3.2. Chemical Synthesis
Trisodium (
R)-homocitrate, dimethyl (S)-methylenehomocitrate and (
R)-homocitric lactone were synthesized as described previously [
15]. (2
R,3
S)-Homoisocitric acid was synthesized according to the procedure of Ma and Palmer [
9].
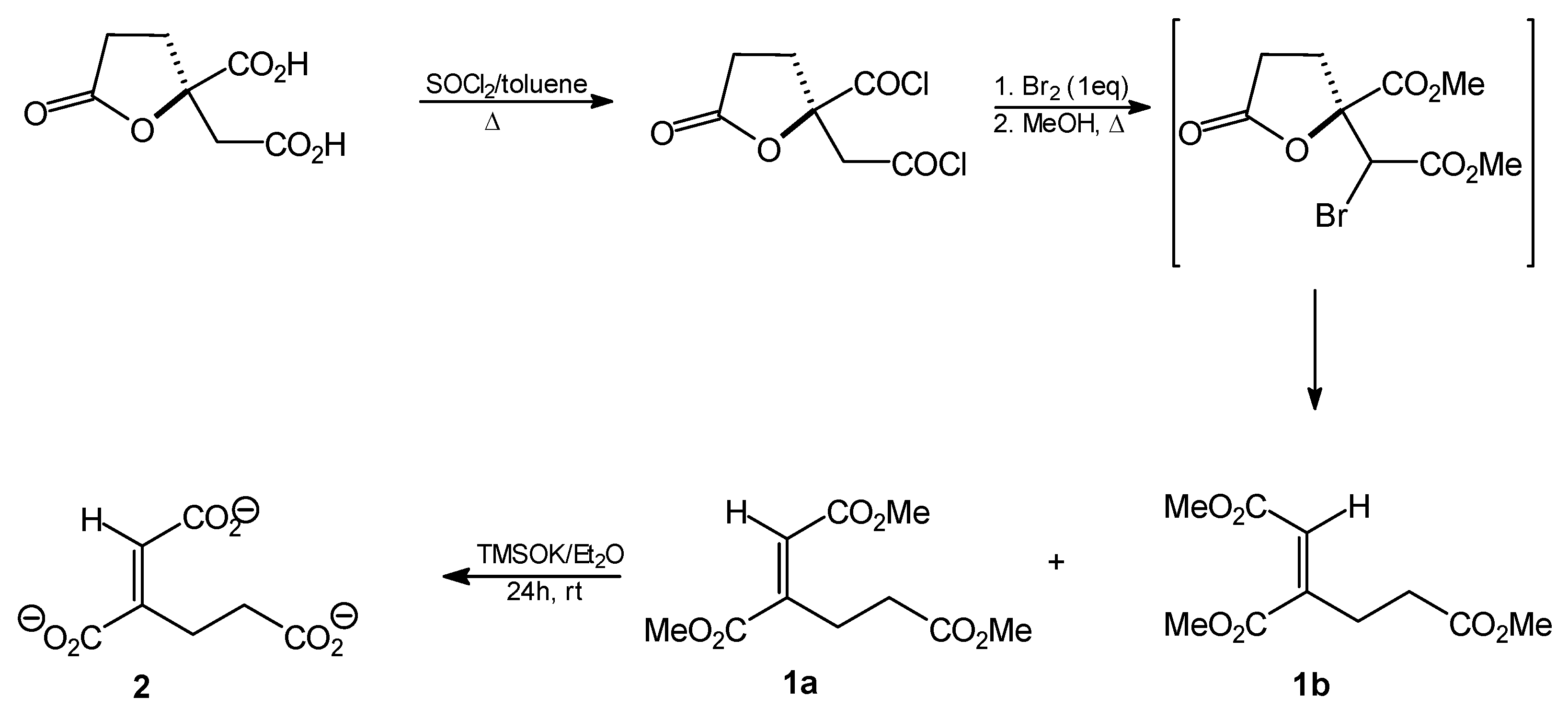
Trimethyl homoaconitate (1a and 1b). Thionyl chloride (9.6 mL; 132 mmol) was added dropwise to a solution of (R)-homocitric lactone (13.5 g; 66 mmol) in toluene (400 mL) and the resulting mixture was heated under reflux for 48 h. The solvent was then removed by evaporation under reduced pressure to give the corresponding dichloride as an oily residue (13 g; 54 mmol; 82%). Anhydrous bromine (3 mL; 54 mmol) was then added dropwise and the mixture was heated under reflux for 1 h. An additional portion of bromine (1 mL) was then added and heating was continued overnight. Anhydrous methanol (500 mL) was added to the cooled reaction mixture and the solution was heated under reflux for about 4 h. The solvent was then distilled off, the residue was dissolved in ethyl acetate and sequentially washed with aqueous 2% Na2CO3 solution, water, 3% Na2CO3 solution and water. The organic layer was then dried over anhydrous MgSO4 and the solvent was subsequently removed by evaporation in vacuo. The crude oily product (5.27 g, 37%, mixture of cis and trans isomers) was resolved by silica gel (70–230 mesh) column chromatography. The column was developed with hexane/AcOEt (5:1). The following oily products were isolated: trimethyl trans-homoaconitate (1a, 1.12 g, 8%) and trimethyl cis-homoaconitate (1b, 3.54 g, 24.5%).
Trimethyl trans-homoaconitate [trimethyl (1E)-but-1-ene-1,2,4-tricarboxylate, 1a]. Rf = 0.54 (solvent system A). 1H-NMR (CDCl3) δ [ppm]: 2.54 (t, J = 7.9 Hz, 2H, CH2CH2CO); 3.09 (t, J = 7.7 Hz, 2H, CH=CCH2); 3.67 (s, 3H, OCH3); 3.78 (s, 3H, OCH3); 3.82 (s, 3H, OCH3); 6.82 (s, 1H, =CH-); 13C-NMR (CDCl3) δ[ppm]: 23.58; 33.28; 51.92; 52.14; 52.91; 127.94; 146.12; 166.01; 167.10; 173.08. Elemental anal. (%), calcd. for C10H14O6: C, 52.17; H, 6.09; found: C, 52.14; H, 5.93.
Trimethyl cis-homoaconitate [trimethyl (1Z)-but-1-ene-1,2,4-tricarboxylate 1b]. Rf = 0.47 (solvent system A). 1H-NMR (CDCl3) δ [ppm]: 2.55 (t, J = 7.2 Hz, 2H, CH2CH2CO); 2.69 (t, J = 7.5 Hz, 2H, CH=CCH2); 3.69 (s, 3H, OCH3); 3.73 (s, 3H, OCH3); 3.83 (s, 3H, OCH3); 5.90 (s, 1H, =CH-); 13C-NMR (CDCl3) δ [ppm]: 29.16; 31.49; 51.86; 51.91; 52.44; 120.86; 147.67; 165.21; 168.51; 172.08. Elemental anal. (%), calcd. for C10H14O6: C, 52.17; H, 6.09; found: C, 52.07; H, 5.97.
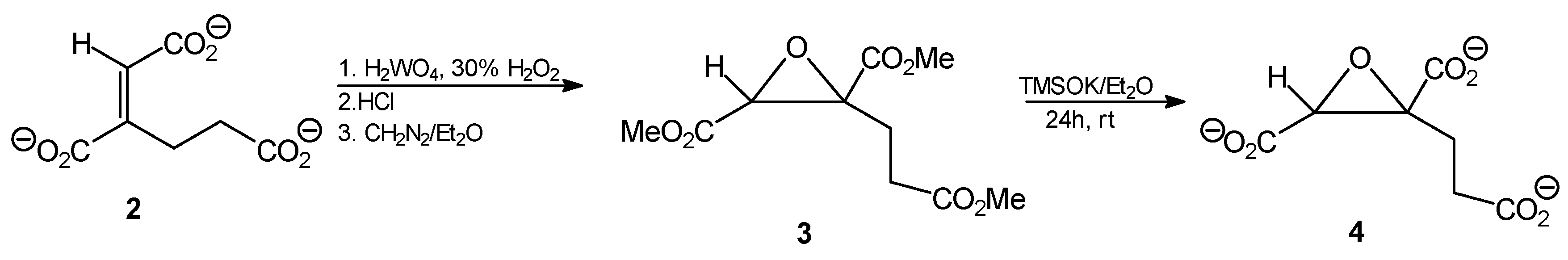
Tripotassium trans-homoaconitate [tripotassium (1E)-but-1-ene-1,2,4-tricarboxylate 2]. Trimethyl trans-homoaconitate (1a, 276 mg, 1.2 mmol) was dissolved in anhydrous diethyl ether (20 mL). Potassium trimethylsilanolate (510 mg, 4 mmol) was added to the solution and the mixture was stirred for 24 h. The solvent was removed by evaporation under reduced pressure, the residue was washed with anhydrous diethyl ether and finally dried under reduced pressure. The product was obtained as a slightly brownish powder (322 mg, 89%). 1H-NMR (D2O) δ [ppm]: 2.54 (t, J = 7.9 Hz, 2H, CH2,); 2.74 (t, J = 8.1 Hz, 2H, CH2,); 6.51 (s, CH=C, 1H). 13C-NMR (D2O) δ [ppm]: 23.54; 33.32; 51.82; 52.18; 52.99; 127.90; 146.02. Elemental anal. (%), calcd. for C7H5O6K3: C, 27.63; H, 2.30; found: C, 27.69; H, 2.27.
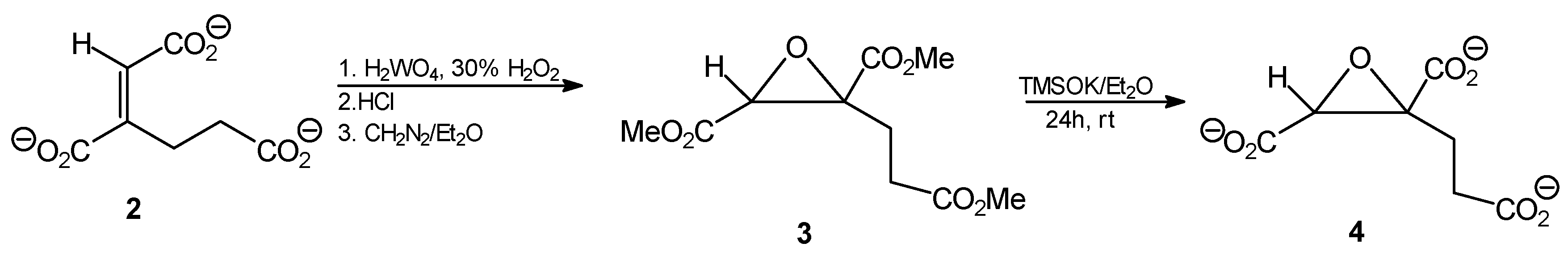
Trimethyl (1E)1,2-epoxy-butane-1,2,4-tricarboxylate (3). Tripotassium trans-homoaconitate (2, 302 mg; 1 mmol) was dissolved in water (5 mL) and tungstic acid (35 mg; 0.14 mmol) and 30% H2O2 (300 μL) were added. The mixture was stirred for 2 h at 85 °C. Then 10 M HCl (0.35 mL) was added and the reaction mixture was subjected to continuous extraction with diethyl ether for several hours. The organic solvent was removed from the extract by evaporation, to give 172 mg of a slightly yellowish oil. The ethereal solution of diazomethane was then added dropwise at 0 °C until pH of the solution reached 7.0, then the mixture was left overnight at room temperature. After solvent evaporation, 216 mg of the crude oily product was obtained, that was further purified by column silica gel chromatography eluting with hexane/AcOEt (5:1), followed by AcOEt. Finally, after solvent evaporation, a slightly yellow oily product was obtained (202 mg; 82%); Rf = 0.57 (solvent system B). 1H-NMR (CDCl3) δ [ppm]: 2.39–2.33 (m, 1H, CH2CH2CO); 2.63–2.53 (m, 2H, CH2CH2CO); 2.75–2.67 (m, 1H, CH2CH2CO); 3.67 (s, 3H, OCH3); 3.69 (s, 3H, OCH3); 3.70 (s, 3H, OCH3); 3.74 (s, 1H, CH); 13C-NMR (CDCl3) δ [ppm]: 31.57; 45.64; 52.19; 52.92; 52.97; 53.21; 60.05; 168.16; 170.82; 173.23. Elemental anal. (%), calcd. for C10H14O7: C, 48.78; H, 5.69; found: C, 48.67; H, 5.63.
Tripotassium (1E)1,2-epoxy-butane-1,2,4-tricarboxylate (4). Trimethyl (1E)1,2-epoxybutane-1,2,4-tricarboxylate (3, 148 mg, 0.6 mmol) was dissolved in anhydrous diethyl ether (10 mL). Potassium trimethylsilanolate (255 mg, 2 mmol) was added to the solution and the mixture was stirred for 24 h. The solvent was removed by evaporation under reduced pressure, the residue was washed with anhydrous diethyl ether and finally dried under reduced pressure. The product was obtained as a yellow amorphous powder (156 mg, 82%). 1H-NMR (D2O) δ [ppm]: 2.29–2.35 (m, 1H, CH2CH2CO); 2.57–262 (m, 2H, CH2CH2CO); 2.66–2.71 (m, 1H, CH2CH2CO); 3.78 (s, 1H, CH); 13C-NMR (CDCl3) δ [ppm]: 31.32; 45.68; 51.88; 53.15; 53.24; 53.48; 61.60. Elemental anal. (%), calcd. for C7H5O7K3: C, 27.27; H, 1.62; found: C, 27.27; H, 1.65.
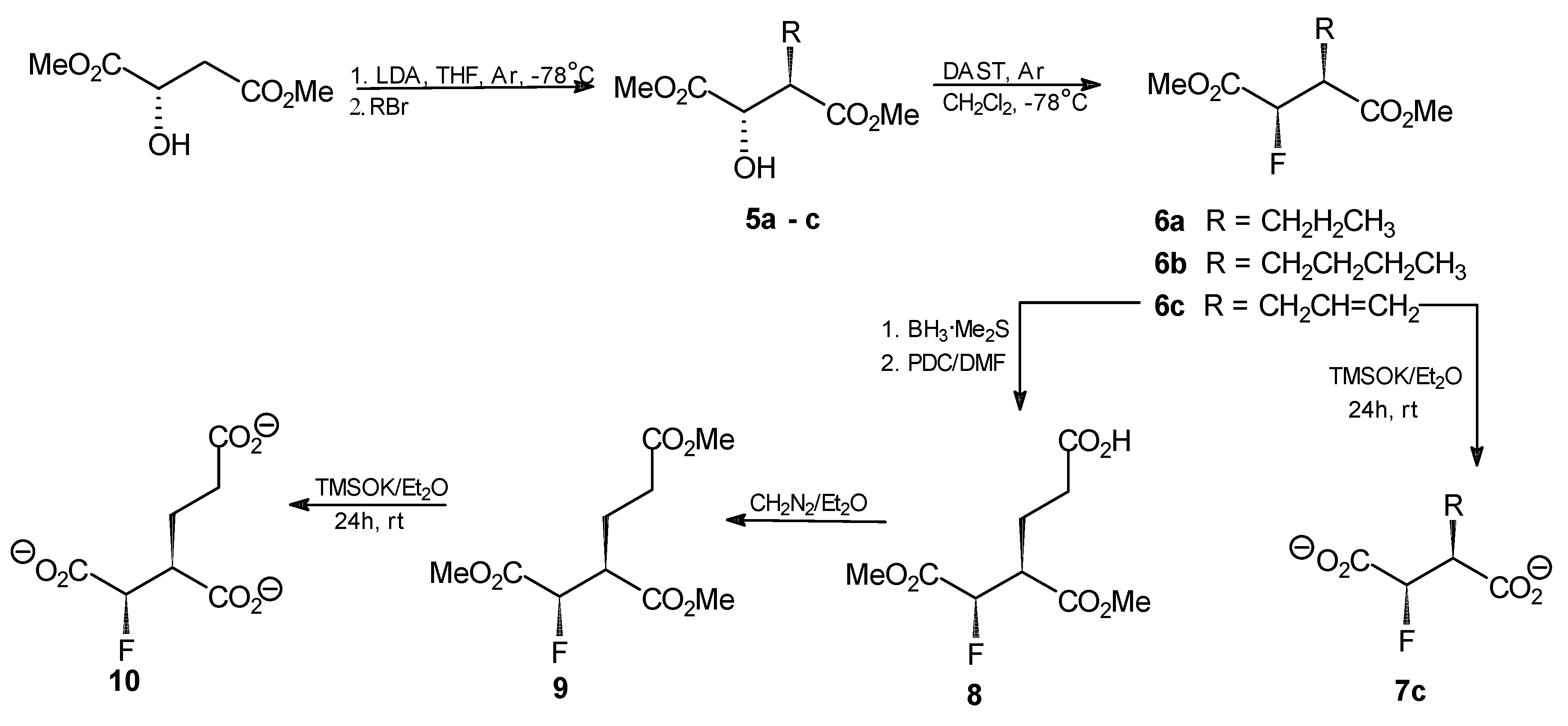
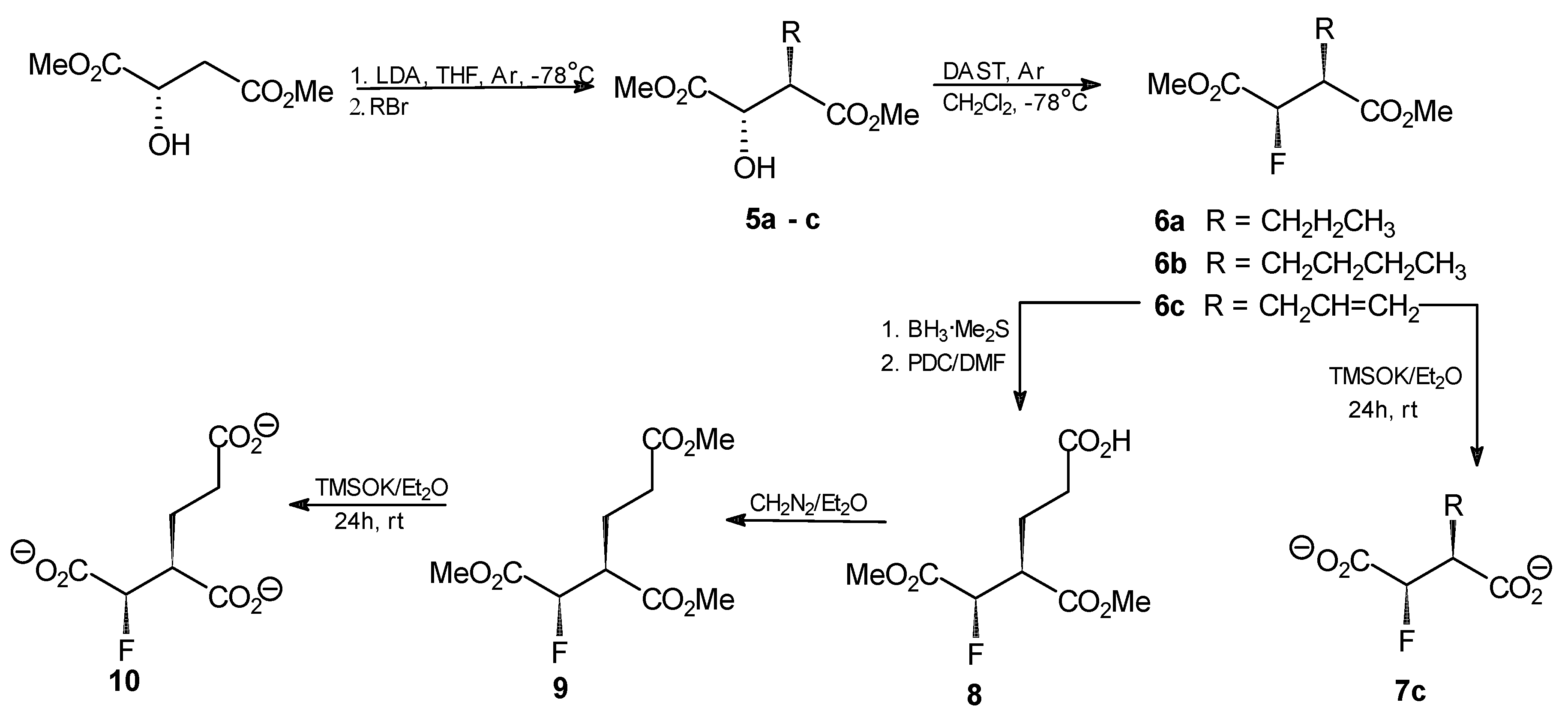
General procedure for the synthesis of compounds 5a–c. n-BuLi (20.8 mL, 52.2 mmol, 2.5 M in hexane) was added dropwise to a stirred solution of diisopropylamine (62.6 mmol) in THF (38 mL) at −78 °C under argon. After 1 h, a solution of dimethyl (S)-malate (3.38 g, 20.9 mmol) in THF (6 mL) was added, while the temp. was kept at −78 °C. The mixture was then warmed to −25 °C and stirred for 1 h. After that time, the mixture was again chilled to −78 °C and propyl, butyl or allyl bromide (53 mmol) was added dropwise. The resulting solution was stirred for 2 h at −25 °C and then left overnight at 4 °C. The mixture was chilled to −78 °C, acetic acid (6.4 mL in 10 mL of Et2O) was added and the resulting mixture was combined with Et2O (250 mL) and water (30 mL). The organic layer was washed with the saturated solution of sodium carbonate and with saline. Aqueous layers were combined and extracted three times with Et2O. Combined ethereal layers were dried over anhydrous MgSO4 and the solvent was evaporated. The crude products were purified by flash silica-gel chromatography with hexane/AcOEt (5:1) as an eluent to afford the final products.
Dimethyl (2S,3R)-2-hydroxy-3-propylsuccinate (5a). Oil, final yield 58%. [α +19.7 (c1.33; CHCl3). 1H-NMR (CDCl3) δ [ppm]: 0.95 (t, 3H, CH2CH2CH3), 1.41 (m, 2H, CH2CH2CH3), 1.66 (m, 1H, CH2CH2CH3), 1.83 (m, 1H, CH2CH2CH3), 3.69 (s, 3H, OCH3); 3.80 (s, 3H, OCH3); 4.29 (d, J = 3.9 Hz, 1H, CHOH); 13C-NMR (CDCl3) δ [ppm]: 14.08; 20.79; 30.36; 48.54; 52.18; 52.93; 71.22; 173.74; 174.18. Elemental anal. (%), calcd. for C9H16O5: C, 52.94; H, 7.84; found: C, 52.87; H, 7.90.
Dimethyl (2S,3R)-2-hydroxy-3-butylsuccinate (5b). Oil, final yield 65%. [α +12.8 (c1.15; CHCl3). 1H-NMR (CDCl3) δ [ppm]: 0.94 (t, 3H, CH2CH2CH2CH3), 1.39 (m, 2H, CH2CH2CH2CH3), 1.68 (m, 2H, CH2CH2CH2CH3), 1.89 (m, 1H, CH2CH2CH2CH3), 3.70 (s, 3H, OCH3); 3.77 (s, 3H, OCH3); 4.29 (d, J = 4.2 Hz, 1H, CHOH); 13C-NMR (CDCl3) δ [ppm]: 12.01; 18.45; 21.01; 30.63; 48.91; 51.65; 52.98; 70.88; 173.11; 174.55. Elemental anal. (%), calcd. for C10H18O5: C, 55.05; H, 8.26; found: C, 54.98; H, 8.30.
Dimethyl (2S,3R)-2-hydroxy-3-allylsuccinate (5c). Oil, final yield 62%. [α +16.1 (c1.24; CHCl3). 1H-NMR (CDCl3) δ [ppm]: 2.45 (m, 1H, CH2CH=CH2); 2.63 (m, 1H, CH2CH=CH2); 2.99 (m, 1H, CH(OH)CHCO2CH3); 3.69 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 4.31 (d, J = 3 Hz, 1H, CH(OH)CHCO2CH3); 5.19–5.11 (dd, J = 17 Hz and 10 Hz, 2H, CH2CH=CH2); 5.82 (m, 1H, CH2CH=CH2); 13C-NMR (CDCl3) δ [ppm]: 32.36; 48.38; 52.27; 52.99; 70.36; 118.30; 134.91; 172.78; 174.21. Elemental anal. (%), calcd. for C9H14O5: C, 53.47; H, 6.93; found: C, 53.37; H, 7.01.
General procedure for the synthesis of compounds 6a–c. DAST (0.76 mL, 5.9 mmol) was dissolved in anhydrous methylene chloride (30 mL) and the resulting solution was chilled to −78 °C under argon. An analogously chilled solution of one of the compounds 5a–c (4.4 mmol) in methylene chloride (10 mL) was added dropwise using a cannulated syringe and the resulting mixture was stirred for 1 h at −78°C and for 2 h at room temperature. Chloroform (40 mL) was added and the resulting solution was washed with 5% NaHCO3, water and saline. The solvent was evaporated and the crude products were purified by flash silica-gel chromatography with hexane/AcOEt (9:1) to afford the final products.
Dimethyl (2R,3S)-2-fluoro-3-propylsuccinate (6a). Oil, final yield 92%. [α +6.2 (c1.5; CHCl3). 1H-NMR (CDCl3) δ [ppm]: 0.93 (t, 3H, CH2CH2CH3), 1.32 (m, 1H, CH2CH2CH3), 1.43 (m, 1H, CH2CH2CH3), 1.62 (m, 1H, CH2CH2CH3), 1.82 (m, 1H, CH2CH2CH3), 2.98 (m, 1H, FCHCHCO2CH3), 3.74 (s, 3H, OCH3); 3.82 (s, 3H, OCH3); 5.16 (dd, 2JHF = 47.4 Hz, 3JHH = 5.9 Hz, 1H, FCH); 13C-NMR (CDCl3) δ [ppm]: 14.07; 20.67; 29.30; 47.91 (2JFC = 23.8 Hz); 52.43; 52.85; 88.92 (JFC = 197.3 Hz); 169.04; 172.21. 19F-NMR (CDCl3) δ[ppm]: −197.51 (dd, 3JHF = 23 Hz, 2JHF = 47 Hz). Elemental anal. (%), calcd. for C9H15O4F: C, 52.43; H, 7.28; found: C, 52.34; H, 7.35.
Dimethyl (2R,3S)-2-fluoro-3-butylsuccinate (6b). Oil, final yield 88%. [α +4.9 (c1.3; CHCl3). 1H-NMR (CDCl3) δ [ppm]: 0.93 (t, 3H, CH2CH2CH2CH3), 1.24 (m, 1H, CH2CH2CH2CH3), 1.32 (m, 1H, CH2CH2CH2CH3), 1.41 (m, 1H, CH2CH2CH2CH3), 1.53 (m, 1H, CH2CH2CH2CH3), 1.62 (m, 1H, CH2CH2CH2CH3); 1.82 (m, 1H, CH2CH2CH2CH3); 2.99 (m, 1H, FCHCHCO2CH3), 3.74 (s, 3H, OCH3); 3.82 (s, 3H, OCH3); 5.17 (dd, 1H, 2JHF = 42 Hz, 3JHH = 5.9 Hz, FCH); 13C-NMR (CDCl3) δ [ppm]: 12.01; 18.45; 21.01; 30.63; 48.91; 51.65; 52.98; 70.88; 173.11; 174.5514.07; 20.67; 29.30; 47.91 (2JFC = 23.8 Hz); 52.43; 52.85; 88.92 (JFC = 197.3 Hz); 169.04; 172.21. 19F-NMR (CDCl3) δ [ppm]: −197.46 (dd, 3JHF = 19.2 Hz and 2JHF = 46.8 Hz). Elemental anal. (%), calcd. for C10H17O4F: C, 54.55; H, 7.73; found: C, 54.48; H, 7.73.
Dimethyl (2R,3S)-2-fluoro-3-allylsuccinate (6c). Oil, final yield 89%. [α +2.5 (c1.7; CHCl3). 1H-NMR (CDCl3) δ [ppm]: 2.46 (m, 1H, CH2CH=CH2); 2.60 (m, 1H, CH2CH=CH2); 3.10 (m, 1H, CH(OH)CHCO2CH3), 3.73 (s, 3H, OCH3); 3.83 (s, 3H, OCH3); 5.11 (m, 1H, FCHCHCO2CH3); 5.18 (m, 1H, CH2CH=CH2); 5.24 (m, 1H, CH2CH=CH2); 5.76 (m, 1H, CH2CH=CH2); 13C-NMR (CDCl3) δ [ppm]: 38.21; 47.74 (2JFC = 21.5 Hz); 52.13; 52.62; 88.20 (JFC = 190.2 Hz); 119.57; 133.08; 165.68; 169.06; 19F NMR (CDCl3) δ[ppm]: -198.16 (dd, 3JHF = 21 Hz; 2JHF = 47 Hz). Elemental anal. (%), calcd. for C9H13O4F: C, 52.94; H, 6.37; found: C, 53.02; H, 6.40.
Dipotassium (2R,3S)-2-fluoro-3-allylsuccinate (7c). Dimethyl (2R,3S)-2-fluoro-3-allylsuccinate (6c, 41 mg; 0.2 mmol) was dissolved in anhydrous diethyl ether (20 mL). Potassium trimethylsilanolate (56 mg, 0.44 mmol) was added to the solution and the mixture was stirred for 24 h. The solvent was removed by evaporation under reduced pressure, the residue was washed with anhydrous diethyl ether and finally dried under reduced pressure. The product was obtained as a slightly yellow amorphous powder (35 mg; 70%). 1H-NMR (CDCl3) δ [ppm]: 2.44 (m, 1H, CH2CH=CH2); 2.59 (m, 1H, CH2CH=CH2); 3.14 (m, 1H, CH(OH)CHCO2CH3), 5.11 (m, 1H, FCHCHCO2CH3); 5.20 (m, 1H, CH2CH=CH2); 5.25 (m, 1H, CH2CH=CH2); 5.75 (m, 1H, CH2CH=CH2); 13C-NMR (CDCl3) δ[ppm]: 38.44; 47.07 (2JFC = 23 Hz); 52.35; 53.62; 88.20 (JFC = 192 Hz); 118.22; 135.69; 19F-NMR (CDCl3) δ [ppm]: −197.55 (dd, 3JHF = 18 Hz; 2JHF = 44 Hz). Elemental anal. (%), calcd. for C7H7O4FK2: C, 33.33; H, 2.78; found: C, 33.39; H, 2.79.
Dimethyl (2R,3S)-2-fluoro-2-deoxyhomoisocitrate (8). BH3 × SMe2 (0.51 mL, 5.05 mmol) was added dropwise to a solution of dimethyl (2R,3S)-2-fluoro-3-allyl succinate (6c, 1.03 g, 5.05 mmol) in methylene dichloride (8 mL) at 0 °C. The reaction mixture was left at room temperature with stirring for 2 h. Then pyridinium dichromate (PDC, 14.6 g, 38 mmol) in anhydrous DMF (30 mL) was slowly added and stirring was continued for 48 h. After that time, the mixture was transferred to the flask containing water (300 mL) and the whole solution was stirred for another 16 h. The resulting mixture was extracted with diethyl ether (3 × 250 mL) and the aqueous layer was further extracted with ethyl acetate (2 × 170 mL). The organic layers were combined and dried over anhydrous MgSO4. The drying agent was removed and the organic solvents were evaporated under reduced pressure to give 1.2 g of the crude product that was further purified by flash chromatography on silica gel with stepwise elution with hexane/AcOEt (5:1), AcOEt and MeOH to give 0.87 g (73%) of the purified product. 1H-NMR (CDCl3) δ [ppm]: 2.02 (m, 1H, CH2CH2COOH); 2.21 (m, 1H, CH2CH2COOH); 2.48 (m, 2H, CH2CH2COOH ); 3.03 (m, 1H, FCHCOOMe); 3.76 (s, 3H, OCH3); 3.83 (s, 3H, OCH3); 5.32–5.22 (dd, 3JHH = 4.4 Hz, 2JHF = 47.4 Hz, CHF); 13C-NMR (CDCl3) δ [ppm]: 23.59; 31.80; 47.77; 51.98; 52.34; 53.09; 172.66; 173.52. 19F-NMR (CDCl3) δ [ppm]: −196.23 (dd, 3JHF = 21 Hz; 2JHF = 47 Hz). Elemental anal. (%), calcd. for C9H13O6F: C, 45.76; H, 5.50; found: C, 45.71; H, 5.47.
Trimethyl (2R,3S)-2-fluoro-2-deoxyhomoisocitrate [trimethyl (1R,2S)-1-fluorobutane-1,2,4-tricarboxylate, 9]. Dimethyl (2R,3S)-2-fluoro-2-deoxyhomoisocitrate (8, 203 mg, 0.9 mmol) was treated with diazomethane solution in diethyl ether under argon. The mixture was left overnight at room temperature. The solvent was then removed by evaporation under reduced pressure. The residue was washed twice with diethyl ether. The crude oily product was purified by flash chromatography on silica gel with hexane/AcOEt (2:1) as an eluent. The final product was obtained as a colorless oil (228 mg, 95%). [α +12.5 (c2.0; CHCl3). 1H-NMR (CDCl3) δ [ppm]: 2.04 (m, 1H, CH2CH2COOMe); 2.21 (m, 1H, CH2CH2COOMe); 2.48 (m, 2H, CH2CH2COOMe); 3.03 (m, 1H, FCHCOOMe); 3.71 (s, 3H, OCH3); 3.72 (s, 3H, OCH3); 3.83 (s, 3H, OCH3); 4.63 (d, J = 3.4 Hz, 1H, FCH); 13C-NMR (CDCl3) δ [ppm]: 23.59; 31.80; 47.77; 51.98; 52.34; 53.09; 71.34; 172.66; 173.52; 173.84. 19F-NMR (CDCl3) δ [ppm]: −193.44 (dd, 3JHF = 22 Hz; 2JHF = 49 Hz). Elemental anal. (%), calcd. for C10H15O6F: C, 50.00; H, 6.25; found: C, 50.07; H, 6.22.
Tripotassium (2R,3S)-2-fluoro-2-deoxyhomoisocitrate [tripotassium (1R,2S)-1-fluorobutane-1,2,4-tricarboxylate, 10]. Conversion of the trimethyl ester 9 into the respective tripotassium salt was achieved upon treatment with potassium trimethylsilanolate under condition described above for preparation of compound 4. The product was obtained as a yellowish amorphous powder (309 mg, 93%). 1H-NMR (D2O) δ [ppm]: 2.02 (m, 1H, CH2CH2COOK); 2.25 (m, 1H, CH2CH2COOK); 2.52 (m, 2H, CH2CH2COOK); 3.01 (m, 1H, FCHCOOK); 4.68 (d, J = 3.4 Hz, 1H, FCH); 13C-NMR (CDCl3) δ [ppm]: 23.48; 31.85; 47.72; 51.95; 52.38; 53.00; 71.31. 19F-NMR (CDCl3) δ[ppm]: −192.58 (dd, 3JHF = 20 Hz; 2JHF = 51 Hz). Elemental anal. (%), calcd. for C7H6O6FK3: C, 48.58; H, 4.86; found: C, 48.51; H, 4.92.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}