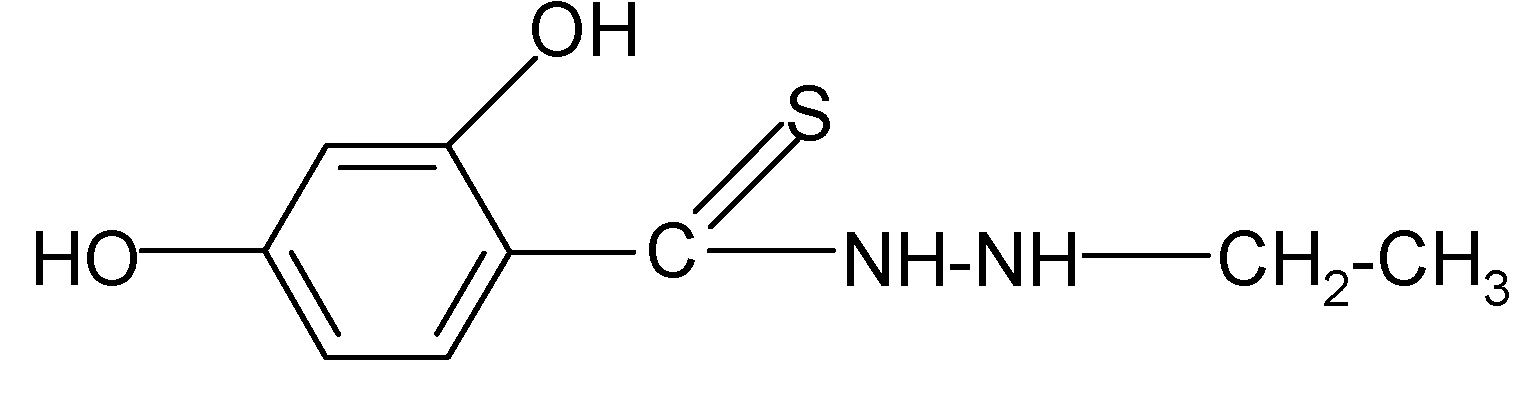 | 2.13 | 3.88 |
| 3 |  | 3.56 | 3.98 |
| 4 |  | 2.57 | 3.68 |
| 5 |  | 3.05 | 3.09 |
| 6 |  | 3.78 | 4.54 |
| 7 |  | 3.05 | 3.78 |
| 8 |  | 2.39 | 3.61 |
| 9 |  | 2.22 | 3.91 |
| 10 |  | 3.47 | 4.02 |
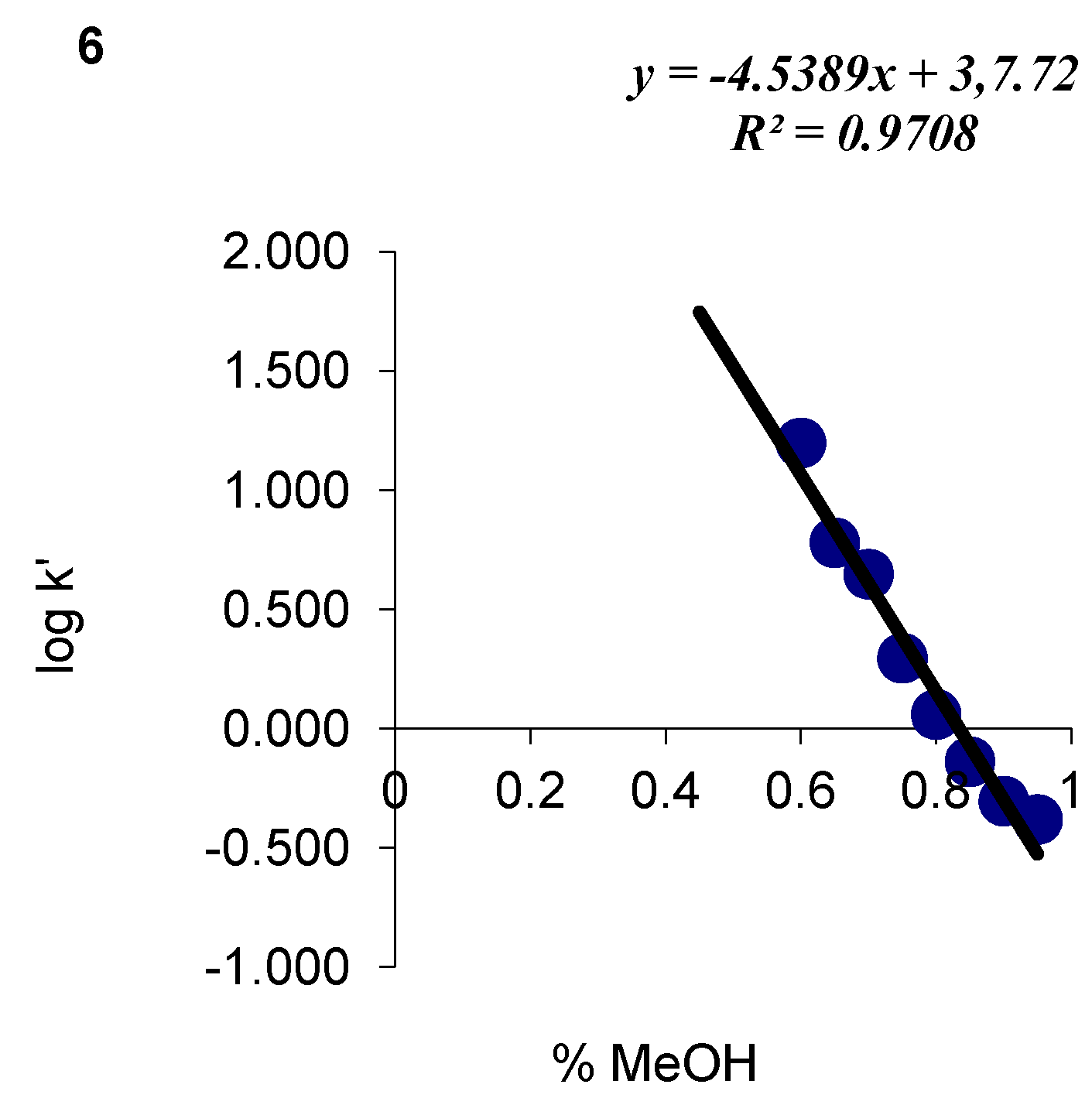
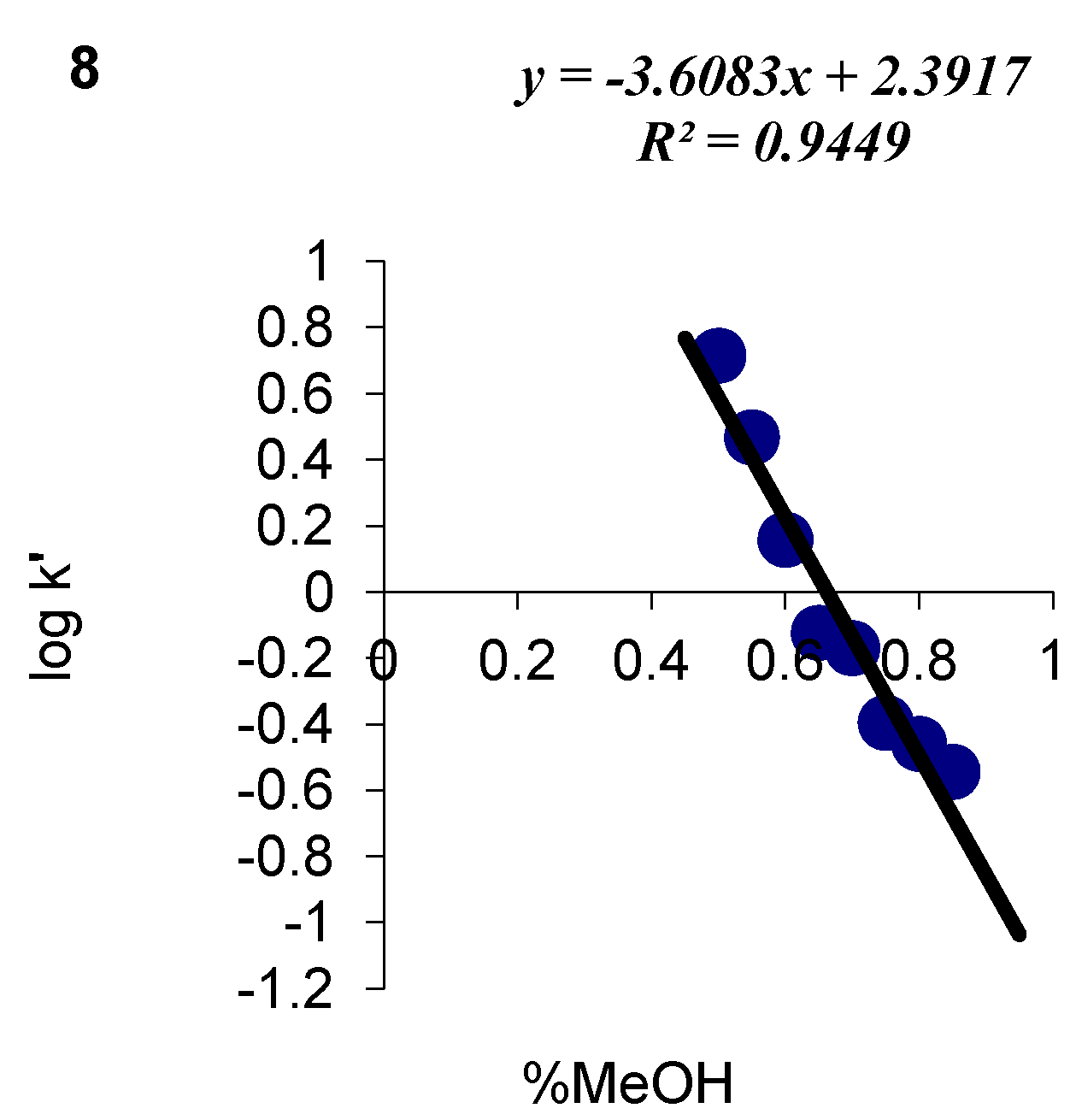
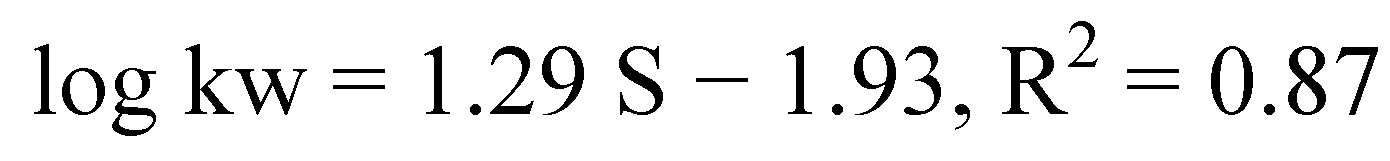
| Compound number | Fusarium solani | Fusarium oxysporum | ||
|---|---|---|---|---|
| 200 μg/mL | 100 μg/mL | 50 μg/mL | 200 μg/mL | |
| 1 | 3 | 3 | 3 | 3 |
| 2 | 3 | 2 | 1 | 1 |
| 3 | 3 | 3 | 2 | 3 |
| 4 | 3 | 3 | 1 | 1 |
| 5 | 3 | 2 | 2 | 3 |
| 6 | 3 | 3 | 3 | 3 |
| 7 | 3 | 3 | 2 | 3 |
| 8 | 3 | 3 | 2 | 1 |
| 9 | 2 | 1 | 0 | 1 |
| 10 | 3 | 3 | 3 | 1 |
| Azoxystrobin | 3 | 3 | 3 | 3 |
| Compound number | Aspergillus fumigatus | |||
|---|---|---|---|---|
| 200 μg/mL | 100 μg/mL | 50 μg/mL | 25 μg/mL | |
| 1 | 3 | 3 | 2 | 2 |
| 2 | 3 | 3 | 3 | 3 |
| 3 | 2 | 2 | 2 | 0 |
| 4 | 2 | 1 | 1 | 0 |
| 5 | 3 | 1 | 1 | 1 |
| 6 | 3 | 3 | 3 | 3 |
| 7 | 3 | 2 | 2 | 2 |
| 8 | 2 | 2 | 0 | 0 |
| 9 | 1 | 1 | 0 | 0 |
| 10 | 3 | 3 | 3 | 3 |
| Azoxystrobin | 3 | 3 | 2 | 2 |
3. Experimental
3.1. General
3.2. Synthesis
3.3. Chromatography (HPLC)
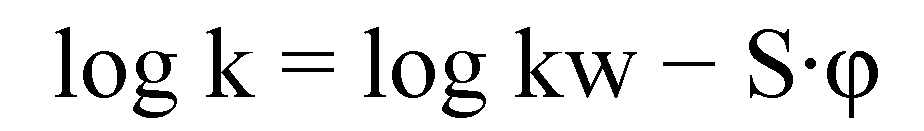
3.4. Biological Assays
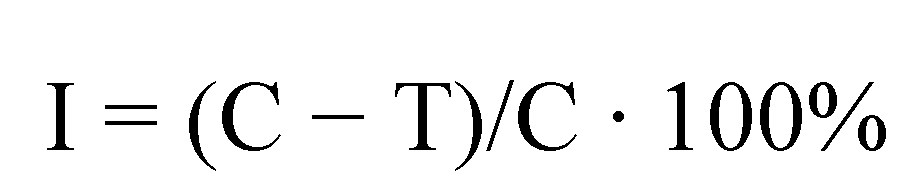
| Effective inhibition of linear growth in % | Evaluation of fungicidal activity | |
|---|---|---|
| Degree on scale | Activity | |
| Above 80 | 3 | satisfactory |
| 50–79.9 | 2 | average |
| 20–49.9 | 1 | weak |
| Below 20 | 0 | no activity |
4. Conclusions
Acknowledgements
- Samples Availability: Samples of the compound 4 are available from the authors.
References
- Danish, I.A.; Prasad, K.J. Syntheses and characterization of N,N'-biscarbazolyl azine and N,N'- hydrazine derivatives and their antimicrobial studies. Acta Pharm. 2004, 54, 133–142. [Google Scholar]
- Suthakaran, R.; Kavimani, S.; Venkappayya, D.; Jayasree, P.; Deepthi, V.; Tehseen, F.; Suganthi, K. Microwave assisted rapid synthesis and anti-microbial activity of 4-oxothiazolidine derivatives. Rasayan J. Chem. 2008, 1, 30–38. [Google Scholar]
- Juszczak, M.; Matysiak, J.; Brzana, W.; Niewiadomy, A.; Rzeski, W. Evaluation of the antiproliferative activity of 2-(monohalogenophenyloamino)-5-(2,4-dihydroxyphenyl)-1,3,4-thiadiazoles. Arzneimittelforschung 2008, 58, 353–357. [Google Scholar]
- Niewiadomy, A.; Krajewska-Kułak, E.; Łukaszuk, C.; Matysiak, J.; Kostecka, M. In vitro antifungal activity of N-3-(1,2,4-dithiazole-5-thione)-beta-resorcylcarbothioamide. Rocz. Akad. Med. Białymst. 2005, 50, 31–35. [Google Scholar]
- Bredihhin, A.; Groth, U.M.; Mäeorg, U. Formation and use of a nitrogen dianion for selective hydrazine alkylation provides a fast and easy access to substituted hydrazines, which are widely used as drugs, pesticides, and precursors for a variety of compounds in organic synthesis. Org. Lett. 2007, 9, 1097–1099. [Google Scholar]
- Tšubrik, O.; Sillard, R.; Mäeorg, U. Excellent regioselectivity is observed in the addition of diverse organometallic nucleophiles to unsymmetrical azo compounds. Primary/secondary/tertiary alkyl, aryl and heteroaryl substituents were introduced this way in high yields. Synthesis 2006, 843–846. [Google Scholar]
- Bredihhin, A.; Mäeorg, U. . Alkylation of hydrazine using a polyanion strategy provides a fast and convenient access to multialkylated hydrazine derivatives. Scope and limitations of the new method are also investigated. Org. Lett. 2007, 9, 4975–4977. [Google Scholar] [CrossRef]
- Manivel, P.; Mohana Roopan, S.; Sathish Kumar, R.; Nawazkhan, F. Synthesis of 3 substituted isoquinolin-1-yl-2-(cycloalk-2-enyldiene) hydrazines and their antimicrobial properties. J. Chil. Chem. Soc. 2009, 54, 183–185. [Google Scholar]
- Sherine, N.K. Synthesis and biological activity of novel amino acid-(N'-Benzoyl) hydrazide and amino Acid-(N'-nicotinoyl)hydrazide derivatives. Molecules 2005, 10, 1218–1228. [Google Scholar] [CrossRef]
- Sechi, M.; Azeena, U.; Delussu, M.P.; Dallochio, R.; Dessi, A.; Cosseddu, A.; Pala, N.; Neamati, N. Design and synthesis of bis-amide and hydrazide containing derivatives of malonic acid as potential HIV-1 integrase inhibitors. Molecules 2008, 13, 2442–2461. [Google Scholar] [CrossRef]
- Mohareb, R.M.; Mohamed, A.A. The reaction of cyanoacetylhydrazine with ω-bromo-(4-methyl)acetophenone: Synthesis of heterocyclic derivatives with antitumor activity. Molecules 2010, 15, 3602–3617. [Google Scholar] [CrossRef]
- Al-Abdullah, E.S. Synthesis and anticancer activity of some novel tetralin-6-yl-pyrazoline, 2-thioxopyrimidine, 2-oxopyridine,2-thioxo-pyridine and 2-iminopyridinederivative. Molecules 2011, 16, 3410–3419. [Google Scholar] [CrossRef]
- Umelsalma, S.; Sudhandiran, G. Ellagic acid prevents rat colon carcinogenesis induced by 1,2- dimethyl hydrazine through inhibition of AKT-phosphoinosotide-3-kinase pathway. Eur. J. Pharmacol. 2011, 660, 249–258. [Google Scholar] [CrossRef]
- Wandall, H.H.; Tarp, M.A. Therapeutic cancer vaccines: Clinical trials and applications. In In Carbohydrate-Based Vaccines and Immunotherapies; Guo, Z., Boons, G.-J., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Kaur, R.; Singh, A.; Budhlakoti, P.; Singh, A.; Sanwal, R. Synthesis, characterization and antifungal activity of certain(E)-1-(1-(substitutedphenyl) ethylidene)-2-(6-methylbenzo [d] thiazol-2-yl) hydrazine analogues. Int. J. Pharm. Biol. Arch. 2010, 1, 56–61. [Google Scholar]
- Dagenais, R.T.; Keller, N.P. Pathogenesis of Aspergillus fumigatus in invasive aspergillosis. Clin. Microbiol. Rev. 2009, 22, 447–465. [Google Scholar] [CrossRef]
- Wild, C.P. Aflatoxin exposure in developing countries: The critical interface of agriculture and health. Food Nutr. Bull. 2007, 28, S372–S380. [Google Scholar]
- Kostecka, M.; Niewiadomy, A.; Czeczko, R. Evaluation of N-substituted 2,4-dihydroxyphenylthioamide fungicide lipophilicity using the chromatographic techniques HPLC and HPTLC. Chromatographia 2005, 62, 121–126. [Google Scholar] [CrossRef]
- Legocki, J.; Matysiak, J.; Niewiadomy, A.; Kostecka, M. Synthesis and fungistatic activity of new groups of 2,4-dihydroxythiobenzoyl derivatives against phytopathogenic fungi. J. Agric. Food Chem. 2003, 51, 362–368. [Google Scholar] [CrossRef]
- Kostecka, M. Search for an efficient compound with antifungal properties inhibiting Fusarium genus fungi. Ecol. Chem. Eng. A 2010, 17, 1331–1338. [Google Scholar]
- Niewiadomy, A.; Matysiak, J.; Mącik-Niewiadomy, G. Nowe tioamidy, produkt poÅśredni do otrzymywania nowych tioamidów. Patent No. P- 330263, 2000.
- Gerlach, W.; Nirenberg, H. The Genus Fusarium—A Pictorial Atlas. In In Mitteilungen aus der Biologischen Bundesanstalt fur Land- und Forstwirtschaft (in Germany); Institute for Plant Virology, Microbiology and Biological Safety: Berlin-Dahlem, Germany, 1982; pp. 1–209. [Google Scholar]
- Muthomi, J.W.; Schutze, A.; Dehne, H.W.; Mutitu, E.W.; Oerke, E.C. Characterization of Fusarium culmorum isolates by mycotoxin production and aggressiveness to winter wheat. J. Plant Dis. Prot. 2000, 107, 113–123. [Google Scholar]
- Nelson, P.E.; Toussoun, T.A.; Marasas, W.F.O. Fusarium Species-An Illustrated Manual for Identification; The Pennsylvania State University Press: University Park, PA, USA, 1983; pp. 1–193. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kostecka, M. Synthesis of a New Group of Aliphatic Hydrazide Derivatives and the Correlations between Their Molecular Structure and Biological Activity. Molecules 2012, 17, 3560-3573. https://doi.org/10.3390/molecules17033560
Kostecka M. Synthesis of a New Group of Aliphatic Hydrazide Derivatives and the Correlations between Their Molecular Structure and Biological Activity. Molecules. 2012; 17(3):3560-3573. https://doi.org/10.3390/molecules17033560
Chicago/Turabian StyleKostecka, Małgorzata. 2012. "Synthesis of a New Group of Aliphatic Hydrazide Derivatives and the Correlations between Their Molecular Structure and Biological Activity" Molecules 17, no. 3: 3560-3573. https://doi.org/10.3390/molecules17033560
APA StyleKostecka, M. (2012). Synthesis of a New Group of Aliphatic Hydrazide Derivatives and the Correlations between Their Molecular Structure and Biological Activity. Molecules, 17(3), 3560-3573. https://doi.org/10.3390/molecules17033560