Solid-Phase Synthesis of a New Diphosphate 5-Aminoimidazole-4-carboxamide Riboside (AICAR) Derivative and Studies toward Cyclic AICAR Diphosphate Ribose

 ,
,
Abstract
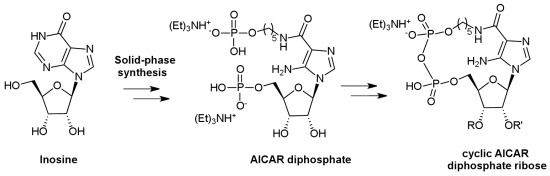
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
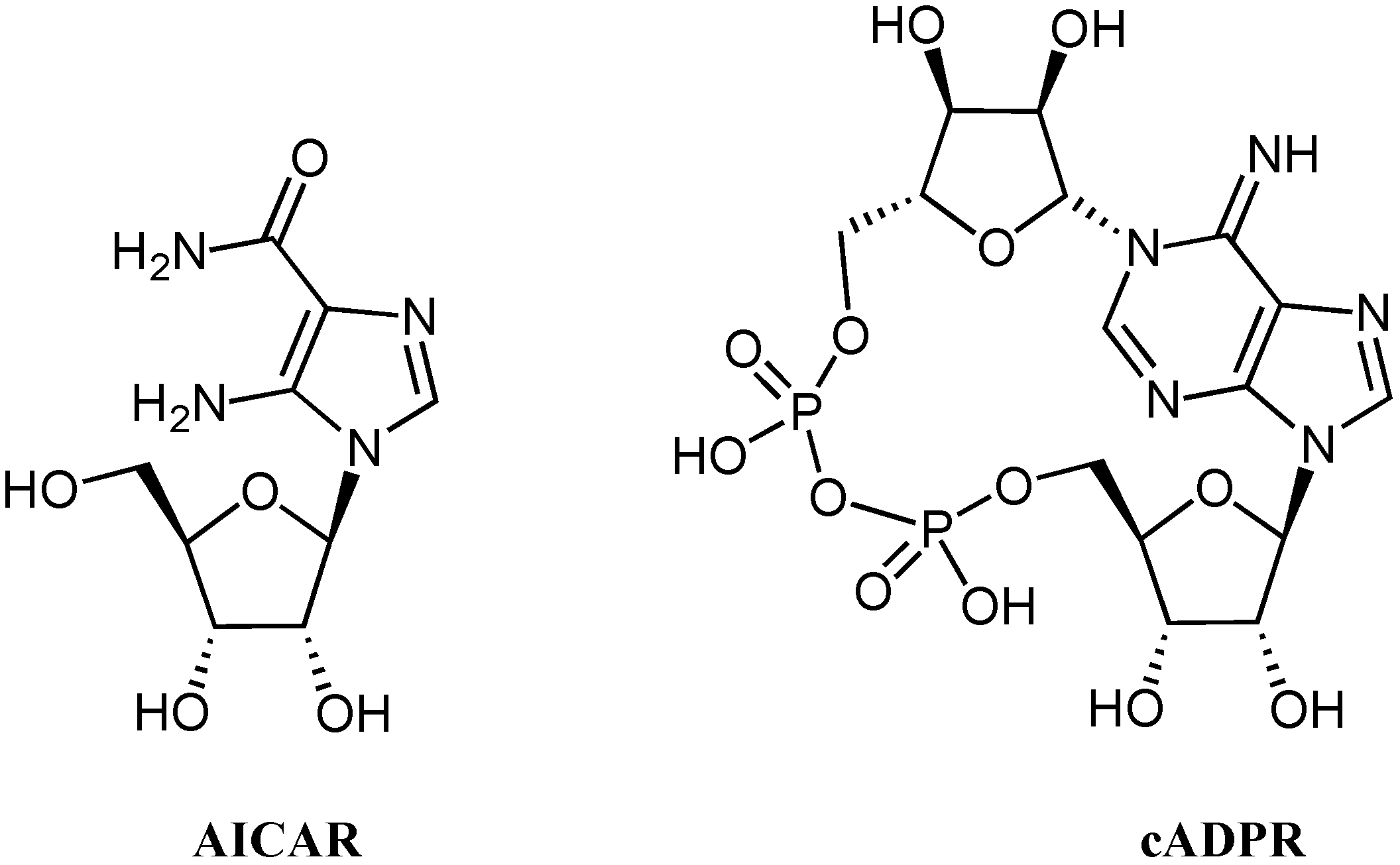
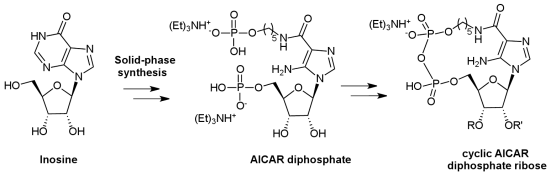
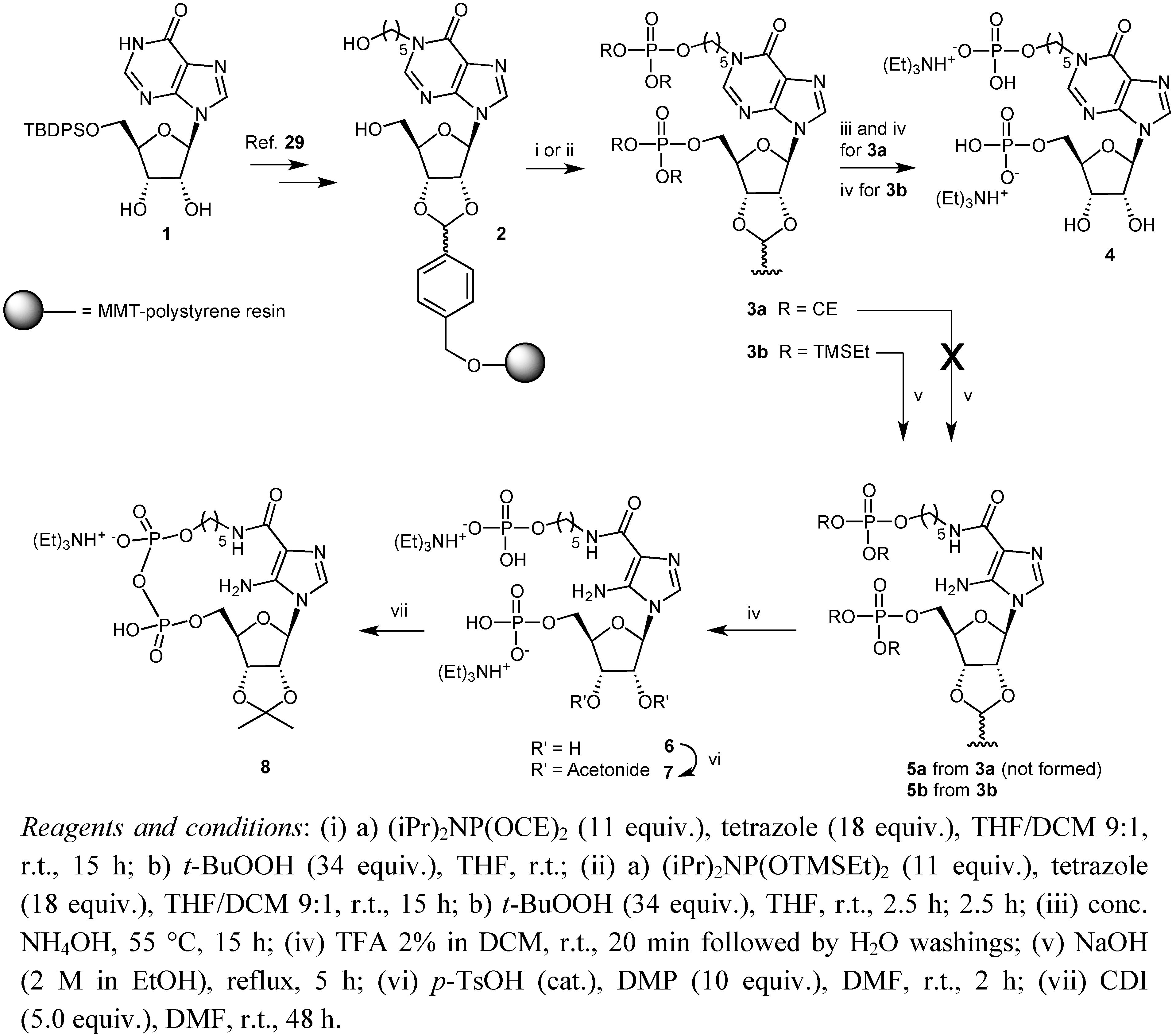
2. Results and Discussion
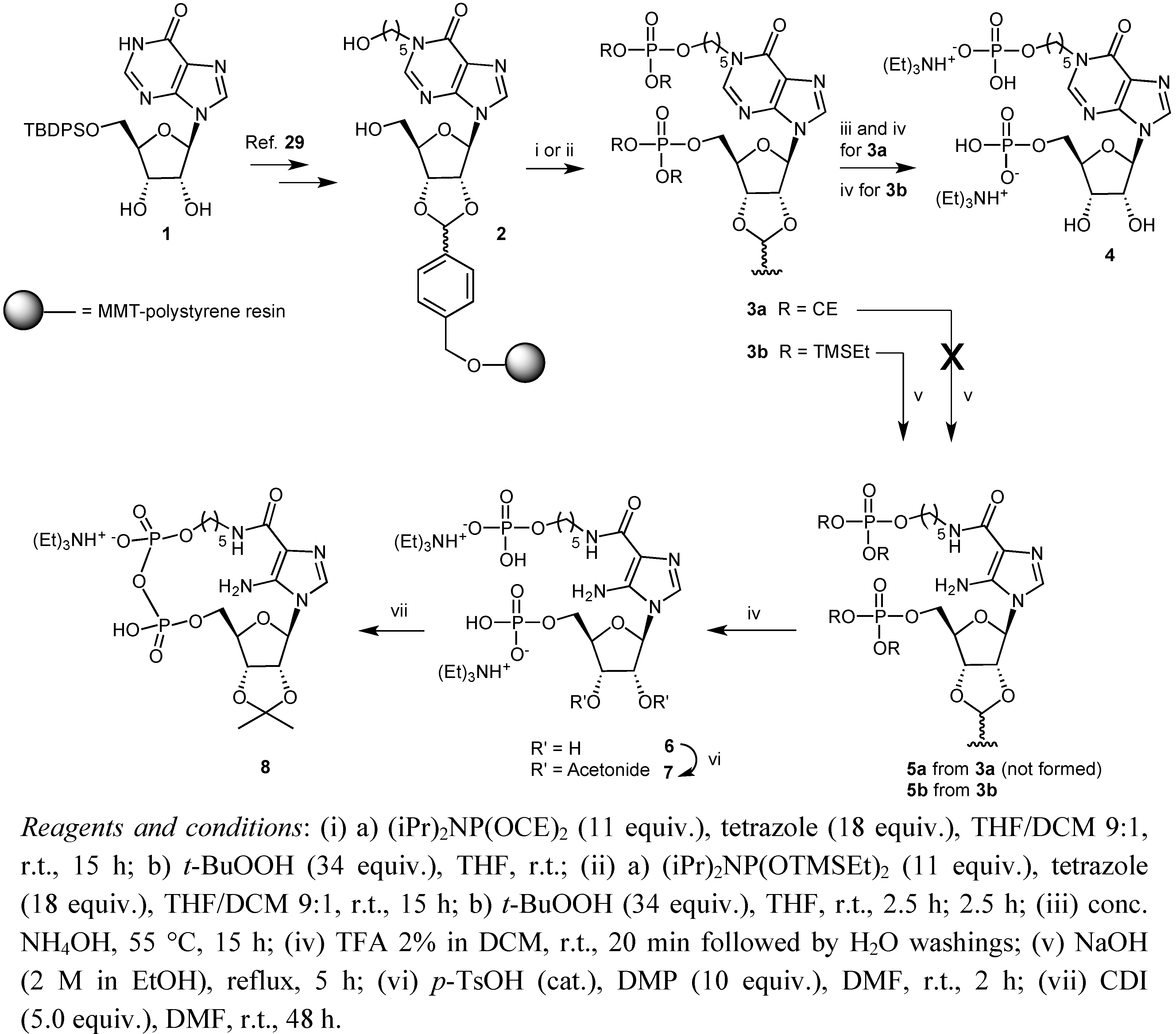
3. Experimental
3.1. General
3.2. Preparation of Solid Supports 3b
3.3. Preparation of Solid Support 5b
3.4. Synthesis of Compound 7
3.5. Pyrophosphate Bond Formation: Synthesis of Compound 8
4. Conclusions
Acknowledgments
- Sample Availability: Samples of the compounds 4, 6, 7, 8 are available from the authors.
References
- Antiviral Nucleosides: Chiral Synthesis and Chemotherapy; Chu, C.K. (Ed.) Elsevier: Amsterdam, The Netherlands, 2003.
- Simons, C.; Wu, Q.; Htar, T.T. Recent Advances in Antiviral Nucleoside and Nucleotide Therapeutics. Curr. Top. Med. Chem. 2005, 5, 1191–1203. [Google Scholar] [CrossRef]
- Lagoja, I.M. Pyrimidine as constituent of natural biologically active compounds. Chem. Biodivers. 2005, 2, 1–50. [Google Scholar] [CrossRef]
- Kimura, K.; Bugg, T.D.H. Recent advances in antimicrobial nucleoside antibiotics targeting cell wall biosynthesis. Nat. Prod. Rep. 2003, 20, 252–273. [Google Scholar] [CrossRef]
- Rachakonda, S.; Cartee, L. Challenges in antimicrobial drug discovery and the potential of nucleoside antibiotics. Curr. Med. Chem. 2004, 11, 775–793. [Google Scholar] [CrossRef]
- Knapp, S. Synthesis of Complex Nucleoside Antibiotics. Chem. Rev. 1995, 95, 1859–1876. [Google Scholar] [CrossRef]
- Miura, S.; Izuta, S. DNA polymerases as targets of anticancer nucleosides. Curr. Drug Targets 2004, 5, 191–195. [Google Scholar] [CrossRef]
- Parker, W.B.; Secrist, J.A.; Waud, W.R. Purine nucleoside antimetabolites in development for the treatment of cancer. Curr. Opin. Investig. Drugs 2004, 5, 592–596. [Google Scholar]
- Szafraniec, S.I.; Stachnick, K.J.; Skierski, J.S. New nucleoside analogs in the treatment of hematological disorders. Acta Pol. Pharm. 2004, 61, 223–232. [Google Scholar]
- Kitanaka, A.; Ohta, Y.; Mihara, K.; Imataki, O.; Ohnishi, H.; Tanaka, T.; Taminato, T.; Kubota, Y. Expression of CD38 with intracellular enzymatic activity: A possible explanation for the insulin release induced by intracellular cADPR. Mol. Cell. Biochem. 2011, 352, 293–299. [Google Scholar] [CrossRef]
- Rutter, G.A.; Silva Xavier, G.; Lecler, I. Roles of 5'-AMP-activated protein kinase (AMPK) in mammalian glucose homoeostasis. Biochem. J. 2003, 375, 1–16. [Google Scholar] [CrossRef]
- Lihn, A.S.; Pedersen, S.B.; Lund, S.; Richelsen, B. The anti-diabetic AMPK activator AICAR reduces IL-6 and IL-8 in human adipose tissue and skeletal muscle cells. Mol. Cell. Endocrinol. 2008, 292, 36–41. [Google Scholar] [CrossRef]
- Lang, C.C.; Wong, A.K.F.; Howie, J.; Petrie, J.R. AMP-activated protein kinase pathway: A potential therapeutic target in cardiometabolic disease. Clin. Sci. 2009, 116, 607–620. [Google Scholar] [CrossRef]
- Boon, H.; Bosselaar, M.; Praet, S.F.E.; Blaak, E.E.; Saris, W.H.M.; Wagenmakers, A.J.M.; Mcgee, S.L.; Tack, C.J.; Smits, P.; Hargreaves, M.; van Loon, L.J.C. Intravenous AICAR administration reduces hepatic glucose output and inhibits whole body lipolysis in type 2 diabetic patients. Diabetologia 2008, 51, 1893–1900. [Google Scholar] [CrossRef]
- Lara, D.R.; Dall’Igna, O.P.; Ghisolfi, E.S.; Brunstein, M.G. Involvement of adenosine in the neurobiology of schizophrenia and its therapeutic implications. Prog. Neuro-psychopharmacol. Biol. Psychiatry 2006, 30, 617–629. [Google Scholar] [CrossRef]
- Karagounis, L.G.; Hawley, J.A. The 5' adenosine monophosphate-activated protein kinase: Regulating the ebb and flow of cellular energetics. Int. J. Biochem. Cell. Biol. 2009, 41, 2360–2363. [Google Scholar] [CrossRef]
- Piccialli, G.; Oliviero, G.; Amato, J.; Borbone, N.; D’Errico, S.; Bucci, E.; Piccialli, V.; Mayol, L. Synthesis of 4-N-alkyl and ribose-modified AICAR analogues on solid support. Tetrahedron 2008, 64, 6475–6481, and references cited in. [Google Scholar]
- Piccialli, G.; Oliviero, G.; D’Errico, S.; Borbone, N.; Amato, J.; Piccialli, V.; Mayol, L. Facile Solid-Phase Synthesis of AICAR 5'-Monophosphate (ZMP) and Its 4-N-Alkyl Derivatives. Eur. J. Org. Chem. 2010, 1517–1524. [Google Scholar]
- Potter, B.V.L.; Walseth, T.F. Medicinal chemistry and pharmacology of cyclic ADP-ribose. Curr. Mol. Med. 2004, 4, 303–311. [Google Scholar] [CrossRef]
- Wagner, G.K.; Guse, A.H.; Potter, B.V.L. Rapid synthetic route toward structurally modified derivatives of cyclic adenosine 5'-diphosphate ribose. J. Org. Chem. 2005, 70, 4810–4819. [Google Scholar] [CrossRef]
- Xu, J.; Yang, Z.; Dammermann, W.; Zhang, L.; Guse, A.H.; Zhang, L. Synthesis and agonist activity of cyclic ADP-ribose analogues with substitution of the northern ribose by ether or alkane chains. J. Med. Chem. 2006, 49, 5501–5512. [Google Scholar] [CrossRef]
- Kudoh, T.; Murayama, T.; Matsuda, A.; Shuto, S. Substitution at the 8-position of 3''-deoxy-cyclic ADP-carbocyclic-ribose, a highly potent Ca2+-mobilizing agent, provides partial agonists. Bioorg. Med. Chem. 2007, 15, 3032–3040. [Google Scholar]
- Zhang, L.R.; Wu, H.M.; Yang, Z.J.; Zhang, L.H. Concise synthesis of novel acyclic analogues of cADPR with an ether chain as the northern moiety. New J. Chem. 2010, 34, 956–966. [Google Scholar]
- Huang, X.; Min, D.; Liu, J.; Zhang, K.; Yang, Z.; Zhang, L.; Zhang, L. Concise syntheses of trifluoromethylated cyclic and acyclic analogues of cADPR. Molecules 2010, 15, 8689–8701. [Google Scholar]
- Dong, M.; Kirchberger, T.; Huang, X.; Yang, Z.J.; Zhang, L.R.; Guse, A.H.; Zhang, L.H. Trifluoromethylated cyclic-ADP-ribose mimic: Synthesis of 8-trifluoromethyl-N(1)-[(5''-O-phosphorylethoxy)methyl]-5'-O-phosphorylinosine-5',5''-cyclic pyrophosphate (8-CF(3)-cIDPRE) and its calcium release activity in T cells. Org. Biomol. Chem. 2010, 8, 4705–4715. [Google Scholar] [CrossRef]
- Moreau, C.; Ashamu, G.A.; Bailey, V.C.; Galione, A.; Guse, A.H.; Potter, B.V.L. Synthesis of cyclic adenosine 5'-diphosphate ribose analogues: A C2'endo/syn "southern" ribose conformation underlies activity at the sea urchin cADPR receptor. Org. Biomol. Chem. 2011, 9, 278–290. [Google Scholar] [CrossRef]
- Catalanotti, B.; De Napoli, L.; Galeone, A.; Mayol, L.; Oliviero, G.; Piccialli, G.; Varra, M. Syntheses of [1-N-15]-2'-deoxyinosine, [4-N-15]-2'-deoxyAICAR, and [1-N-15]-2'-deoxyguanosine. Eur. J. Org. Chem. 1999, 2235–2239. [Google Scholar]
- Piccialli, G.; Oliviero, G.; Amato, J.; Borbone, N.; D’Errico, S.; Mayol, L. Synthesis of N-1 and ribose modified inosine analogues on solid support. Tetrahedron Lett. 2007, 48, 397–400. [Google Scholar] [CrossRef]
- Piccialli, G.; Oliviero, G.; D’Errico, S.; Borbone, N.; Amato, J.; Piccialli, V.; Varra, M.; Mayol, L. A solid-phase approach to the synthesis of N-1-alkyl analogues of cyclic inosine-diphosphate-ribose (cIDPR). Tetrahedron 2010, 66, 1931–1936. [Google Scholar] [CrossRef]
- Kohyama, N.; Yamamoto, Y. A facile synthesis of AICAR from inosine. Synthesis 2003, 17, 2639–2642. [Google Scholar] [CrossRef]
- Ross, K.C.; Rathbone, D.L.; Thomson, W.; Freeman, S. Use of bis[2-(trialkylsilyl)ethyl]N,N-dialkylphosphoramidites for the synthesis of phosphate monoesters. J. Chem. Soc. Perkin Trans. 1995, 4, 421–426. [Google Scholar]
- Galeone, A.; Mayol, L.; Oliviero, G.; Piccialli, G.; Varra, M. Synthesis of a new N-1-pentyl analogue of cyclic inosine diphosphate ribose (cIDPR) as a stable potential mimic of cyclic ADP ribose (cADPR). Eur. J. Org. Chem. 2002, 4234–4238. [Google Scholar]
- Blackburn, M. Nucleic Acids in Chemistry and Biology; Blackburn, M., Gait, M.J., Loakes, D., Williams, D.M., Eds.; RSC Publishing: Cambridge, UK, 2006. [Google Scholar]
- Fukuoka, M.; Shuto, S.; Minakawa, N.; Ueno, Y.; Matsuda, A. An Efficient Synthesis of Cyclic IDP- and Cyclic 8-Bromo-IDP-Carbocyclic-Riboses Using a Modified Hata Condensation Method to Form an Intramolecular Pyrophosphate Linkage as a Key Step. An Entry to a General Method for the Chemical Synthesis of Cyclic ADP-Ribose Analogues. J. Org. Chem. 2000, 65, 5238–5248. [Google Scholar] [CrossRef]
- Fortt, S.M.; Potter, B.V.L. An approach to a carbocyclic analogue of cyclic adenosine 5′-diphosphate ribose. The synthesis and bisphosphorylation of N1-[(1S, 3R)-3-(Hydroxymethyl)cyclopent-1-yl]inosine. Tetrahedron Lett. 1997, 38, 5371–5374. [Google Scholar] [CrossRef]
- Maeda, M.; Patel, A.D.; Hampton, A. Formation of ribonucleotide 2',3'-cyclic carbonates during conversion of ribonucleoside S-phosphates to diphosphates and triphosphates by the phosphorimidazolidate procedure. Nucleic Acids Res. 1977, 4, 2843–2853. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
D’Errico, S.; Oliviero, G.; Borbone, N.; Amato, J.; Piccialli, V.; Varra, M.; Mayol, L.; Piccialli, G. Solid-Phase Synthesis of a New Diphosphate 5-Aminoimidazole-4-carboxamide Riboside (AICAR) Derivative and Studies toward Cyclic AICAR Diphosphate Ribose. Molecules 2011, 16, 8110-8118. https://doi.org/10.3390/molecules16098110
D’Errico S, Oliviero G, Borbone N, Amato J, Piccialli V, Varra M, Mayol L, Piccialli G. Solid-Phase Synthesis of a New Diphosphate 5-Aminoimidazole-4-carboxamide Riboside (AICAR) Derivative and Studies toward Cyclic AICAR Diphosphate Ribose. Molecules. 2011; 16(9):8110-8118. https://doi.org/10.3390/molecules16098110
Chicago/Turabian StyleD’Errico, Stefano, Giorgia Oliviero, Nicola Borbone, Jussara Amato, Vincenzo Piccialli, Michela Varra, Luciano Mayol, and Gennaro Piccialli. 2011. "Solid-Phase Synthesis of a New Diphosphate 5-Aminoimidazole-4-carboxamide Riboside (AICAR) Derivative and Studies toward Cyclic AICAR Diphosphate Ribose" Molecules 16, no. 9: 8110-8118. https://doi.org/10.3390/molecules16098110
APA StyleD’Errico, S., Oliviero, G., Borbone, N., Amato, J., Piccialli, V., Varra, M., Mayol, L., & Piccialli, G. (2011). Solid-Phase Synthesis of a New Diphosphate 5-Aminoimidazole-4-carboxamide Riboside (AICAR) Derivative and Studies toward Cyclic AICAR Diphosphate Ribose. Molecules, 16(9), 8110-8118. https://doi.org/10.3390/molecules16098110