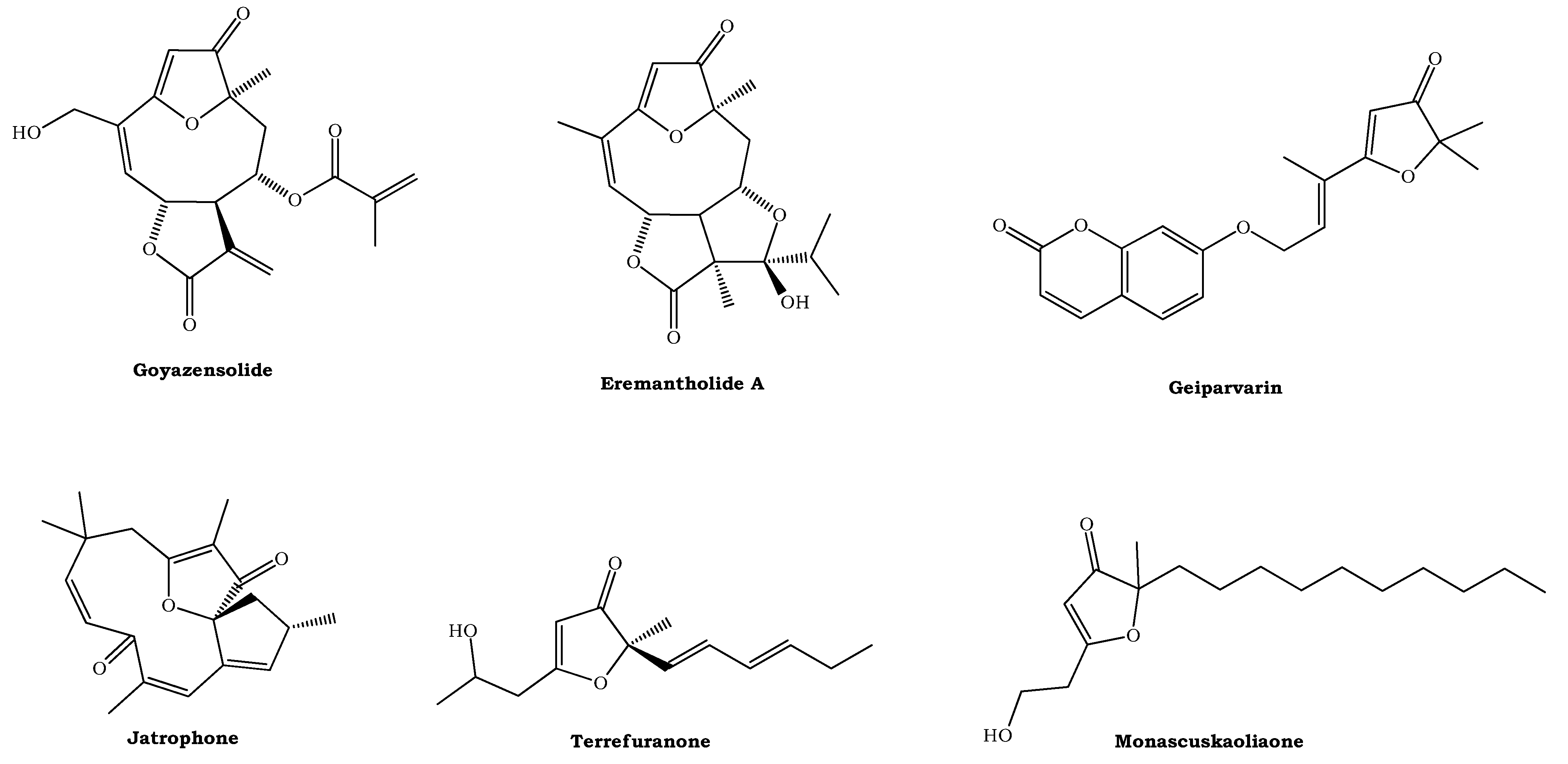
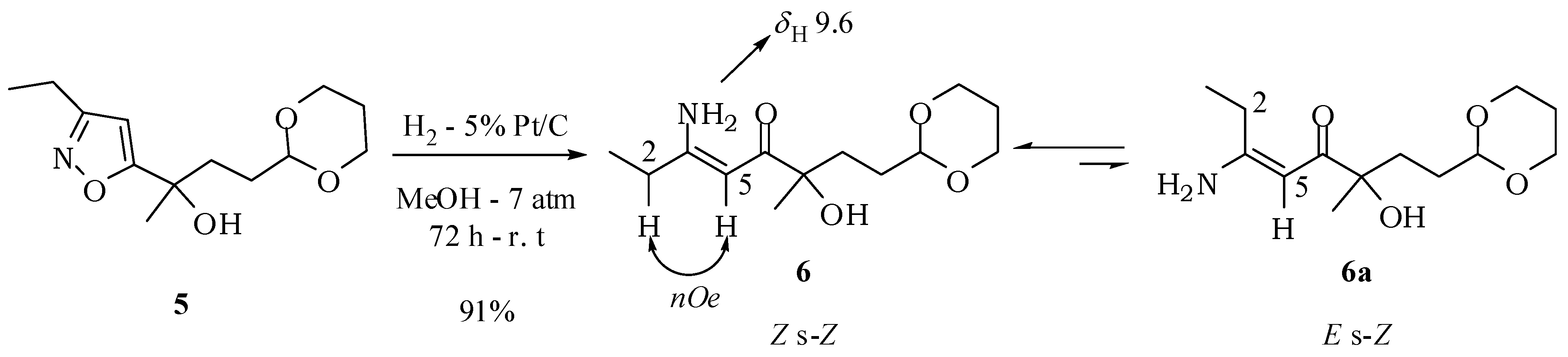
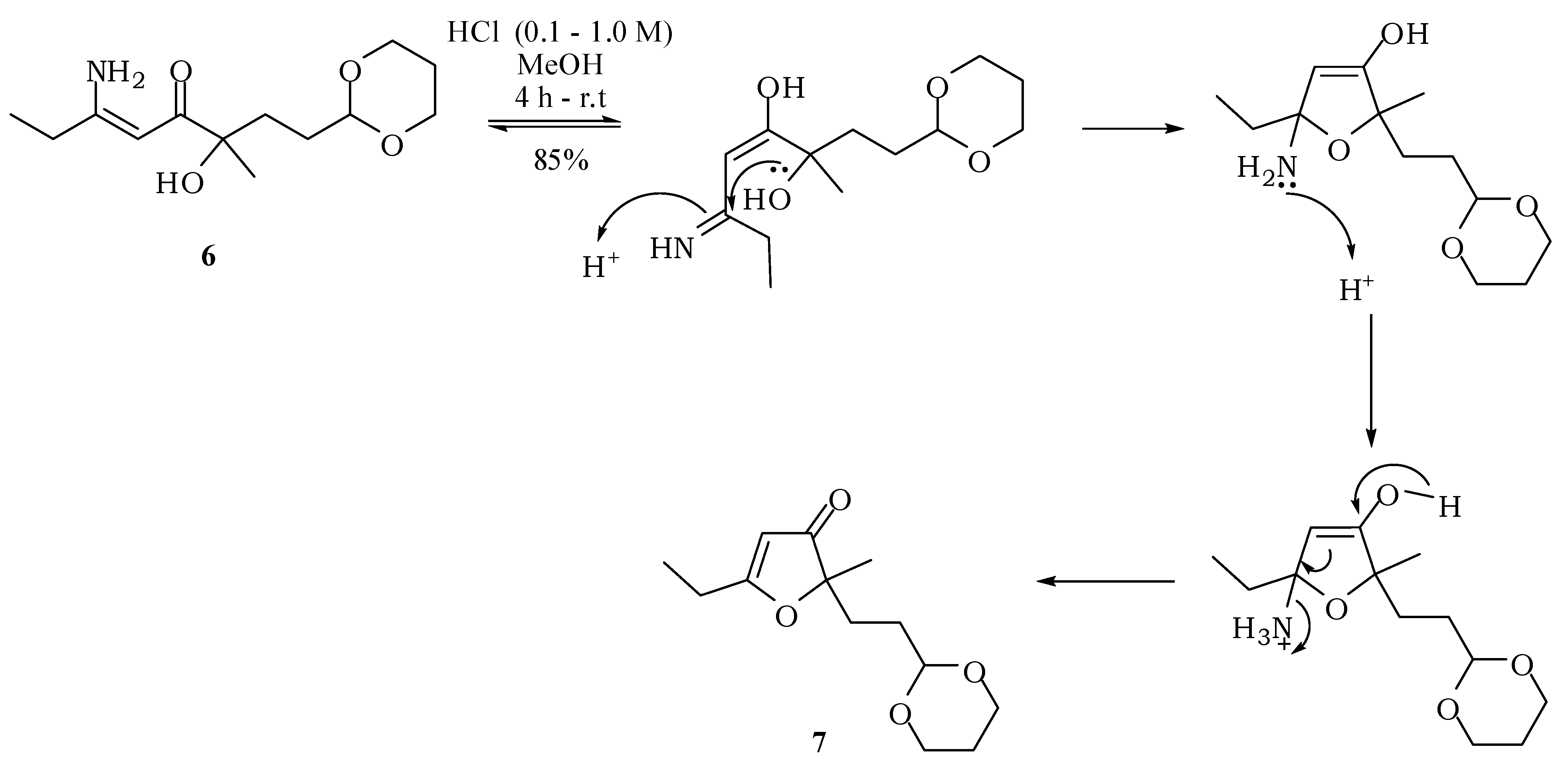
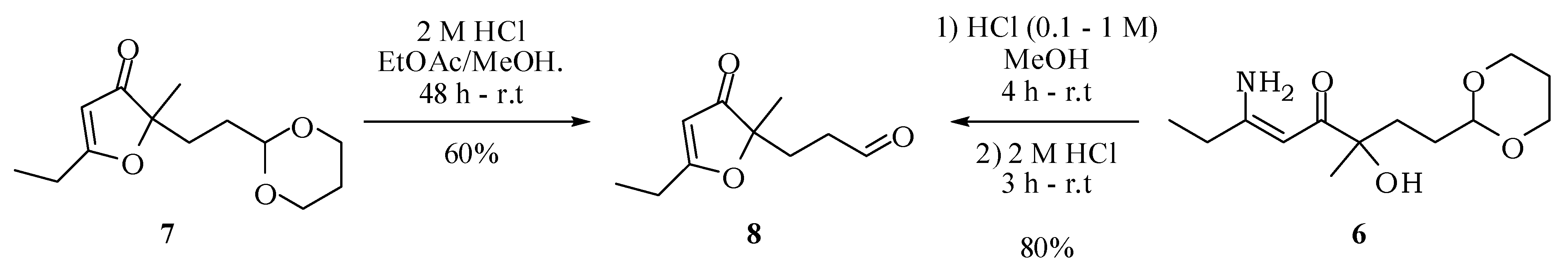
3.2. Experimental Procedures
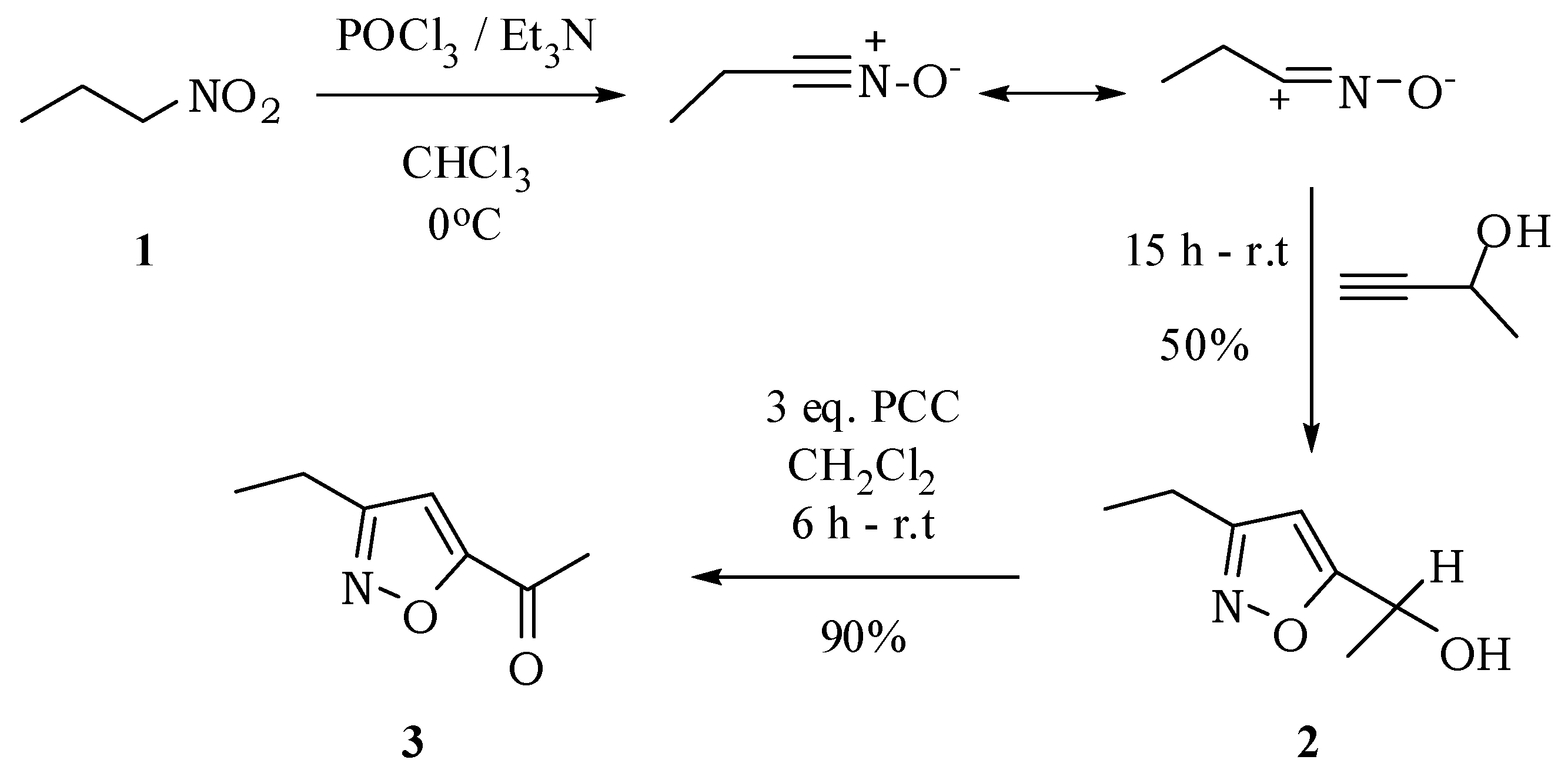
1-(3-Ethylisoxazol-5-yl)ethanol (
2): The preparation of isoxazolylalcohol
2 was described in reference [
8]. IR ν
max (liquid film): 811, 899, 1079, 1107, 1144, 1426, 1463, 1601, 3377 cm
−1.
1H-NMR, 500 MHz, (CDCl
3), δ(ppm): 1.26 (t, 3H,
J = 7.6 Hz); 1.56 (d, 3H,
J = 6.7 Hz); 2.52 (br s, 1H); 2.67 (q, 2H,
J = 7.6 Hz); 4.97 (q, 1H,
J = 6.7 Hz); 6.06 (s, 1H).
13C {
1H} NMR, 125 MHz, (CDCl
3), δ(ppm): 12.6; 19.5; 21.8; 63.1; 99.4; 165.1; 175.9. HRESIMS: calcd for C
7H
12NO
2+ (MH+) 142.0868; found 142.0862.
1-(3-Ethylisoxazol-5-yl)ethanone (3): A solution of PCC (460 mg, 2.13 mmol) in CH2Cl2 (15 mL) was added to a solution of alcohol 2 (100 mg, 0.71 mmol) in CH2Cl2 (5 mL). After 6 h, the mixture was filtered through a sintered glass funnel with a silica gel and Celite® bed, dried and washed several times with ethyl ether. The solvent of the filtrate was evaporated at reduced pressure and compound 3 was obtained with 90% yield (88.8 mg; 0.64 mmol). The crude product crystallized spontaneously at 0 °C. mp 31–32 °C. IR νmax (liquid film): 610, 661, 849, 907, 963, 1090, 1188, 1295, 1368, 1470, 1581, 1698 cm−1. 1H-NMR, 500 MHz, (CDCl3), δ(ppm): 1.30 (t, 3H, J = 7.6 Hz); 2.60 (s, 3H); 2.77 (q, 2H, J = 7.6 Hz); 6.77 (s, 1H). 13C-NMR {1H}, 125 MHz, (CDCl3), δ(ppm): 12.5; 19.6; 27.2; 106.6; 166.0; 166.5; 187.1.
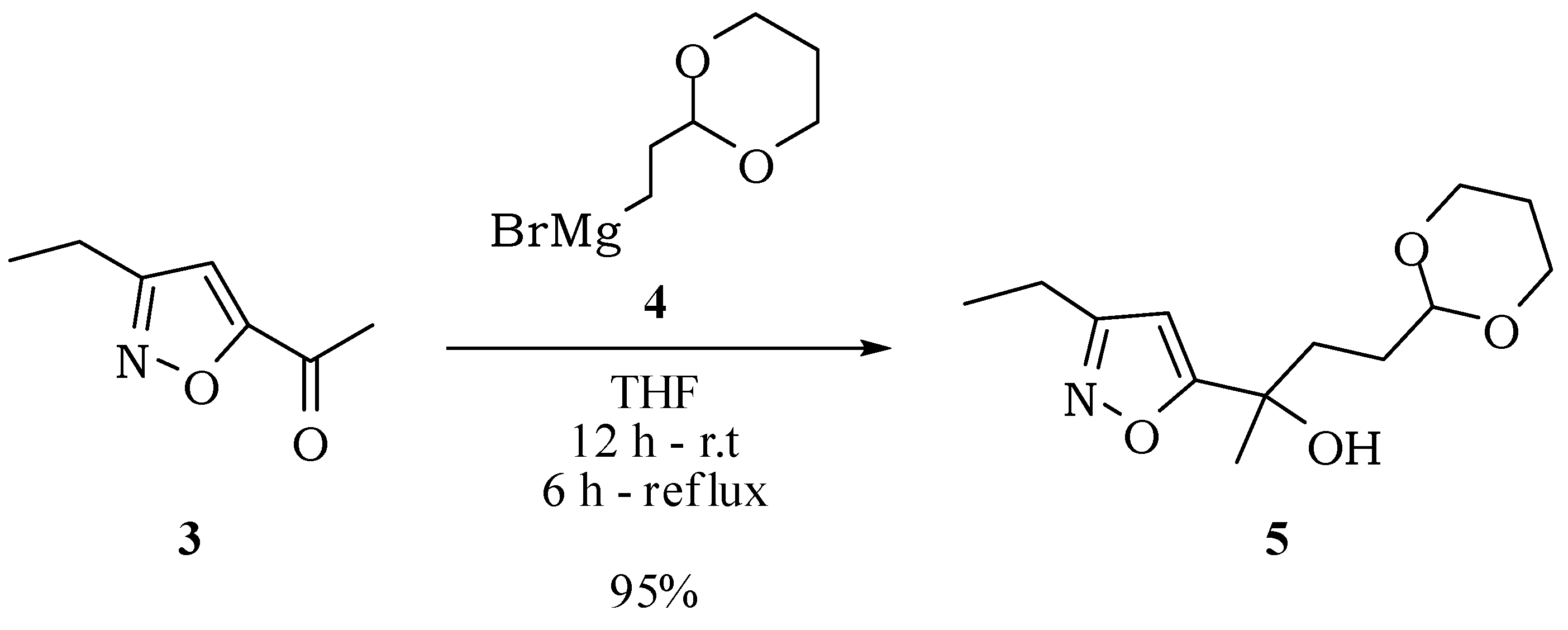
4-(1,3-Dioxan-2-yl)-2-(3-ethylisoxazol-5-yl)butan-2-ol (5): Under an atmosphere of nitrogen, magnesium turnings (120 mg, 4.95 mmol) activated with iodine and THF (5 mL) were heated to reflux. Then, a solution of 2-(2-bromoethyl)-1,3-dioxane (967 mg, 4.95 mmol) in THF (5 mL) was added dropwise by addition funnel and the mixture was kept under reflux and magnetic stirring for about one hour, until the total consumption of magnesium. The temperature was lowered to 0 °C and a solution of compound 3 (230 mg, 1.65 mmol) in THF (5 mL) was added dropwise. The reaction mixture was kept stirring for 12 h at room temperature and further 6 h under reflux. Then, water (10 mL) was added and the mixture was extracted with diethyl ether (3 × 15 mL). Organic extracts were dried with anhydrous magnesium sulfate, filtered and the solvent removed at reduced pressure. The compound 5 was obtained with 95% yield (400.0 mg; 1.57 mmol) after purification by column chromatography on silica gel using hexane/ethyl acetate (60:40). IR νmax (liquid film): 811, 896, 1004, 1145, 1241, 1287, 1378, 1418, 1464, 1597, 1858, 2942, 2973, 3417 cm−1 1H-NMR, 500 MHz, (C6D6), δ(ppm): 0.55 (dtt, 1H, J = 13.2; 11.2; 4.9 Hz); 1.01 (t, 3H, J = 7.6 Hz); 1.46 (s, 3H); 1.69 (dtt, 1H, J = 13.2; 3.1; 2.7 Hz); 1.77 (dtd, 1H, J = 13.3; 7.9; 4.7 Hz); 1.74 (dtd; 1H; J = 13.3; 7.9; 4.7 Hz); 2.01 (ddd, 1H, J = 14.5; 7.9; 6.8 Hz); 2.12 (ddd, 1H, J = 14.5; 7.9; 6.8 Hz); 2.40 (dq; 2H; J = 7.6; 15.5 Hz); 3.23 (ddd; 1H; J = 12.4; 11.2; 2.7 Hz); 3.20 (ddd; 1H; J = 12.4; 11.2; 2.7 Hz); 3.59 (br s, 3H) 3.71 (dddd; 1H; J = 12.4; 4.9; 3.1; 1.5 Hz); 3.69 (dddd; 1H; J = 12.4; 4.9; 3.1; 1.5 Hz); 4.22 (t, 1H, J = 4.7 Hz); 5.91 (s, 1H). 13C-NMR {1H}, 125 MHz, (C6D6), δ(ppm): 12.6; 19.8; 25.7; 28.2; 30.1; 35.8; 66.6; 71.6; 99.9; 101.9; 164.6; 178.2. HRESIMS: calcd for C13H22NO4+ (MH+) 256.1549; found 256.1543.
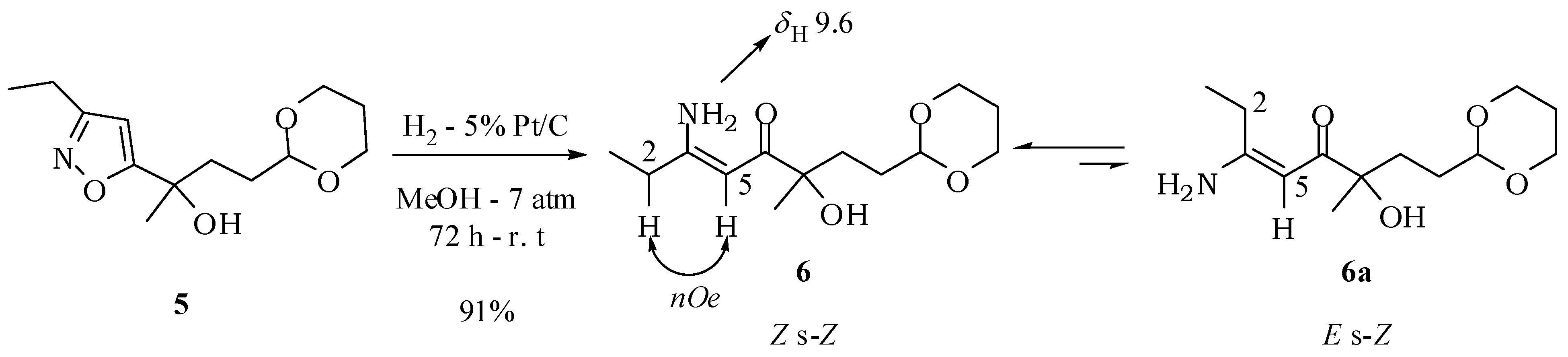
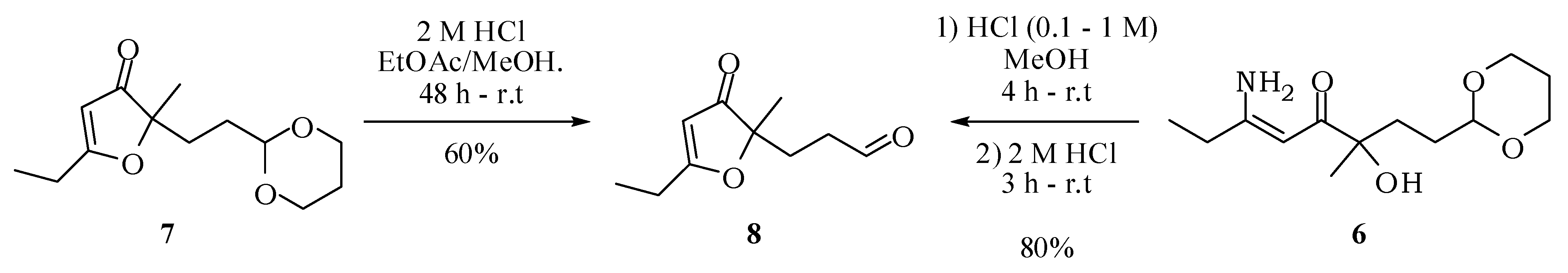
(5Z)-6-Amino-1-(1,3-dioxan-2-yl)-3-hydroxy-3-methyloct-5-en-4-one (6): A solution of compound 5 (100 mg; 0.39 mmol) in methanol (1.5 mL) was added to a suspension of 15% platinum on carbon (15 mg) in methanol (1.5 mL). The reaction mixture was kept under hydrogen at 7 atm for 72 h. The catalyst was filtered off and washed with ethyl acetate. Removal of the solvent under vacuum left a white solid with 91% yield (91.2 mg; 0.35 mmol). mp 93–94 °C. IR νmax (liquid film): 640, 808, 814, 999, 1147, 1264, 1537, 1624, 2848, 2938, 2973, 3201, 3358, 3417 cm−1 1H-NMR, 500 MHz, (CDCl3), δ(ppm): 1.18 (t, 3H, J = 7.6 Hz); 1.30 (dtt, 1H, J = 13.2; 11.2; 4.9 Hz); 1.33 (s, 3H); 1.50 (m, 1H); 1.69 (m, 1H); 1.72 (dd, 2H, J = 6.9; 9.4 Hz); 2.05 (dtt, 1H, J = 13.2; 3.1; 2.7 Hz); 2.24 (dq, 2H, J = 7.6; 1.7 Hz); 3.76 (ddd, 1H, J = 12.4; 11.2; 2.7 Hz); 3.74 (ddd, 1H, J = 12.4; 11.2; 2.7 Hz); 4.10 (dddd, 1H, J = 12.4; 4.9; 3.1; 1.5 Hz); 4.08 (dddd, 1H, J = 12.4; 4.9; 3.1; 1.5 Hz); 4.50 (dd, 1H, J = 9.4; 6.0 Hz); 4.57 (br s, 1H); 5.14 (d, 1H, J = 1.7 Hz); 5.19 (br s, 1H); 9.59 (br s, 1H). 13C-NMR {1H}, 125 MHz, (CDCl3), δ(ppm): 12.0; 26.9; 29.6; 34.9; 66.8; 75.4; 88.2; 102.5; 168.7; 200.3. HRESIMS: calcd for C13H24NO4+ (MH+) 258.1705; found 258.1670.
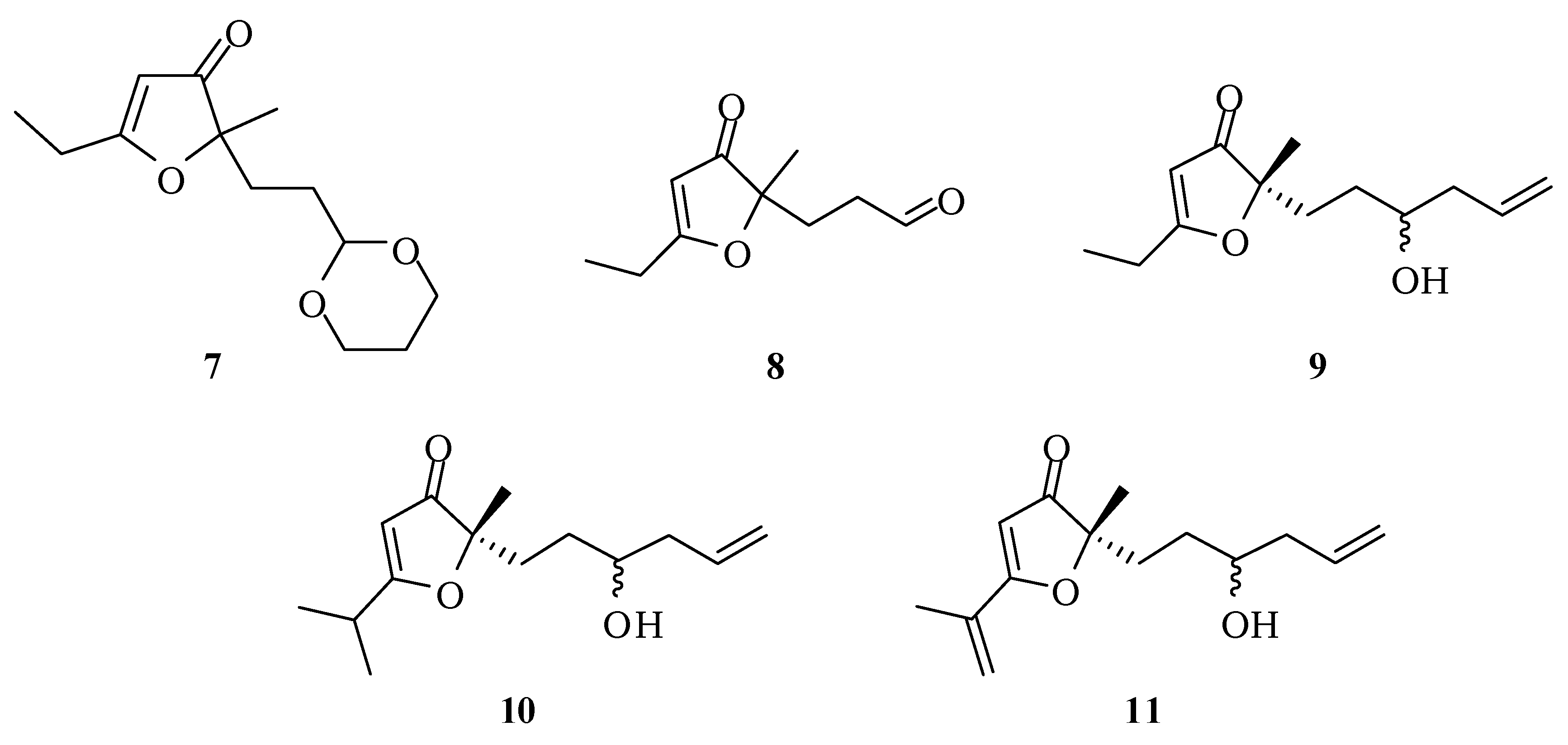
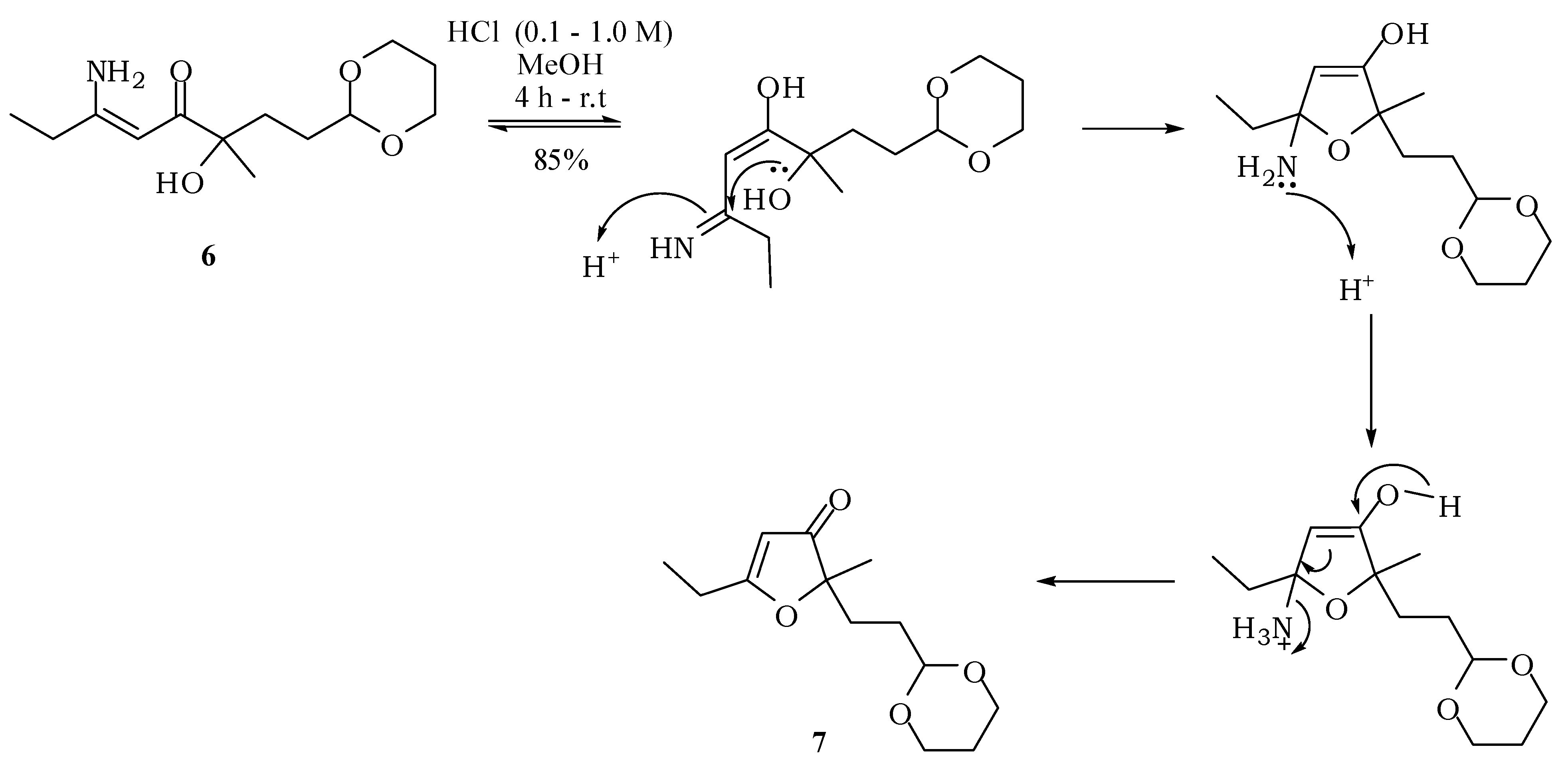
2-[2-(1,3-Dioxan-2-yl)ethyl]-5-ethyl-2-methylfuran-3(2H)-one (7): A solution of 6 (100 mg; 0.39 mmol) in methanol (3 mL) was treated under stirring with aqueous hydrochloric acid (0.1 M; 0.50 mL). After one hour, a more concentrated solution of hydrochloric acid (1 M; 0.50 mL) was added and this treatment was repeated twice. The mixture was then neutralized with saturated solution of NaHCO3 and saturated with NaCl. The extraction with ethyl acetate (3 × 15 mL), drying with anhydrous magnesium sulfate and removal of the solvent gave 7 with 85% (79.6 mg; 0.33 mmol). IR νmax (liquid film): 811, 926, 1007, 1145, 1241, 1287, 1383, 1450, 1591, 1703, 2853, 2927, 2972 cm−1 1H-NMR, 500 MHz, (CDCl3), δ(ppm): 1.15 (t, 3H, J = 7.5 Hz); 1.24 (dtt, 1H, J = 10.4; 4.9 Hz); 1.27 (s, 3H); 1.45 (dt, 2H, J = 8.4; 5.1 Hz); 1.77 (t, 2H, J = 8.4 Hz); 1.99 (dtt, 1H, J = 13.2; 4.9; 2.1 Hz); 2.42 (q, 2H, J = 7.5 Hz); 3.65 (ddd, 2H, J = 11.0; 10.4; 2.1 Hz); 4.01 (dt; 2H; J = 11.0; 4.9 Hz); 4.40 (t, 1H, J = 5.1 Hz); 5.30 (s, 1H). 13C-NMR {1H}, 125 MHz, (CDCl3), δ(ppm): 10.2; 21.8; 24.1; 25.7; 28.8; 30.8; 66.8; 90.3; 101.5; 101.6; 193.7; 206.9. HRESIMS: calcd for C13H21O4+ (MH+) 241.1440; found 241.1433.
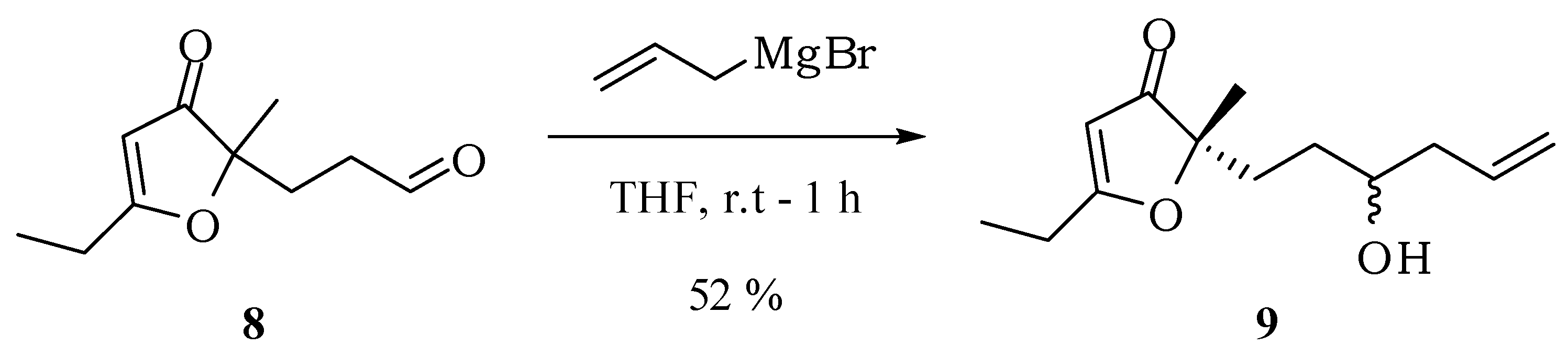
3-(5-Ethyl-2-methyl-3-oxo-2,3-dihydrofuran-2-yl)propanal (8): Hydrochloric acid (2 M, 1.5 mL) was added to a solution of compound 7 (153 mg, 0.64 mmol) in methanol (3 mL) and ethyl acetate (1 mL). After 24 h, another portion of hydrochloric acid (2 M, 1.5 mL) was added. The mixture was kept stirring for 2 days at room temperature. Then, methanol was evaporated under reduced pressure, a saturated solution of NaHCO3 was added until complete neutralization of the acid and the mixture extracted with ethyl acetate (3 × 15 mL). The combined organic extracts were dried with anhydrous magnesium sulfate, filtered and the solvent removed at reduced pressure. The compound 8 was obtained in 60% (70 mg; 0.38 mmol) after purification by column chromatography on silica gel using hexane–ethyl acetate–methanol (60:30:10) as eluent. IR νmax (liquid film): 811, 925, 1057, 1126, 1389, 1593, 1703, 2932, 2978 cm−1 1H-NMR, 500 MHz, (CDCl3), δ(ppm): 1.30 (t, 3H, J = 7.5 Hz); 1.38 (s, 3H); 2.11 (t, 2H, J = 6.0 Hz); 2.28 (qd; 2H; J = 7.5; 1.4 Hz); 2.38 (td, 2H, J = 6.0; 1.8 Hz); 5.43 (s, 1H); 9.71 (t, 1H, J = 1.8 Hz). 13C-NMR {1H}, 125 MHz, (CDCl3), δ(ppm): 10.2; 21.9; 24.1; 28.8; 37.8; 89.6; 101.8; 193.9; 200.6; 206.2. HRESIMS: calcd for C10H15O3+ (MH+) 183.1021; found 183.1016.
3-(5-Ethyl-2-methyl-3-oxo-2,3-dihydrofuran-2-yl)propanal (8): Hydrochloric acid (0.1 M, 0.50 mL) was added to a solution of compound 6 (100 mg, 0.39 mmol) in methanol (3 mL). Keeping the mixture stirring at room temperature, additional portions of hydrochloric acid were added at one hour intervals (3 × 0.5 mL of 1 M HCl and then 4 × 0.5 mL of 2 M HCl). To end the reaction, a saturated solution of sodium bicarbonate was added until complete neutralization of the acid and the mixture extracted with ethyl acetate (3 × 15 mL). The combined organic extracts were dried with anhydrous magnesium sulfate, filtered and the solvent removed at reduced pressure. The compound 8 was obtained in 80% yield (85 mg; 0.47 mmol, 2 steps) after purification by column chromatography on silica gel using hexane/ethyl acetate/methanol (60:30:10) as eluent. IR νmax (liquid film): 811, 925, 1057, 1126, 1389, 1593, 1703, 2932, 2978 cm−1 1.30 (t, 3H, J = 7.5 Hz); 1.38 (s, 3H); 2.11 (t, 2H, J = 6.0 Hz); 2.28 (qd; 2H; J = 7.5; 1.4 Hz); 2.38 (td, 2H, J = 6.0; 1.8 Hz); 5.43 (s, 1H); 9.71 (t, 1H, J = 1.8 Hz). 13C-NMR {1H}, 125 MHz, (CDCl3), δ(ppm): 10.2; 21.9; 24.1; 28.8; 37.8; 89.6; 101.8; 193.9; 200.6; 206.2. HRESIMS: calcd for C10H15O3+ (MH+) 183.1021; found 183.1016.
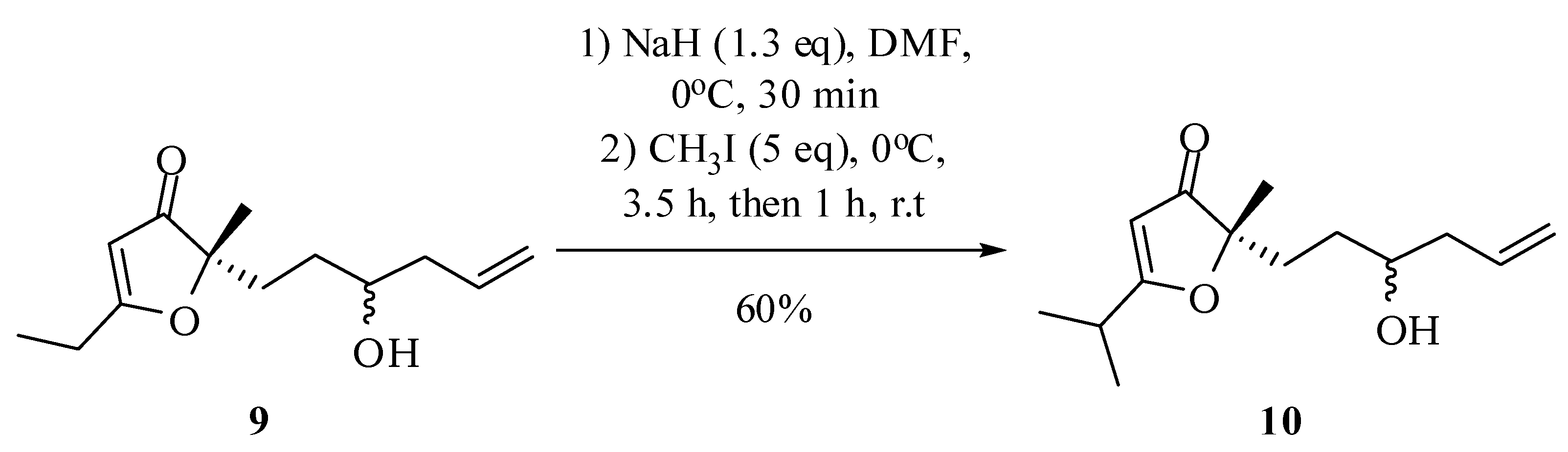
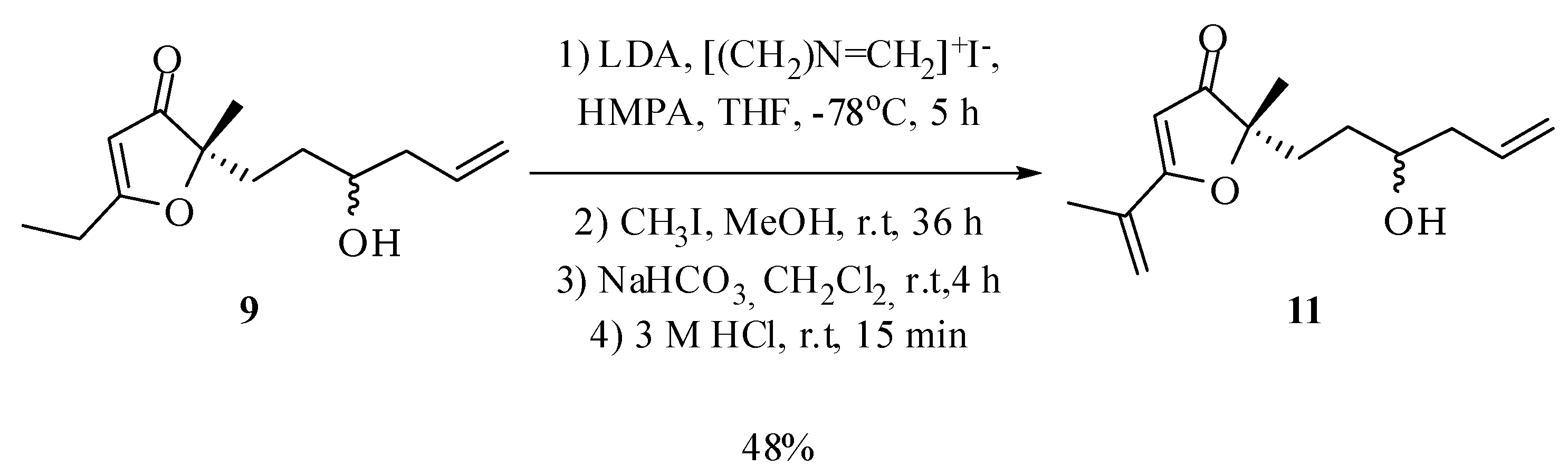
5-Ethyl-2-(3-hydroxyhex-5-en-1-yl)-2-methylfuran-3(2H)-one (9): Preparation of the Grignard reagent: A solution of allyl bromide (0.46 g, 0.33 mL, 3.8 mmol) in diethyl ether (2 mL) was added slowly to magnesium turnings (0.28 g, 11.4 mmol) activated with iodine and diethyl ether (1.2 mL), at 0 °C under an atmosphere of nitrogen, keeping the stirring for an additional hour. A portion of this solution (1.6 mL) was added dropwise, at room temperature, into a flask containing a solution of compound 8 (173.6 mg, 0.95 mmol) in THF (30 mL). Immediately there was formation of a white solid. After 1 h a saturated solution of ammonium chloride (15 mL) was added to dissolve the precipitate and the mixture was extracted with dichloromethane (4 × 15 mL) and ethyl acetate (4 × 15 mL). The combined organic extract was dried with anhydrous magnesium sulfate, filtered and the solvent removed at reduced pressure. The compound 9 in 52% yield (110.8 mg; 0.49 mmol) was obtained by column chromatography on silica gel using hexane/ethyl acetate/methanol (75:25:5) as eluent. IR νmax (liquid film): 812, 923, 997, 1370, 1393, 1450, 1589, 1691, 2927, 2977, 3435 cm−1 1H-NMR, 500 MHz, (CDCl3), δ(ppm): 1.24 (t, 3H, J = 7.6 Hz); 1.25 (t, 3H, J = 7.6 Hz); 1.36 (s, 3H); 1.36 (m overlapped on the s, 1H); 1.50 (ddt, 1H, J = 14.3; 11.5; 4.9 Hz); 1.78 (ddd, 1H, J = 13.7; 11.5; 4.9 Hz); 1.95 (ddd, 1H, J = 13.7; 11.5; 4.9 Hz); 2.12 (dtt, 1H, J = 14.0; 10.4; 1.8 Hz); 2.26 (dddt, 1H, J = 14.5; 6.8; 5.2; 1.8 Hz); 2.51 (qd, 2H, J = 7.6; 1.8 Hz); 3.59 (tt, 1H, J = 7.5; 4.5 Hz); 5.11 (ddt, 1H, J = 17.0; 2.5; 1.8 Hz); 5.14 (ddt, 1H, J = 11.2; 2.5; 1.8 Hz); 5.40 (s, 1H); 5.78 (dddd, 1H, J = 17.0; 11.2; 10.4; 6.8 Hz). 13C-NMR {1H}, 125 MHz, (CDCl3), δ(ppm): 10.3; 22.0 (21.8); 24.2; 30.0; 32.6 (32.5); 41.9 (41.7); 70.2 (70.4); 90.7; 101.6 (101.6); 118.3; 134.4; 193.9 (193.7); 207.3 (207.2). The number in parenthesis refers to duplicated signal assigned to the other diastereoisomer. HRESIMS: calcd for C13H21O3+ (MH+) 225.1491; found 225.1484.
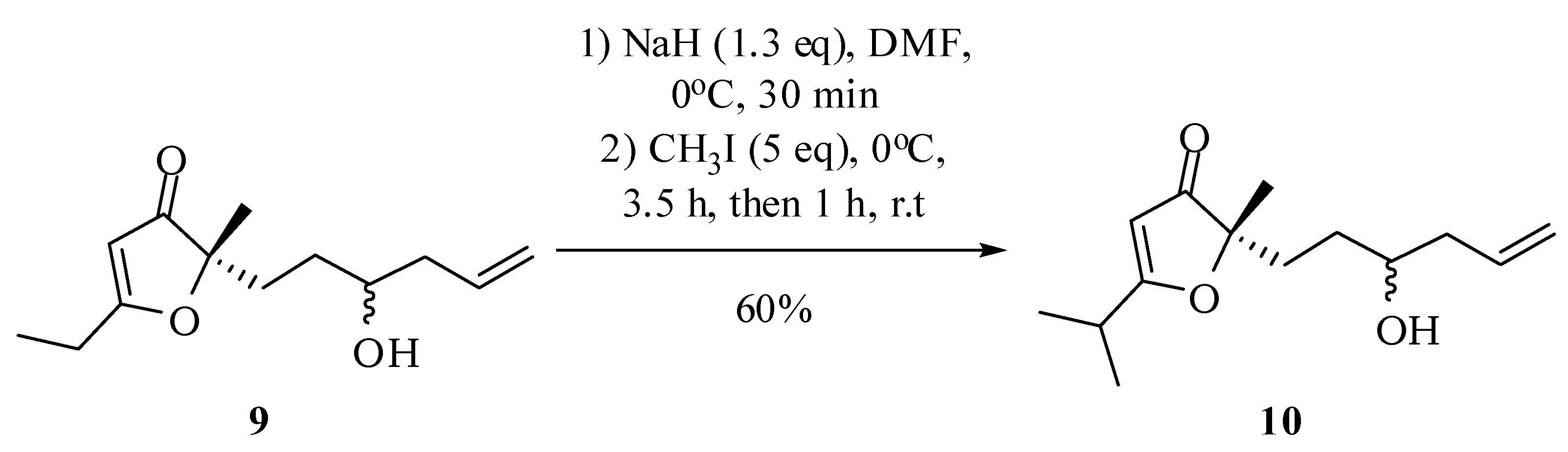
2-(3-Hydroxyhex-5-en-1-yl)-5-isopropyl-2-methylfuran-3(2H)-one (10): Under nitrogen atmosphere and 0 °C, sodium hydride (31.8 mg, 79.4 mmol, 60% in mineral oil) was dissolved in dimethylformamide (2 mL). Then, the compound 9 (137 mg, 0.61 mmol) in dimethylformamide (7 mL) was added and maintained for 30 minutes under these conditions. Then methyl iodide was added (0.43 g, 0.19 mL, 3.05 mmol) at 0 °C and the reaction was maintained under these conditions for 3 h and then another 1 h at room temperature. The compound 10 was obtained after column chromatographic purification on silica gel using hexane–ethyl acetate–methanol (75:25:5) as eluent. IR νmax (liquid film): 806, 1070, 1287, 1585, 1699, 2877, 2932, 2976, 3432 cm−1 1H-NMR, 500 MHz, (CDCl3), δ(ppm): 1.26 (2 d overlapped, 6H, J = 9.0 Hz); 1.36 (s, 3H); 1.36 (m overlapped on the s, 1H); 1.47 (ddt, 1H, J = 14.3; 12.5; 5.9 Hz); 1.78 (ddd, 1H, J = 14.5; 12.5 e 5.9 Hz); 1.96 (ddd, 1H, J = 14.5; 13.0; 5.9 Hz); 2.02 (br s, 1H); 2.11 (dtt, 1H, J = 14.5; 9.3; 2.0 Hz); 2.25 (dddt, 1H, J = 14.5; 7.7; 5.9; 2.0 Hz); 2,74 (sept, 1H, J = 9.0 Hz); 3.59 (tt, 1H, J = 9.3; 5.9 Hz); 5.11 (ddt, 1H, J = 17.2; 2.5; 2.0 Hz); 5.13 (ddt, 1H, J = 11.6; 2.5; 2.0 Hz); 5.38 (s, 1H); 5.80 (dddd, 1H, J = 17.2; 11.6; 9.3; 7.7 Hz). 13C-NMR {1H}, 125 MHz, (CDCl3), δ(ppm): (19.6) 19.5; 22.1 (21.9); 27.4; 30.0; 30.3; 32.6 (32.5); 41.9 (41.7); (70.4) 70.1; 90.5; 100.3 (101.6); 118.3 (118.2); 134.4; 197.5 (197.3); 207.5 (207.3). The number in parenthesis refers to duplicated signal assigned to the other diastereoisomers. HRESIMS: calcd for C14H23O3+ (MH+) 239.3306; found: 239.1642.
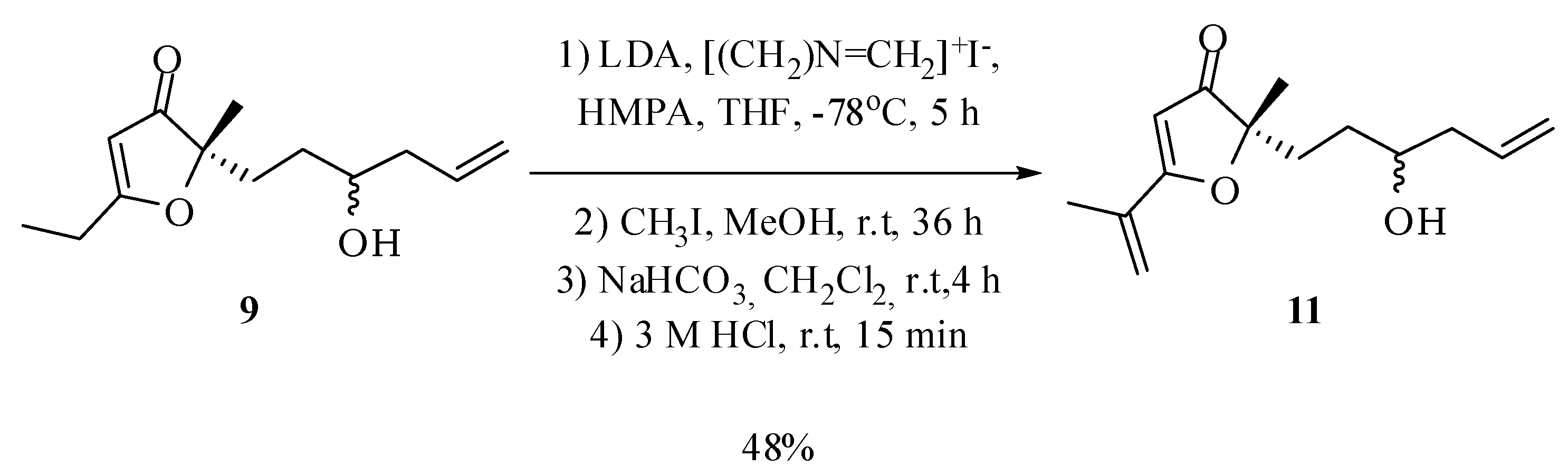
2-(3-Hydroxyhex-5-en-1-yl)-5-isopropenyl-2-methylfuran-3(2H)-one (11): Under nitrogen atmosphere, n-butyllithium (1.39 M, 2.1 mL, 3 mmol) was added to a solution of diisopropylamine (0.30 g, 0.42 mL, 3 mmol) in THF (0.92 mL) at 0 °C. After stirring for 5 minutes under these conditions, the temperature of the reaction mixture was lowered to −78 °C and a solution of compound 9 in THF (2.5 mL) and HMPA (0.45 mL) was added (66.6 mg, 0.3 mmol). The mixture was kept under stirring for 45 minutes at −78 °C and dimethylmethyleneammonium iodide (1.2 g, 6.5 mmol) was added rapidly to the mixture, keeping the stirring for 5 h at −78 °C. Then the temperature was elevated to room temperature and the solvent removed at reduced pressure. Methanol (4.6 mL) and methyl iodide (3.5 mL) were added and the mixture was kept under stirring for 36 h at room temperature. Then the solvent was removed under reduced pressure and to the residue was added a 5% NaHCO3 aqueous solution (14.6 mL) and CH2Cl2 (11.6 mL). The mixture was kept stirring at room temperature for 4 h. Then the reaction mixture was acidified with hydrochloric acid (3 M, 10 mL) and stirred for 15 minutes at room temperature. Finally, the phases were separated and the aqueous phase was extracted with dichloromethane (4 × 15 mL) and ethyl acetate (4 × 15 mL). The combined organic extract was dried with anhydrous magnesium sulfate, filtered and the solvent removed at reduced pressure. The compound 10 in 48% yield (34 mg; 0.14 mmol; 4 steps) was obtained after purification by column chromatography on silica gel using hexane–ethyl acetate–methanol (70:25:5) as eluent. IR νmax (liquid film): 812, 924, 1369, 1448, 1558, 1635, 1686, 2868, 2930, 2977, 3437 cm−1 1H-NMR, 500 MHz, (CDCl3), δ(ppm): 1.40 (s, 3H); 1.40 (m overlapped on the s, 1H); 1.51 (ddt, 1H, J = 14.5; 11.2; 4.9 Hz); 1.91 (ddd, 1H, J = 13.5; 11.2; 4.9 Hz); 1.98 (ddd, 1H, J = 13.5; 11.4; 4.9 Hz); 2.02 (dd, 3H, J = 1.7; 0.9 Hz); 2.10 (dtt, 1H, J = 14.5; 9.0; 2.0 Hz); 2.26 (dddt, 1H, J = 14.5; 6.2; 4.9; 2.0 Hz); 3.59 (tt, 1H, J = 9.0; 4.9 Hz); 5.11 (ddt, 1H, J = 15.7; 2.5; 2.0 Hz); 5.14 (ddt, 1H, J = 10.0; 2.5; 2.0 Hz); 5.44 (m, 1H); 5.59 (s, 1H); 5.78 (dddd, 1H, J = 15.7; 10.0; 9.0; 6.2 Hz). 13C-NMR {1H}, 125 MHz, (CDCl3), δ(ppm): 18.9; 22.1 (21.9); 30.1; 33.0 (32.9); 41.8 (41.7); 70.2 (70.4); 90.6; 101.3 (101.6); 118.3; 121.5 (121.4); 133.6; 134.5; 184.1 (184.0); 207.4 (207.2). The number in parenthesis refers to duplicated signal assigned to the other diastereoisomer. HRESIMS: calcd for C14H21O3+ (MH+) 237.1491; found 237.1485.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}