Abstract
The diastereotopy of the methylene protons at positions 2 and 6 in 1,4-dihydropiridine derivatives with various substituents has been investigated. NMR spectroscopy and quantum chemistry calculations show that the CH···O intramolecular hydrogen bond is one of the factors amplifying the chemical shift differences in the 1H-NMR spectra.
1. Introduction
The 1,4-dihydropyridine (1,4-DHP) scaffold is a common component of pharmacologically active molecules which possess a variety of biological activities [1]. Several 1,4-DHP derivatives play an important role in regulating the decrease or increase of the penetration of calcium ions through membranes into cells. Substituents at the 2,6-, 3,5-, 4- and 1- positions of 1,4-DHP derivatives influence their structure-activity relationships, so structural and conformational investigations of these systems are important to gain insight into the mechanisms of their physiological action [2].
2. Results and Discussion
In recent years many studies attempting to modify the 2,6-methyl groups of 1,4-DHP by introducing various substituents have been performed [3,4,5,6]. On the other hand cationic amphiphilic 1,4-DHP derivatives have gained significance as useful transport molecules for the delivery of nucleotides into target cells [4,7].
Our attention was drawn to the fact that the CH2X protons of substituents in positions 2 and 6 of symmetrically substituted 1,4-dihydropyridine rings (compounds 1-3, 5, Table 1) become diastereotopic in the presence of different substituents at position 4, thereby, providing an AB system in the corresponding 1H-NMR spectra. The extent of the observed non-equivalence of the methylene protons should be influenced by the spatial conformation of side chains in the molecule and/or the anisotropy of the substituents.

Table 1.
Characterization of compounds 1–5. 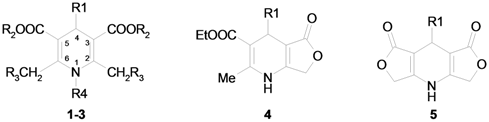
| Comp. | R1 | R2 | R3 | R4 | Yield,% | Reference |
|---|---|---|---|---|---|---|
| 1a | Me | Et | Br | H | 59 | |
| 1b | Ph | Et | Br | Me | 88 | |
| 1c | PhCF3-o | Et | Br | H | 76 | |
| 1d | PhOCHF2-o | (CH2)2OC3H7-n | Br | H | 61 | |
| 1e | COOMe | Et | Cl | H | 87 | |
| 1f | Ph | Et | Br | H | 47 | [3] |
| 1g | PhOCHF2-o | Me | Br | H | 86 | [3] |
| 1h | Ph | C12H25 | Br | H | 72 | [5] |
| 1i | PhOCHF2-o | Me | Br | Me | 89 | |
| 2a | Me | Et | Py+Br− | H | 63 | |
| 2b | Ph | Et | Py+Br− | Me | 66 | |
| 2c | PhCF3-o | Et | Py+Br− | H | 93 | |
| 2d | PhOCHF2-o | (CH2)2OC3H7-n | Py+Br− | H | 74 | |
| 2e | COOMe | Et | Py+Cl− | H | 91 | |
| 2f | Ph | Et | Py+Br− | H | 63 | [6] |
| 2g | PhOCHF2-o | Me | Py+Br− | H | 64 | [4] |
| 2h | PhOCHF2-o | Me | Py+Br− | Me | 84 | |
| 3a | Ph | C10H21 | Py+Br− | H | 63 | [4] |
| 3b | Ph | C12H25 | Py+Br− | H | 82 | [4] |
| 3c | Ph | C14H29 | Py+Br− | H | 35 | [4] |
| 3d | Ph | C16H33 | Py+Br− | H | 33 | [7] |
| 3e | Ph | C12H25 | Py+Br− | Me | 38 | [7] |
| 4a | Ph | 20 | [10] | |||
| 4f | PhOCHF2-o | 33 | [11] | |||
| 5a | Ph | 50 | [12] | |||
| 5f | PhOCHF2-o | 60 | [13] |
The synthetic pathway for obtaining 1,4-dihydropyridine derivatives bearing substituents on the 2,6-methylene groups (Table 1) involved Hantzsch synthesis, followed by bromination of the methyl groups with NBS and nucleophilic substitution of bromine by pyridine giving the target compounds 1a–d, i and 2a–d, h (Scheme 1).
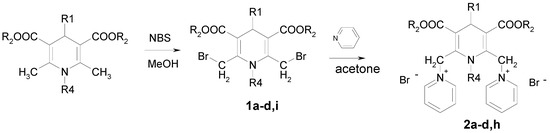
Scheme 1.
Synthesis of compounds 1 and 2.
A different approach was elaborated for the synthesis of 4-methoxycarbonyl derivative 1e (Scheme 2). Thus, the condensation of ethyl 4-chloroacetoacetate with glyoxylic acid monohydrate in methanol at rt in the presence of piperidine/acetate as catalyst provided (E,Z)-2-(2-chloroacetyl)-but-2-enedioic acid 1-ethyl ester, which was used without isolation in the next reaction with ethyl 3-amino-4-chlorobut-2-enoate. The obtained 3,5-diethyl 2,6-bis(chloromethyl)-1,4-dihydropyridine-3,4,5-tri-carboxylate was esterified with methanol using conc. sulfuric acid as catalyst to afford compound 1e. The target compound 2e was successfully obtained via nucleophilic substitution of chlorine by pyridine in the presence of potassium iodide in dry acetone.
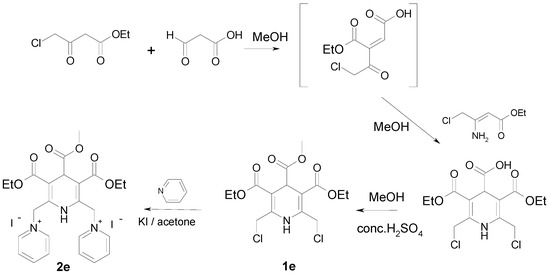
Scheme 2.
Synthesis of compounds 1e, 2e.
Compounds 1f–h, 2f–g, 3a–e, 4a,f and 5a,f were prepared according to the literature, as mentioned in Table 1. The 4-substituent in the molecule is located at a considerable distance from the 2- (6–) methylene group, and the experimentally observed difference in the 1H-NMR chemical shifts of the CH2 groups in cyclic lactones 4–5 does not exceed 0.08 ppm (Table 2); a similar value was observed in [8]. However, its potential anisotropic influence on the 2,6-CH2 methylene proton chemical shifts can’t be ignored. In monocyclic derivatives 1–3 the conformation of the 3,5-ethoxycarbonyl groups should be another important factor in the anisotropy. Indeed in these compounds the chemical shift difference for CH2X protons becomes very significant (Table 2), so we can assume that the difference in chemical shifts of CH2X groups in 1–3 induced by the anisotropy of the 4-substituent may also be influenced by the position of these protons relative to the 3,5-alkoxycarbonyl substituents. In order to study the substituent influence on the magnetic nonequivalence of the mentioned 1H-NMR signals, quantum chemical studies were carried out for some derivatives of 1,4-DHP (compounds 1–5). The obtained data are presented in Table 3.
Table 2.
1H-NMR data of compounds 1–5.
| Comp. | Solvent | δNH | δHB | 1J(C,HB) | δHA | 1J(C,HA) | 2J(H,H) |
|---|---|---|---|---|---|---|---|
| 1a | CDCl3 | 6.35 | 4.89 | 159 | 4.51 | 157 | 11.5 |
| DMSO | 9.44 | 4.69 | 4.40 | 9.6 | |||
| 1b | CDCl3 | 4.96 | 4.82 | 11.0 | |||
| DMSO | 5.17 | 4.87 | 11.3 | ||||
| 1c | CDCl3 | 6.49 | 4.81 | 159 | 4.66 | 157 | 11.6 |
| DMSO | 9.67 | 4.61 | 158 | 4.56 | 157 | 9.9 | |
| 1d | CDCl3 | 6.55 | 4.86 | 4.57 | 11.8 | ||
| DMSO | 9.62 | 4.67 | 4.53 | 9.7 | |||
| 1e | CDCl3 | 7.37 | 5.19 | 160 | 4.74 | 158 | 14.6 |
| DMSO | 9.78 | 4.93 | 158 | 4.48 | 156 | 11.1 | |
| 1f | CDCl3 | 6.45 | 4.91 | 159 | 4.63 | 156.4 | 11.8 |
| DMSO | 9.58 | 4.67 | 160 | 4.57 | 158.7 | 9.6 | |
| 1g | CDCl3 | 6.56 | 4.86 | 4.62 | 11.3 | ||
| DMSO | 9.65 | 4.62 | 4.59 | 9.7 | |||
| 1h | CDCl3 | 6.45 | 4.92 | 158 | 4.63 | 156 | 11.5 |
| DMSO | 9.62 | 4.68 | 159 | 4.59 | 157 | 9.7 | |
| 1i | CDCl3 | 4.91 | 4.84 | 11.0 | |||
| DMSO | 5.18 | 4.85 | 11.4 | ||||
| 2a | CDCl3 | 10.88 | 6.28 | 5.76 | 14.1 | ||
| DMSO | 10.10 | 5.99 | 5.49 | 15.0 | |||
| 2b | CDCl3 | 6.82 | 152.1 | 6.59 | 146.3 | 15.6 | |
| DMSO | 6.45 | 152.2 | 5.77 | 146.2 | 16.0 | ||
| 2c | CDCl3 | 10.98 | 6.35 | 153.1 | 5.97 | 147.1 | 13.6 |
| DMSO | 10.48 | 5.97 | 151.1 | 5.66 | 149.1 | 15.1 | |
| 2d | CDCl3 | 10.80 | 6.16 | 6.11 | 14.1 | ||
| DMSO | 10.15 | 5.90 | 150.1 | 5.62 | 149.2 | 14.9 | |
| 2e | CDCl3 | 10.31 | 6.30 | 6.06 | 14.1 | ||
| DMSO | 10.20 | 5.97 | 5.64 | 15.6 | |||
| 2f | CDCl3 | 10.92 | 6.38 | 150.6 | 5.92 | 146.5 | 13.6 |
| DMSO | 10.34 | 6.08 | 151.9 | 5.63 | 146.4 | 15.3 | |
| 2g | CDCl3 | 10.88 | 6.24 | 6.10 | 13.7 | ||
| DMSO | 10.31 | 5.94 | 5.46 | 14.9 | |||
| 2h | CDCl3 | 6.84 | 6.62 | 15.8 | |||
| DMSO | 6.39 | 5.77 | 16.2 | ||||
| 3a | CDCl3 | 10.96 | 6.40 | 150.7 | 5.89 | 146.5 | 13.9 |
| DMSO | 10.21 | 6.08 | 151.1 | 5.56 | 145.4 | 15.1 | |
| 3b | CDCl3 | 10.95 | 6.40 | 151.4 | 5.89 | 146.3 | 13.9 |
| DMSO | 10.12 | 6.07 | 152.4 | 5.53 | 146.3 | 15.0 | |
| 3c | CDCl3 | 10.92 | 6.32 | 150.1 | 5.86 | 146.2 | 13.8 |
| DMSO | 10.28 | 6.08 | 152.1 | 5.56 | 146.4 | 15.1 | |
| 3d | CDCl3 | 10.96 | 6.40 | 148.3 | 5.89 | 146.2 | 13.9 |
| DMSO | 10.12 | 6.07 | 5.53 | 15.2 | |||
| 3e | CDCl3 | 6.73 | 152.5 | 6.56 | 150.2 | 16.1 | |
| DMSO | 6.48 | 152.5 | 5.78 | 147.5 | 16.5 | ||
| 4a | CDCl3 | 8.30 | 4.52 | and | 4.47 | 16.7 | |
| DMSO | 9.74 | 4.84 | and | 4.74 | 16.5 | ||
| 4f | CDCl3 | 6.42 | 4.69 | and | 4.65 | 16.4 | |
| 5a | DMSO | 9.91 | 4.72 | and | 4.65 | 16.0 | |
| 5f | DMSO | 9.97 | 4.64 | and | 4.60 | 16.2 |
Table 3.
Comparison of H-bond parameters in model compounds 1f, 1b and 2b, 2f and 3b.
| Compound | C2(CH···O) | C6(CH···O) | C2 pyr(CH···O) | C6 pyr(CH···O) |
|---|---|---|---|---|
| 1f | 2.508 Å, | 2.511 Å, | - | - |
| (86.6°) | (86.5°) | |||
| 2.376 Å, | 2.374 Å, | |||
| (93.6°) | (93.7°) | |||
| 1b | 2.032 Å, | 2.073 Å, | - | - |
| (131.5°) | (126.0°) | |||
| 2f | 2.329 Å, | 2.168 Å, | 2.629 Å, | 2.152 Å, |
| (99.6°) | (117.7°) | (89.7°) | (137.3°) | |
| 2b | 2.254 Å, | 2.198 Å, | 2.544 Å, | 2.913 Å, |
| (103.9°) | (107.7°) | (94.0°) | (87.5°) | |
| 3b | 2.341 Å, | 2.366 Å, | 2.591 Å, | 2.849 Å, |
| (98.914°) | (96.979°) | (90.362°) | (82.121°) |
Signals of the AB protons of the 2,6-methylene groups for compounds 1–5 are easily distinguishable in the 1H-NMR spectra by their 11–15 Hz geminal 2J coupling constants. The values of the constants for compounds 1 are less than for compounds 2 and 3. Neighbouring π-bonds of the pyridines in 2, 3 usually result in an increase of the absolute value of the constants 2J.
According to literature data [9], increasing the polarity of the medium usually slightly increases the negative contribution to the geminal coupling, and if its sign is negative, the absolute value increases. Such effect is observed in the N-methyl-substituted derivatives 1b, 1i and in compounds 2, 3 where |2Jcdcl3| < |2Jdmso|. However, for compounds 1a and 1c–h, contrary to the literature data, the 2J values decrease by about 2 Hz on going from CDCl3 to DMSO solution. The unusual behaviour of the geminal couplings for compounds 1a, 1c–h may be explained by the variation of the dihedral angle between the methylene group protons and 1,4-DHP cycle in distribution of rotamers around the C2,6-CH2X bond.
Formation of intermolecular hydrogen bonds between the NH group and solvent DMSO moves the resonance signal of NH proton to a higher-frequency field ~3 ppm (Table 2). Contrary in compounds 2a,c–g and 3a–d, the resonance signal of the NH proton moves to a lower-frequency field ~0.5–1.1 ppm on going from CDCl3 to DMSO. The reason for this anomalous behaviour of the NH proton could be that in CDCl3 solutions of these compounds the CH2Py substituent is strongly oriented and NH proton is located in a zone of negative anisotropy of the pyridinium ring. This is confirmed by calculations (Figure 1).
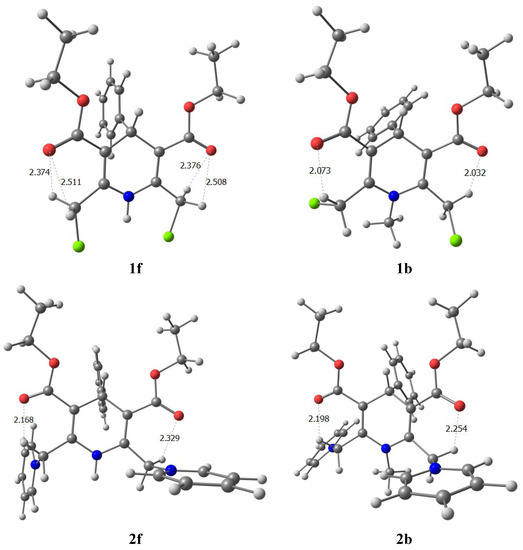
Figure 1.
The calculated optimal conformations in compounds 1f, 1b and 2f, 2b.
Analysis of NOEs in the 1H NOESY spectra of compounds 1–3 allows us to determine the relative position of the AB protons for the methylene group—the NOE from NH to the more shielded proton HA is twice as intense as the HB (Figure 2). This means that the HA proton resonating in a lower-frequency field is located closer to the NH proton by about 0.3 angstroms.
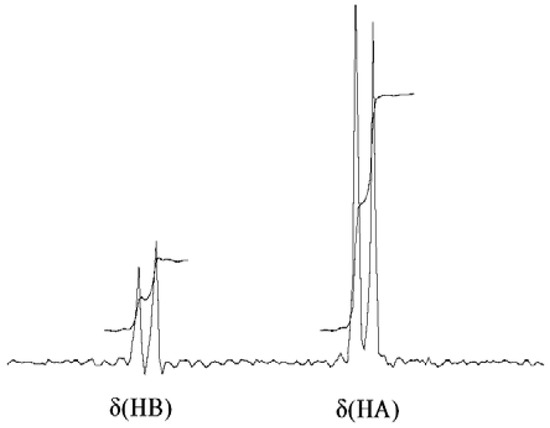
Figure 2.
Slice of 1H-NMR NOESY spectrum for compound 3b. More intense NOE is registered between NH and HA, showing preferable orientation toward the NH.
Such an arrangement of CH2 protons leads to an intramolecular contact between the hydrogen atom HB and the carboxyl group at the C3 carbon, leading to the formation of a CHB···O=C hydrogen bond. It is known that protons involved in hydrogen bonds are less shielded, as compared to free ones [14]. Short interactions of C-H···O play an important role in biology and have been observed in many cases previously [15].
Additional confirmation for the existence of C-H···O hydrogen bonding in compounds 1–3 comes from the different deuteration rates of the 2(6)-CH2 protons (Figure 3). In D2O solution the intensity of the resonance signal of the more shielded proton HA of compound 3b decreases faster than for the more deshielded HB. This is because the hydrogen-bonded protons are usually less influenced by the intermolecular H-D exchange. This fact confirms the formation of a hydrogen bond between HB methylene proton and the carboxyl group.
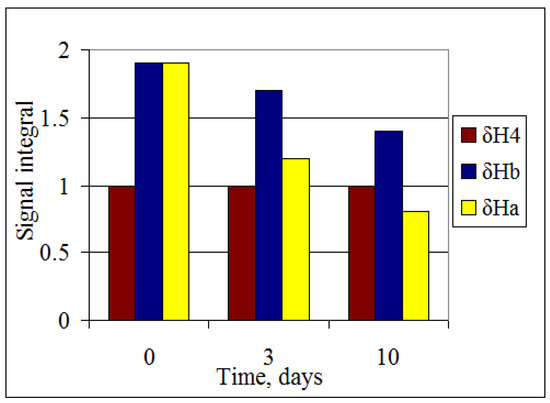
Figure 3.
1H-NMR spectral data of D2O solutions of compound 3b at 25 °C.
The measured coupling constants 1J(13C, 1H) for each of the methylene protons also differ 1J(13C, 1HA) < 1J(13C, 1HB) (see Table 2). The smaller 1J(13C, 1H) value corresponds to the more shielded proton HA. This is consistent with the literature data [16], which states that if the CH-proton is involved in a hydrogen bond, the 1J(13C, 1H) value increases.
1H-13C-HMBC spectra reveal a more intensive magnetization transfer from the low field HB proton to 13C3 carbon as compared to HA. It is known that the intensities of cross peaks in the HMBC spectra are proportional to the value of the vicinal coupling constant 3J(13C, 1H), normally being 3J(C,H)trans > 3J(C,H)cis [17]. The delay value used to generate long-range spin-spin interaction between 13C-1H in HMBC spectrum was 8 Hz. The higher intensity of HA cross peak to the carbon C3 (110.1 ppm) points to its trans-orientation relative to C3. Therefore, the orientation of the less shielded proton HB favours its participation in the CH···O hydrogen bond.
Thus, on the basis of these data, we can unambiguously claim that the intramolecular CH···O hydrogen bond is one of the reasons for the non-equivalence of the methylene protons in these systems. In compounds 1 and 2 the difference in the chemical shifts of the AB methylene protons (ΔδAB = δB − δA) vary in the range 0–0.5 ppm, rather randomly, depending on the nature of substituents and the solvent used. In compounds 3a–d, with bulk aliphatic substituents at C3,5, the difference is relatively constant and is equal to ~0.5 ppm (Table 2). To get more insight for the results obtained by NMR quantum-chemical calculations were carried out.
The results obtained reveal that for compounds 2b, 2f the energy minima correspond to the conformations with s-cis-s-cis carboxyl group orientation relative to the double bond in the dihydropyridine cycle, indicating the dominant stabilizing effect of intramolecular hydrogen bond on the conformation of molecules 2b, 2f the same as in case of 1b, 1f. Structures with s-cis-s-trans and s-trans-s-trans carboxyl groups orientation are with higher energy, consequently the stabilizing effect in solutions has to be attributed to the hydrogen bonds C-H···O. The calculated equilibrium conformations and the internuclear distances R (CH···O) also reveal the formation of a hydrogen bond (Table 3).
In description of hydrogen bonds of the CH···O type geometric characteristics are often used. In our case, the calculated parameters of intramolecular hydrogen bonds for compounds 1b, 1f, 2b, 2f and 3b, presented in Table 3, are consistent with the X-ray diffraction and NMR spectral data. According to calculations, the distance d(CH···O) decreases, and the angle (C-H···O) increases on going from 1f, 1b to 3b, indicating weakening of the hydrogen bond between the methylene HB proton and oxygen of the carboxyl group. At the same time, in the most energetically favourable conformations of 2b, 2f and 3b there exist additional H-bonds between the pyridine ortho protons and the oxygen atoms of the carboxyl group (see Table 3). These additional H-bonds also stabilize the structure of the molecule. In conformations with s-cis-s-trans and s-trans-s-trans orientation of the carboxyl groups relative to the double bonds of DHP cycle there is a possibility to form H-bonds with a length of about 2.400 Å between protons in position 4 and the ethoxycarbonyl oxygen. The latter contact is much weaker than the ones with protons of the methylene groups and does not contribute significantly to the conformation of the molecule.
In summary, some novel derivatives of 1,4-dihydropyridine bearing substituents at 2,6-methylene groups were synthesized. It was shown that one of the reasons for the diastereotopy of the methylene protons at positions 2 and 6 of symmetrically substituted 1,4-dihydropyridine rings is the formation of an intramolecular hydrogen bond of the CH···O type, which was confirmed by NMR spectroscopy and quantum-chemistry calculations.
3. Experimental
3.1. Chemicals
All reagents were purchased from Aldrich, Acros, Fluka or Merck and used without further purification. TLC was performed on 20 × 20 cm silica gel TLC-PET F254 foils (Fluka). Melting points were determined on an SRS OptiMelt apparatus (Stanford Research Systems, Sunnyvale, CA, USA). Elemental analyses were performed on an EA 1106 (Carlo Erba Instruments, Milan, Italy). Compounds were recrystallized from methanol, acetone or purified by column chromatography.
3.2. NMR Experiments (Table 2)
The one-dimensional 1H- and 13C- and two dimensional 1H-1H NOESY, 13C-1H HMBC, 13C-1H HSQC of compounds 1–5 were recorded on a Varian-Mercury 400 MHz BB instrument. The mixing time in the 2D-NOESY spectra was 800 ms. The 13C-HMBC were recorded with the evolution time of 62.5 s delay for the generation of long-range correlations. For all two dimensional spectra a 4,096 × 1,024 data matrix was used. To improve the signal-noise ratio, the data matrix before Fourier transformation was zero-filled twice and multiplied by a cosine function. The chemical shifts of the hydrogen and carbon atoms are presented in ppm and referred to the residual signals of the solvent a 7.25 (1H) and 77.0 ppm (13C) ppm, respectively. Multiplicities are abbreviated as: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. The coupling constants are expressed in Hertz.
3.3. Synthesis
General method for the synthesis of 2,6-di(bromomethyl)-3,5-bis(alkoxycarbonyl)-4-aryl(4-alkyl)-(or N-methyl)-1,4-dihydropyridines 1a–d,i: To a solution of the appropriate 2,6-dimethyl-3,5-bis(alkoxycarbonyl)-4-aryl(4-alkyl)-1,4-dihydro-pyridine (5 mmol) in methanol (50 mL) NBS (10 mmol) was added portionwise at ambient temperature. The reaction mixture was stirred at rt for 24 h. The pale yellow precipitate was filtered and washed with water. The precipitate was crystallized from ethanol giving 1a–d,i.
2,6-Di(chloromethyl)-3,5-bis(ethoxycarbonyl)-4-methoxycarbonyl-1,4-dihydropyridine (1e). A mixture of ethyl 4-chloroacetoacetate (2.72 mL, 20 mmol) and glyoxylic acid monohydrate (1.74 g, 19 mmol) in methanol (40 mL) was stirred at rt for 72 h in the presence of piperidine/acetate as catalyst. Then ethyl (E,Z)-3-amino-4-chlorobut-2-enoate (3.27 g, 20 mmol) was added and the combined mixture was stirred at rt additional 72 h. The solvent was removed under vacuum and the residue was washed with water. The obtained yellow oil was crystallized from methanol yielding 3,5-diethyl 2,6-bis-(chloromethyl)-1,4-dihydropyridine-3,4,5-tricarboxylate (4.98 g, 68%); m.p.164–166 °C; elemental analysis calcd (%) for C14H17Cl2NO6: C 45.92, H 4,68, N 3.82; found: C 45.95, H 4.58, N 3.72. To a solution of 3,5-diethyl 2,6-bis(chloromethyl)-1,4-dihydropyridine-3,4,5-tricarboxylate (1.83 g, 5 mmol) in methanol (30 mL) conc. sulfuric acid (0.1 mL) was added and the reaction mixture was refluxed for 2 h. The solvent was removed under vacuum and the residual crude product was triturated with water. After cooling the precipitate was filtered off and crystallized from methanol giving compound 1e (1.65 g, 87%); m.p. 99–101 °C; p elemental analysis calcd. (%) for C15H19Cl2NO6: C 47.38, H 5.04, N 3.68; found: C 47.41; H 4.95; N 3.68.
General method for the synthesis of 1,1`-{[3,5-bis(alkoxycarbonyl)(3,5-bis(2-propoxyethoxycarbonyl)-4-aryl(4-alkyl) (or N-methyl)-1,4-dihydropyridine-2,6-diyl] dimethylene} bispyridinium dibromides 2a–d,h: To a solution of compounds 1a–d,i (2 mmol) in dry acetone (20 mL) pyridine (0.20 mL, 2.2 mmol) was added and the reaction mixture was stirred at rt for 24 h. After cooling the precipitate was filtered off, washed with dry acetone and crystallized from ethanol giving compounds 2a–d,h as pale yellow powders.
1,1`-{[3,5-Bis(ethoxycarbonyl)-4-methoxycarbonyl-1,4-dihydropyridine-2,6-diyl]dimethylene} bis-pyridinium diiodide (2e). To a solution of compound 1e (0.38 g, 1 mmol) in dry acetone (4 mL) pyridine (0.18 mL, 2 mmol) and potassium iodide (0.33 g, 2 mmol) were added and the mixture was stirred at rt for 48 h. The mixture was diluted with acetone (20 mL), and the potassium chloride formed was filtered off. Acetone was removed under vacuum, and the residue was crystallized from methanol yielding compound 2e as a pale yellow powder, (0.65 g, 91%); m.p.137–139 °C; elemental analysis calcd (%) for C25H29I2N3O6: C 41.63; H 4.05; N 5.83; found: C 41.49; H 3.98; N 5.65.
3.4. Computional Methods
The Firefly (7.1.G) software package [18] was used for calculation/optimisation of structures/structures’ geometry. The structures of 1d, 1e, 1d and 2a were optimised using a combined RHF/DFT (b3lyp) method and 6-31g basis set. In the calculation of compounds 1 and 2 the basis set was supplemented by two polarization functions (**) and a diffuse (++) - 6–31g ++ ** (hybrid DFT/RHF B3LYP/6-31++G**). For 3b the same method and theory level was used, only simplified by using one polarization function and one diffuse - (hybrid DFT/RHF B3LYP/6-31+G*).
4. Conclusions
Some novel derivatives of 1,4-dihydropyridine were synthesized. Due to the presence of a prochiral centre at C-4 in the heterocyclic ring magnetic non-equivalence of the diastereotopic methylene protons in positions 2 and 6 was observed. NMR spectroscopy and quantum-chemistry calculations confirmed that the formation of a CH···O intramolecular hydrogen bond amplifies the extent of the anisochrony of the methylene protons.
Supplementary Materials
Supplementary File 1Acknowledgments
This work was supported by the ESF project No.2009/0197/1DP/1.1.1.2.0./09/APIA/VIAA/014 “Design of new pharmacomodulators and studies of their nanoassociates as transport forms”, VPP-4-02.2 and VPP-4-07-3.
References
- Triggle, D.J. The chemist as astronaut: Searching for biologically useful space in the chemical universe. Biochem. Pharmacol. 2009, 78, 217–223. [Google Scholar] [CrossRef]
- Edraki, N.; Mehdipour, A.R.; Khoshneviszadeh, M.; Miri, R. Dihydropyridines: Evaluation of their current and future pharmacological applications. Drug Discov. Today 2009, 14, 1058–1066. [Google Scholar] [CrossRef]
- Skrastin’sh, I.P.; Kastron, V.V.; Chekavichus, B.S.; Sausin’sh, A.é.; Zolotoyabko, R.M.; Dubur, G.Y. Bromination of 4-aryl-3,5-dialkoxycarbonyl-2,6-dimethyl-1,4-dihydropyridines. Chem. Heterocycl. Comp. 1991, 27, 989–994. [Google Scholar] [CrossRef]
- Urtti, A.; Hyvonen, Z.; Plotniece, A.; Makarova, N.; Reine, I.; Tirzitis, G.; Vigante, B.; Cekavicus, B.; Shmidlers, A.; Krauze, A.; Zhalubovskis, R.; Duburs, G.; Turunen, M.; Yla-Herttuala, S.; Jaaskelainen, I.; Toppinen, M.R. Cationic amphiphilic 1,4-dihydropyridine derivatives useful for delivery of nucleotide containing compounds. WO 01/62946 A1, 70, 8. 2001.
- Pajuste, K.; Plotniece, A.; Kore, K.; Intenberga, L.; Cekavicus, B.; Kaldre, D.; Duburs, G.; Sobolev, A. Use of pyridinium ionic liquids as catalysts for the synthesis of 3,5-bis(dodecyloxy- carbonyl)-1,4-dihydropyridine derivatives. Cent. Eur. J. Chem. 2011, 9, 143–148. [Google Scholar] [CrossRef]
- Plotniece, A.; Pajuste, K.; Kaldre, D.; Cekavicus, B.; Vigante, B.; Turovska, B.; Belyakov, S.; Sobolev, A.; Duburs, G. Oxidation of cationic 1,4-dihydropyridine derivatives as model compounds for putative gene delivery agents. Tetrahedron 2009, 65, 8344–8349. [Google Scholar]
- Hyvönen, Z.; Plotniece, A.; Reine, I.; Chekavichus, B.; Duburs, G.; Urtti, A. Novel cationic amphiphilic 1,4-dihydropyridine derivatives for DNA delivery. Biochim. Biophys. ActaBiomembr. 2000, 1509, 451–466. [Google Scholar]
- Görlitzer, K.; Bartke, U.; Schmidt, E. Zur reaktion von nifedipin mit pyridiniumbromidperbromid. Arch. Pharm. 1991, 324, 105–109. [Google Scholar] [CrossRef]
- Bystrov, V.F. Spin–Spin coupling between geminal and vicinal protons. Russ. Chem. Rev. 1972, 41, 281–304. [Google Scholar] [CrossRef]
- Pfister, J.R. Rapid, high-yield oxidation of hantzsch-type 1,4-dihydropyridines with ceric ammonium nitrate. Synthesis 1990, 8, 689–690. [Google Scholar] [CrossRef]
- Skrastin`sh, I.P.; Kastron, V.V.; Vitolin, R.O.; Dubur, G.Ya.; Stivrinya, M.I.; Kaidaka, K.A. Synthesis and pharmacological activity of furo-1,4-dihydropyridines. Pharm. Chem. J. 1989, 23, 893–896. [Google Scholar] [CrossRef]
- Martin, N.; Segura, J.L.; Seanone, C.; Soto, J.L.; Morales, M.; Suarez, M. A novel one-step synthesis of 4H-furo[3,4-b]pyrans and a transformation into a difuro[3,4-b: 3′,4′-e]pyridine. Liebigs Anna. Chem. 1991, 8, 827–830. [Google Scholar]
- Skrastin`sh, I.P.; Kastron, V.V.; Dubur, G.Ya.; Mazheika, I.B.; Liepin`sh, E.E. Bromination of 2,6-dimethyl-3,5-dimethoxycarbonyl-4-(2,-difluoromethoxyphenyl)-1,4-dihydropyridine (foridone). Chem. Heterocycl. Compd. 1989, 25, 791–795. [Google Scholar] [CrossRef]
- Schaefer, T.; Peeling, J.; Sebastian, R. Relation between hydroxyl proton chemical shifts and torsional frequencies in some ortho-substituted phenol derivatives. J. Phys. Chem. 1975, 79, 1888–1890. [Google Scholar] [CrossRef]
- Leroy, I.L.; Snoussi, K.; Gueron, M. Investigation of the energetics of C-H···O hydrogen bonds in the DNA i-motif via the equilibrium between alternative intercalation topologies. Magn. Reson. Chem. 2001, 39, 171–176. [Google Scholar] [CrossRef]
- Afonin, A.V.; Ushakov, I.A.; Simonenko, D.E.; Shmidt, E.Y.; Zorina, N.V.; Mikhaleva, A.I.; Trofimov, B.A. 1H and 13C NMR Study of bifurcated intramolecular hydrogen bonds in 2,6-bis(2-pyrrolyl)pyridine and 2,6-bis(1-vinyl-2-pyrrolyl)pyridine. Russ. J. Org. Chem. 2005, 41, 1516–1521. [Google Scholar] [CrossRef]
- Marshall, J.L. Carbon-Carbon and Carbon-Proton NMR Coupling. Methods in Stereochemical Analysis 2; Verlag Chemie International: Weinheim, Germany, 1983; p. 241. [Google Scholar]
- Granovsky, A.A. Firefly version 7.1.G. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 7 September 2011).
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).