Gold-Catalyzed Cyclizations of Alkynol-Based Compounds: Synthesis of Natural Products and Derivatives



Abstract
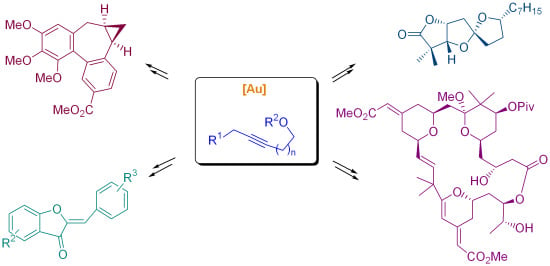
:
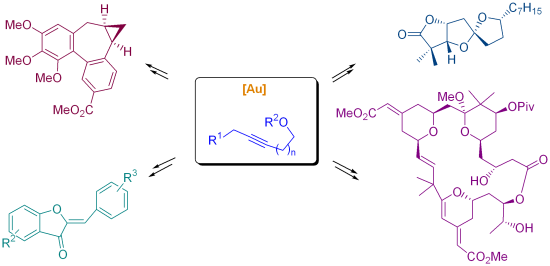{kind=link}
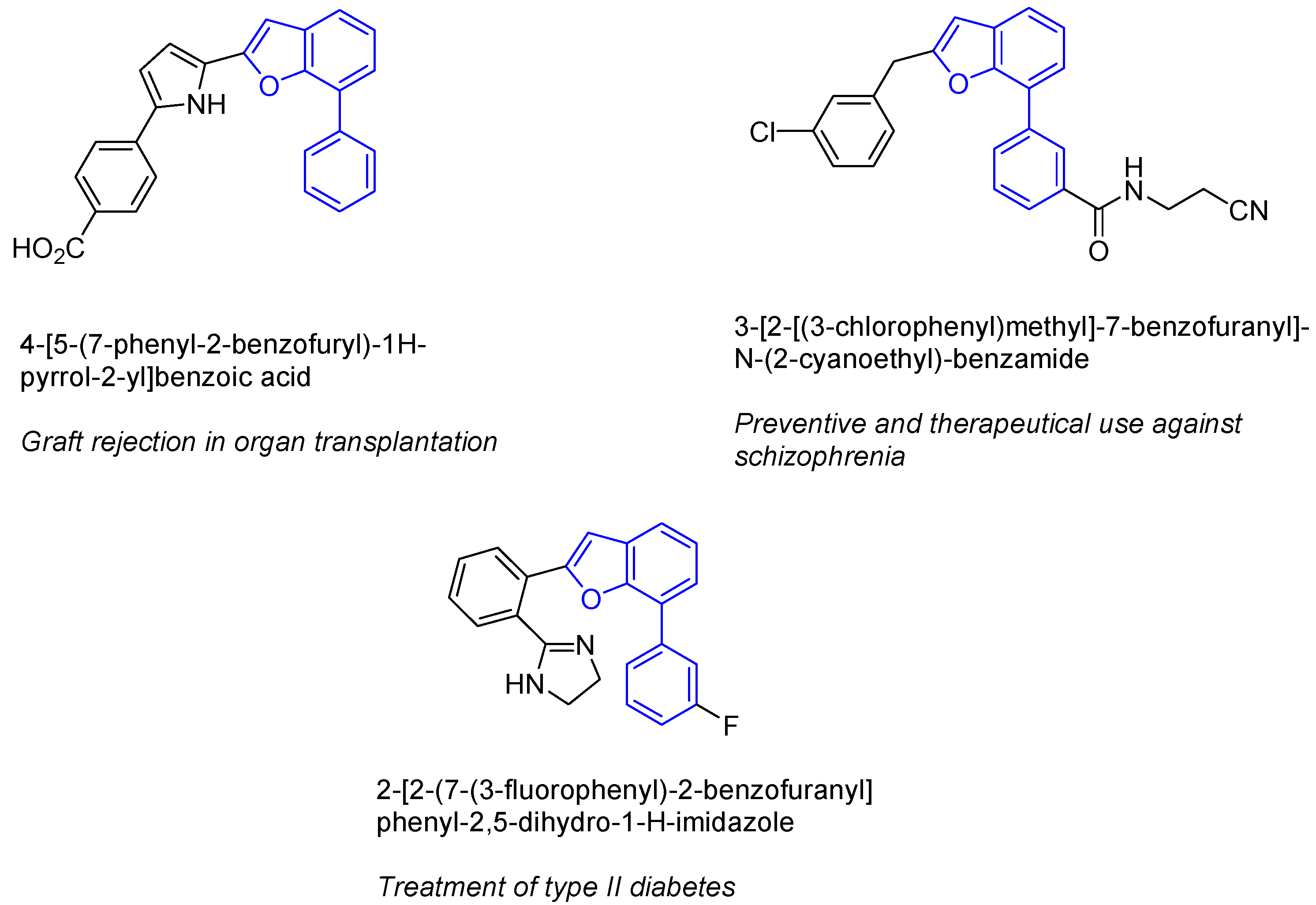{kind=link}
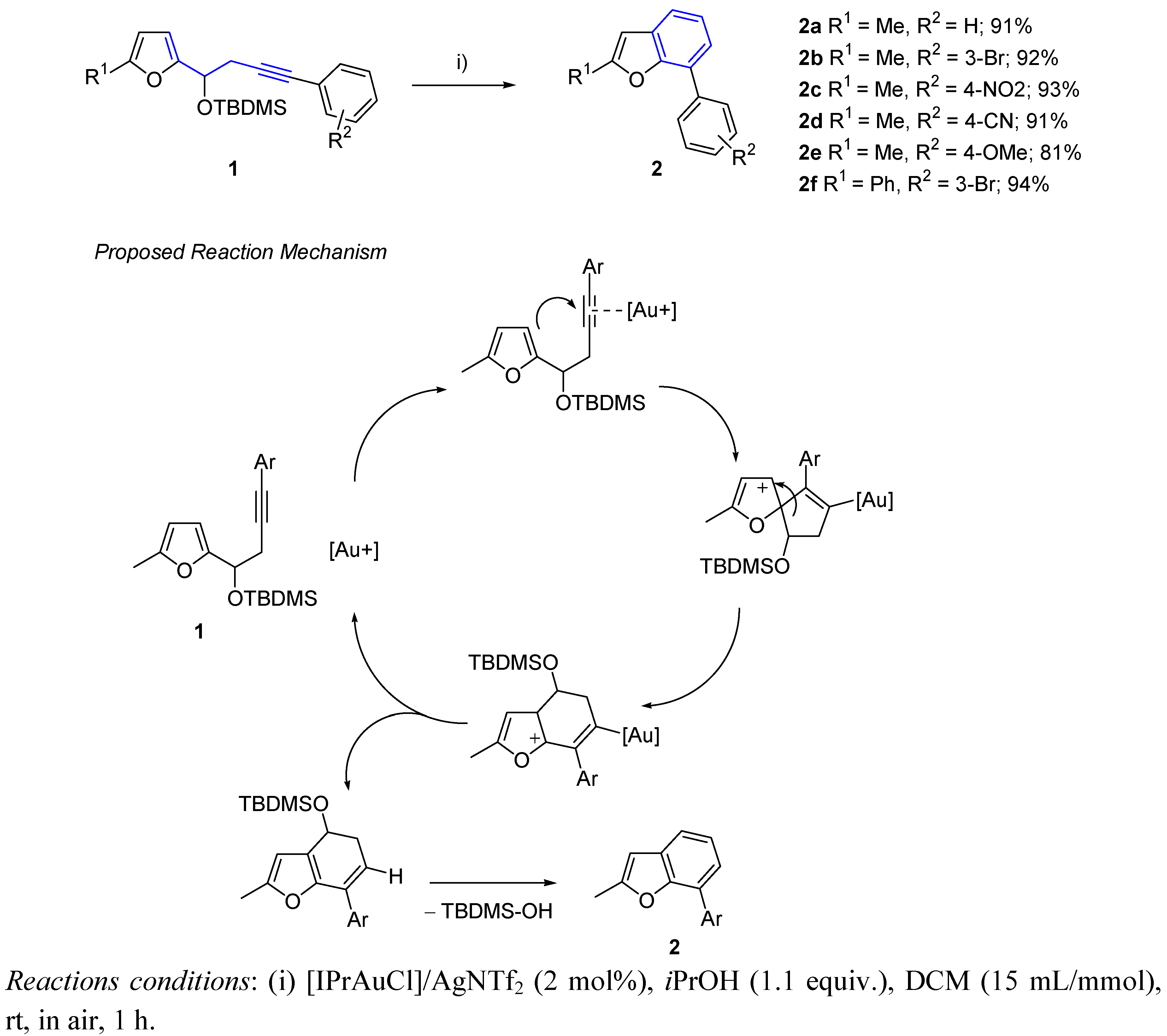{kind=link}
{kind=link}
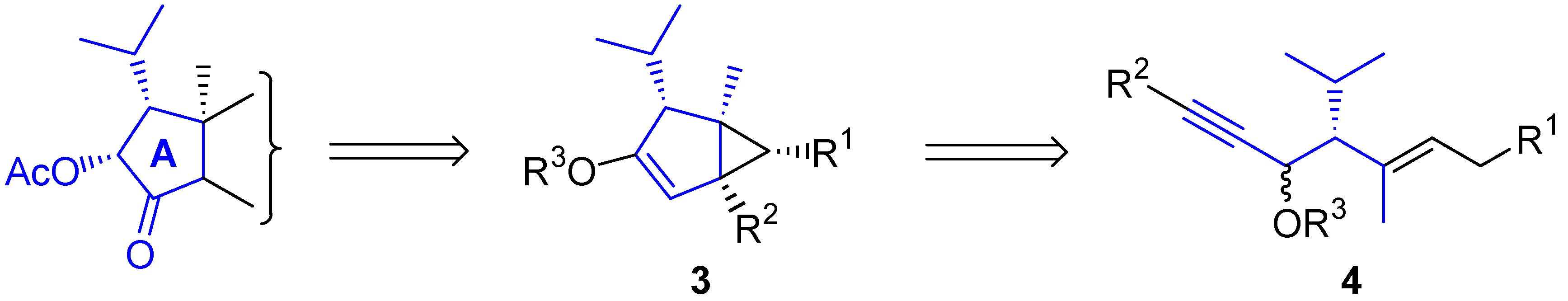{kind=link}
{kind=link}
{kind=link}
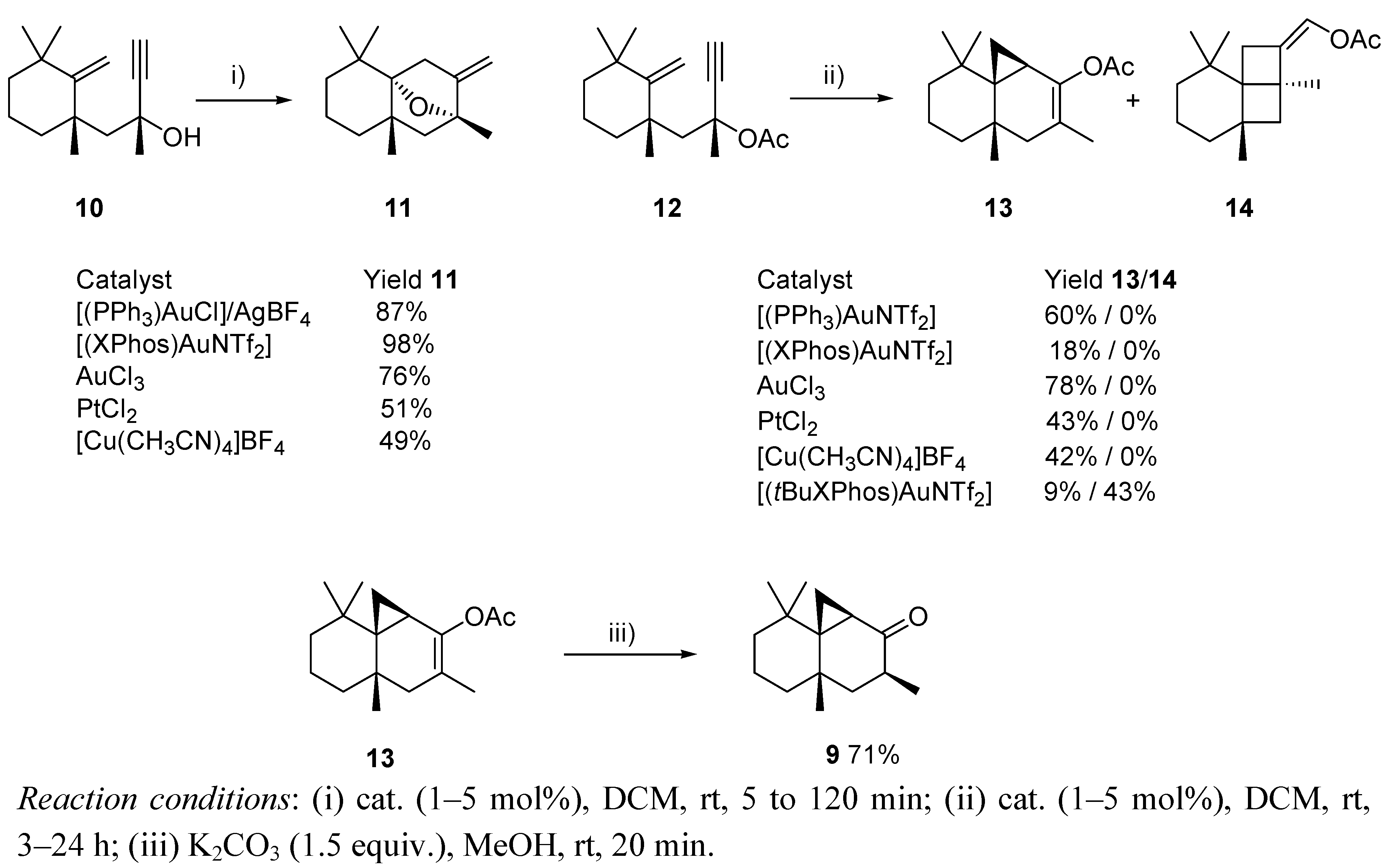{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
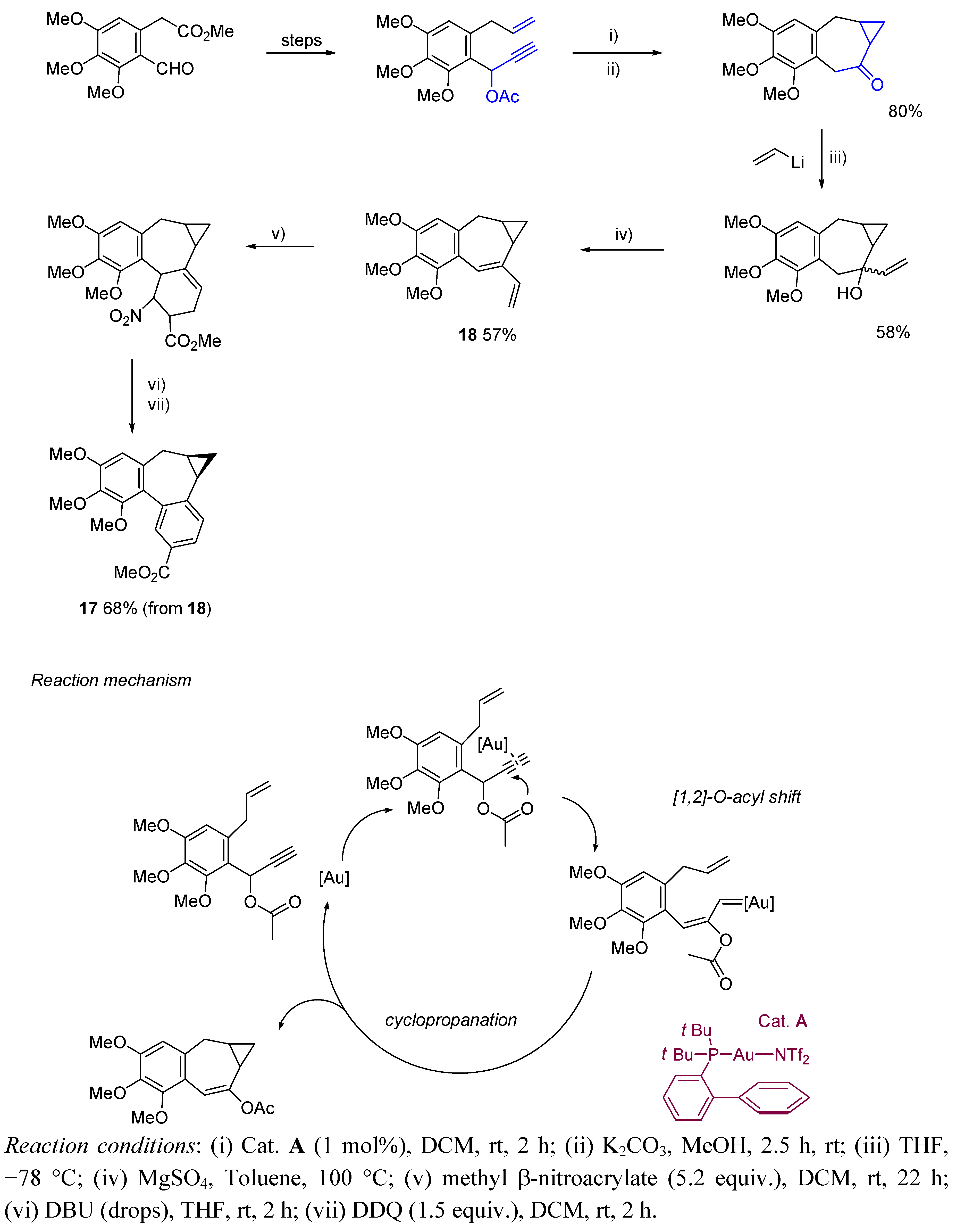
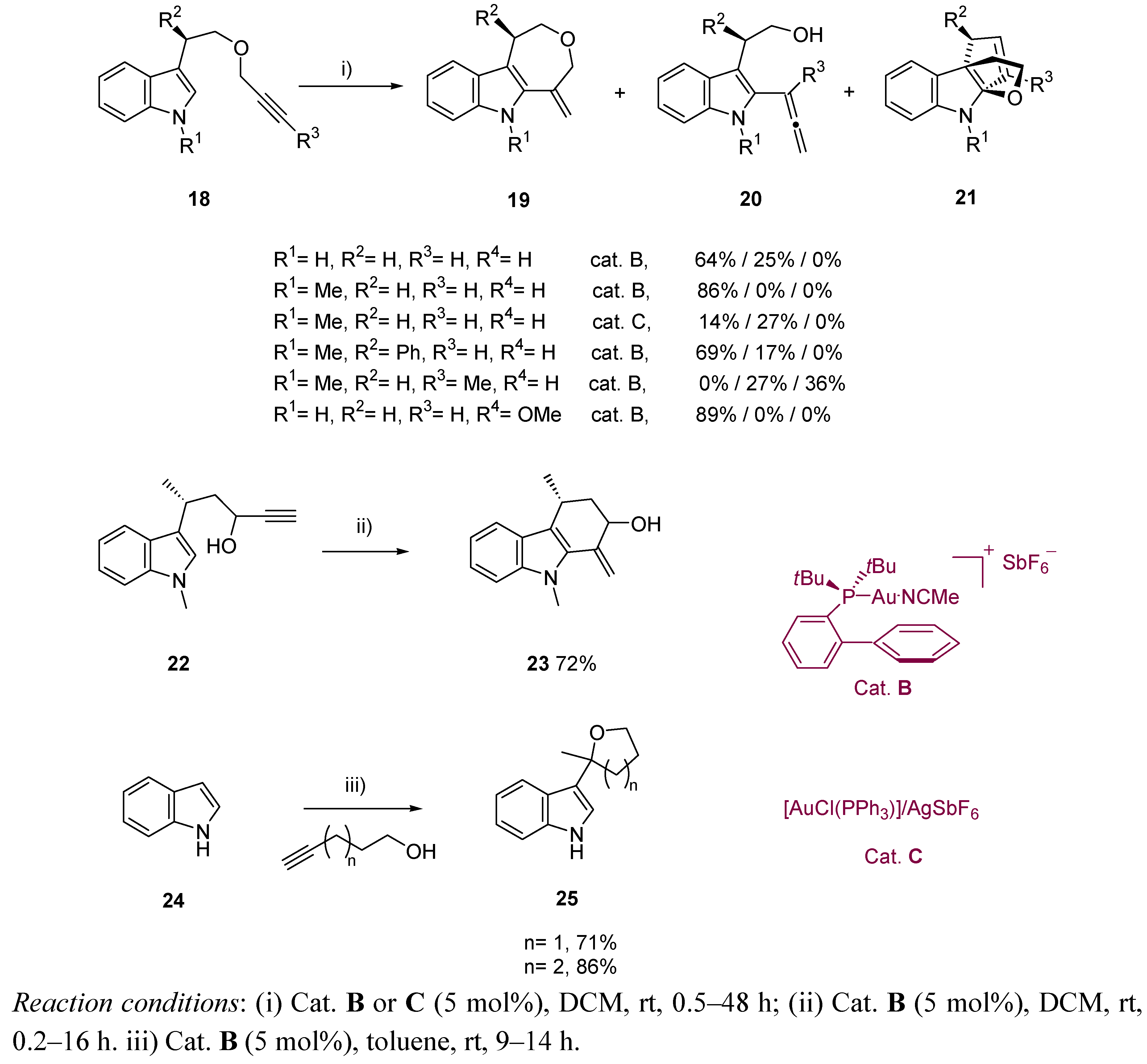
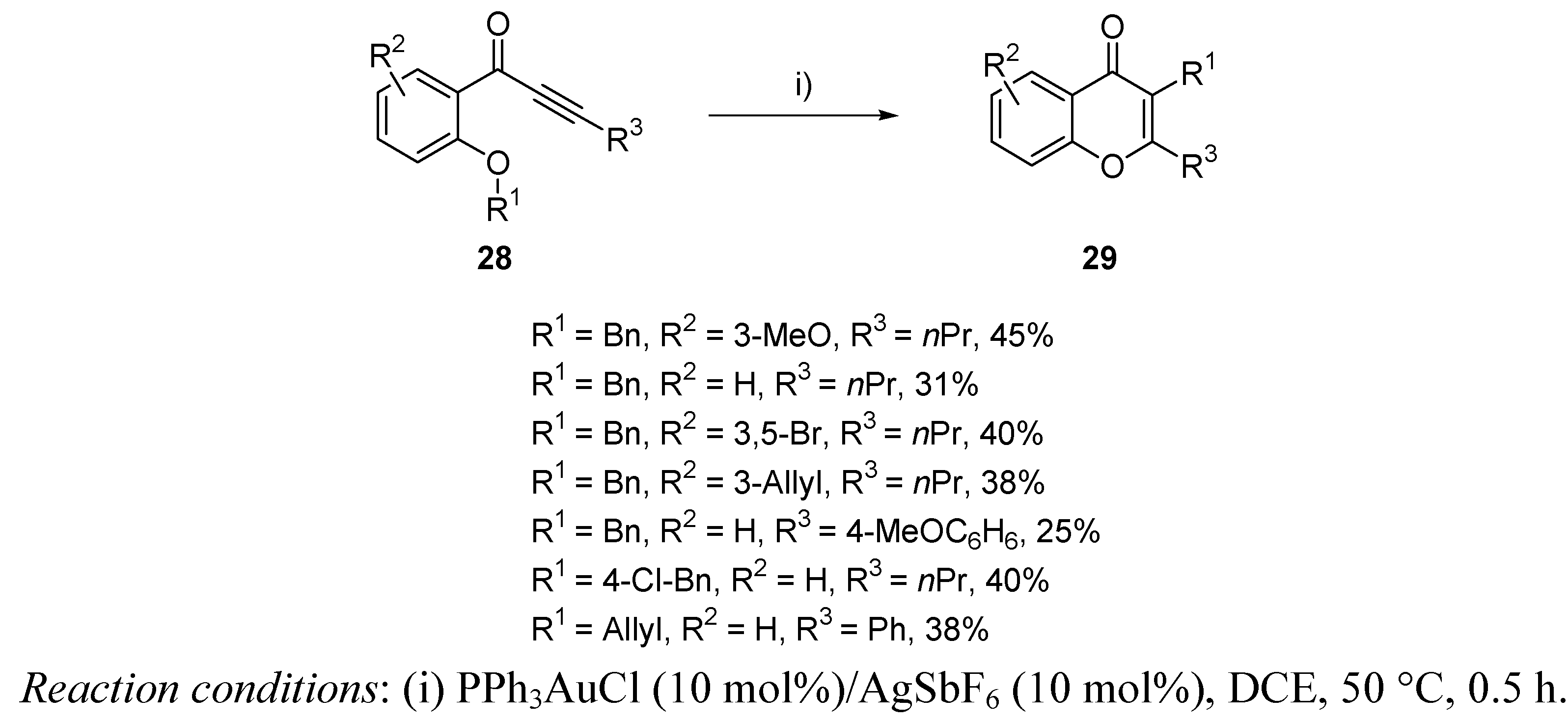
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Cycloisomerization Processes Involving Carbon-Carbon Bond Formation
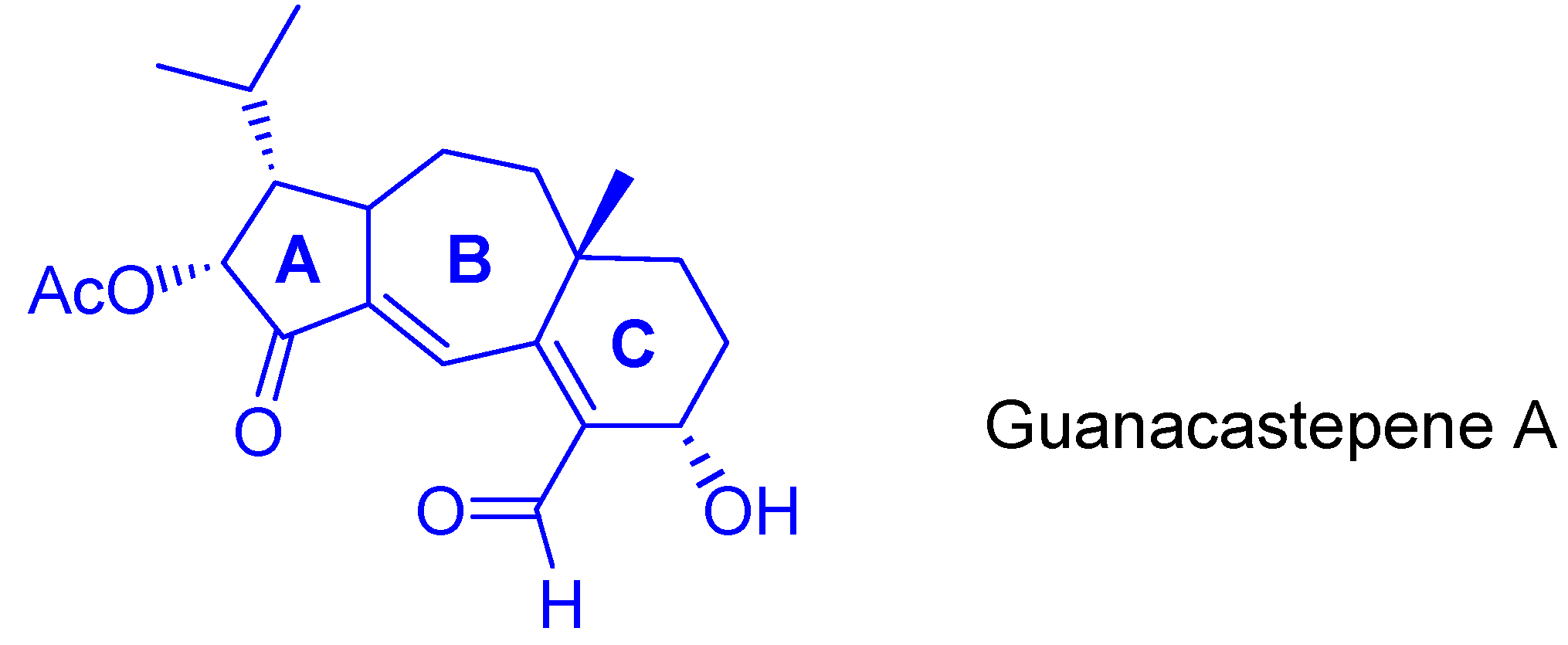
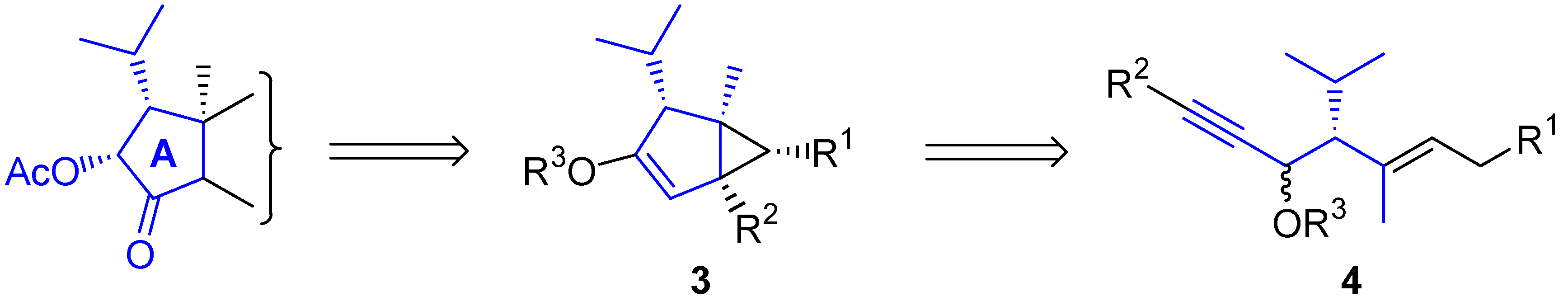
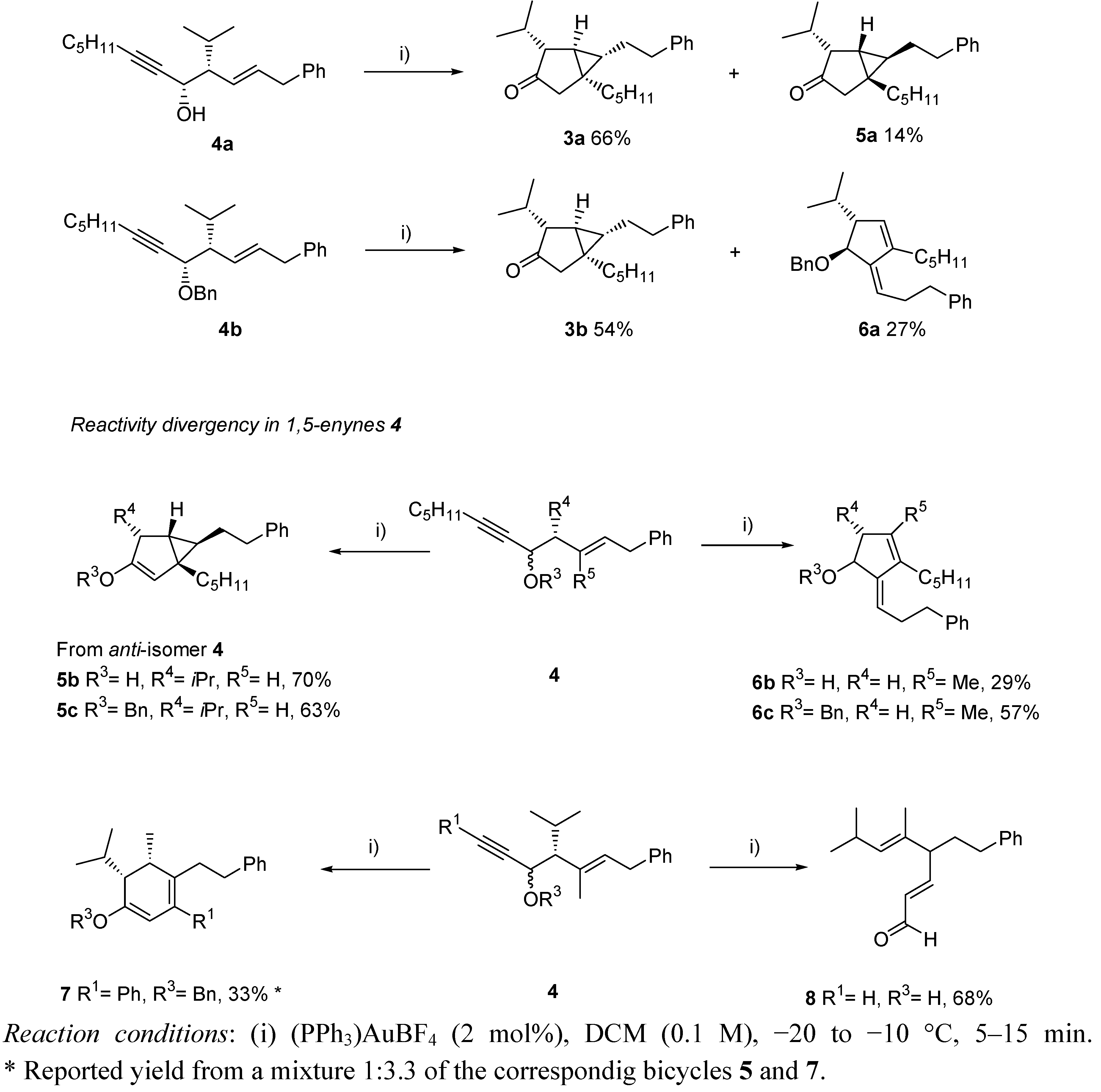
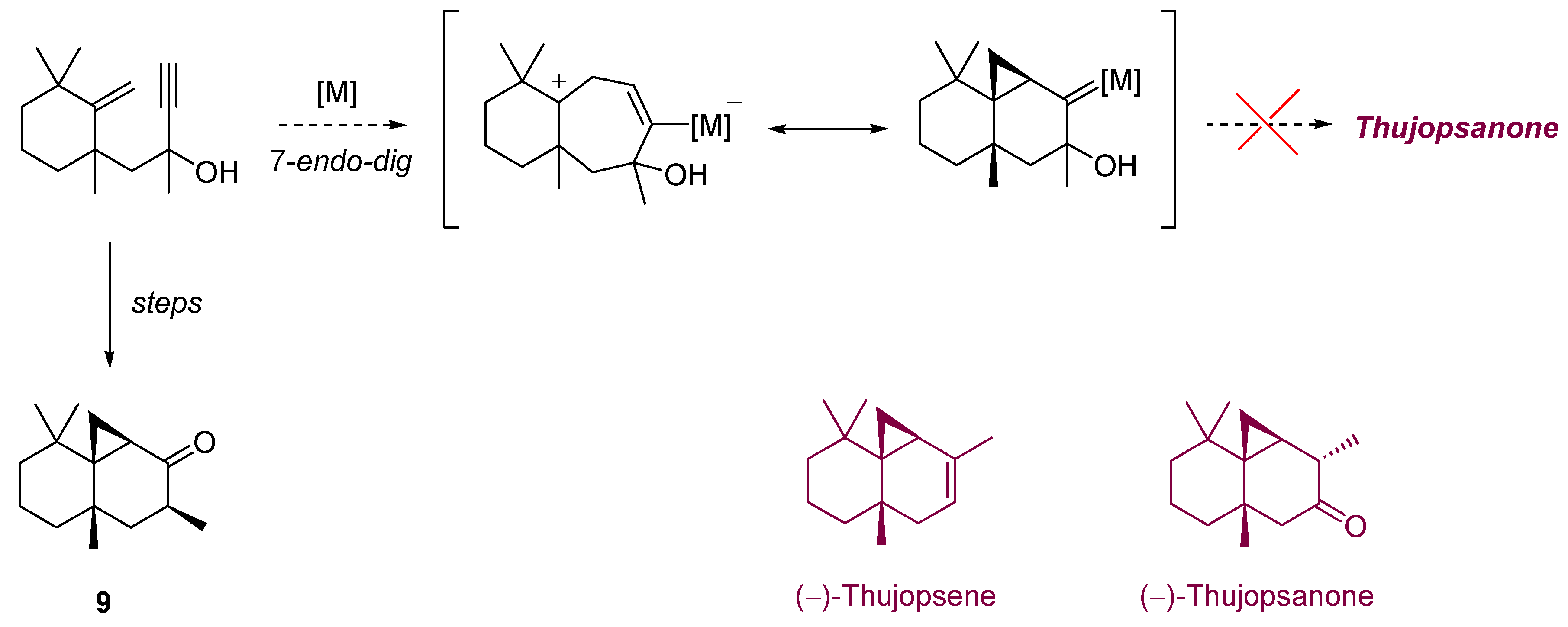
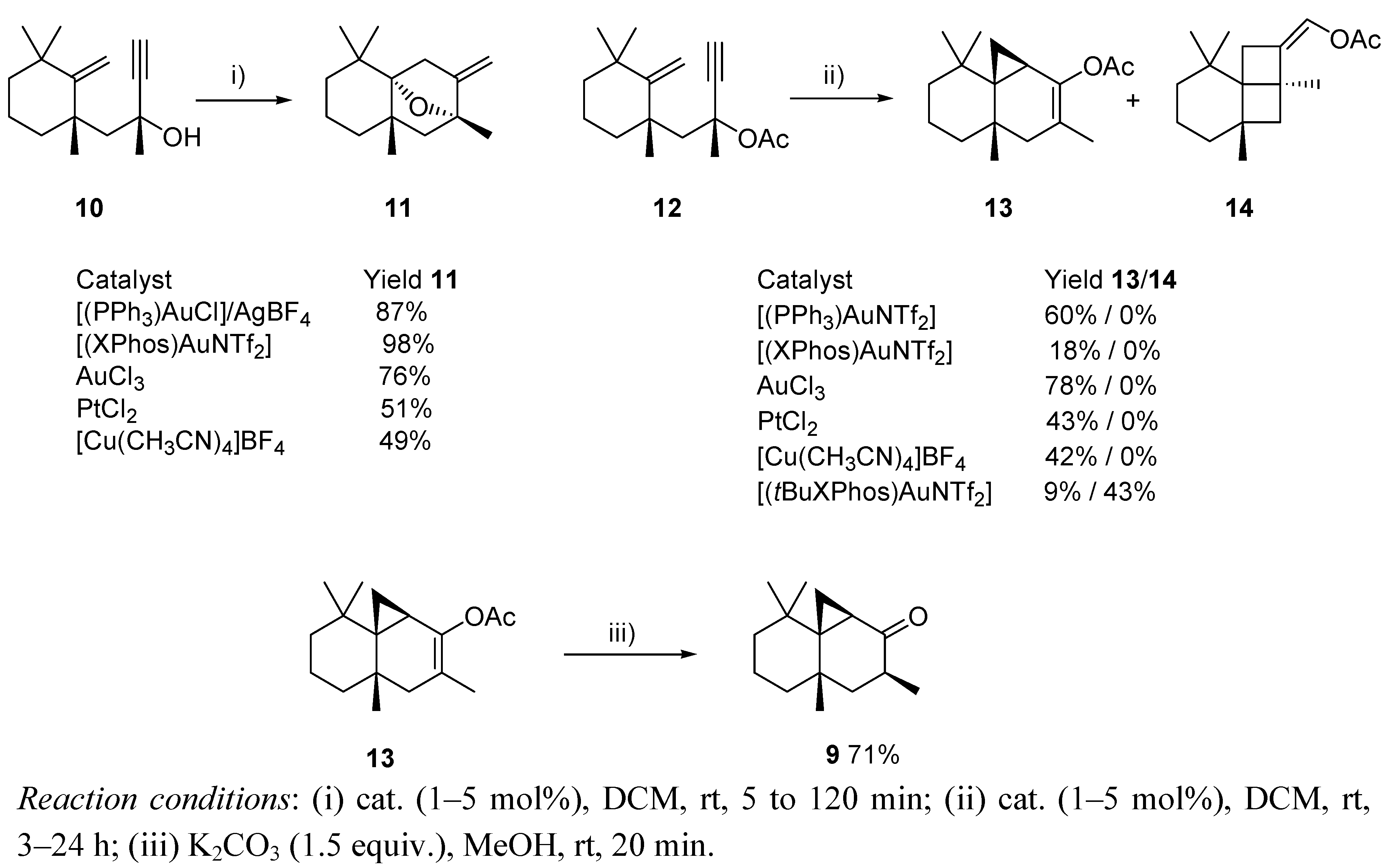
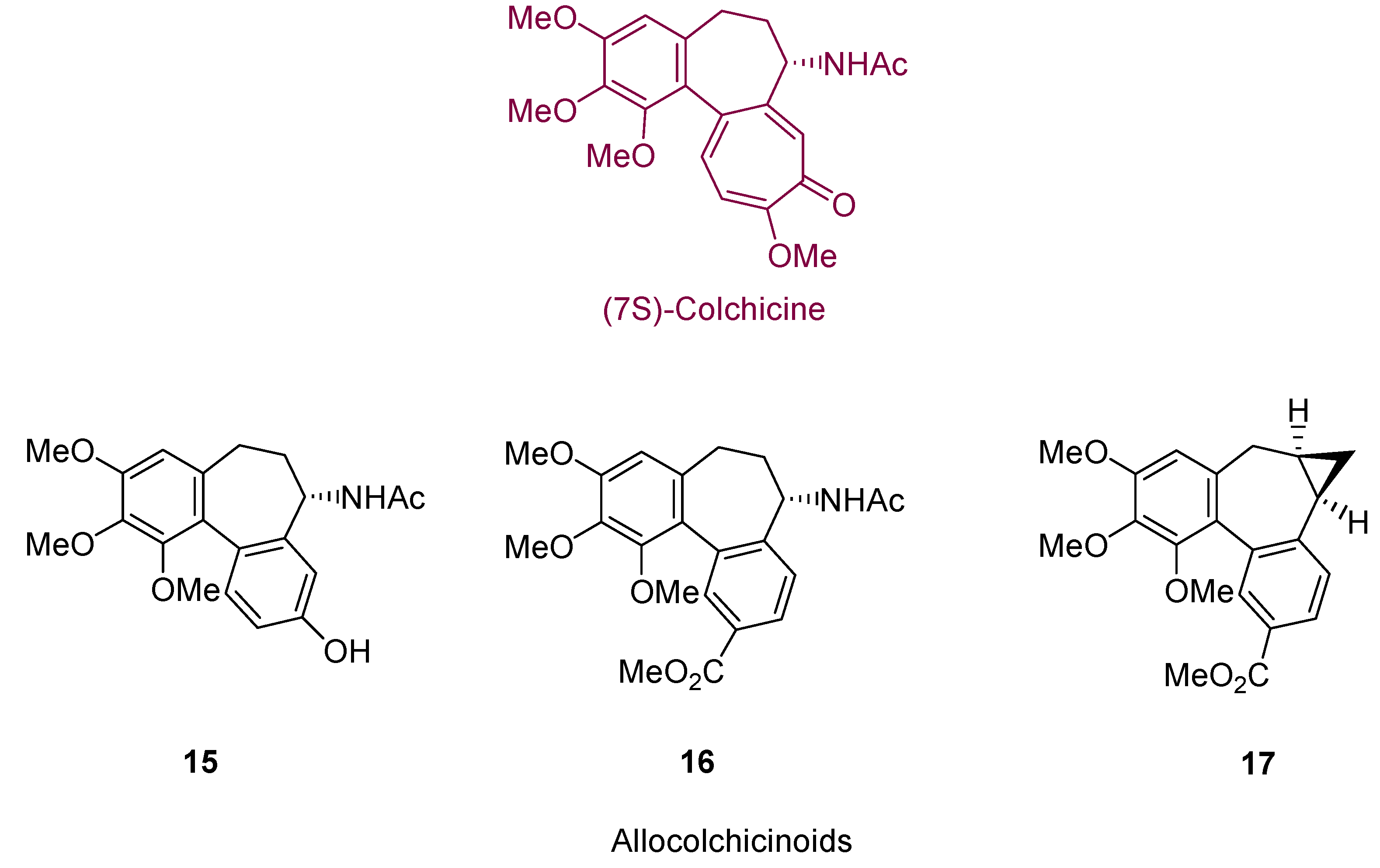
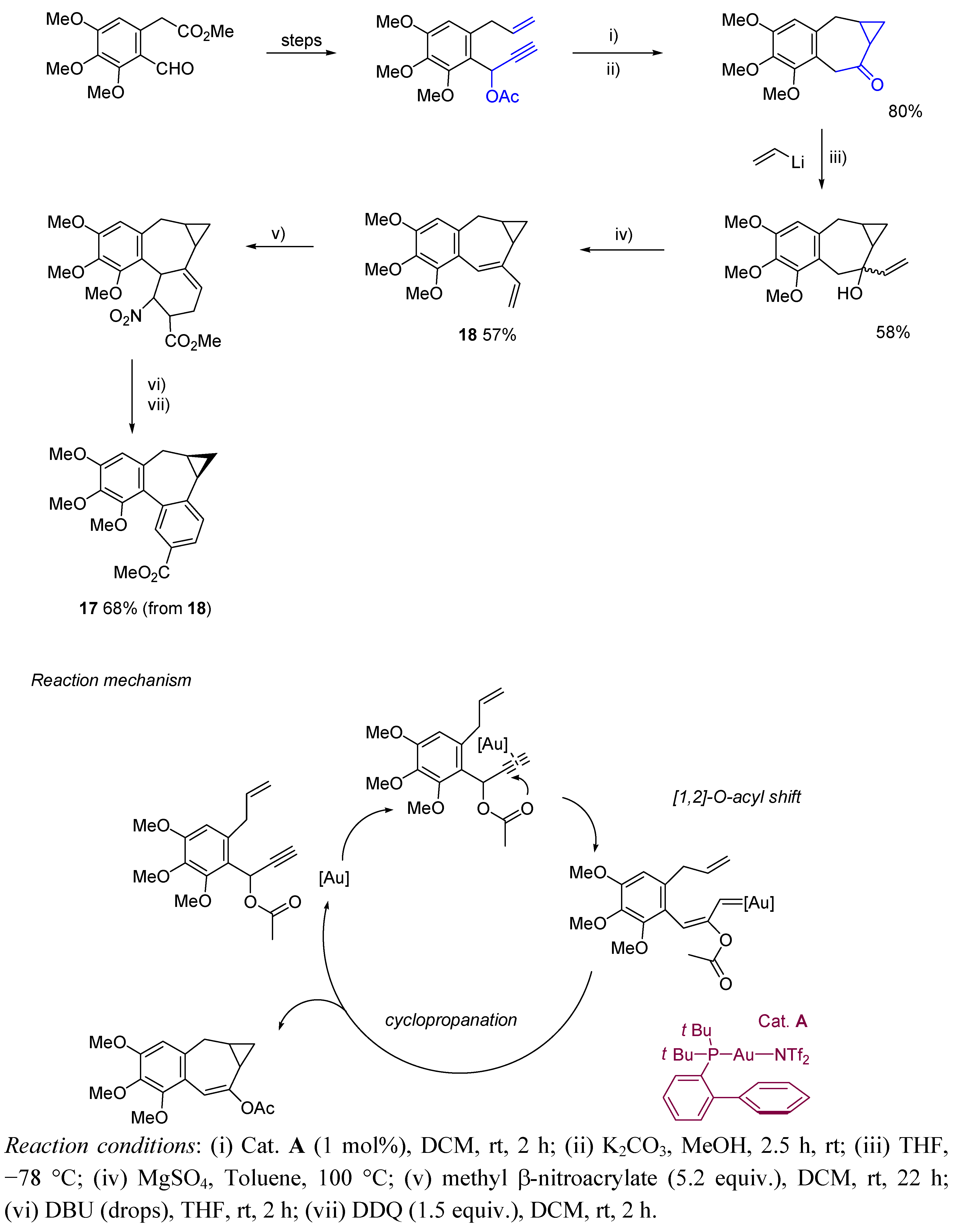
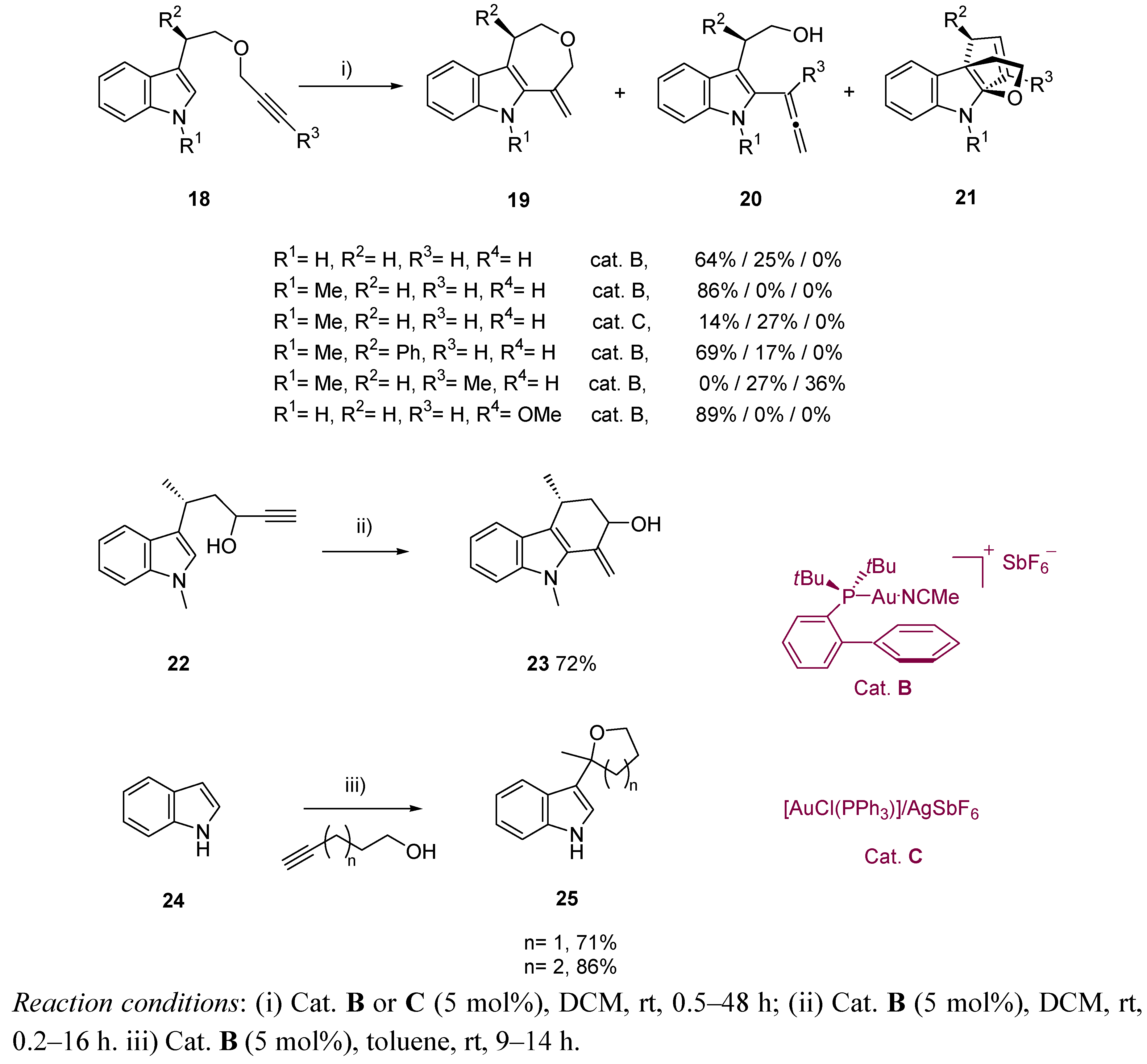
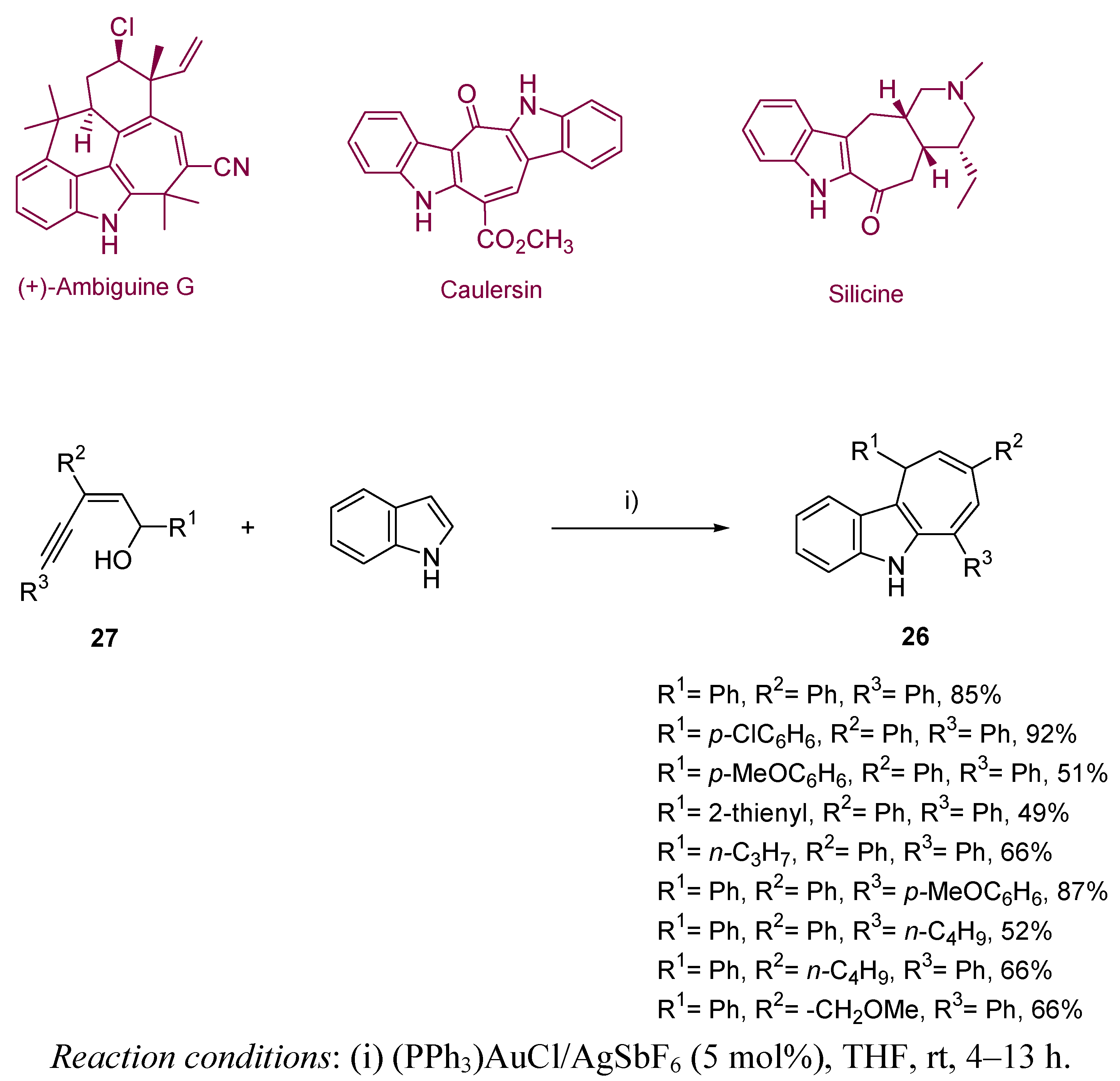
3. Cycloisomerization Processes Involving Carbon-Heteroatom Bond Formation
3.1. Cycloisomerization on Alkynol-Based Systems
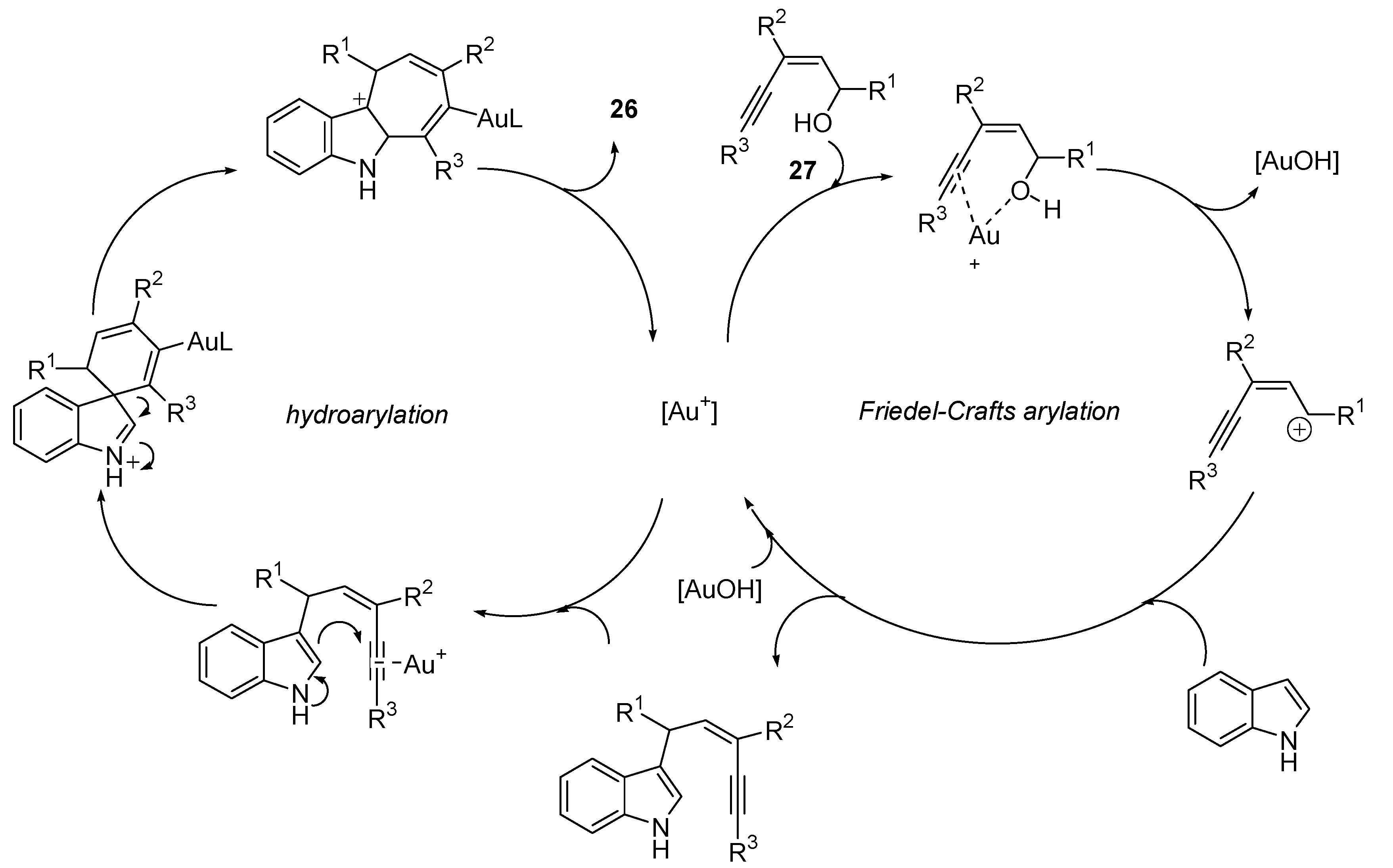
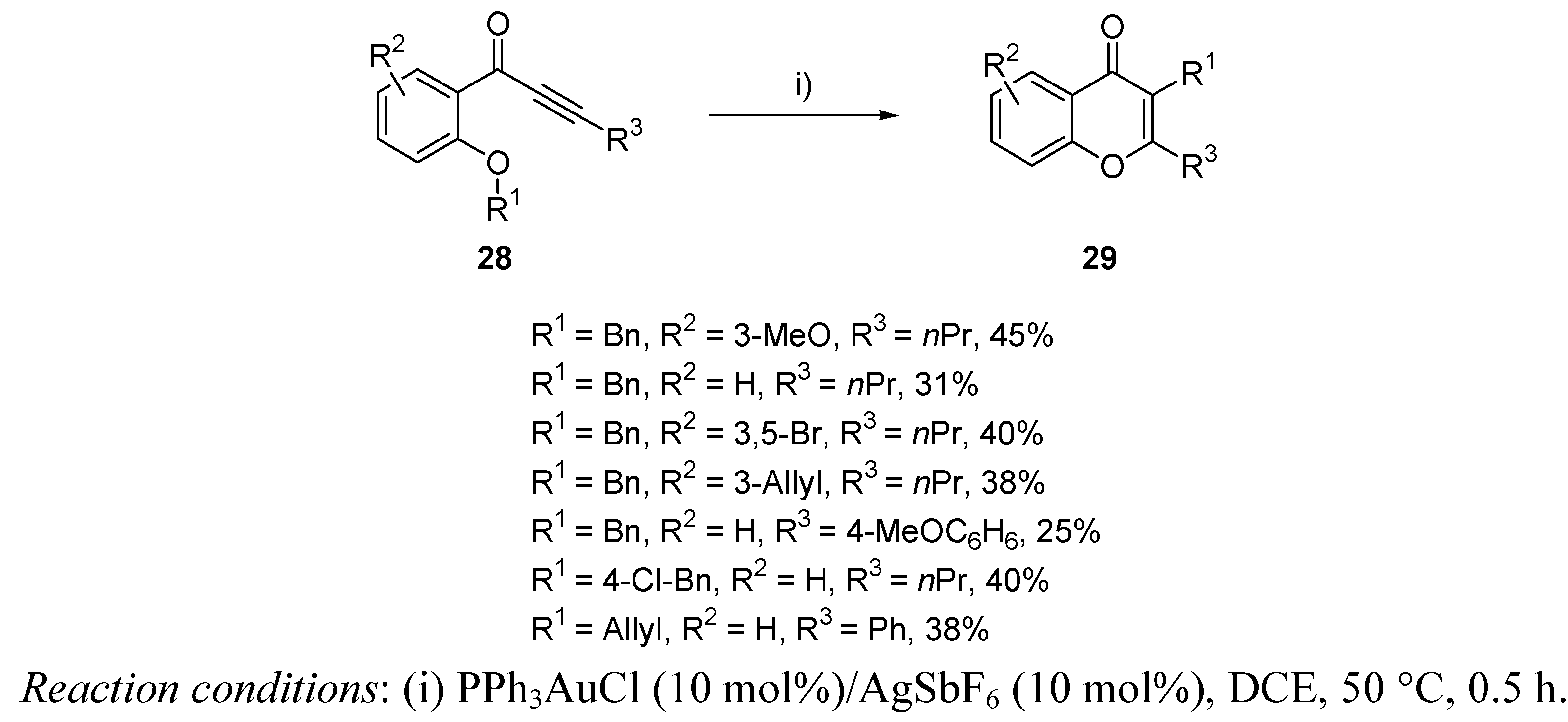
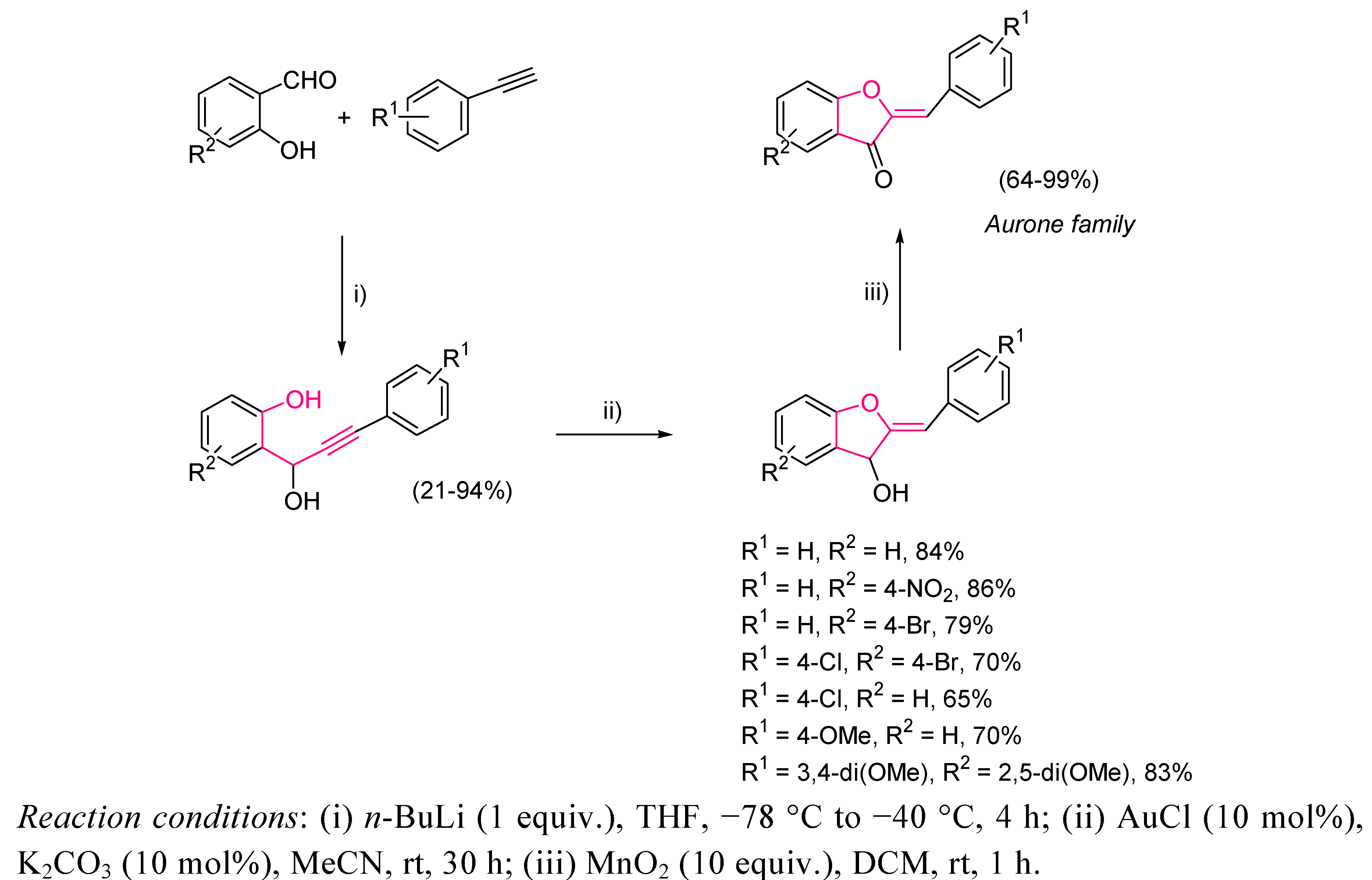
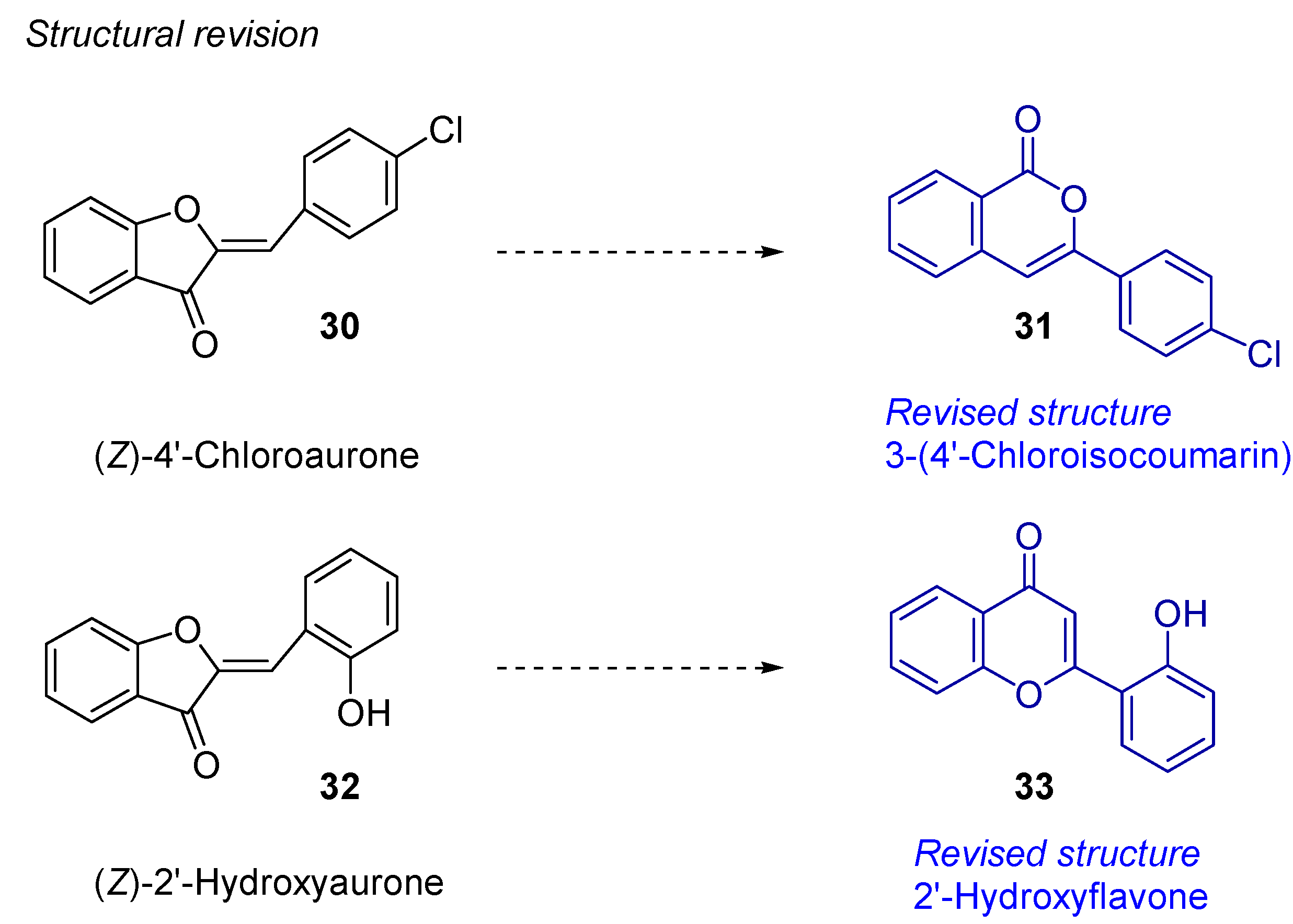

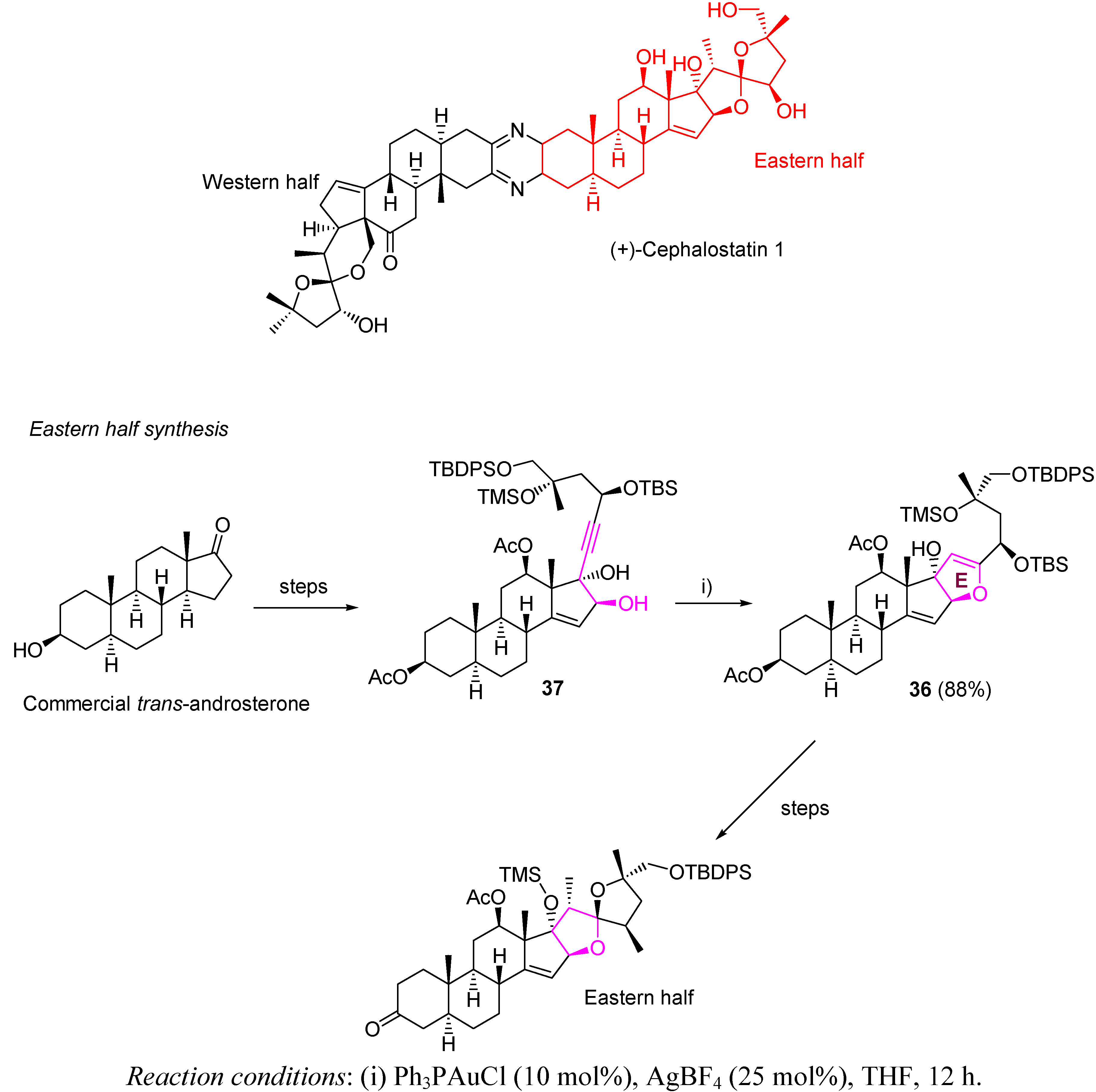
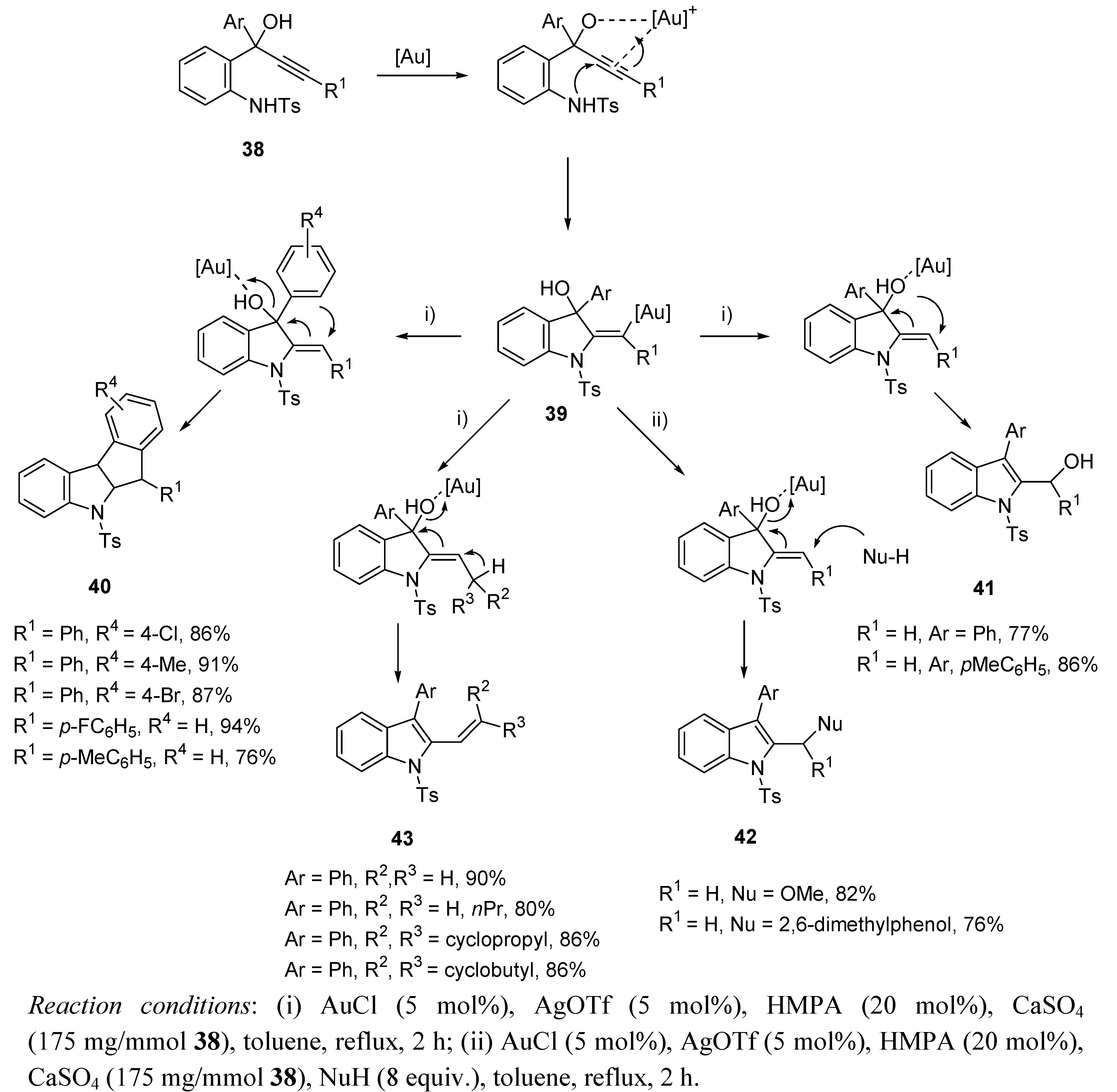
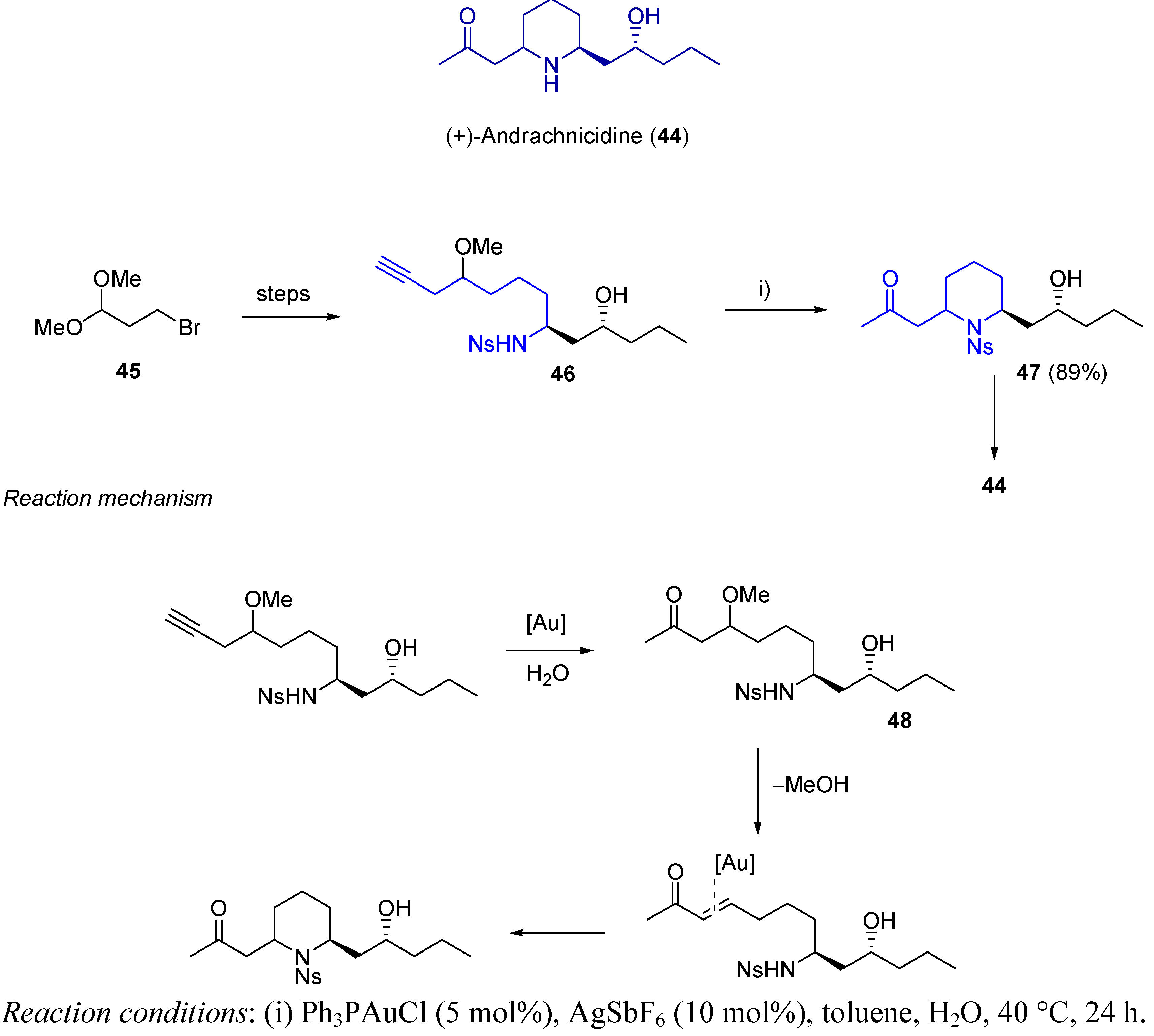
3.2. Cycloisomerization on Alkynediol-Based Systems
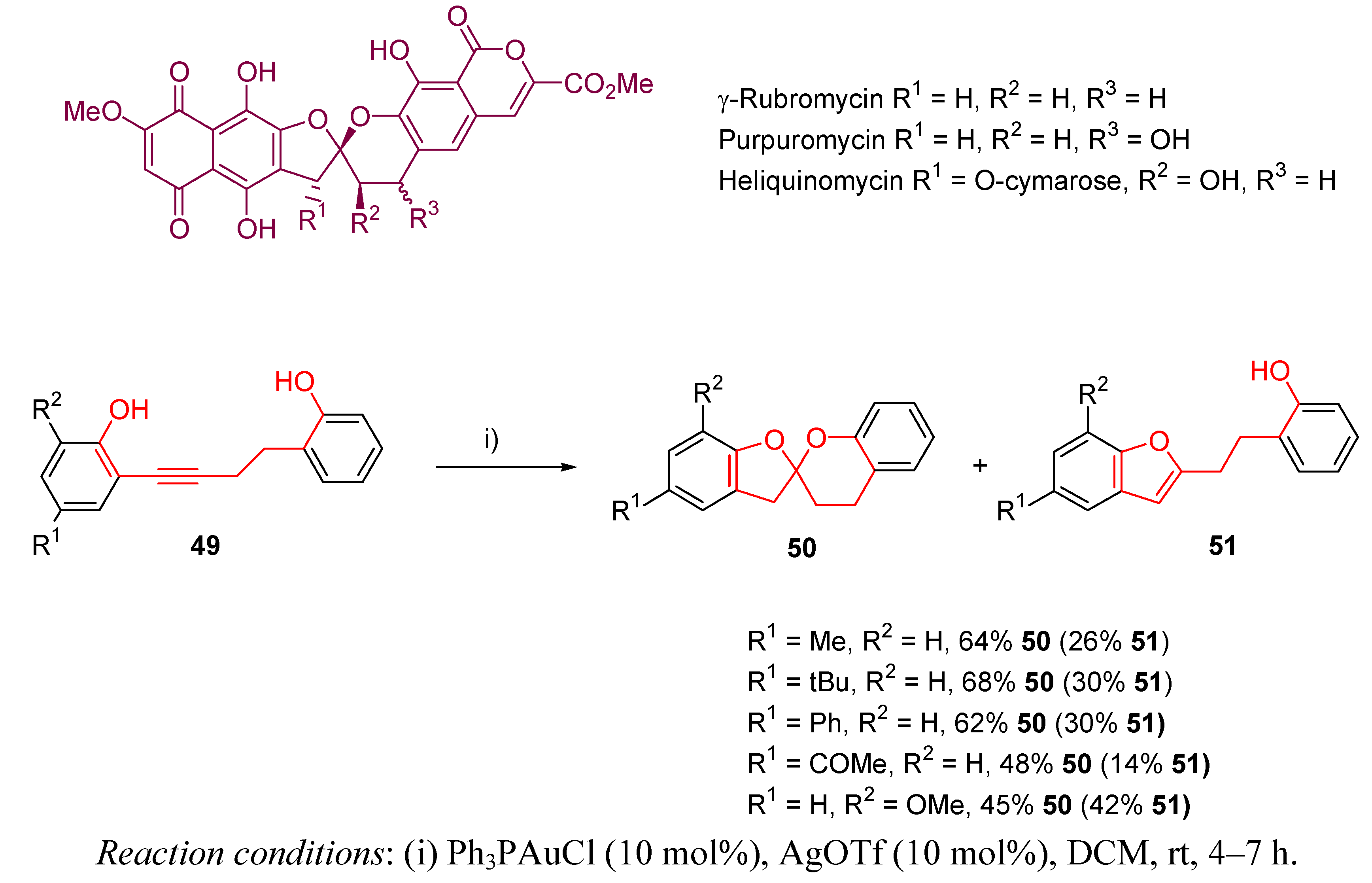
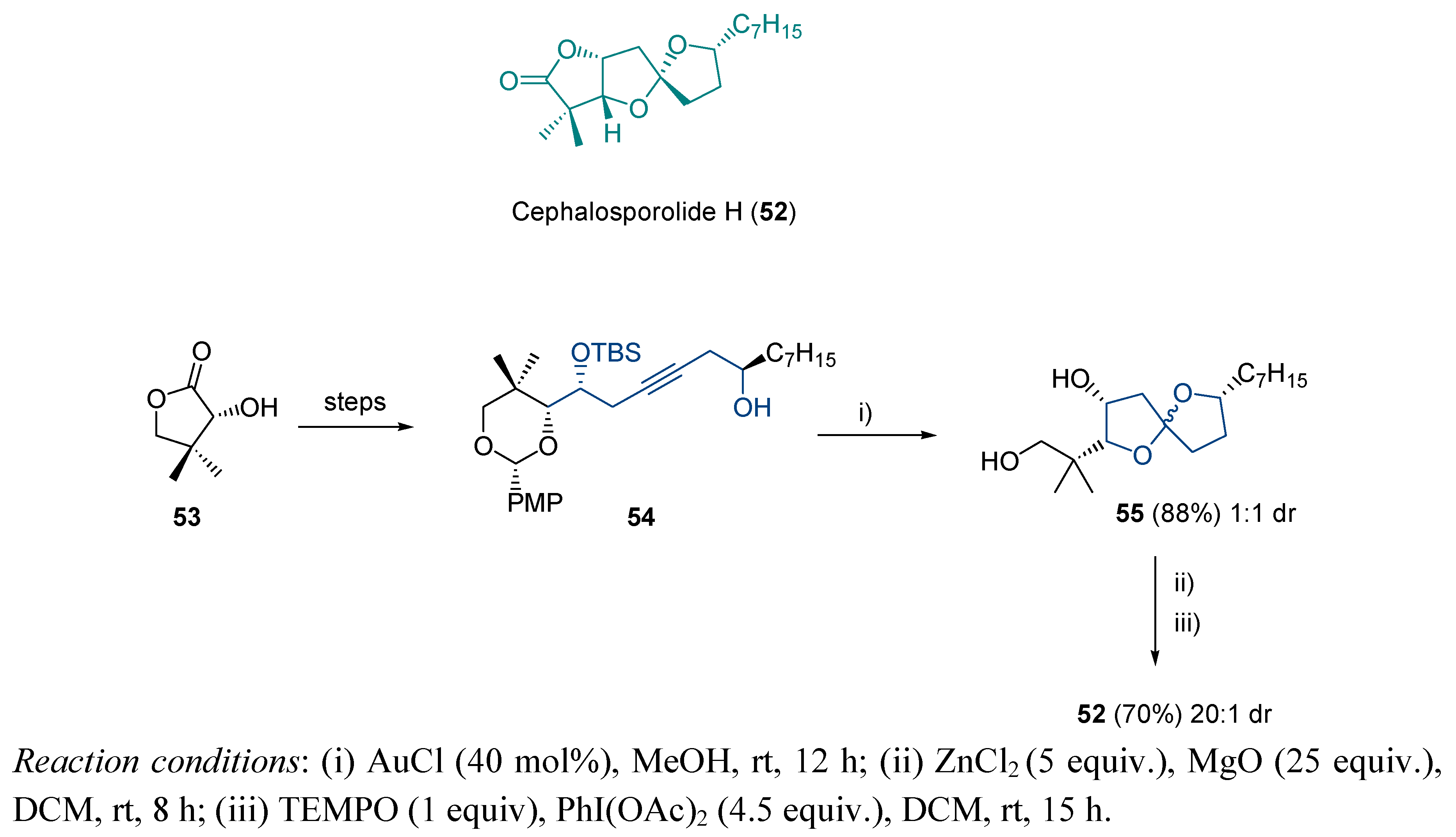
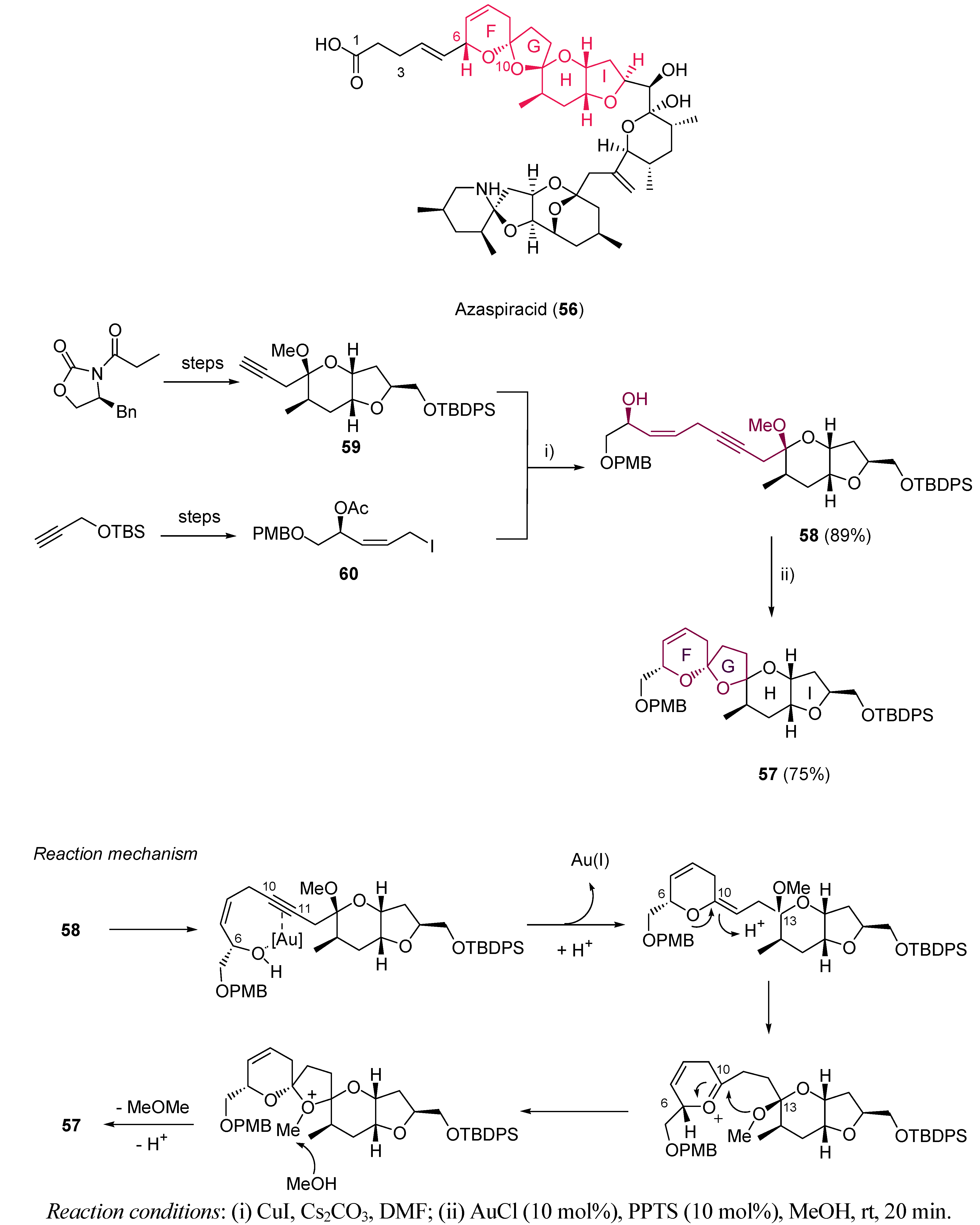
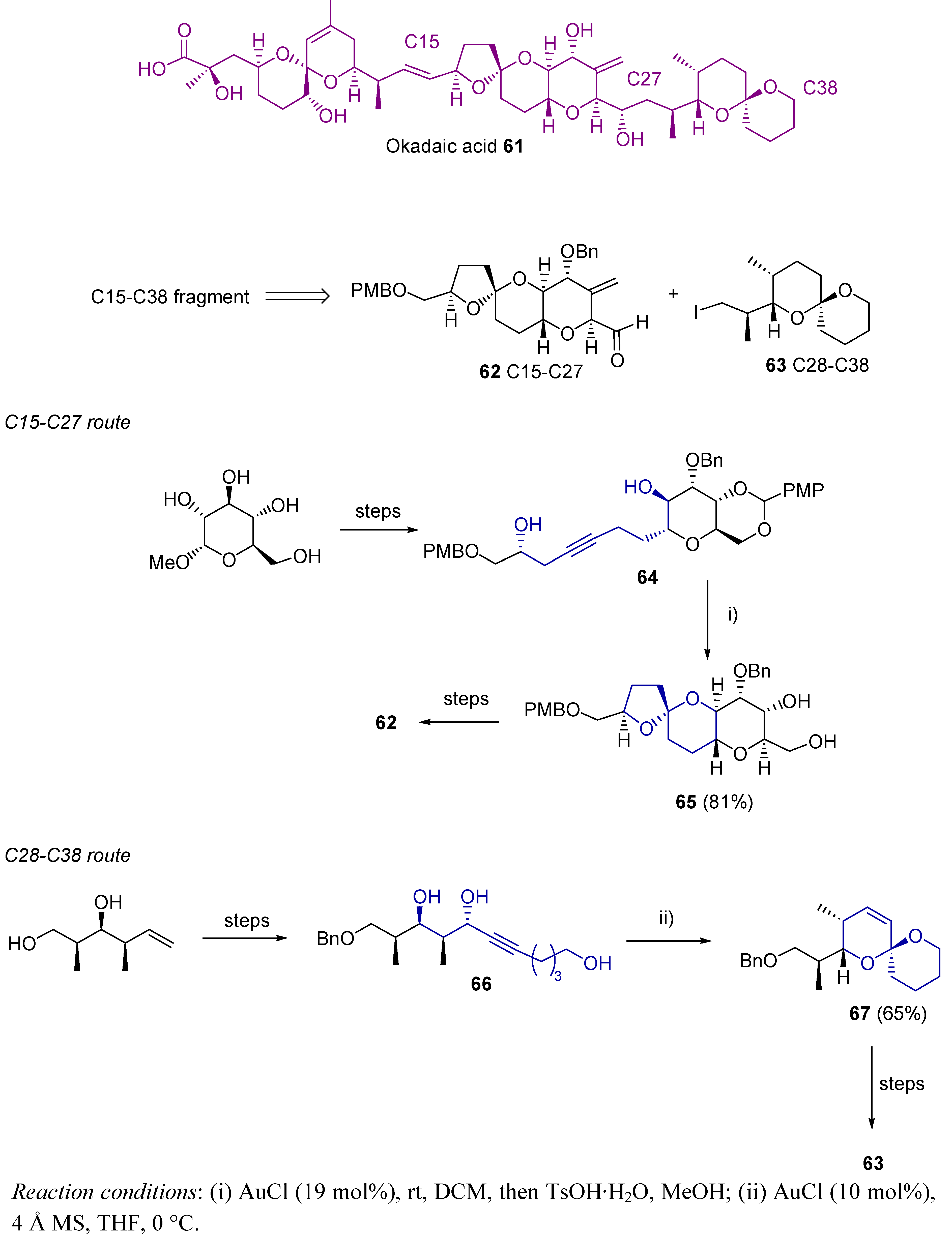
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Hashmi, A.S.K.; Rudolph, M. Gold Catalysis in Total Synthesis. Chem. Soc. Rev. 2008, 37, 1766–1775. [Google Scholar] [CrossRef]
- Fürstner, A. Gold and Platinum Catalysis-A Convenient Tool for Generating Molecular Complexity. Chem. Soc. Rev. 2009, 28, 3208–3221. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Alonso, J.M. Gold Catalyzed Oxycyclizations of Alkynols and Alkynediols. Org. Biomol. Chem. 2011, 9, 4405–4416. [Google Scholar] [CrossRef]
- Zeni, G.; Larock, R.C. Synthesis of Heterocycles via Palladium n-Olefin and n-Alkyne Chemistry. Chem. Rev. 2004, 104, 2285–2309. [Google Scholar] [CrossRef]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition-Metal-Catalyzed Addition of Heteroatom-Hydrogen Bonds to Alkynes. Chem. Rev. 2004, 104, 3079–3159. [Google Scholar] [CrossRef]
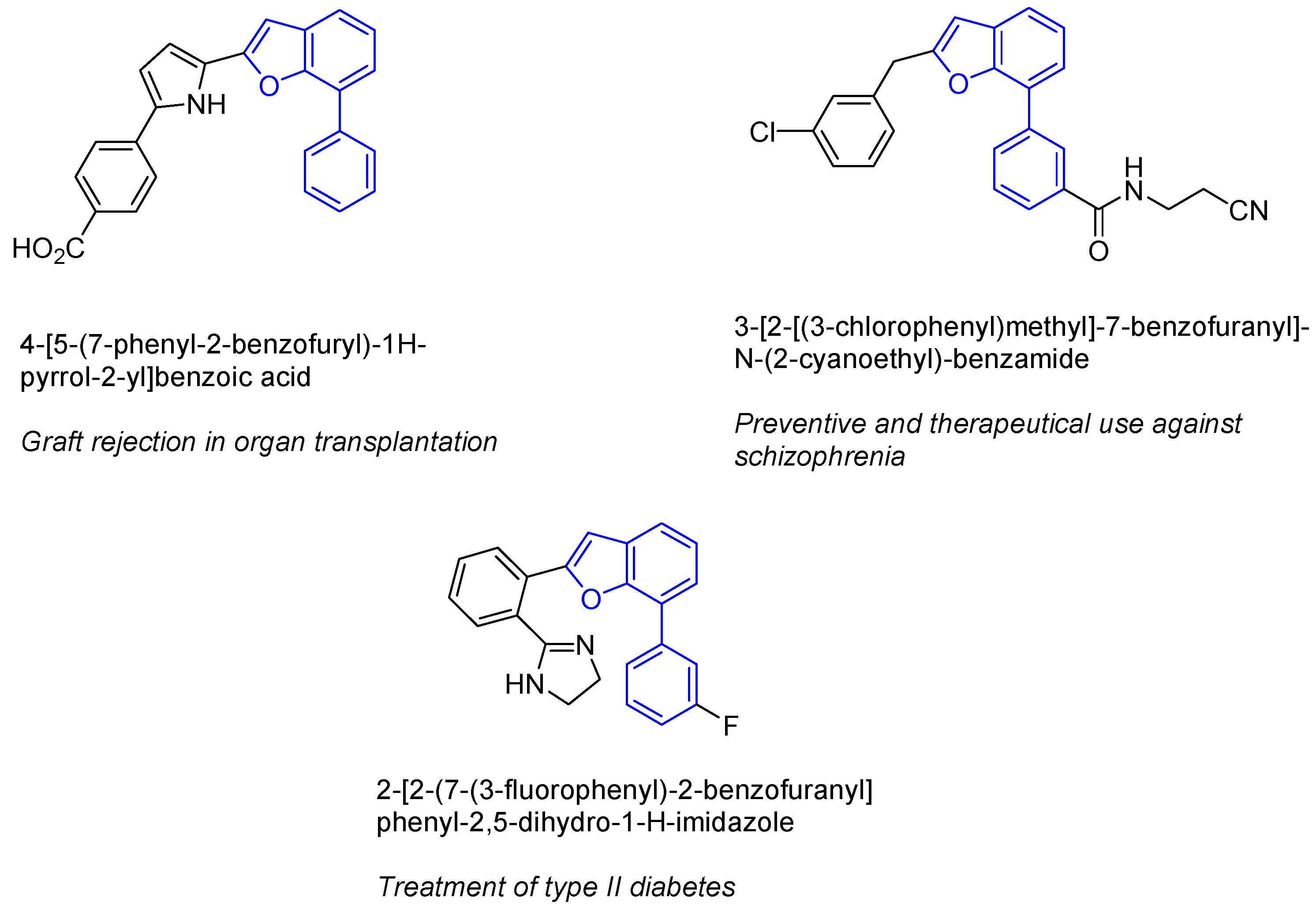
- Michael, P.; Gerd, R.; Theo, S. (Eli Lilly Co.) Imidazoline Derivatives for the Treatment of Diabetes, Especially Type II Diabetes. WO Patent 2000078726 A1, 28 December 2000. [Google Scholar] [Green Version]
- Tagami, K.; Yoshimura, H.; Nagai, M.; Hibi, S.; Kikuchi, K.; Sato, T.; Okita, M.; Okamoto, Y.; Nagasaka, Y.; Kobayashi, N.; Hida, T.; Tai, K.; Tokuhara, N.; Kobayashi, S. (Eisai Co.,Ltd.) Preparation of Fused-ring Carboxylic Acid Compounds as Retinoic Acid Receptor Agonists. WO Patent 9734869 A1, 25 September 1997. [Google Scholar] [Green Version]
- Masaki, S.; Mitsunori, K.; Yuhei, M.; Masakuni, K. (Takeda Pharmaceutical) Amide Compound. WO Patent 2010018874 A1, 18 February 2010. [Google Scholar] [Green Version]
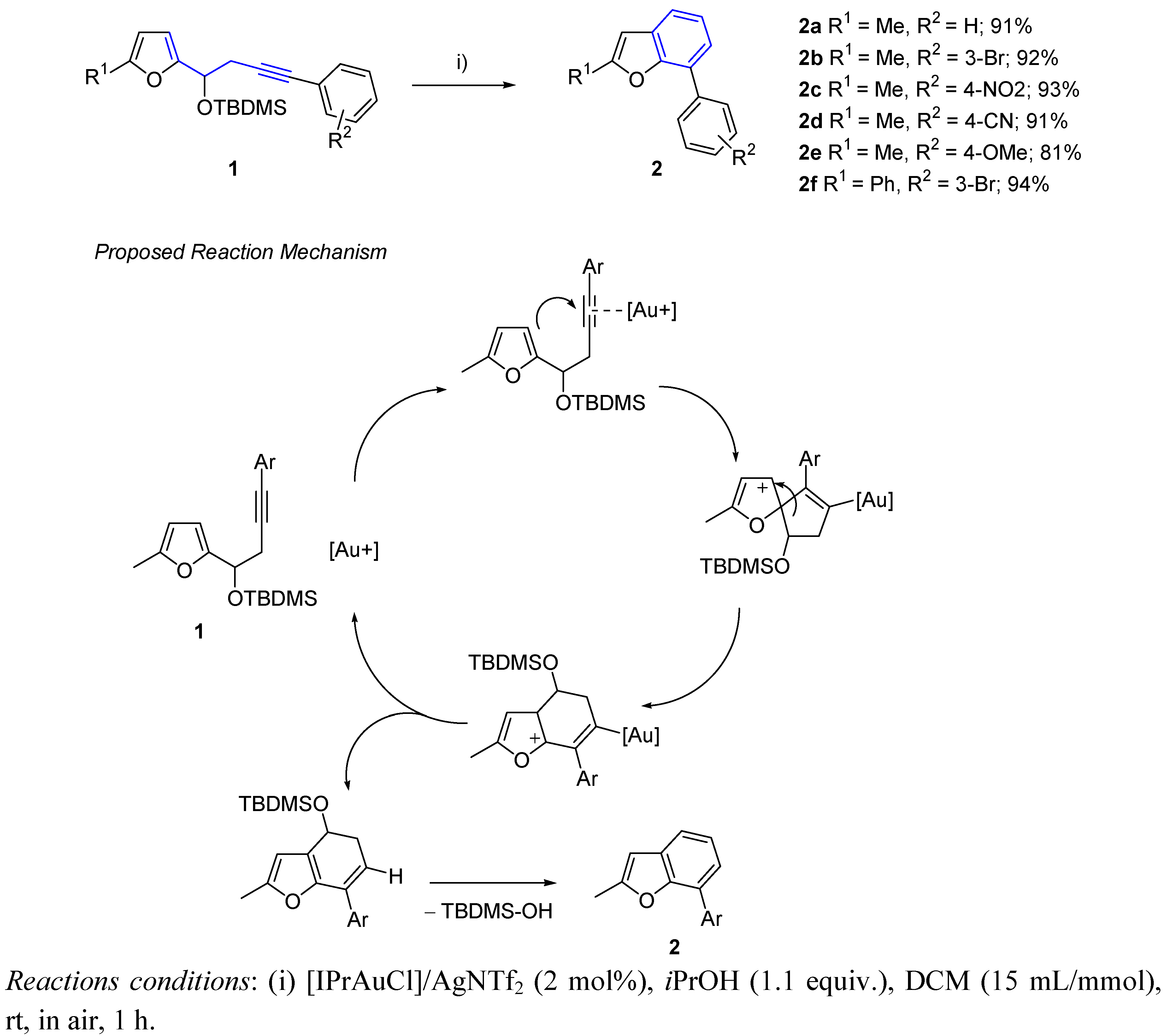
- Hashmi, A.S.K.; Yang, W.; Rominger, F. Gold(I)-Catalyzed Formation of Benzo[b]furans from 3-Silyloxy-1,5-enynes. Angew. Chem. Int. Ed. 2011, 50, 5762–5765. [Google Scholar] [CrossRef]
- Henkel, T.; Brunne, R.M.; Reichel, F.; Muller, H. Statiscal Investigation into the Structural Complementarity of Natural Products and Synthetic Compounds. Angew. Chem. Int. Ed. 1999, 38, 643–647. [Google Scholar] [CrossRef]
- Pearce, C. Biologically Active Fungal Metabolites. Adv. Appl. Microbiol. 1997, 44, 1–80. [Google Scholar] [CrossRef]
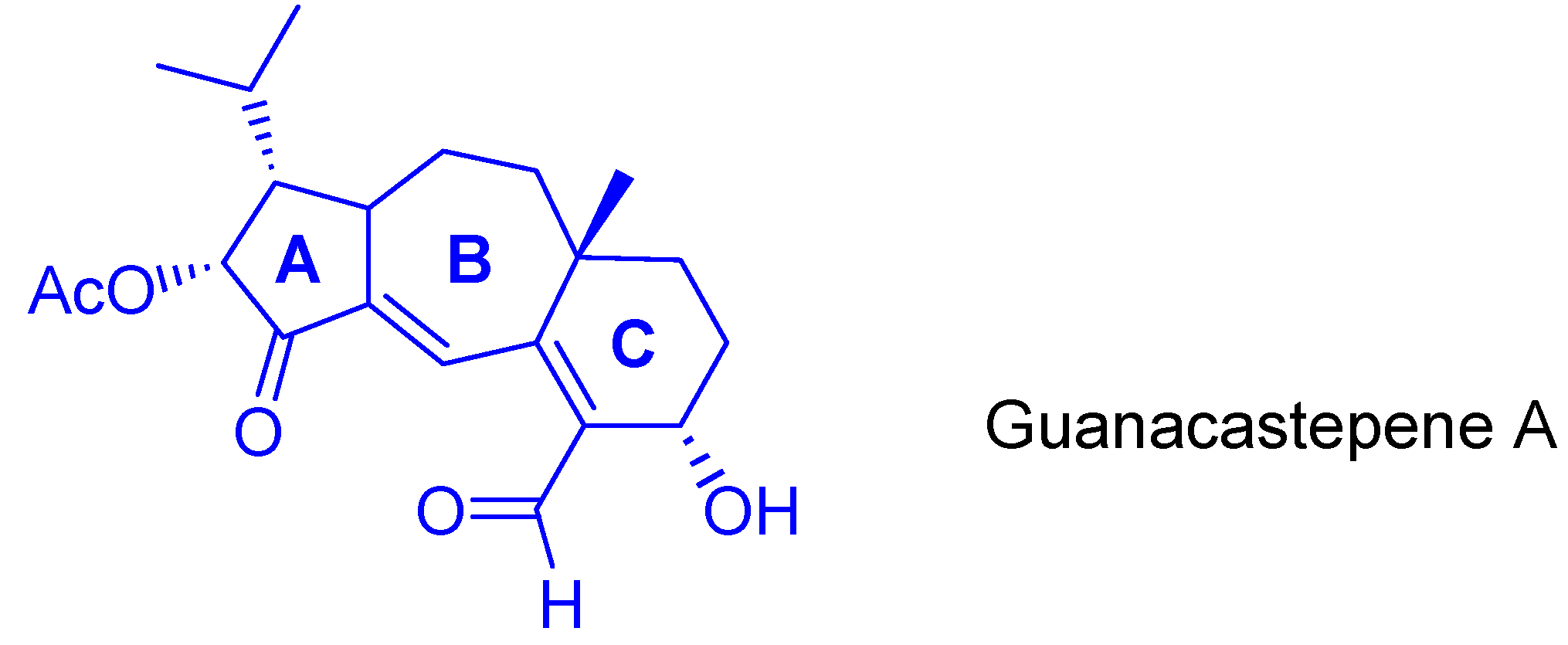
- For isolation and structural determination of guanacastepene, see: Brady, S.F.; Singh, M.P.; Janso, J.E.; Clardy, J. Guanacastepene, a Fungal-Derived Diterpene Antibiotic with a New Carbon Skeleto. J. Am. Chem. Soc. 2000, 122, 2116–2117. [Google Scholar] [CrossRef]
- For selected studies about the mentioned pathogens, see: Neu, H.C. The Crisis in Antibiotic Resistance. Science 1992, 257, 1064–1073. [Google Scholar]
- Swartz, M.N. Hospital-Acquired Infections: Diseases with Increasingly Limited Therapies. Proc. Natl. Acad. Sci. USA 1994, 91, 2420–2427. [Google Scholar] [CrossRef]
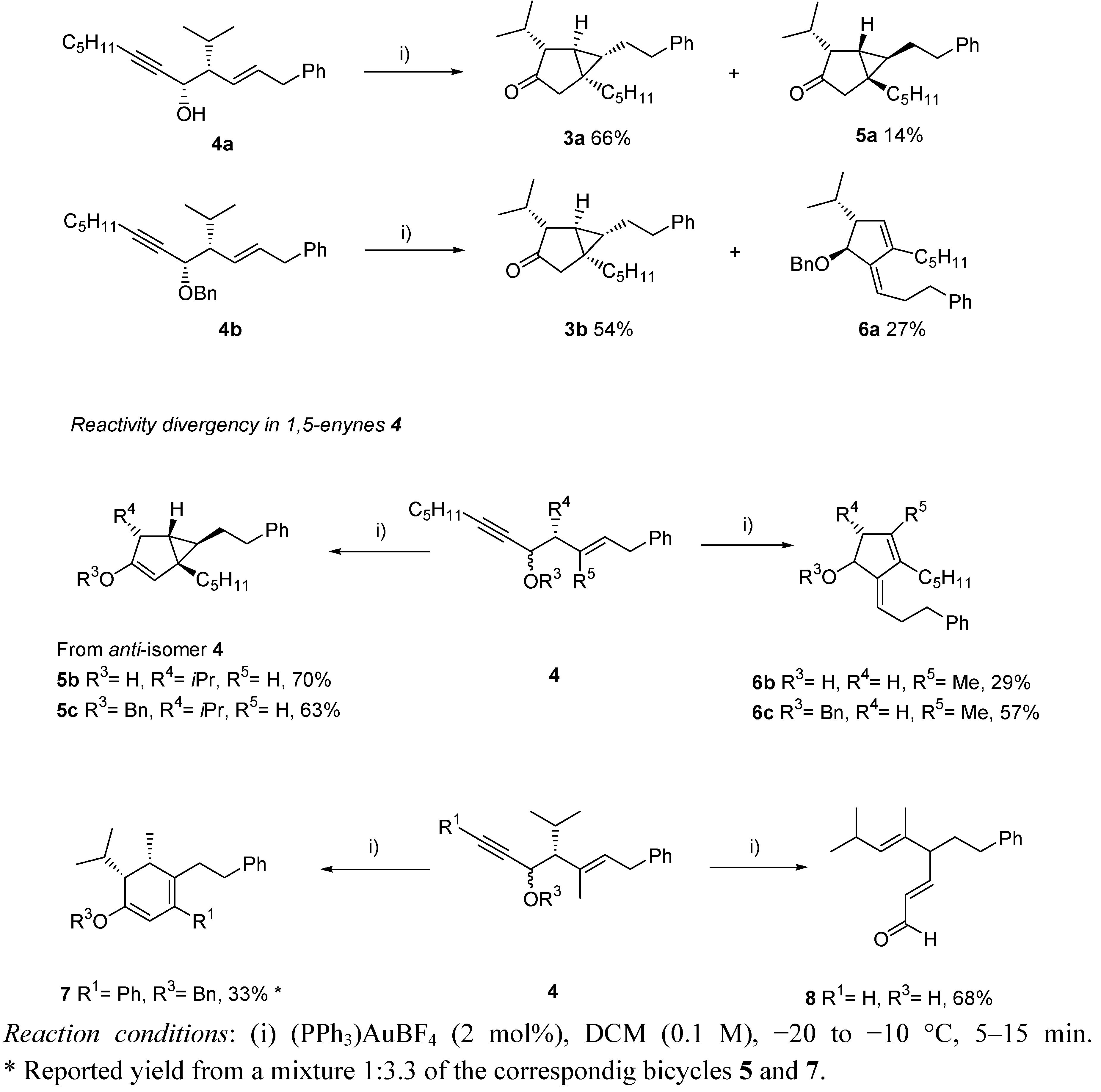
- Gagosz, F. Unusual Gold(I)-Catalyzed Isomerization of 3-Hydroxylated 1,5-Enynes: Highly Substrate-Dependent Reaction Manifolds. Org. Lett. 2005, 7, 4129–4132. [Google Scholar] [CrossRef]
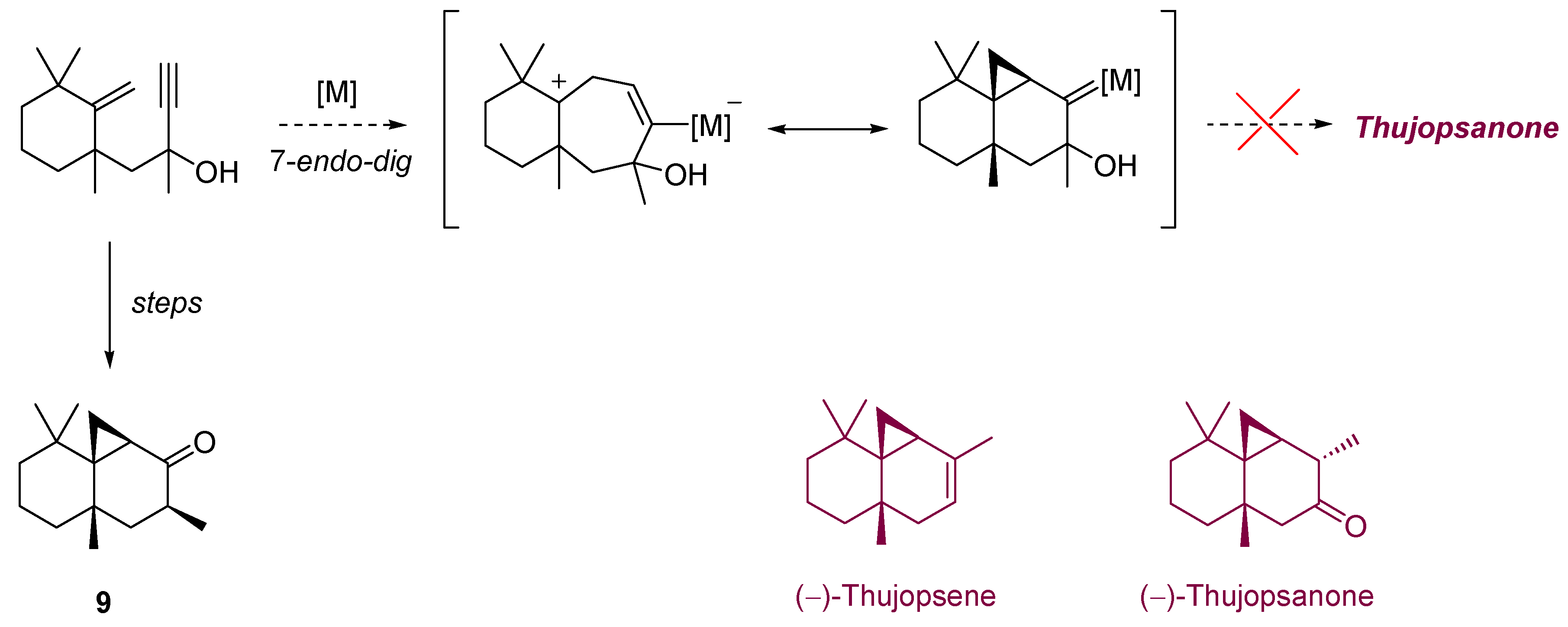
- Ohloff, G.; Strickler, H.; Willhalm, B.; Borer, C.; Hinder, M. En-Syntheses with Singlet Oxygen. II. Dye-Sensitized Photooxygenation of (−)-Thujopsene and Stereochemistry of the Prepared Thujopsanols. Helv. Chim. Acta 1970, 53, 623–637. [Google Scholar] [CrossRef]
- Hatsui, T.; Suzuki, N.; Takeshita, H. Dicyanoanthracene-Sensitized Photooxygenation of Thujopsene. Chem. Lett. 1985, 639–642. [Google Scholar] [Green Version]
- Fehr, C.; Vuagnoux, M.; Buzas, A.; Arpagaus, J.; Sommer, H. Gold- and Copper-Catalyzed Cycloisomerizations towards the Synthesis of Thujopsanone-Like Compounds. Chem. Eur. J. 2011, 17, 6214–6220. [Google Scholar] [CrossRef]
- Graening, T.; Schmalz, H.-G. Total Synthesis of Colchicine in Comparison: A Journey through 50 Years of Synthetic Organic Chemistry. Angew. Chem. Int. Ed. 2004, 43, 3230–3256. [Google Scholar] [CrossRef]
- Boyé, O.; Brossi, A. The Alkaloids; Brossi, A., Cordell, G.A., Eds.; Academic Press: New York, NY, USA, 1992; Volume 41, p. 125. [Google Scholar] [Green Version]
- Jordan, M.A.; Wilson, L. Microtubules as a Target for Anticancer Drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Vorogushin, A.V.; Wulff, W.D.; Hansen, H.-J. Central-to-Axial Chirality Transfer in the Benzannulation Reaction of Optically Pure Fischer Carbene Complexes in the Synthesis of Allocolchicinoids. Tetrahedron 2008, 64, 949–968. [Google Scholar] [CrossRef]
- Besong, G.; Jarowski, K.; Kocienski, P.J.; Sliwinski, E.; Boyle, F.T. Synthesis of (S)-(−)-N-Acetylcolchinol Using Intramolecular Biaryl Oxidative Coupling. Org. Biomol. Chem. 2006, 4, 2193–2207. [Google Scholar] [CrossRef]
- Leblanc, M.; Fagnou, K. Allocolchicinoid Synthesis via Direct Arylation. Org. Lett. 2005, 7, 2849–2852. [Google Scholar] [CrossRef]
- Büttner, F.; Bergemann, S.; Guénard, D.; Gust, R.; Seitz, G.; Thoret, S. Two Novel Series of Allocolchicinoids with Modified Seven Membered B-Rings: Design, Synthesis, Inhibition of Tubulin Assembly and Citotoxicity. Bioorg.Med. Chem. 2005, 13, 3497–3511. [Google Scholar] [CrossRef]
- Wu, T.R.; Chong, J.M. Asymmetric Synthesis of Propargylamines via 3,3’-Disubstituted Binaphtol-Modified Alkynylboronates. Org. Lett. 2006, 8, 15–18. [Google Scholar] [CrossRef]
- Vorogushin, A.V.; Predeus, A.V.; Wulff, W.D.; Hansen, H.-J. Diels-Alder Reaction-Aromatization Approach towards Functionalized Ring C Allocolchicinoids. Enantioselective Total Synthesis of (−)-7S-Allocolchicine. J. Org. Chem. 2003, 64, 5826–5831. [Google Scholar]
- For a reported synthesis of 16 see:Boyer, F.-D.; Hanna, I. Synthesis of Allocolchicines Using Sequential Ring-Closing Enyne Methathesis-Diels-Alder Reactions. Org. Lett. 2007, 9, 715–718. [Google Scholar] [CrossRef]
- Boyer, F.-D.; Le Goff, X.; Hanna, I. Gold(I)-Catalyzed Cycloisomerization of 1,7- and 1,8-Enynes: Application to the Synthesis of a New Allocolchicinoid. J. Org. Chem. 2008, 74, 5163–5166. [Google Scholar]
- Carbone, M.; Yan, L.; Irace, C.; Mollo, E.; Castelluccio, F.; Di Pascale, A.; Cimino, G.; Santamaria, R.; Guo, Y.-W.; Gavagnin, M. Structure and Cytotoxicity of Phidianidines A and B: First Finding of 1,2,4-Oxadiazole System in a Marine Natural Product. Org. Lett. 2011, 13, 2516–2519. [Google Scholar]
- Sunderhaus, J.D.; Sherman, D.H.; Williams, R.M. Studies on the Biosynthesis of the Stephadin and Notoamide Natural Products: A Stereochemical and Genetic Coundrum. Israel J. Chem. 2011, 51, 442–452. [Google Scholar] [CrossRef]
- Wu, M.; Wu, P.; Xie, H.; Wu, G.; Wei, X. Monoterpenoid Indole Alkaloid Mediating DNA Strand Scission from Turpina Arguta. Planta Medica 2011, 77, 284–286. [Google Scholar] [CrossRef]
- Ozcelik, B.; Kartal, M.; Orhan, I. Cytotoxicity, Antiviral and Antimicrobial Activities of Alkaloids, Flavonoids and Phenolic Acid. Pharm. Biol. 2011, 49, 396–402. [Google Scholar] [CrossRef]
- Yap, W.-S.; Gan, C.-Y.; Low, Y.-Y.; Choo, Y.-M.; Etoh, T.; Hayashi, M.; Komiyama, K.; Kam, T.-S. Grandilodines A-C, Biologically Active Indole Alkaloids from Kopsia Grandifolia. J. Nat. Prod. 2011, 74, 1309–1312. [Google Scholar] [CrossRef]
- Chung, Y.-M.; Lan, Y.-H.; Hwang, T.-L.; Leu, Y.-L. Anti-Inflammatory and Antioxidant Components from Hygroyza Aristata. Molecules 2011, 16, 1917–1927. [Google Scholar] [CrossRef]
- Foo, K.; Newhouse, T.; Mori, I.; Takayama, H.; Baran, P.S. Total Synthesis Guided Structure Elucidation of (+)-Psychotetramine. Angew. Chem. Int. Ed. 2011, 50, 2716–2719. [Google Scholar] [CrossRef]
- Finlayson, R.; Pearce, A.; Norrie, A.; Page, M.J.; Kaiser, M.; Bourguet-Kondracki, M.-L.; Harper, J.; Webb, V.; Copp, B. Didemnidines A and B, Indole Spermidine Alkaloids from the New Zealand Ascidian Didemnum sp. J. Nat. Prod. 2011, 74, 888–892. [Google Scholar] [CrossRef]
- Nakadate, S.; Nozawa, K.; Horie, H.; Fuji, Y.; Yaguchi, T. New Type Indole Diterpene, Eujindoles, From Eupenicillium Javanicum. Heterocycles 2011, 83, 351–356. [Google Scholar] [CrossRef]
- Ruiz-Sanchis, P.; Savina, S.; Albericio, F.; Alvarez, M. Structure, Bioactivity and Synthesis of Natural Products with Hexahydropyrrolo[2,3-b]Indole. Chem. Eur. J. 2011, 17, 1388–1408. [Google Scholar] [CrossRef]
- Yamada, Y.; Kitajima, M.; Kogure, N.; Wongseripipatana, S.; Takayama, H. Seven New Monoterpenoid Indole Alkaloids from Gelsemium Elegans. Chem. Asian J. 2011, 6, 166–173. [Google Scholar] [CrossRef]
- Palmisano, G.; Penoni, A.; Sisti, M.; Tiblietti, F.; Tollari, S.; Nicholas, K. Synthesis of Indole Derivatives with Biological Activity by Reactions between Unsaturated Hydrocarbons and N-Aromatic Precursors. Curr. Org. Chem. 2010, 14, 2409–2441. [Google Scholar] [CrossRef]
- Nakajima, T.O.; Satoshi, Y.; Fukuyama, T. Total Synthesis of (−)-Mersicarpine. J. Am. Chem. Soc. 2010, 132, 1236–1237. [Google Scholar] [CrossRef]
- Zhang, L. Tandem Au-catalyzed 3,3-Rearrangement-[2+2] Cycloadditions of Propargylic Esters: Expeditious Access to Highly Functionalized 2,3-Indoline-Fused Cyclobutanes. J. Am. Chem. Soc. 2005, 127, 16804–16805. [Google Scholar] [CrossRef]
- Ferrer, C.; Amijs, C.H.M.; Echavarren, A.M. Intra- and Intermolecular Reactions of Indoles with Alkynes Catalyzed by Gold. Chem. Eur. J. 2007, 13, 1358–1373. [Google Scholar] [CrossRef]
- Lu, Y.; Du, X.; Jia, X.; Liu, Y. Gold Catalyzed Intermolecular Reactions of (Z)-Enynols with Indoles for the Construction of Dihydrocyclohepta[b]indole Skeletons through a Cascade Friedel-Crafts/Hydroarylation Sequence. Adv. Syth. Catal. 2009, 351, 1517–1522. [Google Scholar] [CrossRef]
- Raffa, G.; Belot, S.; Balme, G.; Monteiro, N. Iodocyclization versus Diiodination in the Reaction of 3-Alkynyl-4-methoxycoumarins with Iodine: Synthesis of 3-Iodofuro[2,3-b]chromones. Org. Biomol. Chem. 2011, 9, 1474–1478. [Google Scholar] [CrossRef]
- Wang, L.; Peng, S.; Wang, J. Palladium-Catalyzed Cascade Reactions of Coumarins with Alkynes: Synthesis of Highly Substituted Cyclopentadiene Fused Chromones. Chem. Commun. 2011, 47, 5422–5424. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, Y.; Fu, H. Transition-Metal-Free Intramolecular Ullmann-Type O-Arylation: Synthesis of Chromone Derivatives. Angew. Chem. Int. Ed. 2011, 50, 3769–3773. [Google Scholar] [CrossRef]
- Renault, J.; Qian, Z.; Uriac, P.; Goualt, N. Electrophilic Carbon Transfer in Gold Catalysis: Synthesis of Substituted Chromones. Tetrahedron Lett. 2011, 52, 2476–2479. [Google Scholar] [CrossRef]
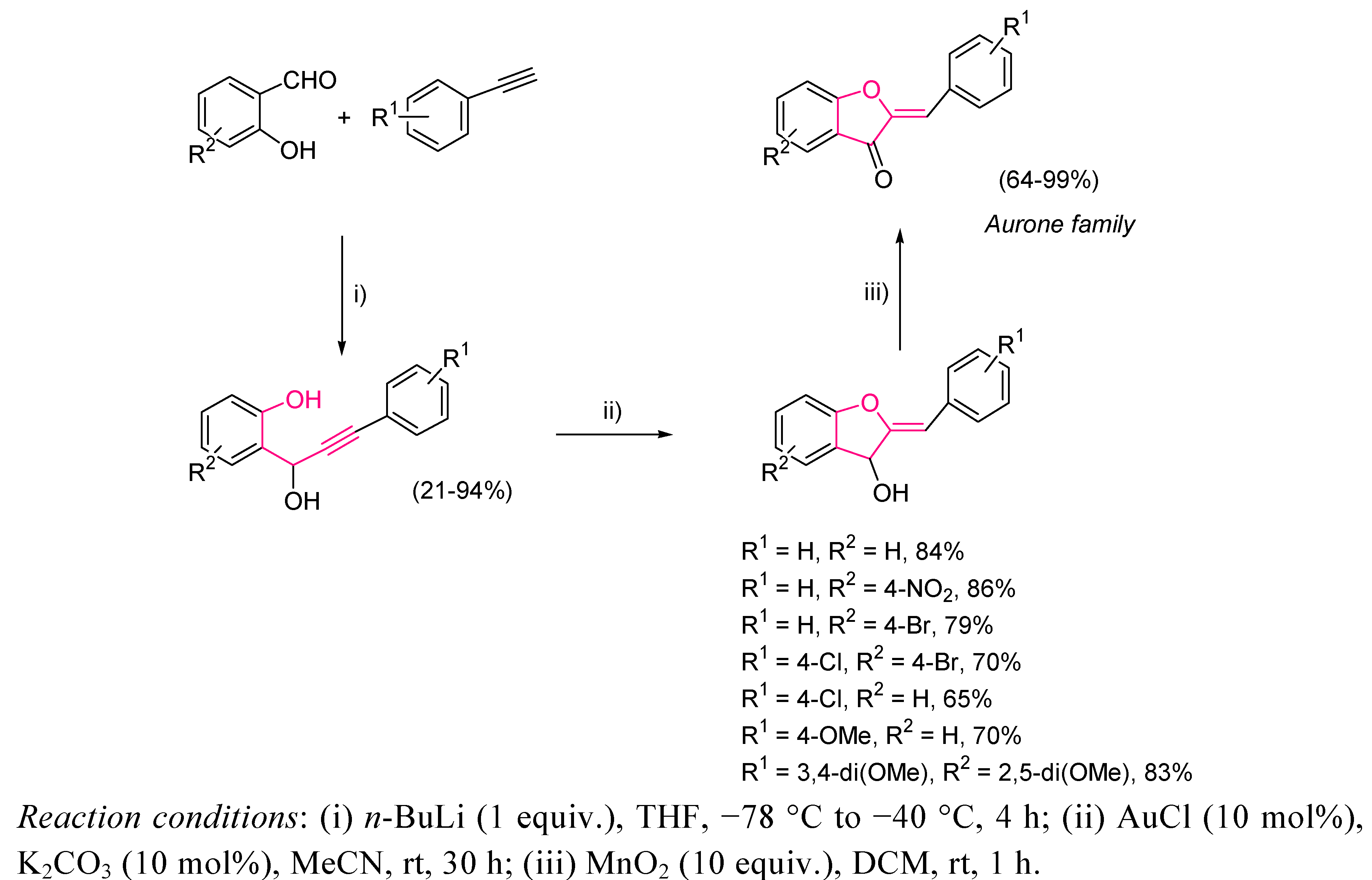
- For a review, see: Boumendjel, A. Aurones: A Subclass of Flavone with Promising Biological Potential. Curr. Med. Chem. 2003, 10, 2621–2330. [Google Scholar] [CrossRef]
- Brooks, C.J.; Watson, D.G. Phytoalexins. Nat. Prod. Rep. 1985, 427–459. [Google Scholar] [Green Version]
- Morimoto, M.; Fukumoto, H.; Nozoe, T.; Hagiwara, A.; Komai, K. Synthesis and Insect Antifeedant Activity of Aurones against Spodoptera Litura Larvae. J. Agric. Food. Chem. 2007, 55, 700–705. [Google Scholar] [CrossRef]
- Okombi, S.; Rival, D.; Bonnet, S.; Mariotte, A.-M.; Perrier, E.; Boumendjel, A. Discovery of Benzylidenezofuran-3(2H)-one (Aurones) as Inhibitors of Tyrosinase Derived from Human Melanocytes. J. Med. Chem. 2006, 49, 329–333. [Google Scholar] [CrossRef]
- Venkateswarlu, S.; Panchagnula, G.K.; Subbaraju, G.V. Synthesis and Antioxidative Activity of 3’,4’,6,7-tetrahydroxyaurone, a Metabollite of Biden Frondosa. Biosci. Biotechnol. Biochem. 2004, 68, 2183–2185. [Google Scholar] [CrossRef]
- Auf’mkolk, M.; Koerhle, J.; Hesch, R.D.; Cody, V. Inhibition of Rat Liver Iodothyronine Deiodinase. Interaction of Aurones with the Iodothyronine Ligand-Binding Site. Biol. Chem. 1986, 261, 11623–11630. [Google Scholar]
- Donnelly, J.A.; Fox, M.J.; Sharma, T.C. α-Halo Ketones. XI Generation of the Wheeler Aurone Synthesis. Tetrahedron 1979, 35, 875–879. [Google Scholar] [CrossRef]
- Bose, G.; Mondal, E.; Khan, A.T.; Bordoloi, M.J. An Environmentally Benign Synthesis of Aurones and Flavones from 2’-Acetoxychalcones using n-Tetrabutylammonium Tribromide. Tetrahedron Lett. 2001, 42, 8907–8909. [Google Scholar] [CrossRef]
- Lévai, A.; Tökés, A.L. Synthesis of Aurones by the Oxidative Rearrangement of 2’-Hydroxychalcones with Thallium(III) Nitrate. Synth. Commum. 1982, 12, 701–707. [Google Scholar]
- Imafuku, K.; Honda, M.; McOmie, J.F.W. Cyclodehydrogenation of 2’-Hydroxychalcones with 2,3-Dichloro-5,6-dicyano-p-benzoquinone: A Simple Route for Flavones and Aurones. Synthesis 1987, 199–201. [Google Scholar] [Green Version]
- Sekizaki, H. Synthesis of 2-Benzylidene-3(2H)-benzofuran-3-ones (Aurones) by Oxidation of 2’-Hydroxychalcones with Mercury(II) Acetate. Bull. Chem. Soc. Jpn. 1988, 61, 1407–1409. [Google Scholar] [CrossRef]
- Thakkar, K.; Cushman, M. A Novel Oxidative Cyclization of 2’-Hydroxychalcones to 4,5-Dialkoxyaurones by Thallium(III) Nitrate. J. Org. Chem. 1995, 60, 6499–6510. [Google Scholar] [CrossRef]
- An, Z.-W.; Catellani, M.; Chiusoli, G.P. Palladium-catalyzed Synthesis of Aurone from Salicyloyl Chloride and Phenylacetylene. J. Organomet. Chem. 1990, 397, 371–373. [Google Scholar] [CrossRef]
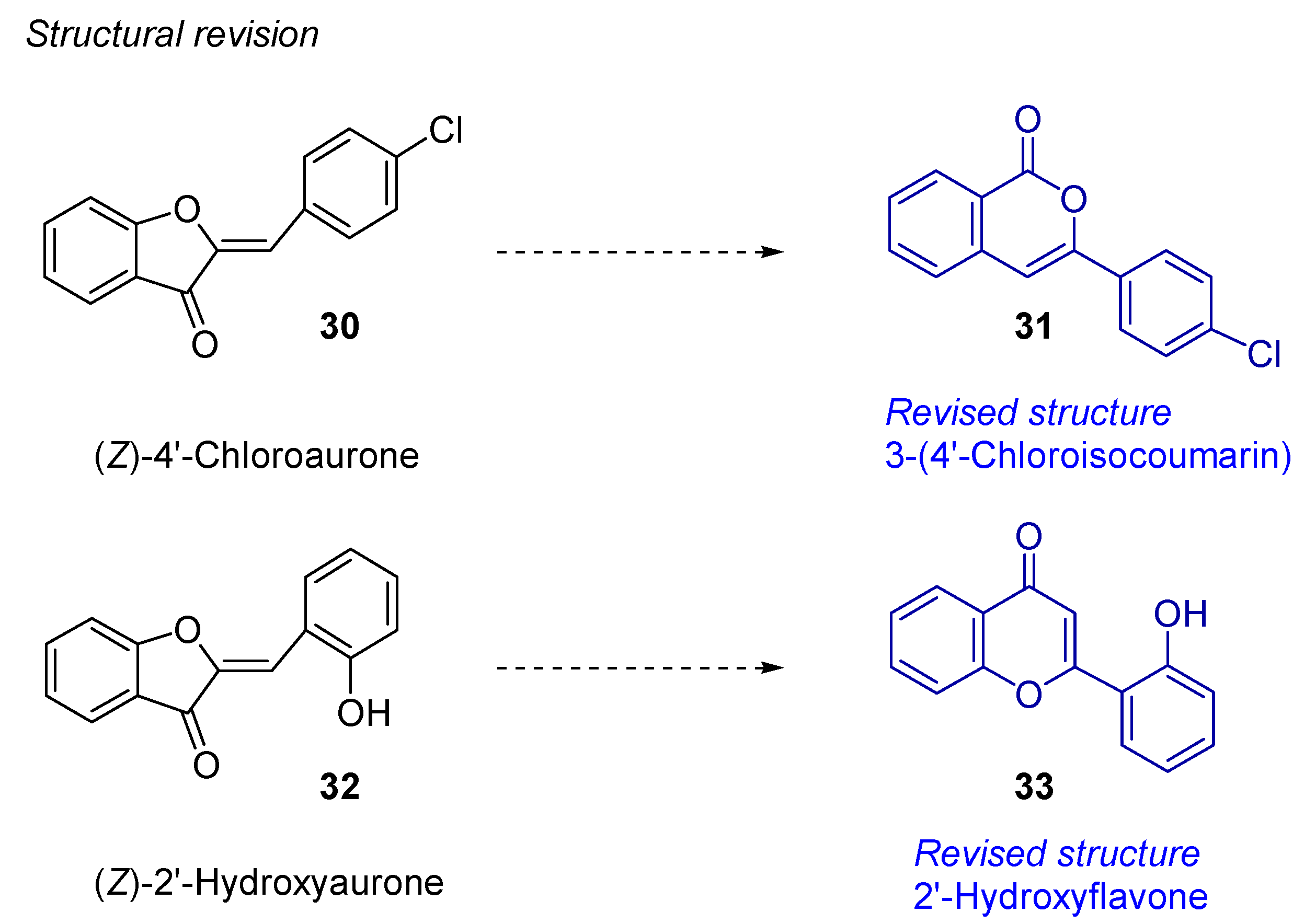
- Harkat, H.; Blanc, A.; Weibel, J.-M.; Pale, P. Versatile and Expeditious Synthesis of Aurones via AuI-Catalyzed Cyclization. J. Org. Chem. 2008, 73, 1620–1623. [Google Scholar] [CrossRef]
- Atta-ur-Rahman; Choudhary, M.I.; Hayat, S.; Khan, A.; Ahmed, A. Two New Aurones from Marine Brown Alga Spatoglossum Variable. Chem. Pharm. Bull. 2001, 49, 105–107. [Google Scholar] [CrossRef]
- Kobayashi, S.; Miyase, T.; Noguchi, H. Polyphenolic Glycosides and Oligosacharide Multiesters from the Roots of Polygala Dalmaisiana. J. Nat. Prod. 2002, 65, 319–328. [Google Scholar] [CrossRef]
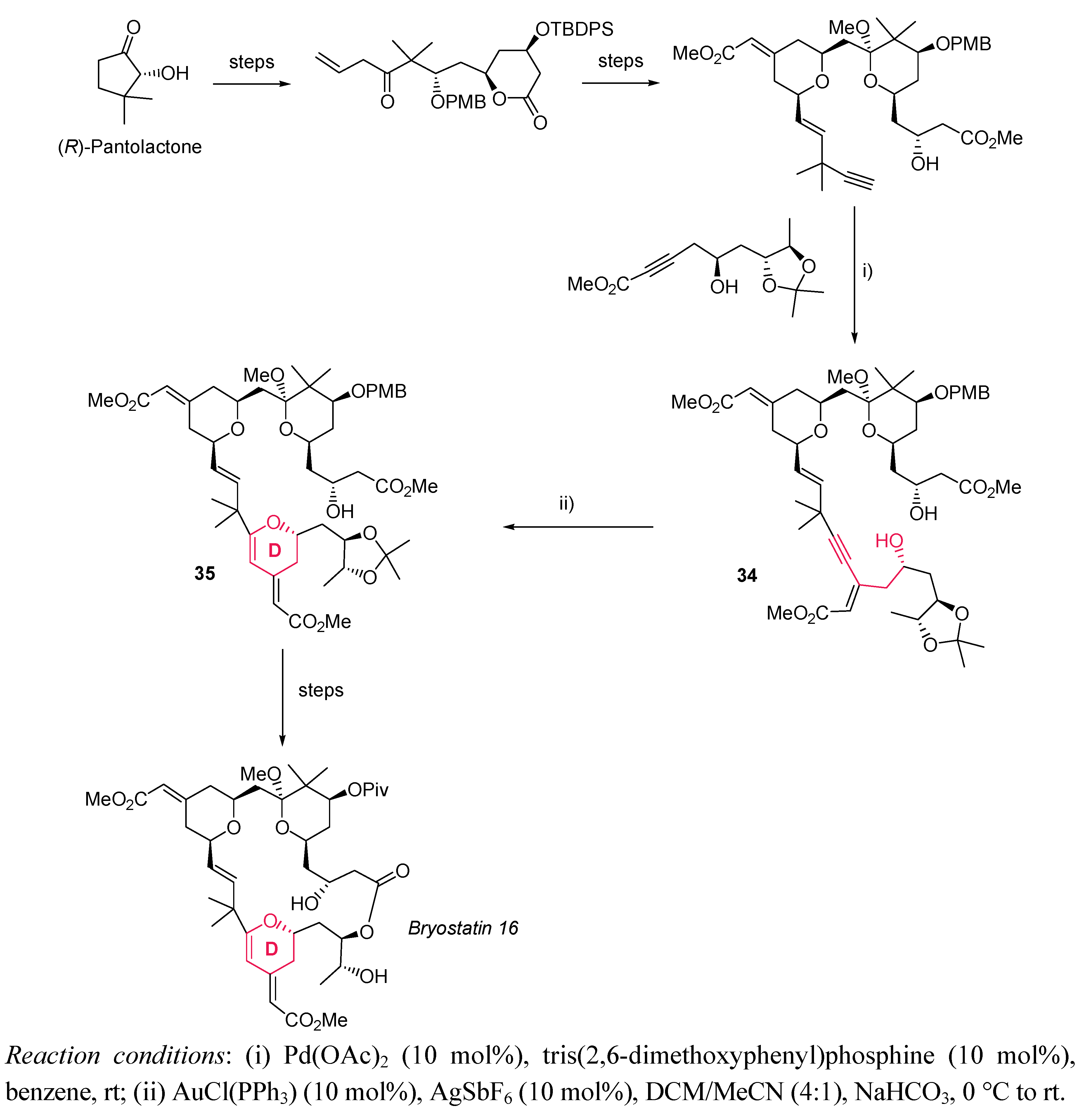
- Trost, B.M.; Dong, G. Total Synthesis of Bryostatin 16 Using a Pd-Catalyzed Diyne Coupling as Macrocyclization Method and Synthesis of C20-epi-Bryostatin 7 as a Potent Anticancer Agent. J. Am. Chem. Soc. 2010, 132, 16403–16416. [Google Scholar] [CrossRef]
- Pettit, G.R.; Gao, F.; Blumberg, P.M.; Herald, C.L.; Coll, J.C.; Kamano, Y.; Lewin, N.E.; Schmidt, J.M.; Chapuis, J.-C. Antineoplastic Agents. 340. Isolation and Structural Elucidation of Bryostatins 16-18. J. Nat. Prod. 1996, 59 For more information of bryostatin 16, see:, 286–289. [Google Scholar] [CrossRef]
- Hale, K.J.; Hummersome, M.G.; Manaviazar, S.; Frigerio, M. The Chemistry and Biology of the Bryostatin Antitumour Macrolides. Nat. Prod. Rep. 2002, 19, 413–453. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Marine Natural Products and related Compounds in Clinical and Advanced Preclinical Trials. J. Nat. Prod. 2004, 67, 1216–1238. [Google Scholar] [CrossRef]
- Hale, K.J.; Manaviazar, S. New Approaches to the Total Synthesis of Bryostatin Antitumor Macrolides. Chem. Asian J. 2010, 5, 704–754. [Google Scholar] [CrossRef]
- Manaviazar, S.; Hale, K.J. Total Synthesis of Bryostatin 1: A Short Route. Angew. Chem. Int. Ed. 2011. [Google Scholar] [CrossRef]
- Serrano, M. The Tumour Suppressor Protein p16INK4a. Exp. Cell. Res. 1997, 237, 7–13. [Google Scholar] [CrossRef]
- Pettit, G.R.; Inoue, M.; Kamano, Y.; Herald, D.L.; Arm, C.; Dufresne, C.; Christie, N.D.; Schmidt, J.M.; Doubek, D.L.; Krupa, T.S. Antineoplastic Agent. 174. Isolation and Styructure of the Cytostatic Depsipeptide Dolastatin 13 from the Sea Hare Dolabella Auricularia. J. Am. Chem. Soc. 1989, 110, 2006–2007. [Google Scholar]
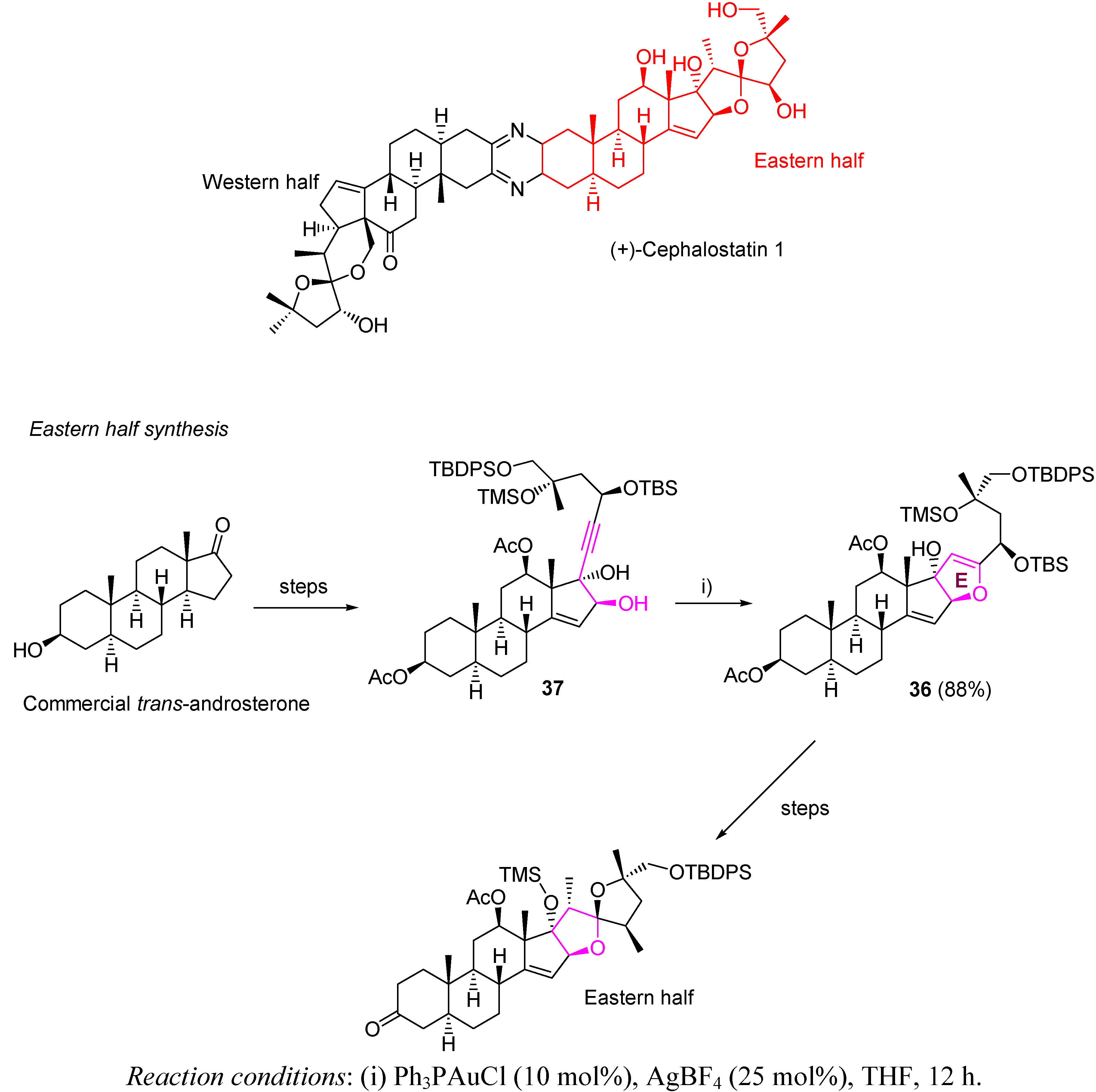
- Jeong, J.U.; Sutton, S.C.; Kim, S.; Fuchs, P.L. Biomimetic Total Syntheses of (+)-Cephalostatin 12, and (+)-Ritterazine K. J. Am. Chem. Soc. 1995, 117, 10157–10158. [Google Scholar] [CrossRef]
- LaCour, T.G.; Guo, C.; Bhandatu, S.; Fuchs, P.L.; Boyd, M.R. Interphylal Product Splicing: The First Total Syntheses of Cephalostatin 1, the North Hemisphere of Ritterazine G, and the High Active Hybrid Analog, Ritterostatin GN1N. J. Am. Chem. Soc. 1998, 120, 692–707. [Google Scholar]
- Jeong, J.U.; Guo, C.; Fuchs, P.L. Synthesis of the South Unit of Cephalostatin. 7. Total Syntheses of (+)-Cephalostatin 7, (+)-Cephalostatin 12, and (+)-Ritterazine K. J. Am. Chem. Soc. 1999, 121, 2071–2084. [Google Scholar] [CrossRef]
- Kim, S.; Sutton, S.C.; Guo, C.; LaCour, T.G.; Fuchs, P.L. Synthesis of the North 1 Unit of the Cephalostatin Family from Hecogenin Acetate. J. Am. Chem. Soc. 1999, 121, 2056–2070. [Google Scholar]
- Lee, S.; Fuchs, P.L. The First Total Synthesis of (Corrected) Ritterazine M. Org. Lett. 2002, 4, 317–318. [Google Scholar] [CrossRef]
- Fortner, K.C.; Kato, D.; Tanaka, Y.; Shair, M.D. Enantioselective Synthesis of (+)-Cephalostatin 1. J. Am. Chem. Soc. 2010, 132, 275–280. [Google Scholar] [CrossRef]
- Ueda, M.; Sato, A.; Ikeda, Y.; Miyoshi, T.; Naito, T.; Miyata, O. Direct Synthesis of Trisubstituted Isoxazoles through Gold-Catalyzed Domino Reaction of Alkynyl Oxime Ethers. Org. Lett. 2010, 11, 2594–2597. [Google Scholar]
- He, W.; Li, C.; Zhang, L. An Efficient [2+2+1] Synthesis of 2,5-Disubstututed Oxazoles via Gold-Catalyzed Intermolecular Alkyne Oxidation. J. Am. Chem. Soc. 2011, 133, 8482–8485. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Schuster, A.M.; Zimmer, M.; Rominger, F. Synthesis of 5-Halo-4H-1,3-oxazine-6-amines by Copper-Mediated Domino Reaction. Chem. Eur. J. 2011, 17, 5511–5515. [Google Scholar] [CrossRef]
- Gao, X.; Pan, Y.-M.; Li, M.L.; Chen, L.; Zhan, Z.-P. Facile One-Pot Synthesis of Three Different Substituted Thiazoles from Propargylic Alcohols. Org. Biomol. Chem. 2010, 8, 3259–3266. [Google Scholar] [CrossRef]
- Yoshimatsu, M.; Matsui, M.; Yamamoto, T.; Sawa, A. Convinient Preparation of 4-Arylmethyl- and 4-Hetarylmethyl Thiazoles by Regioselective Cycloaddition Reactions of 3-Sulfanyl- and Selenylpropargyl Alcohols. Tetrahedron Lett. 2010, 66, 7975–7987. [Google Scholar]
- Asanuma, Y.; Fujiwara, S.-I.; Shin-Ike, T.; Kambe, N. Selenoimidoylation of Alcohols with Selenium and Isocyanides and its Application to the Synthesis of Selenium-Containing Heterocycles. J. Org. Chem. 2004, 69, 4845–4848. [Google Scholar] [CrossRef]
- Wilckens, K.; Uhlemann, M.; Czekelius, C. Gold-Catalyzed endo-Cyclizations of 1,4-Diynes to Seven-Membered Ring Heterocycles. Chem. Eur. J. 2009, 15, 13323–13326. [Google Scholar]
- Yan, B.; Zhou, Y.; Zhang, H.; Chen, J.; Liu, Y. Highly Efficient Synthesis of Functionalized Indolizines and Indolizinones by Copper-Calayzed Cycloisomerization of Propargylic Pyridines. J. Org. Chem. 2007, 72, 7783–7786. [Google Scholar] [CrossRef]
- Bunnelle, E.M.; Smith, C.R.; Lee, S.K.; Singaram, S.W.; Rhodes, A.J.; Sarpong, R. Pt-Catalyzed Cyclization/Migration of Propargylic Alcohols for the Synthesis of 3(2H)-Furanones, Pyrrolones, Indolizines, and Indolizinones. Tetrahedron Lett. 2008, 64, 7008–7014. [Google Scholar]
- For some examples of non-metal catalyzed cycloisomerizations see: Ji, K.-G.; Zhu, H.-T.; Yang, F.; Shu, X.-Z.; Zhao, S.-C.; Liu, X.-Y.; Shaukat, A.; Liang, Y.-M. A Novel Iodine-Promoted Tandem Cyclization: An Efficient Synthesis of Substituted 3,4-Diiodoheterocyclic Compounds. Chem. Eur. J. 2010, 16, 6151–6154. [Google Scholar] [CrossRef]
- Zang, X.; Teo, W.T.; Chan, S.W.H.; Chan, P.W.H. Brønsted Acid Catalyzed Cyclization of Propargylic Alcohols wiyh Thioamides. Facile Synthesis of Di- and Trisubstituted Thiazoles. J. Org. Chem. 2010, 75, 6290–6293. [Google Scholar] [CrossRef]
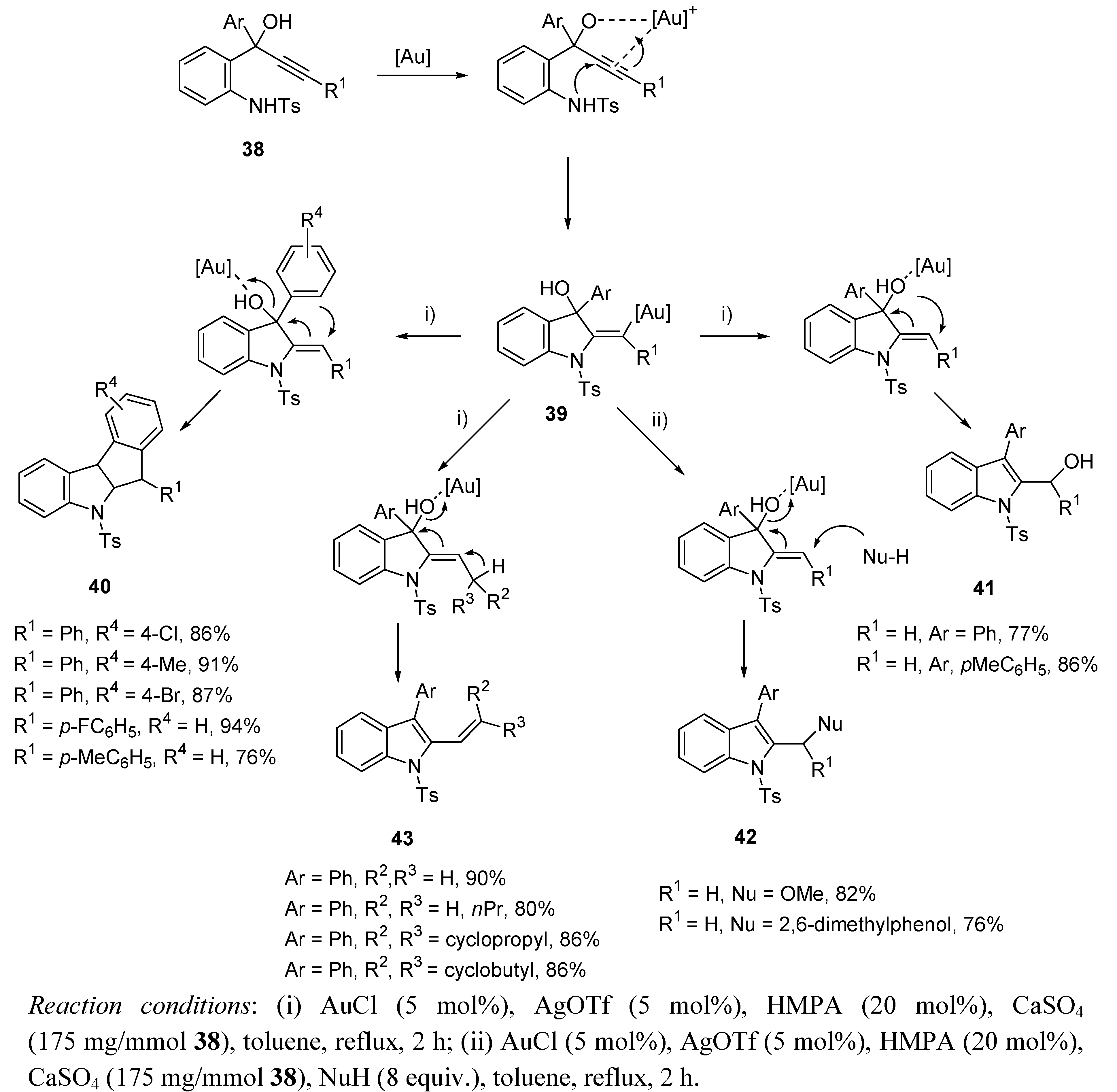
- Kothandaraman, P.; Rao, W.; Foo, S.J.; Chan, P.W.H. Gold-Catalyzed Cycloisomerization Reaction of 2-Tosylamino-phenylprop-1-yn-3-ols via a Versatile Approach for Indole Synthesis. Angew. Chem. Int. Ed. 2010, 49, 4619–4623. [Google Scholar] [CrossRef]
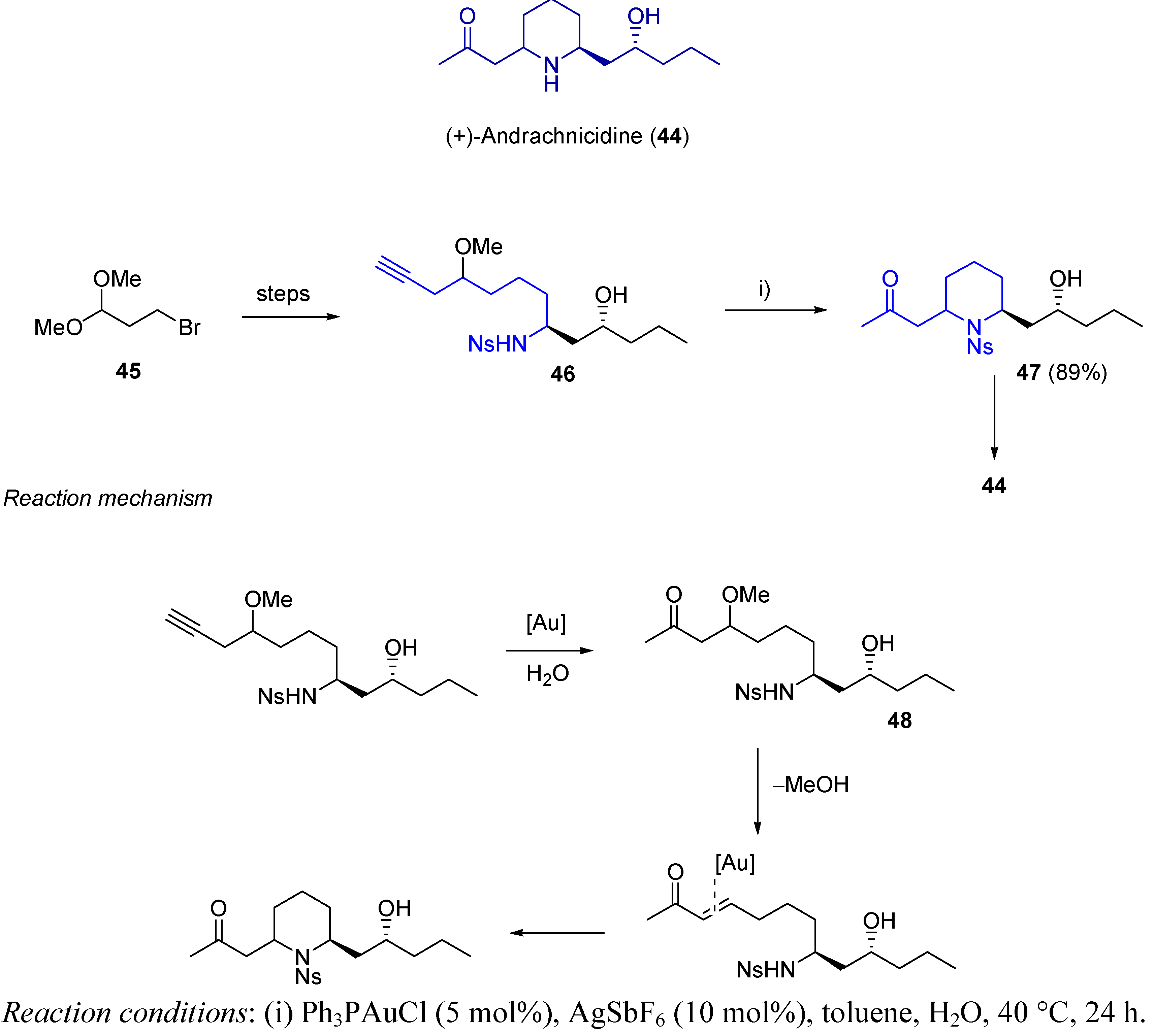
- Jung, H.H.; Floreancig, P.E. Gold-Catalyzed Synthesis of Oxygen- and Nitrogen-Containing Heterocycles from Alkynyl Ethers: Application to the Total Synthesis of Andrachcinidine. J. Org. Chem. 2007, 72, 7359–7366. [Google Scholar] [CrossRef]
- Mill, S.; Hootelé, C. Alkaloids of Andrachne Aspera. J. Nat. Prod. 2000, 63, 762–764. [Google Scholar] [CrossRef]
- Forsyth, C.J. Asymmetric Synthesis, 2nd; Christmann, M., Braese, S., Eds.; Wiley-VCH Verlag Gmbh & Co.: Weinheim, Germany, 2008; pp. 271–276. [Google Scholar]
- Ballini, R.; Petrini, M. Nitroalkanes as Key Building Blocks for the Synthesis of Heterocyclic Derivatives. ARKIVOC 2008, 9, 195–223. [Google Scholar]
- Forsyth, C.J. Asymmetric Synthesis; Christmann, M., Braese, S., Eds.; Wiley-VCH Verlag Gmbh & Co.: Weinheim, Germany, 2007; pp. 256–261. [Google Scholar]
- Sherry, B.D.; Maus, L.; Laforteza, B.N.; Toste, D.F. Gold(I)-Catalyzed Synthesis of Dihydropyrans. J. Am. Chem. Soc. 2006, 128, 8132–8133. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Carrascosa, R.; Torres, M.R. Gold/Acid-Cocatalyzed Regiodivergent Preparation of Bridged Ketals via Direct Bis-Oxycyclization of Alkynic Acetonides. Adv. Synth. Catal. 2010, 352, 1277–1283. [Google Scholar] [CrossRef]
- Perron, F.; Albizati, K.F. Chemistry of Spiroketals. Chem. Rev. 1989, 89, 1617–1661. [Google Scholar] [CrossRef]
- Aho, J.E.; Pihko, P.M.; Rissa, T.K. Nanometric Spiroketals in Natural Products: Structures, Sources, and Synthetic Strategies. Chem. Rev. 2005, 105, 4406–4440. [Google Scholar] [CrossRef]
- Rama Raju, B.; Saikia, A.K. Asymmetric Synthesis of Naturally Ocurring Spiroketals. Molecules 2008, 13, 1942–2038. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Hu, W.P.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2009, 26, 170–244. [Google Scholar] [CrossRef]
- Brimble, M.A.; Bryant, C.J. Synthesis of the Spiroketal-Containing Anti-Helicobacter Pylori Agents CJ-12,954 and CJ-13,014. Chem. Commun. 2006, 4506–4508. [Google Scholar] [CrossRef]
- Izquierdo-Cubero, I.; Plaza Lopez-Espinosa, M.T.; Kari, N. Synthesis of Optically Active Chalcogran from L-Sorbose. Carbohydr. Res. 1994, 261, 231–242. [Google Scholar] [CrossRef]
- Antoniotti, S.; Genin, E.; Michelet, V.; Genêt, J.-P. Highly Efficient Access to Strained Bicyclic Ketals via Gold-catalyzed Cycloisomerization of Bis-Homopropargylic Diols. J. Am. Chem. Soc. 2005, 127, 9976–9977. [Google Scholar] [CrossRef]
- Liu, L.-P.; Hammond, G.B. Highly Efficient and Tunable Synthesis of Dioxabicyclo[4.2.1] Ketals and Tetrahydropyrans via Gold-Catalyzed Cycloisomerization of 2-Alkynyl-1,5-diols. Org. Lett 2009, 11, 5090–5092. [Google Scholar] [CrossRef]
- Liu, B.; De Brabander, J.K. Metal-catalyzed Regioselective Oxy-Functionalization of Internal Alkynes: An Entry into Ketones, Acetals, and Spiroketals. Org. Lett. 2006, 8, 4907–4910. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Bührle, M.; Wölfe, M.; Rudolph, M.; Wieteck, M.; Rominger, F.; Frey, W. Gold Catalysis: Tandem reactions of Diyne-Diols and External Nuchleophiles as an Easy Access to Tricyclic Cage-Like Structures. Chem. Eur. J. 2010, 16, 9846–9854. [Google Scholar]
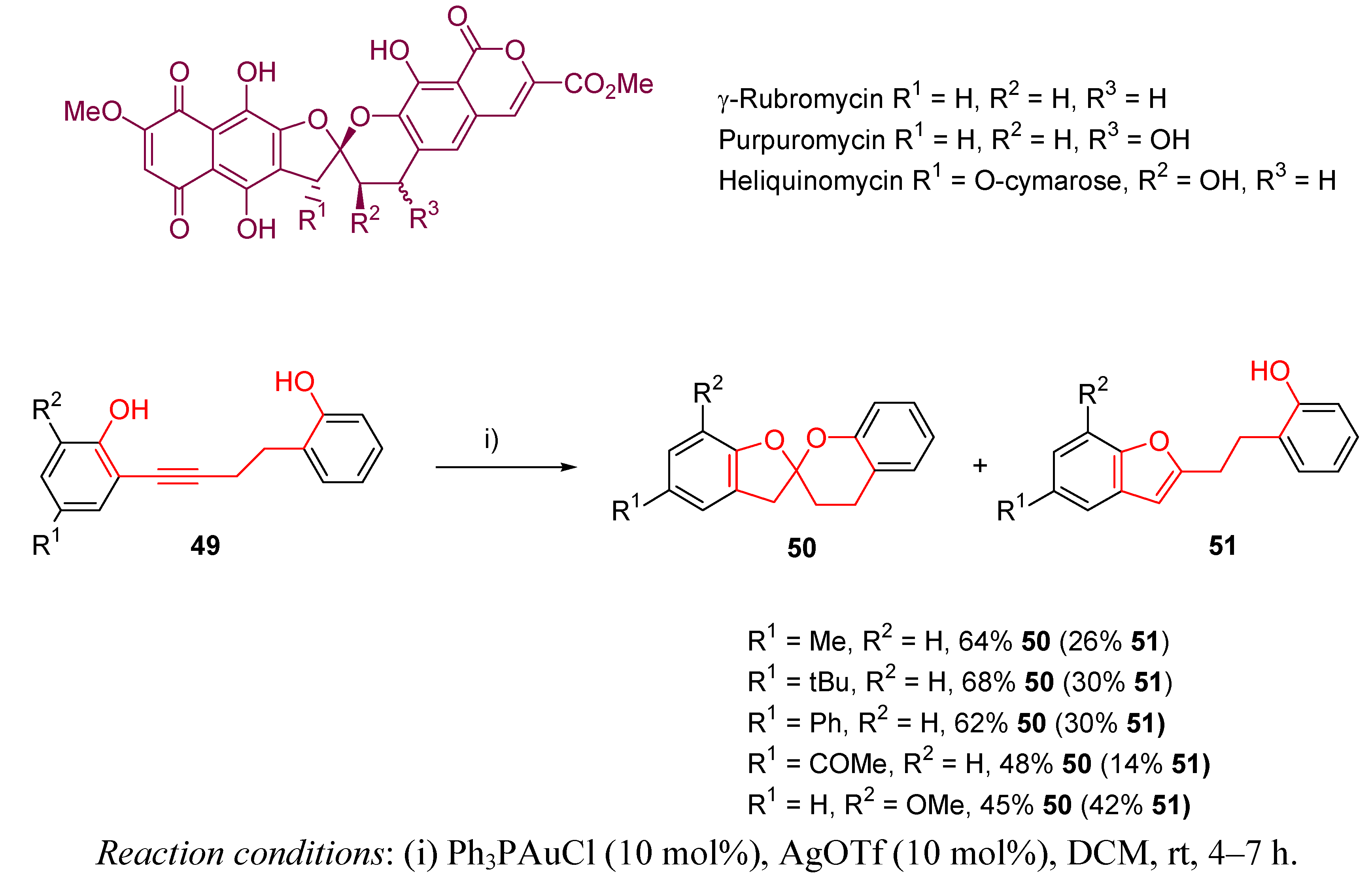
- Zhang, Y.; Xue, J.; Xin, Z.; Xie, Z.; Li, Y. Gold-Catalyzed Double Intramolecular Alkyne Hydroalkoxylation: Synthesis of the Bisbenzannulated Spiroketal Core of Rubromycins. Synlett 2008, 6, 0940–0944. [Google Scholar]
- Brockmann, H.; Lenk, W.; Scwantje, G.; Zeeck, A. Robromycins. Tetrahedron Lett. 1966, 3525–3530. [Google Scholar]
- Goldman, M.E.; Salituro, G.S.; Bowen, J.A.; Williamson, J.M.; Zink, L.; Scleif, W.A.; Emini, E.A. Inhibition of Human Immunodefiency Virus-1 reverse Transcriptase Activity by Robromycins: Competitive Interaction at the Template Primer Site. Mol. Pharmacol. 1990, 38, 20. [Google Scholar]
- Trani, A.; Dallanoce, C.; Pranzone, G.; Ripamonti, F.; Goldstein, B.P.; Cibiatti, R. Semisynthetic Derivatives of Purpuromycin as Potential Topical Agents for Vaginal Infections. J. Med. Chem. 1997, 40, 967–971. [Google Scholar] [CrossRef]
- Chino, M.; Nishikawa, K.; Umekia, M.; Hayashi, C.; Yamazaki, T.; Tsuchida, T.; Sawa, T.; Hamada, M.; Takeuchi, T. Heliquinomycin, A New Inhibitor of DNA Helicase, Produced by Streptomyces sp. MJ929-SF2. Taxonomy, Production, Isolation, Physicochemical Properties and Biological Activities. J. Antibiot. 1996, 49, 752. [Google Scholar] [CrossRef]
- Li, X.; Yao, Y.; Zheng, Y.; Satter, I.; Lin, W. Cephalosporolides H and I, Two Novel Lactones from a Marine-Derived Fungus, Penicillium sp. Arch. Phram. Res. 2007, 30, 812–815. [Google Scholar] [CrossRef]
- Penning, T.M. Inhibition of 5β-Dihydrocortisone Reduction in Rat Liver Cytosol: A Rapid Spectrophotometric Screen for Nonsteroidal Antiinflammatory Drug Potency. Pharm. Sci. 1985, 74, 651–654. [Google Scholar] [CrossRef]
- Sami, F.; Dudley, G.B. Stereocontrol of 5,5-Spiroketals in the Synthesis of Cephalosporolide H Epimers. Org. Lett. 2010, 12, 4698–4701. [Google Scholar] [CrossRef]
- Sami, F.; Dudley, G.B. A Gold-Catalyzed Alkyne-Diol Cycloisomerization for the Synthesis of Oxygenated 5,5-Spiroketals. Beilstein J. Org. Chem. 2011, 7, 570–577. [Google Scholar] [CrossRef]
- McMahon, I.; Silke, J. Winter Toxicity of Unknown Aetiology in Mussels. Harmful Algae News 1996, 14, 2. [Google Scholar]
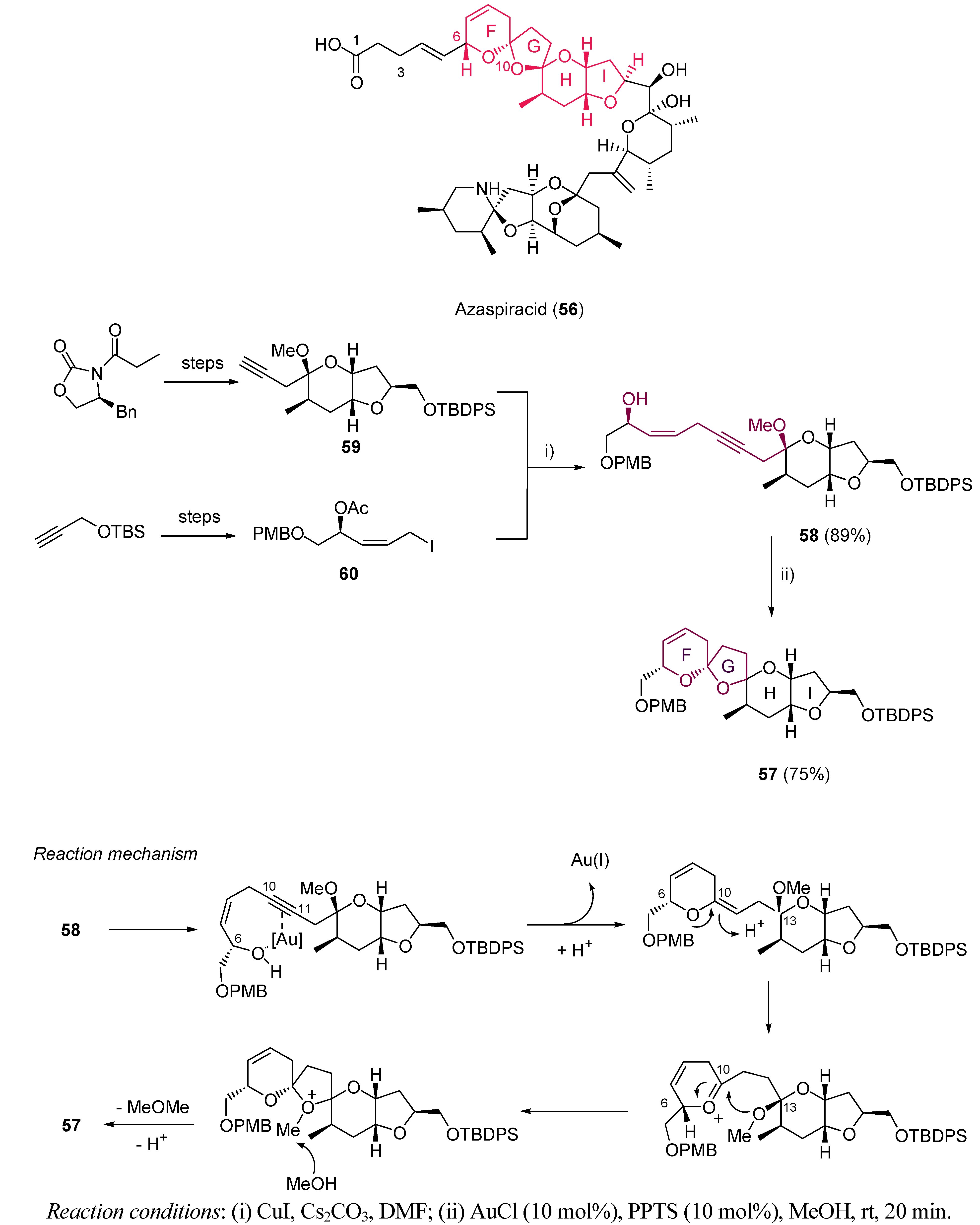
- Ito, E.; Satake, M.; Ofuji, N.; Kurita, N.; McMahon, T.; James, K.; Yasumoto, T. Multiple Organ Damage Caused by a New Toxin Azaspiracid, Isolated from Mussels Produced in Ireland. Toxicon 2000, 38, 917–930. [Google Scholar] [CrossRef]
- Satake, M.; Ofuji, K.; Naoki, H.; James, K.J.; Furey, A.; McMahon, T.; Silke, J.; Yasumoto, T. Azaspiracid, a New Marine Toxin Having Unique Spiro Ring Assemblies, Isolated from Irish Mussels, Myttilus Edulis. J. Am. Chem. Soc. 1998, 120, 9967–9968. [Google Scholar] [CrossRef]
- Nicolau, K.C.; Kotifs, T.V.; Vyskocil, S.; Petrovic, G.; Ling, T.; Yamamda, Y.M.A.; Tang, W.; Frederick, M.O. Structural Revision and Total Synthesis of Azaspiracid-1, part 2: Definition of the ABCD Domain and Total Synthesis. Angew. Chem. Int. Ed. 2004, 43, 4318–4324. [Google Scholar]
- Inoki, S.; Mukaiyama, T. A Convinient Method for the Stereoselective Preparation of trans-(2-Hydroxymethyl)tetrahydrofurans by the Oxidative Cyclization of 5-Hydroxy-1-alkenes with Molecular Oxygen Catalyzed by Cobalt(II) Complex. Chem. Lett. 1990, 67–70. [Google Scholar]
- Li, Y.; Zhou, F.; Forsyth, C.J. Gold(I)-Catalyzed Bis-Spiroketalization: Synthesis of the Trioxadispiroketal-Containing A−D Rings of Azaspiracid. Angew. Chem. Int. Ed. 2007, 46, 279–282. [Google Scholar] [CrossRef]
- Tachibana, K.; Scheuer, P.J.; Tsukitamni, Y.; Kikuchi, H.; Van Engen, D.; Clardy, J.; Gopichand, Y.; Schimtz, F.J. Okadaic Acid, A Cytotoxic Ployether from Two Marine Sponges of the Genus Halichondria. J. Am. Chem. Soc. 1981, 103, 2469–2471. [Google Scholar] [CrossRef]
- Dounay, A.B.; Forsyth, C.J. Okadaic Acid: The Archetypal Serine/Threonine Protein Phosphatase Inhibitor. Curr. Med. Chem. 2002, 9, 1939–1980. [Google Scholar]
- Scheuer, P. Marine Natural Products Research: A Look into the Dive Bag. J. Nat. Prod. 1995, 58, 335–343. [Google Scholar] [CrossRef]
- Yasumoto, T.; Murata, M.; Oshima, Y.; Sano, M.; Matsumoto, G.K.; Clardy, J. Diarrhetic Shellfish Toxins. Tetrahedron 1985, 41, 1019–1022. [Google Scholar] [CrossRef]
- Bialojan, C.; Takai, A. Inhibitory Effect of Marine-Sponge Toxin, Okadaic Acid, on Protein Phosphatases. Specifity and Kinetics. Biochem. J. 1988, 256, 283–290. [Google Scholar]
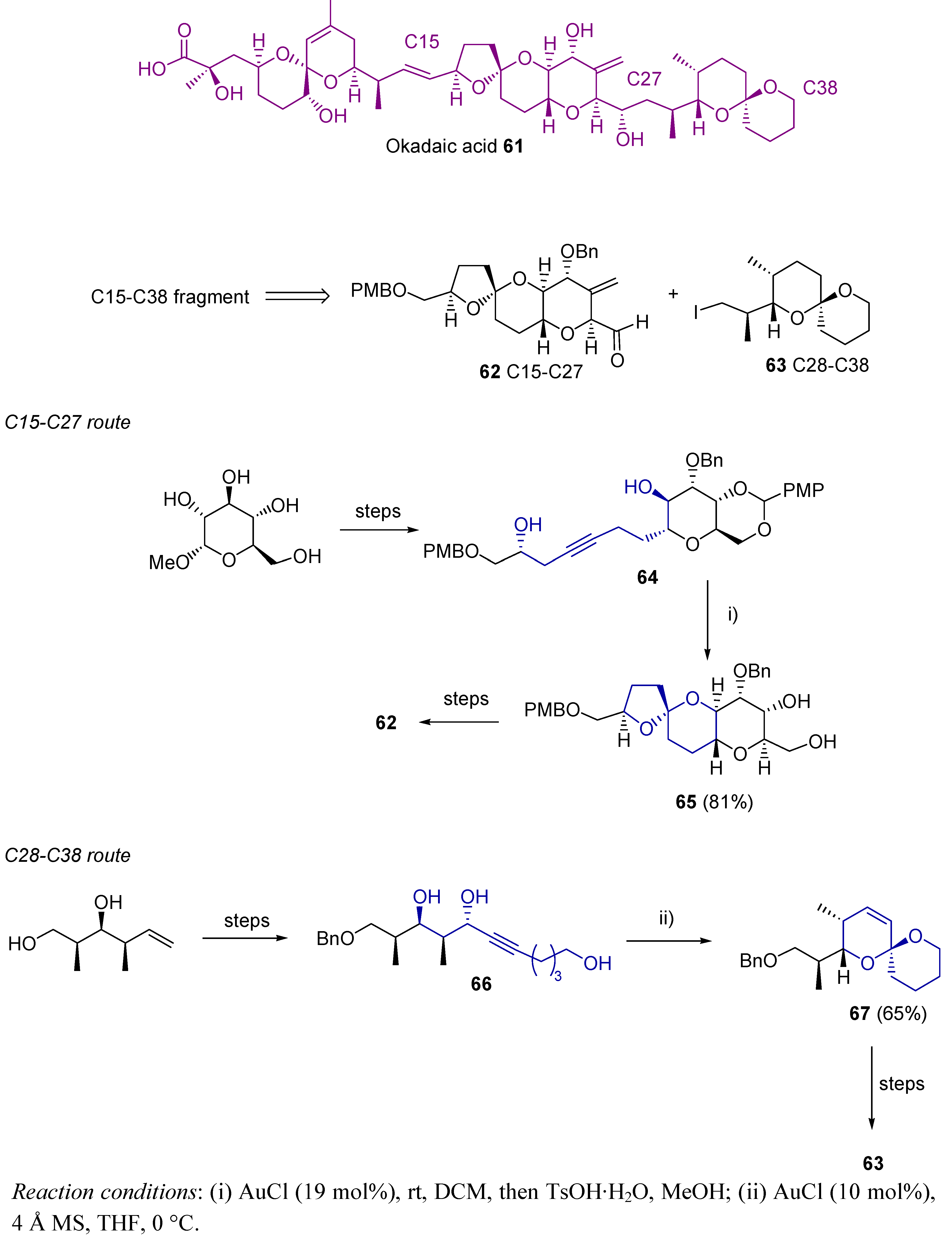
- Fang, C.; Pang, Y.; Forsyth, C.J. Formal Total Synthesis of Okadaic Acid via Regiocontrolled Gold(I)-Catalyzed Spiroketalizations. Org. Lett. 2010, 12, 4528–4531. [Google Scholar] [CrossRef]
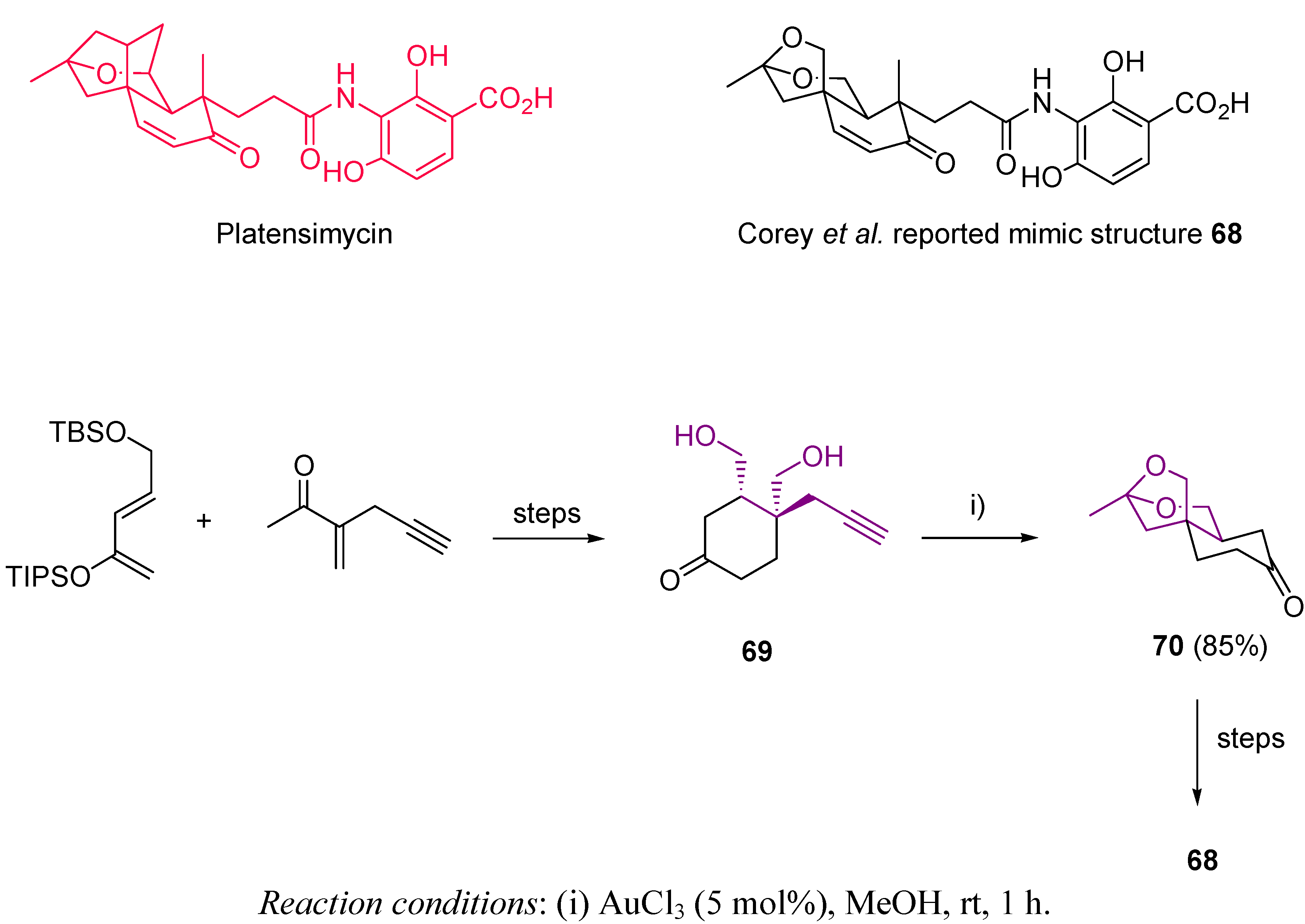
- Wang, J.; Soisson, S.M.; Young, K.; Shoop, W.; Kodali, S.; Galgoci, A.; Painter, R.; Parthasarathy, G.; Tang, Y.S.; Cummings, R.; et al. Platensimycin is a Selective FabF Inhibitor with Potent Antibiotic Properties. Nature 2006, 441, 358–361. [Google Scholar]
- Singh, S.B.; Jayasuriya, H.; Ondeyka, J.G.; Herath, K.B.; Zhang, C.; Zink, D.L.; Tsou, N.N.; Ball, R.G.; Basilio, A.; Genilloud, O.; Diez, M.T.; Vicente, F.; Pelaez, F.; Young, K.; Wang, J. Isolation, Structure, and Absolute Stereochemistry of Platensimycin, a Broad Spectrum Antibiotic Discovered Using an Antisense Differential Sensitivity Strategy. J. Am. Chem. Soc. 2006, 128, 11916–11920. [Google Scholar]
- Häbich, D.; von Nussbaum, F. Platensimycin, A New Antibiotic and “Superbug Challenger” from Nature. ChemMedChem 2006, 1, 951–954. [Google Scholar] [CrossRef]
- Yeung, Y.-Y.; Corey, E.J. A Simple, Efficient, and Enantiocontrolled Synthesis of a Near-Structural Mimic of Platensimycin. Org. Lett. 2008, 17, 3877–3878. [Google Scholar] [CrossRef]
- Nicolau, K.C.; Tang, Y.; Wang, Y.; Stepan, A.F.; Li, A.; Montero, A. Total Synthesis and Antibacterial Properties of Carbaplatensimycin. J. Am. Chem. Soc. 2007, 129, 14850–14851. [Google Scholar] [CrossRef]
- Nicolau, K.C.; Lister, T.; Denton, R.M.; Montero, A.; Edmons, D.J. Adamantaplatensimycin: A Bioactive Analogue of Platensimycin. Angew. Chem. Int. Ed. 2007, 46, 4712–4714. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Alcaide, B.; Almendros, P.; Alonso, J.M. Gold-Catalyzed Cyclizations of Alkynol-Based Compounds: Synthesis of Natural Products and Derivatives. Molecules 2011, 16, 7815-7843. https://doi.org/10.3390/molecules16097815
Alcaide B, Almendros P, Alonso JM. Gold-Catalyzed Cyclizations of Alkynol-Based Compounds: Synthesis of Natural Products and Derivatives. Molecules. 2011; 16(9):7815-7843. https://doi.org/10.3390/molecules16097815
Chicago/Turabian StyleAlcaide, Benito, Pedro Almendros, and José M. Alonso. 2011. "Gold-Catalyzed Cyclizations of Alkynol-Based Compounds: Synthesis of Natural Products and Derivatives" Molecules 16, no. 9: 7815-7843. https://doi.org/10.3390/molecules16097815
APA StyleAlcaide, B., Almendros, P., & Alonso, J. M. (2011). Gold-Catalyzed Cyclizations of Alkynol-Based Compounds: Synthesis of Natural Products and Derivatives. Molecules, 16(9), 7815-7843. https://doi.org/10.3390/molecules16097815