Enzymatic Kinetic Resolution of tert-Butyl 2-(1-Hydroxyethyl)phenylcarbamate, A Key Intermediate to Chiral Organoselenanes and Organotelluranes

Abstract
:
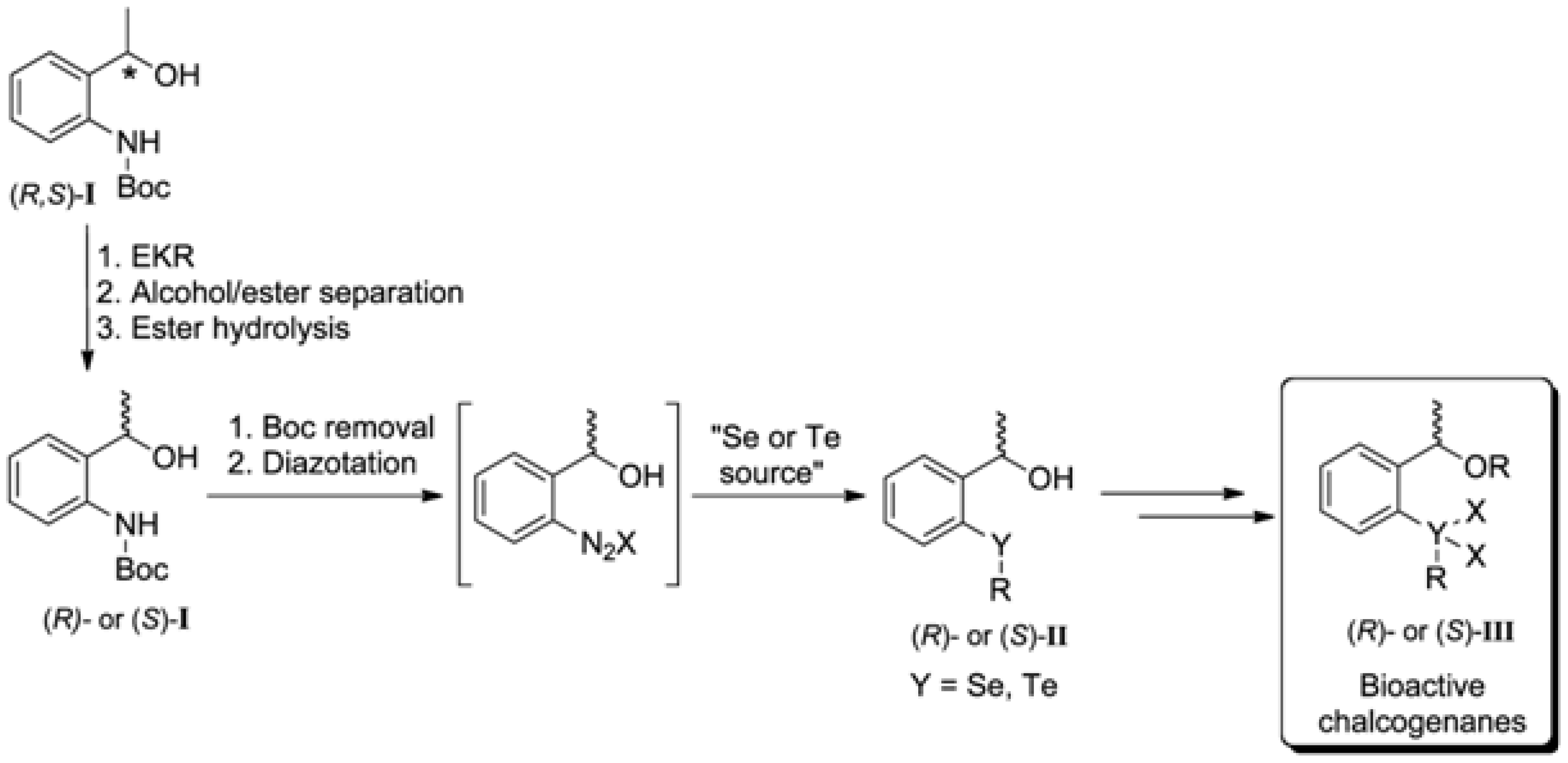
1. Introduction
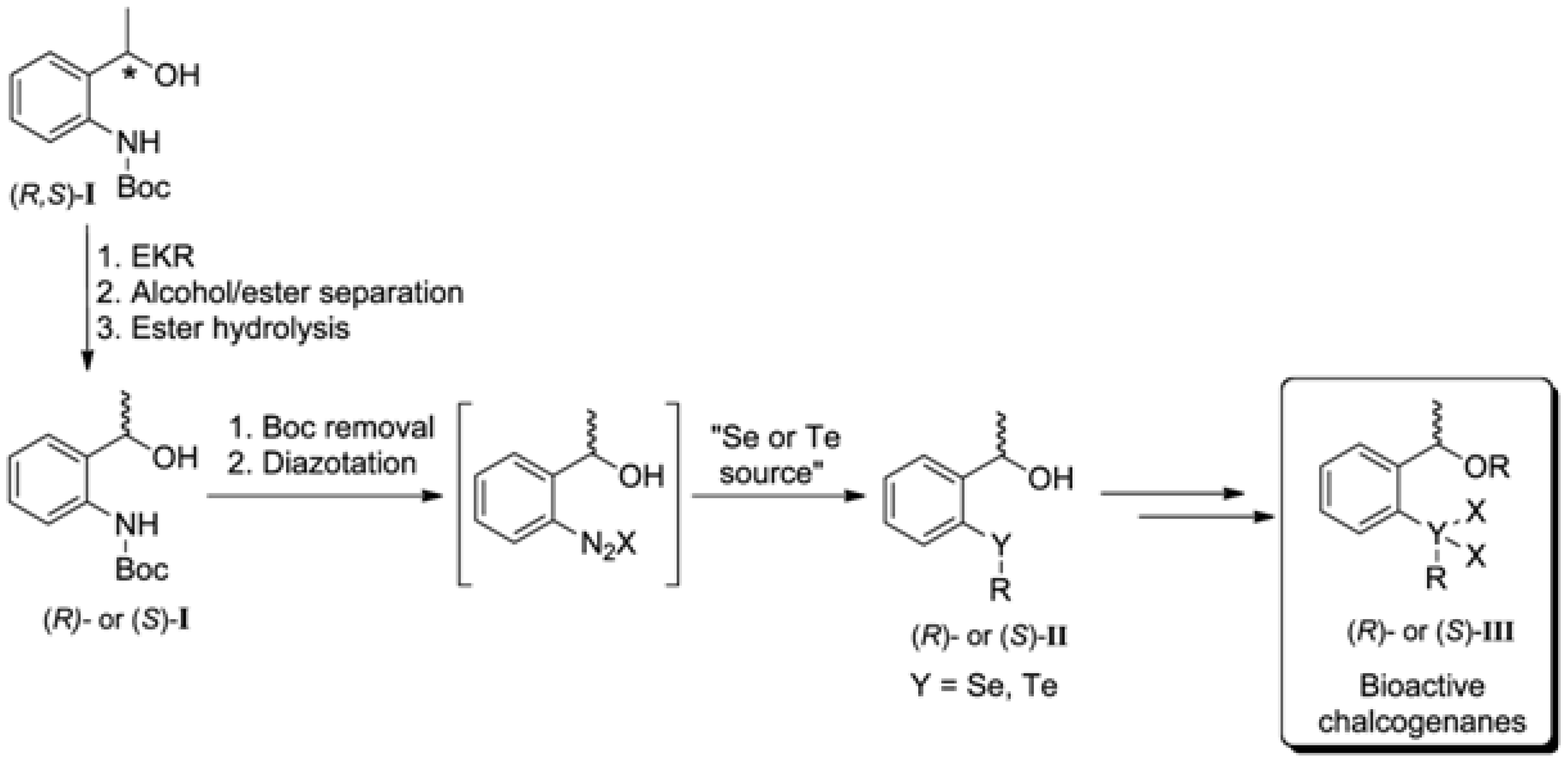
2. Results and Discussion
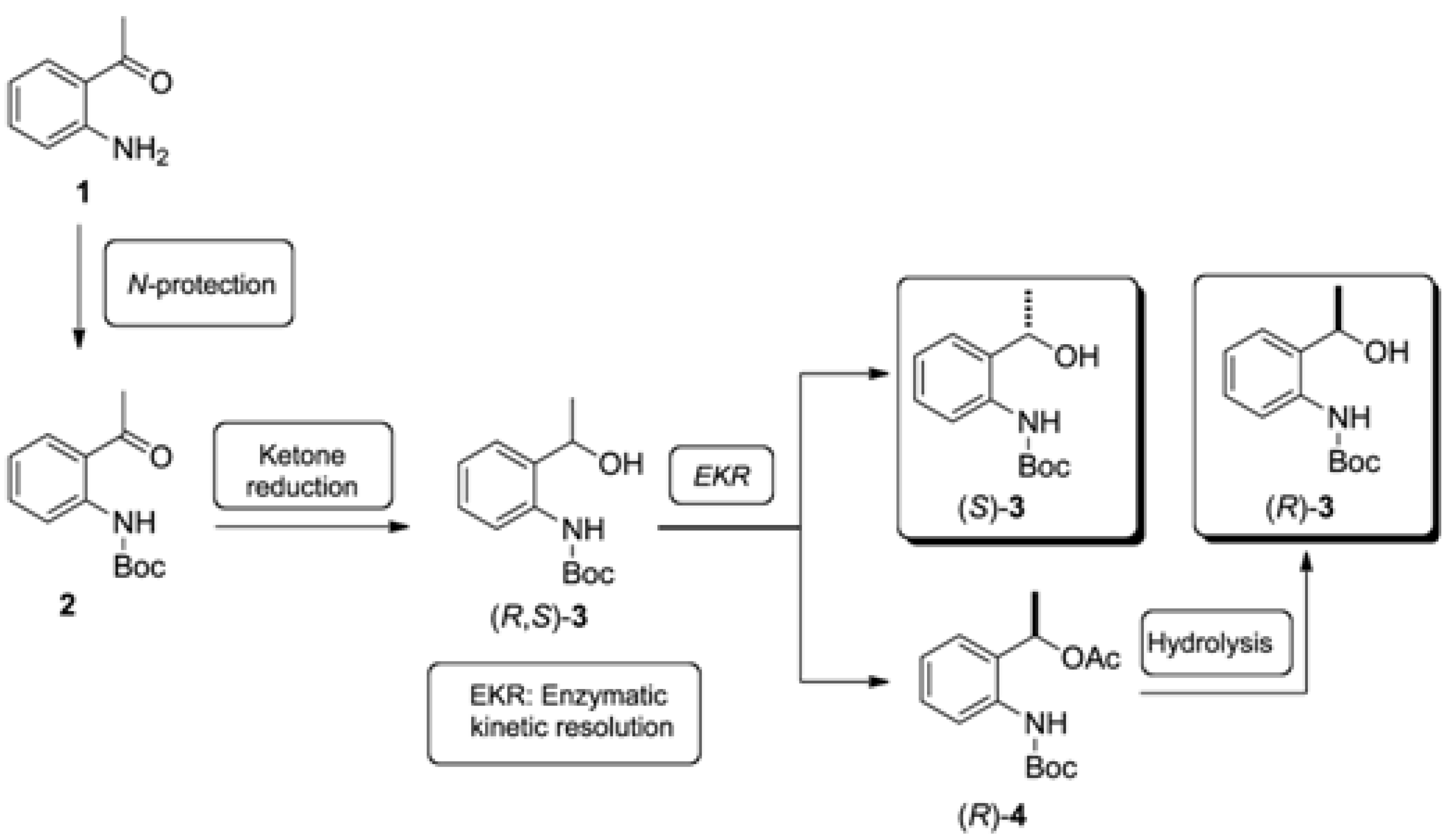
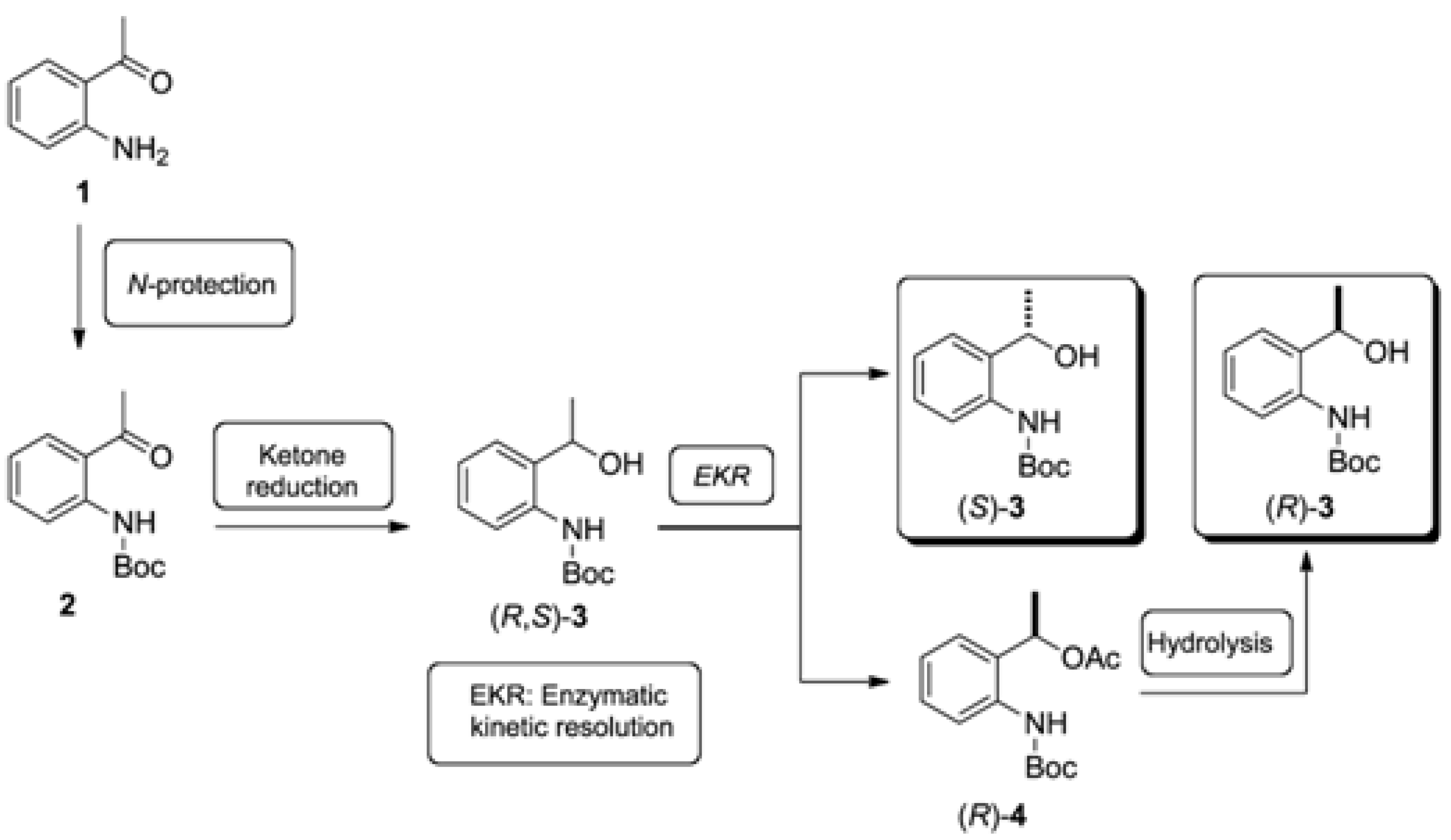
2.1. Synthesis of the (R,S)-tert-butyl 2-(1-Hydroxyethyl)phenylcarbamate (3)

| Entry | Additive (amount) | Solvent | t (°C) | Time (h) | Yield 2 (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | DMAP (1 equiv.) | CH2Cl2 | r.t. | 24 | 60 | [13] |
| 2 | DMAP (1 equiv.) | THF | reflux | 12 | 67 | [13,14] |
| 3 | I2 (2 equiv.) | -- | r.t. | 12 | 37 a | [15] |
| 4 | NaHCO3 (2 equiv.) | Dioxane | r.t. | 12 | traces a | [16] |
| 5 | NaOH (2 equiv.) | Dioxane | 0–r.t. | 12 | traces a | [17] |
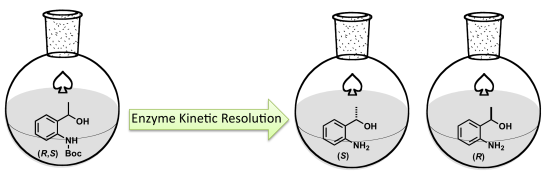
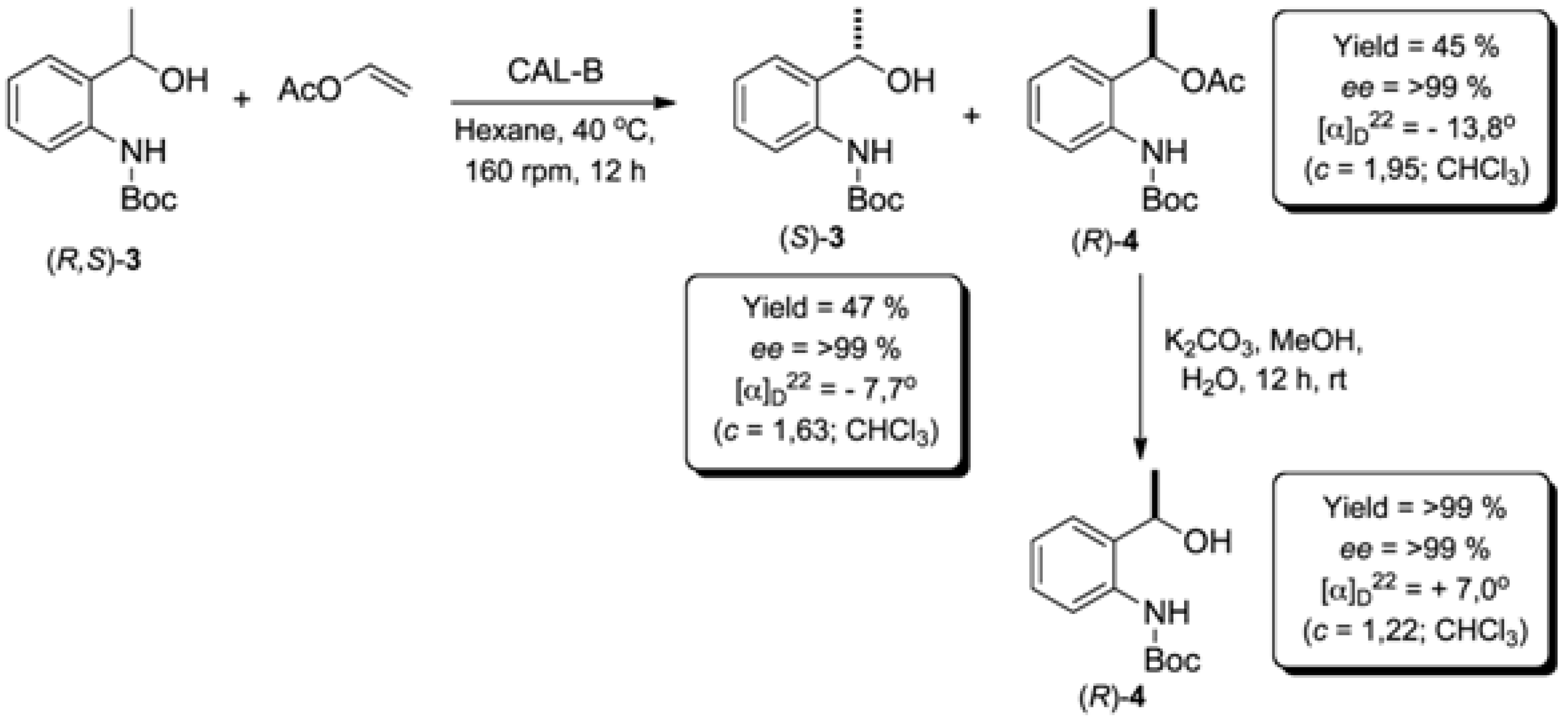
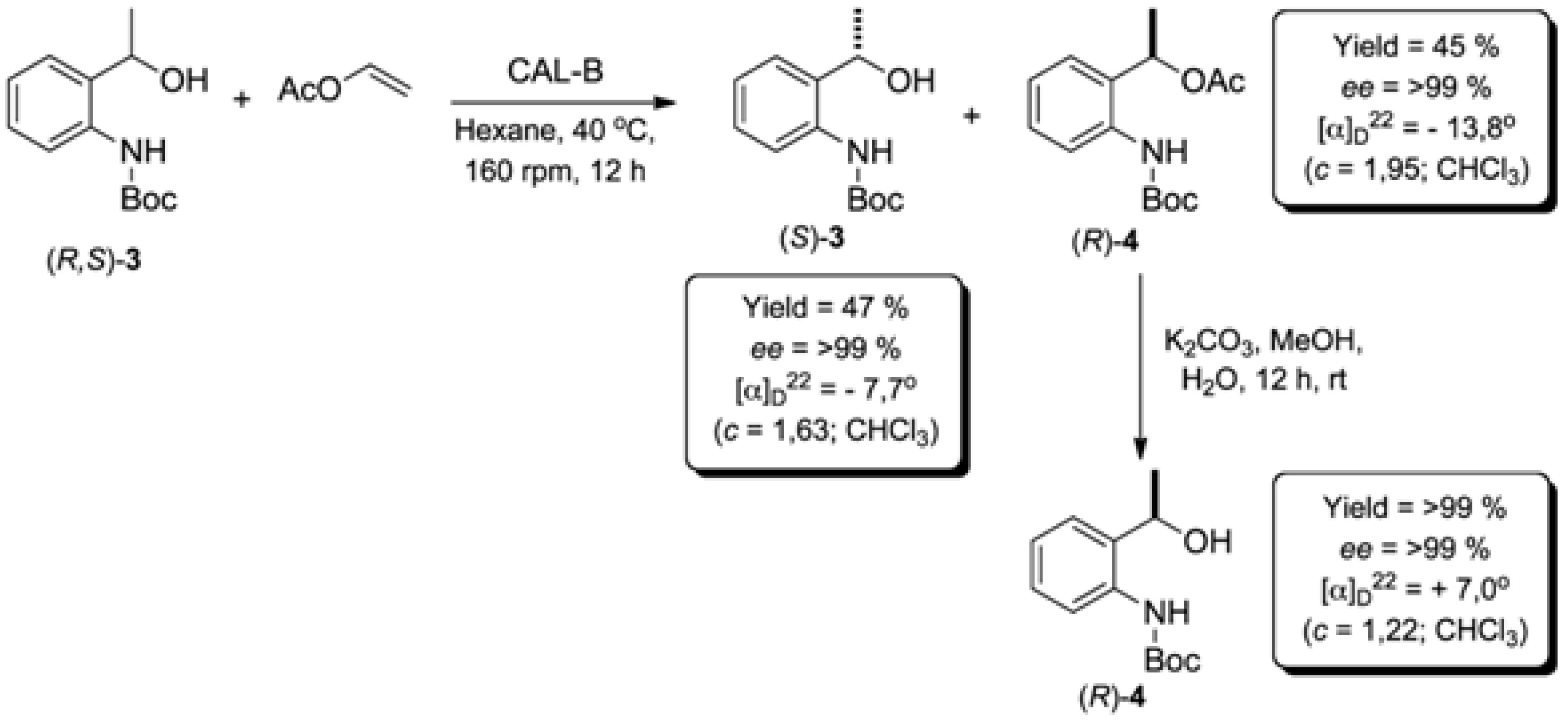
2.2. Enzymatic Kinetic Resolution of the (R,S)-tert-butyl 2-(1-Hydroxyethyl)phenylcarbamate (3)
2.2.1. Screening of Lipases for Kinetic Resolution of (R,S)-3
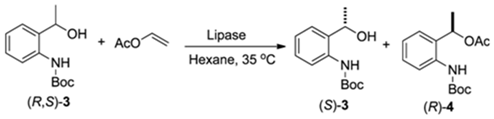
| Entry | Lipase | Time (h) | c a(%) | ee b (%) (S)-3 (R)-4 | E c | |
|---|---|---|---|---|---|---|
| 1 | Candida Antarctica (Novozym® 435; immobilized on acrylic resin) | 12 | 47 | 88 | >99 | >200 |
| 2 | 24 | 50 | >99 | >99 | >200 | |
| 3 | 48 | 51 | >99 | 95 | >200 | |
| 4 | Pseudomonas cepacia (immobilized on ceramics) | 12 | 33 | 49 | >99 | >200 |
| 5 | 24 | 44 | 77 | >99 | >200 | |
| 6 | 48 | 49 | 95 | >99 | >200 | |
| 7 | Pseudomonas cepacia (immobilized on diatomite) | 12 | 16 | 19 | >99 | >200 |
| 8 | 24 | 26 | 34 | >99 | >200 | |
| 9 | 48 | 36 | 56 | >99 | >200 | |
| 10 | Candida rugosa | 12 | 14 | 13 | 81 | 10 |
| 11 | 24 | 17 | 17 | 81 | 11 | |
| 12 | 48 | 21 | 21 | 80 | 11 | |
| 13 | Candida cylindracea | 12 | 12 | 11 | 84 | 12 |
| 14 | 24 | 15 | 14 | 81 | 10 | |
| 15 | 48 | 18 | 17 | 80 | 10 | |
| 16 | Candida sp. (Novozymes® CALB L) | 24 | <5 d | nd | nd | nd |
| 17 | Thermomyces lanuginosus | 24 | <5 d | nd | nd | nd |
| 18 | Rhizomucor miehei | 24 | <5 d | nd | nd | nd |
| 19 | Porcine pancreas lipase | 24 | <5 d | nd | nd | nd |
| 20 | Aspergillus niger | 24 | <5 d | nd | nd | nd |
| 21 | Pseudomonas fluorescens | 24 | <5 d | nd | nd | nd |
| 22 | Penicillium camemberti | 24 | <5 d | nd | nd | nd |
| 23 | Mucor javanicus | 24 | <5 d | nd | nd | nd |
| 24 | Pseudomonas cepacia | 24 | <5 d | nd | nd | nd |
2.2.2. Influence of Solvent in the Kinetic Resolution of (R,S)-3
2.2.3. Influence of Temperature in the Kinetic Resolution of (R,S)-3.
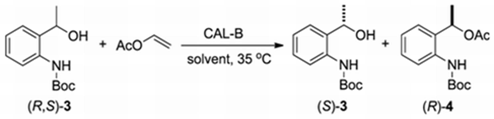
| Entry | Solvent | Time (h) | c a (%) | eeb (%) (S)-3 (R)-4 | E c | |
|---|---|---|---|---|---|---|
| 1 | Hexane | 12 | 47 | 88 | >99 | >200 |
| 2 | 24 | 50 | 99 | >99 | >200 | |
| 3 | Toluene | 12 | 38 | 61 | >99 | >200 |
| 4 | 24 | 47 | 87 | >99 | >200 | |
| 5 | Methyl tert-butyl ether (MTE) | 12 | 42 | 73 | >99 | >200 |
| 6 | 24 | 49 | 94 | >99 | >200 | |
| 7 | Tetrahydrofuran (THF) | 24 | <30 d | nd | nd | nd |
| 8 | Cloroform (CHCl3) | 24 | <30 d | nd | nd | nd |
| 9 | Isobutylic alcohol (i-BuOH) | 24 | <30 d | nd | nd | nd |
| 10 | Diethylic ether (Et2O) | 24 | <30 d | nd | nd | nd |
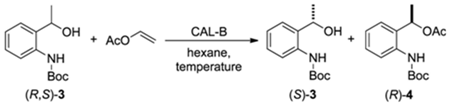
| Entry | Temperature (°C) | Tempo (h) | c a (%) | eeb (%) (S)-3 (R)-4 | Ec | |
|---|---|---|---|---|---|---|
| 1 | 25 | 12 | 45 | 80 | >99 | >200 |
| 2 | 25 | 16 | 46 | 86 | >99 | >200 |
| 3 | 25 | 20 | 49 | 95 | >99 | >200 |
| 4 | 25 | 24 | 50 | >99 | >99 | >200 |
| 5 | 35 | 12 | 48 | 89 | >99 | >200 |
| 6 | 35 | 16 | 50 | >99 | >99 | >200 |
| 7 | 35 | 20 | 50 | >99 | >99 | >200 |
| 8 | 35 | 24 | 50 | >99 | >99 | >200 |
| 9 | 40 | 12 | 50 | >99 | >99 | >200 |
| 10 | 40 | 16 | 50 | >99 | >99 | >200 |
| 11 | 40 | 20 | 51 | >99 | 98 | >200 |
| 12 | 40 | 24 | 52 | >99 | 97 | >200 |
| 13 | 50 | 12 | 50 | >99 | 98 | >200 |
| 14 | 50 | 16 | 50 | >99 | 98 | >200 |
| 15 | 50 | 20 | 52 | >99 | 97 | >200 |
| 16 | 50 | 24 | 53 | >99 | 94 | >200 |
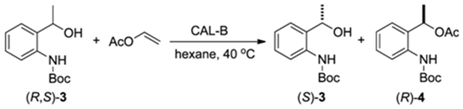
2.2.4. Study of the Ratio of Enzyme to Substrate for Kinetic Resolution of (R,S)-3
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entrada | Massa(mg) | Tempo (h) | c(%) a | ee (%) b 3 4 | E c | |
|---|---|---|---|---|---|---|
| 1 | 10 | 6 | 30 | 63 | >99 | >200 |
| 2 | 10 | 8 | 39 | 75 | >99 | >200 |
| 3 | 10 | 12 | 45 | 90 | >99 | >200 |
| 4 | 20 | 6 | 40 | 84 | >99 | >200 |
| 5 | 20 | 8 | 45 | 93 | >99 | >200 |
| 6 | 20 | 12 | 50 | >99 | >99 | >200 |
| 7 | 40 | 6 | 43 | 86 | >99 | >200 |
| 8 | 40 | 8 | 48 | 97 | >99 | >200 |
| 9 | 40 | 12 | 50 | >99 | >99 | >200 |
| 10 | 80 | 6 | 48 | 97 | >99 | >200 |
| 11 | 80 | 8 | 50 | >99 | >99 | >200 |
| 12 | 80 | 12 | 51 | >99 | 97 | >200 |
| 13 | 100 | 6 | 50 | >99 | >99 | >200 |
| 14 | 100 | 8 | 50 | >99 | >99 | >200 |
| 15 | 100 | 12 | 52 | >99 | 95 | >200 |

| # | ee (%) | [a]D |
|---|---|---|
| Literature [18] | 93 | +52, 5 (c = 1,0; CHCl3) |
| This work | >99 | +63,1 (c = 1,1; CHCl3) |
3. Experimental Section
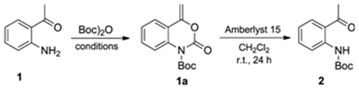
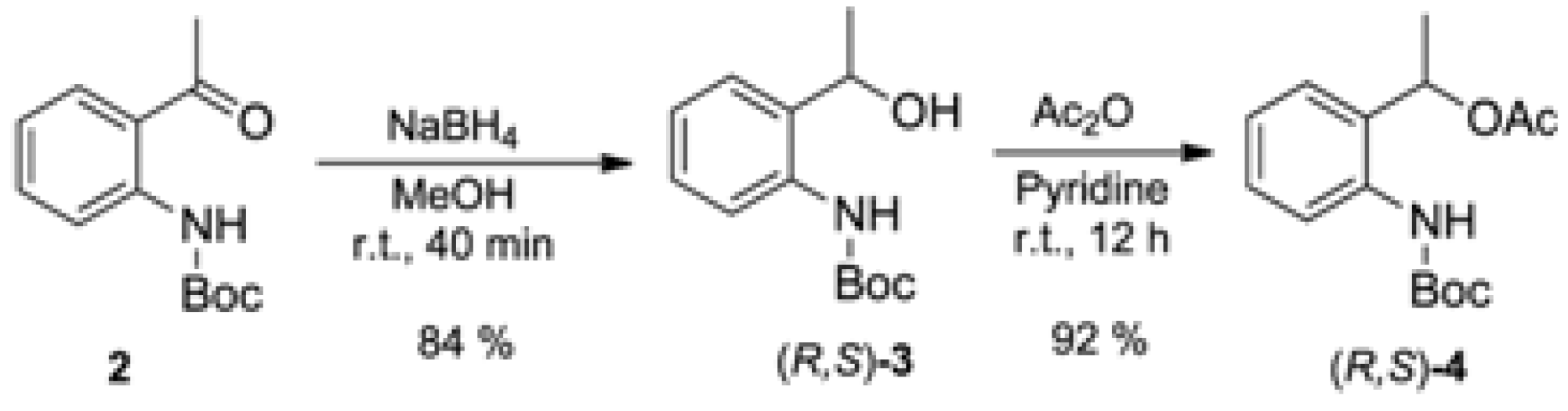
3.1. Synthesis of tert-butyl (2-Acetylphenyl)carbamate (2) (Adapted from References [13,14])
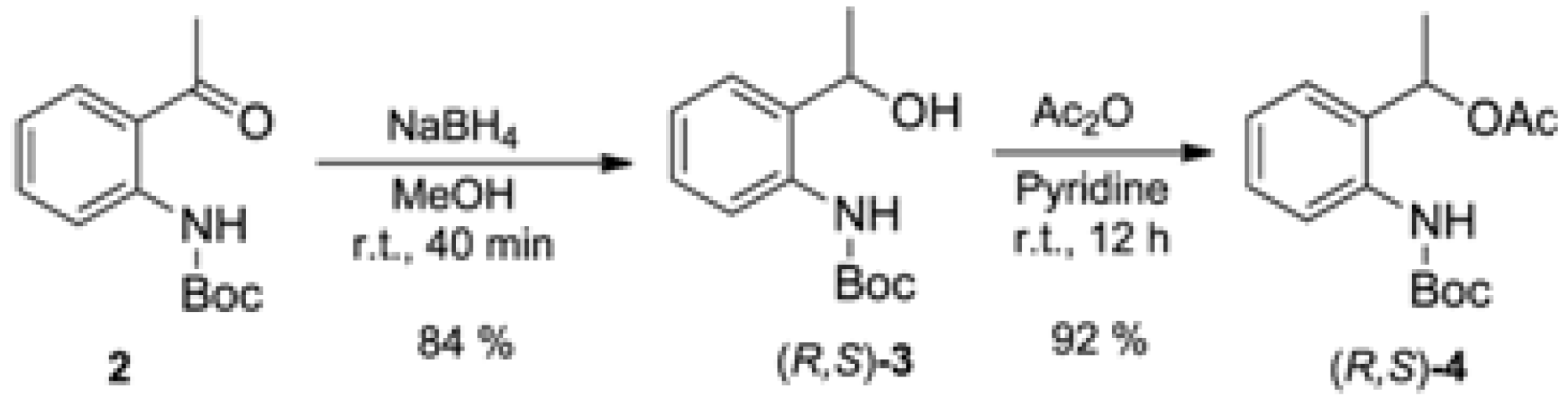
3.2. Synthesis of Racemic tert-butyl (2-(1-Hydroxyethyl)phenyl)carbamate [(R,S)-3]
3.3. Synthesis of Racemic 1-(2-((tert-Butoxycarbonyl)amino)phenyl)ethyl acetate [(R,S)-4]
3.4. Enzymatic Kinetic Resolution of the (R,S)-tert-butyl 2-(1-Hydroxyethyl)phenylcarbamate [(R,S)-3]
3.5. HPLC Analysis of (S)- and (R)-tert-butyl (2-(1-Hydroxyethyl)phenyl)carbamate (3)
3.6. General Procedure to Remove Boc-Protecting Group (Adapted from Reference [19])
4. Conclusions
Acknowledgments
References
- Mugesh, G.; du Mont, W.-W.; Sies, H. Chemistry of biologically important synthetic organoselenium compounds. Chem. Rev. 2001, 101, 2125–2179. [Google Scholar]
- Ba, L.A.; Döring, M.; Jamier, V.; Jacob, C. Tellurium: An element with great biological potency and potential. Org. Biomol. Chem. 2010, 8, 4203–4216. [Google Scholar]
- Brabak, K.P.; Mugesh, G. Functional mimics of glutathione peroxidase bioinspired synthetic antioxidants. Acc. Chem. Res. 2010, 43, 1408–1419. [Google Scholar] [CrossRef]
- Alberto, E.E.; do Nascimento, V.; Braga, A.L. Catalytic application of selenium and tellurium compounds as glutathione peroxidase enzyme mimetics. J. Braz. Chem. Soc. 2010, 21, 2032–2041. [Google Scholar] [CrossRef]
- Piovan, L.; Alves, M.F.M.; Juliano, L.; Bromme, D.; Cunha, R.L.O.R.; Andrade, L.H. Structure-activity relationships of hypervalent organochalcogenanes as inhibitors of cysteine cathepsins V and S. Bioorg. Med. Chem. 2011, 19, 2009–2014. [Google Scholar] [CrossRef]
- Piovan, L.; Alves, M.F.M.; Juliano, L.; Bromme, D.; Cunha, R.L.O.R.; Andrade, L.H. Chemoenzymatic synthesis of organoselenanium(IV) compounds and their evaluation as cysteine proteases inhibitors. J. Braz. Chem. Soc. 2010, 21, 2108–2118. [Google Scholar]
- Cunha, R.L.O.R.; Urano, M.E.; Chagas, J.R.; Almeida, P.C.; Bincoleto, C.; Tersariol, I.L.S.; Comasseto, J.V. Tellurium-based cysteine inhibitors: Evaluation of novel organotellurium(IV) compounds as inhibitors of human cathepsin B. Bioorg. Med. Chem. Lett. 2005, 15, 755–760. [Google Scholar] [CrossRef]
- Cunha, R.L.O.R.; Gouvêa, I.E.; Feitosa, G.P.V.; Alves, M.F.M.; Bromme, D.; Comasseto, J.V.; Tersariol, I.L.S.; Juliano, L. Irreversible inhibition of human cathepsins B, L, S, and K by hypervalent tellurium compounds. Biol. Chem. 2009, 390, 1205–1212. [Google Scholar] [CrossRef]
- Piovan, L.; Wu, L.; Zhang, Z.Y.; Andrade, L.H. Hypervalent organochalcogenanes as inhibitors of protein tyrosine phosphatases. Org. Biomol. Chem. 2011, 9, 1347–1351. [Google Scholar] [CrossRef]
- Gouveia, I.E.; Santos, J.A.N.; Burlandy, F.M.; Tersariol, I.L.S.; da Silva, E.E.; Juliano, M.A.; Juliano, L.; Cunha, R.L.O.R. Poliovirus 3C proteinase inhibition by organotelluranes. Biol. Chem. 2011, 392, 587–591. [Google Scholar]
- Silva, A.; Andrade, L.H. First chemoenzymatic synthesis of organoselenium amines and amides. Tetrahedron: Asymmetry 2008, 19, 1175–1181. [Google Scholar]
- Omori, A.T.; Assis, L.F.; Andrade, L.H.; Comasseto, J.V.; Porto, A.L.M. Enantiomerically pure organoseleno-1-arylethanols by enzymatic resolution with Candida antarctica lipase: Novozym 435. Tetrahedron: Asymmetry 2007, 18, 1048–1053. [Google Scholar] [CrossRef]
- Broutin, P.-E.; Hilty, P.; Thomas, A.W. An efficient synthesis of ortho-N-Boc-arylmethyl ketone derivatives. Tetrahedron Lett. 2003, 44, 6429–6432. [Google Scholar] [CrossRef]
- Darnbrough, S.; Mervic, M.; Condon, S.M.; Burns, C.J. An improved synthesis of N-Boc protected aryl amines. Syn. Commun. 2001, 31, 3273–3280. [Google Scholar]
- Varala, R.; Nuvula, S.; Adapa, S.R. Molecular iodine-catalyzed facile procedure for N-Boc protection of amines. J. Org. Chem. 2006, 71, 8283–8286. [Google Scholar]
- Braga, A.L.; Lüdtke, D.S.; Paixão, M.W.; Alberto, E.E.; Stefani, H.A.; Juliano, H. Straightforward synthesis of non-natural selenium containing amino acid derivatives and peptides. Eur. J. Org. Chem. 2005, 4260–4264. [Google Scholar]
- Chaume, G.; Kuligowski, C.; Benzzenine-Laffolée, S.; Ricard, L.; Pancrazi, A.; Ardisson, J. Zinc-acetic acid reductive cyclisation in a two-step synthesis of the S1-N10 nine-membered lactone core of ent-griseoviridin. Synthesis 2004, 18, 3029–3036. [Google Scholar]
- Mannan, S.; Sekar, G. An enantiopure galactose oxidase model: Synthesis of chiral amino alcohols through oxidative kinetic resolution catalyzed by a chiral copper complex. Tetrahedron: Asymmetry 2009, 20, 497–502. [Google Scholar]
- Stahl, G.L.; Walter, R.; Smith, C.W. General procedure for the synthesis of mono-N-acylated 1,6-diaminohexanes. J. Org. Chem. 1978, 43, 2285–2286. [Google Scholar]
- Sample Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Piovan, L.; Pasquini, M.D.; Andrade, L.H. Enzymatic Kinetic Resolution of tert-Butyl 2-(1-Hydroxyethyl)phenylcarbamate, A Key Intermediate to Chiral Organoselenanes and Organotelluranes. Molecules 2011, 16, 8098-8109. https://doi.org/10.3390/molecules16098098
Piovan L, Pasquini MD, Andrade LH. Enzymatic Kinetic Resolution of tert-Butyl 2-(1-Hydroxyethyl)phenylcarbamate, A Key Intermediate to Chiral Organoselenanes and Organotelluranes. Molecules. 2011; 16(9):8098-8109. https://doi.org/10.3390/molecules16098098
Chicago/Turabian StylePiovan, Leandro, Monica D. Pasquini, and Leandro H. Andrade. 2011. "Enzymatic Kinetic Resolution of tert-Butyl 2-(1-Hydroxyethyl)phenylcarbamate, A Key Intermediate to Chiral Organoselenanes and Organotelluranes" Molecules 16, no. 9: 8098-8109. https://doi.org/10.3390/molecules16098098
APA StylePiovan, L., Pasquini, M. D., & Andrade, L. H. (2011). Enzymatic Kinetic Resolution of tert-Butyl 2-(1-Hydroxyethyl)phenylcarbamate, A Key Intermediate to Chiral Organoselenanes and Organotelluranes. Molecules, 16(9), 8098-8109. https://doi.org/10.3390/molecules16098098