Synthesis and Transformations of di-endo-3-Aminobicyclo-[2.2.2]oct-5-ene-2-carboxylic Acid Derivatives


Abstract
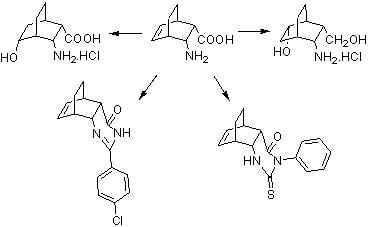
:
1. Introduction
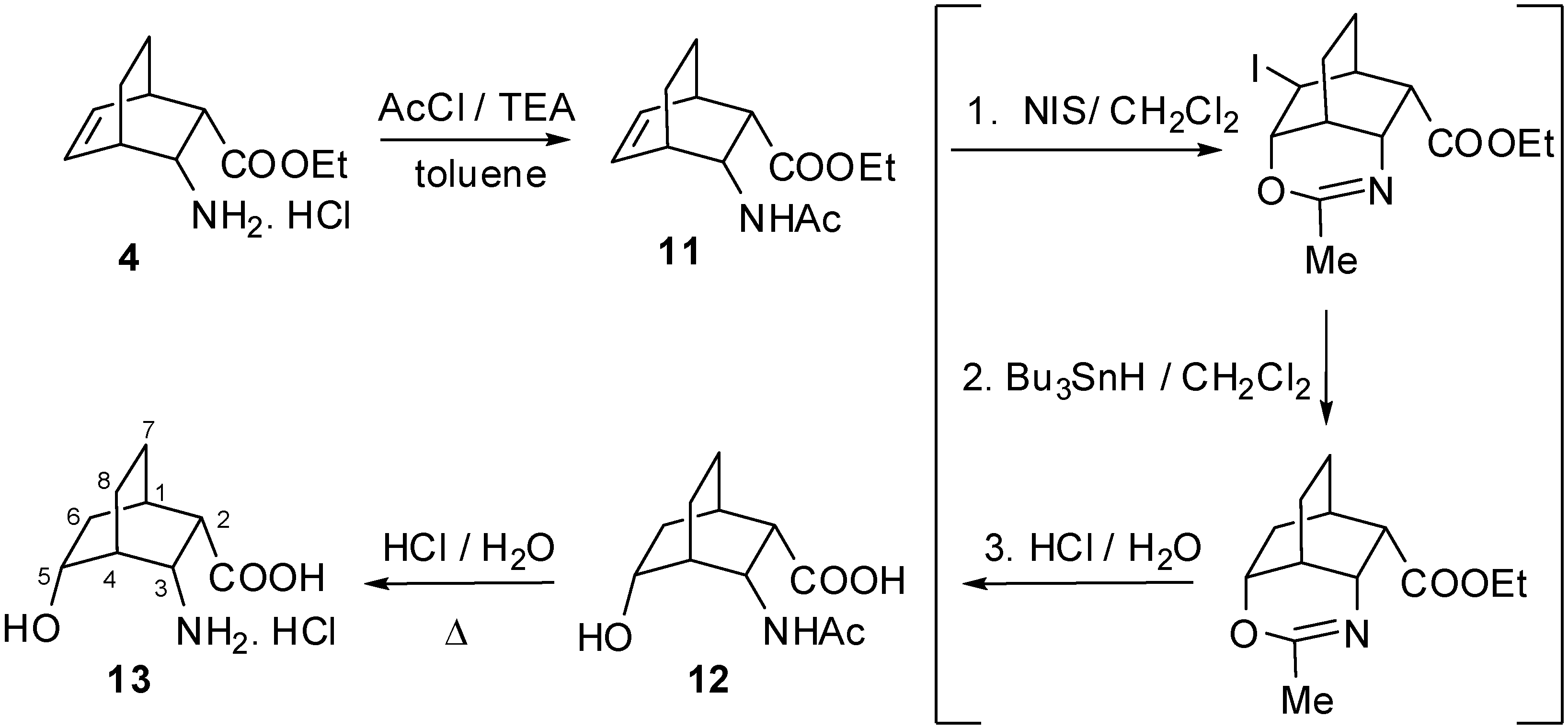
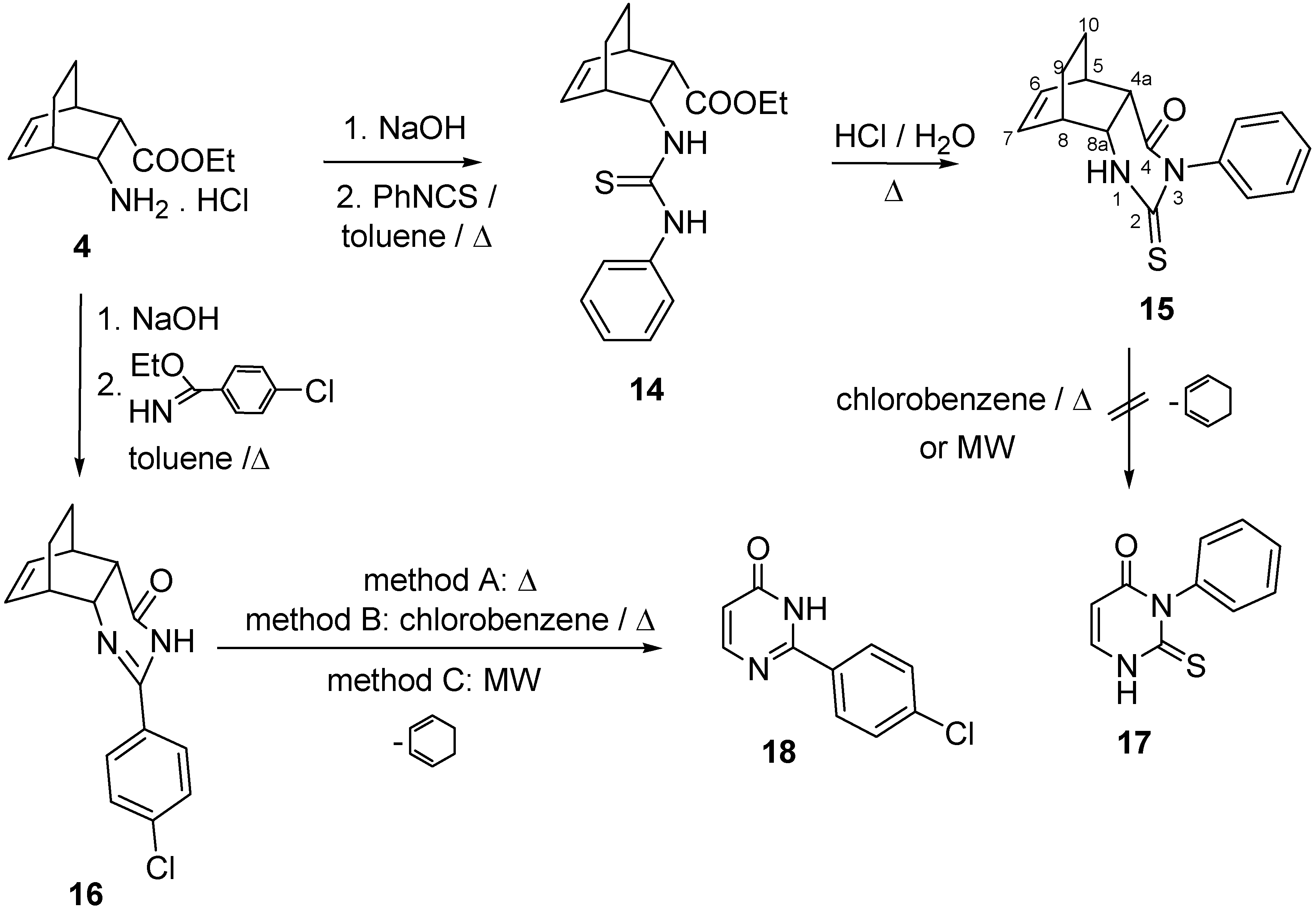
2. Results and Discussion
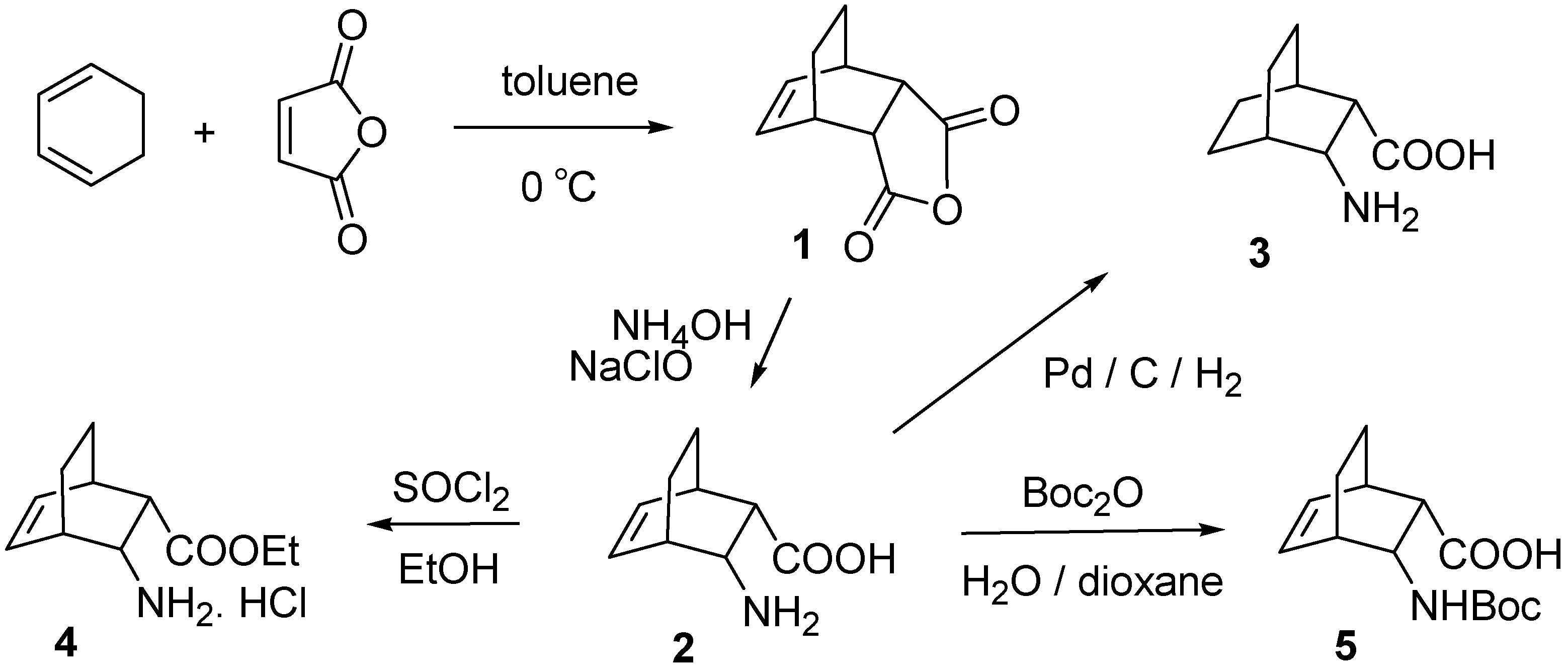
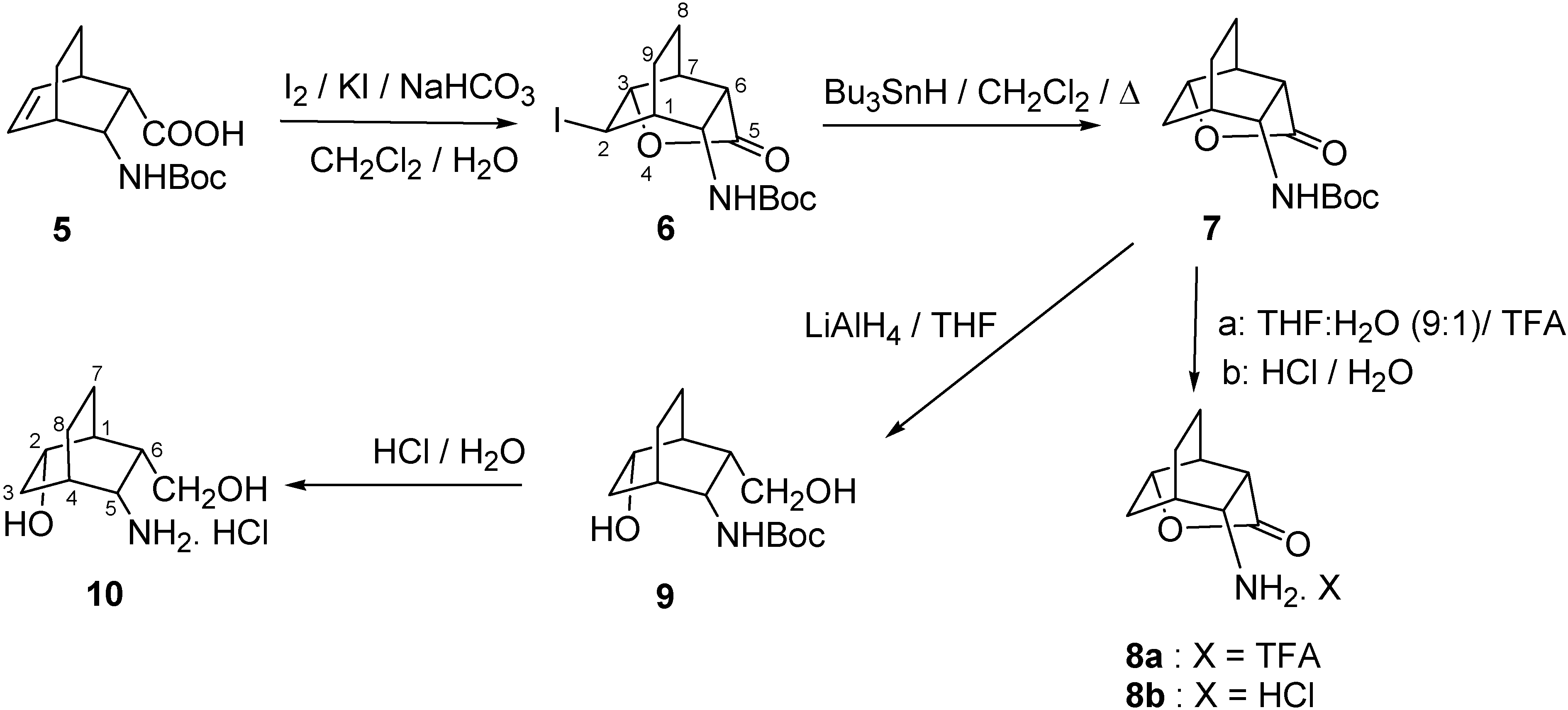
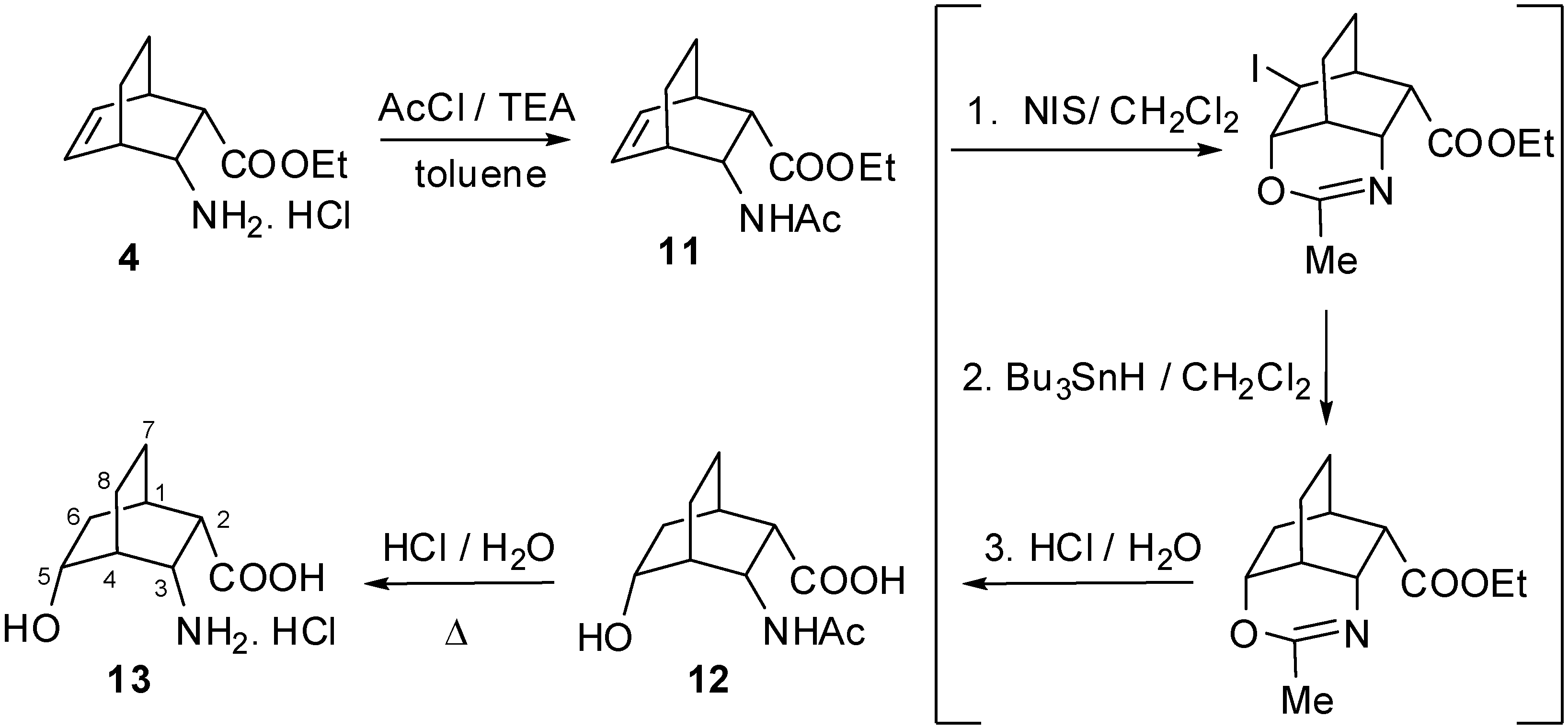
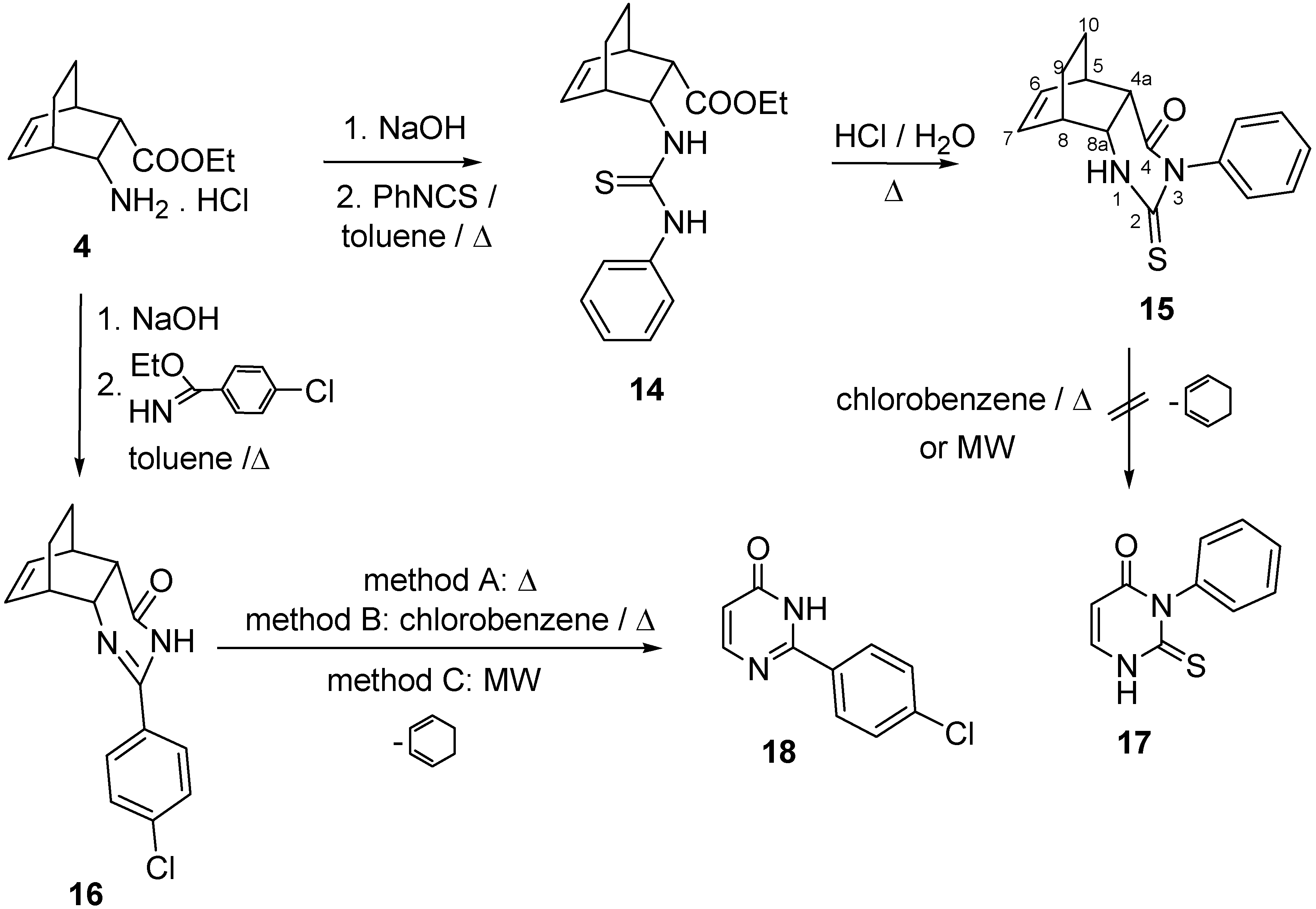
2.1. IR and NMR Results

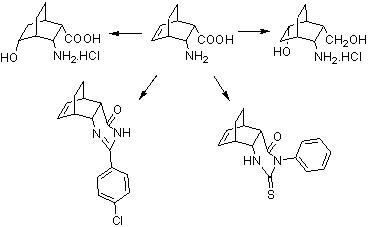{kind=link}
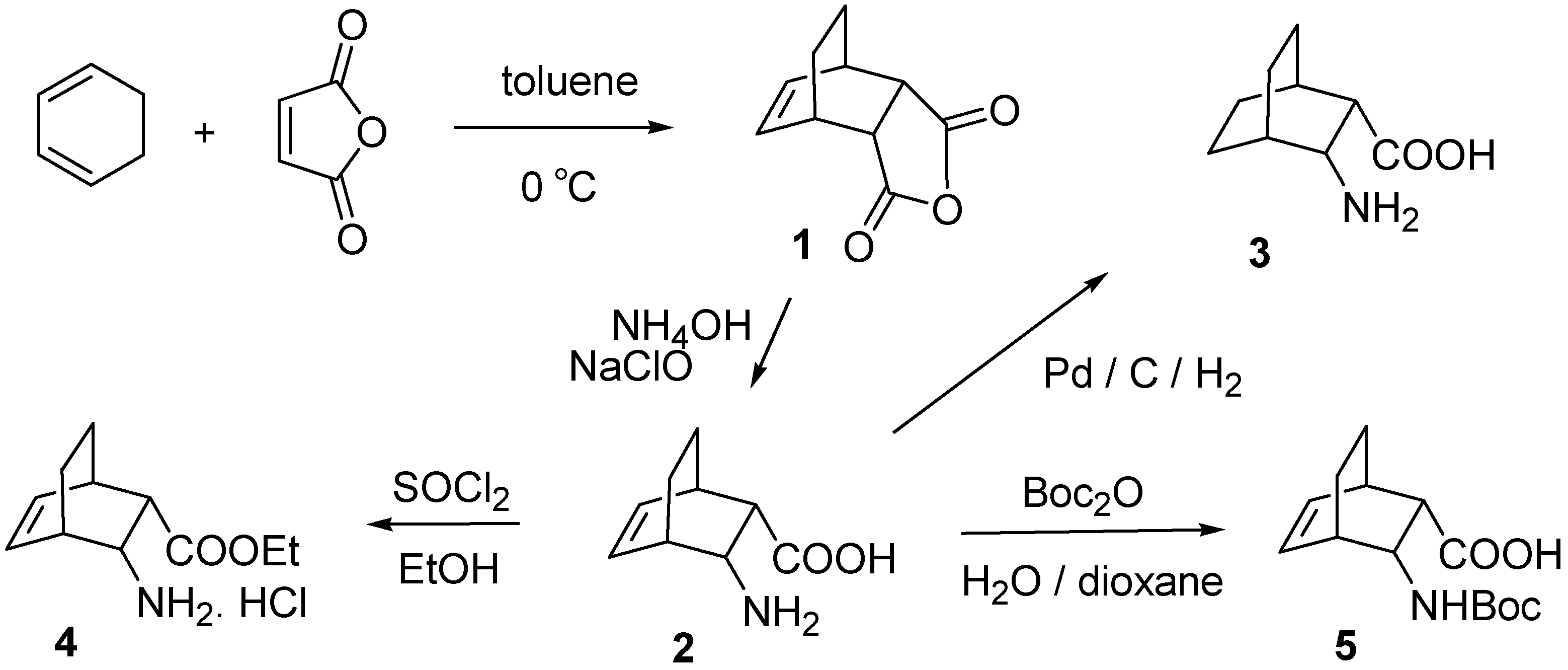{kind=link}
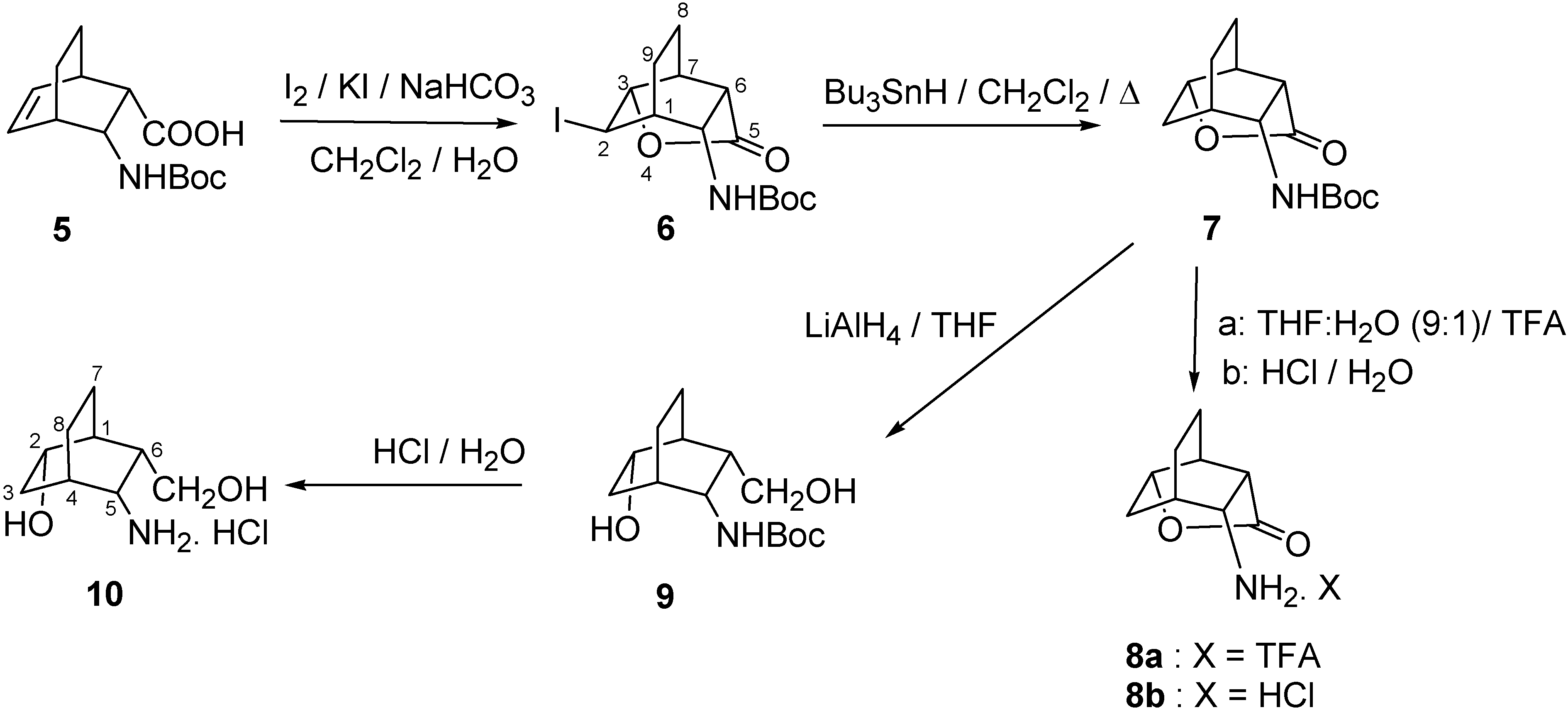{kind=link}
{kind=link}
{kind=link}
| Compound | H–1 c ~ s / br | H–2 d m (1H) | H–3 e m (1H) | H–4 c ~ s / br | H–5 1/2 signal (1/2H) f | H–6 1/2 signal (1/2H) f | NH/NH3+ br (1/3H) g | OH br (1H) |
|---|---|---|---|---|---|---|---|---|
| 2 | 2.65 | 2.33 | 3.38 | 2.96 | 6.08 | 6.33 | ~8.7 | – |
| 3h | 1.94 | 2.39 | 3.31 | 1.67 | ~1.5 m (3H), i 1.73 t (1H) | ~8.9 | – | |
| 4 | 2.80 | 3.2 | 3.56 | 2.96 | 6.16 | 6.33 | 7.95 | – |
| 5 | 2.55 | 2.88 | 4.04 | 2.65 | 6.11 | 6.37 | 5.32 | 12.07 i |
| 6 | ~2.2 j | 2.90 | 4.12 | 2.60 | 4.43 ~ s (1H) | 4.96 k,l | 4.96 l | – |
| 7 | 2.62 | 2.88 | 3.85 | 1.94 j | 1.65, l 1.95 j | 4.60 | 4.88 | – |
| 8a | 2.69 | 2.96 | 3.50 | 1.89 | 1.80, m 2.11 k | 4.75 | ~8.3 | – |
| 8b | 2.64 | 2.84 | 3.42 | 2.04 | ~1.75, j 2.14 k | 4.71 | ~8.55 | – |
| 9n | ~1.6 j | 1.88 | 3.75 l | ~1.6 j | 1.35, ~1.65 j | 3.75 l | 5.37 | 4..63o |
| 10 | 1.68 | 1.95 j | 3.39 | 1.95 j | 1.53, k 1.70 m | 3.78 l | 7.88 | ~5.1p |
| 11 | 2.73 | 2.97 | 4.52 | 2.65 | 6.16 | 6.48 | 5.85 | – |
| 12 | 2.02 | 3.05 | 4.20 | 1.88 | 5.06 q | 1.76, m 2.23 q | 12.85 r | ~3.5 |
| 13 | 2.05 | 2.65 | 3.57 | 2.03 | 3.92 k | 1.58,k 1.89 | ~8.0 | ~5.9 |
| 14 | 2.73 s | 3.05 | 5.06 | 2.94 s | 6.05 | 6.37 | 6.81t | – |
| 15 | 3.32 | 3.15 | 3.91 | 2.88 | 6.44 narrow m (2H) u | 7.75 | – | |
| 16 | 3.23 v | 2.81 | 4.25 | 3.15 v | 6.25 narrow m (2H) u | 8.90 | – | |
| Compound | C–1 | C–2 | C–3 | C–4 | C–5 | C–6 | C–7 | C–8 | C=O |
|---|---|---|---|---|---|---|---|---|---|
| 2 | 36.0 | 46.3 | 50.8 | 35.0 | 130.3 | 137.4 | 25.7 | 23.6 | 175.6 |
| 3c | 29.2 d | 43.5 | 48.9 | 29.1 d | 25.2 e | 25.9 e | 19.2 | 21.9 | 176.3 |
| 4 | 32.92 d | 46.0 | 51.4 | 32.96 d | 130.8 | 135.8 | 24.7 | 22.5 | 171.8 |
| 5 | 36.2 | 50.2 | 52.1 | 33.2 | 130.6 | 136.5 | 25.3 | 22.7 | 174.6 |
| 6 | 39.1 | 42.5 | 48.1 | 37.4 | 25.8 | 86.2 | 14.9 | 25.0 | 176.2 |
| 7 | 37.1 | 43.9 | 48.1 | 30.3 | 30.0 | 78.3 | 15.6 | 26.4 | 177.6 |
| 8a | 37.1 | 42.4 | 48.4 | 28.6 | 29.3 | 78.6 | 15.1 | 26.3 | 177.0 |
| 8b | 37.2 | 42.3 | 48.4 | 28.3 | 29.3 | 78.5 | 15.2 | 26.4 | 176.9 |
| 9f | 34.3 d | 41.2 | 49.8 | 30.5 d | 31.8 | 68.0 | 25.2 | 23.4 | − |
| 10 | 33.8 | 40.4 | 50.1 | 28.4 | 30.6 | 67.3 | 24.1 | 22.9 | − |
| 11 | 33.3 | 49.9 | 50.3 | 35.9 | 130.4 | 136.6 | 25.4 | 22.2 | 173.5 |
| 12 | 25.5 d | 46.7 | 45.4 | 23.0 d | 76.8 | 32.3 | 24.2 | 17.5 | 172.6 e |
| 13 | 33.3 | 47.8 | 49.8 | 28.9 | 67.5 | 38.0 | 19.7 | 21.5 | 174.6 |
| 14 | 33.9 d | 49.2 | 57.0 | 35.3 d | 130.5 | 136.5 | 25.5 | 22.1 | 173.4 |
| 15 | 35.6 | 43.5 | 54.6 | 37.5 | 133.4 | 134.1 | 24.2 | 22.0 | 167.7 |
| 16 | 34.4 d | 44.3 | 61.6 | 37.5 d | 135.0 | 133.4 | 25.3 | 23.4 | 172.6 |
3. Experimental
3.1. General
3.2. (r-1,t-3,t-6,c-7,t-10)-10-Amino-4-oxatricyclo[4.3.1.03,7]decan-5-one trifluoroacetate (8a) and hydrochloride (8b)
3.3. 2-(4-Chlorophenyl)-3H-pyrimidin-4-one (18)
4. Conclusions
Acknowledgments
Conflict of Interest
- Sample Availability: Samples of the compounds 1-18 are available from the authors.
References
- Tager, H.S.; Christensen, H.N. 2-Aminonorbornane-2-carboxylic acid. Preparation, properties and identification of the four isomers. J. Am. Chem. Soc. 1972, 94, 968–972, and references cited therein. [Google Scholar] [CrossRef]
- Abdel-Ghany, M.; Sharp, G.W.C.; Straub, S.G. Glucose stimulation of protein acylation in the pancreatic β-cell. Life Sci. 2010, 87, 667–671. [Google Scholar] [CrossRef]
- Carobbio, S.; Ishihara, H.; Fernandez-Pascual, S.; Bartley, C.; Martin-Del-Rio, R.; Maechler, P. Insulin secretion profiles are modified by overexpression of glutamate dehydrogenase in pancreatic islets. Diabetologia 2004, 47, 266–276. [Google Scholar] [CrossRef]
- Kim, C.S.; Cho, S.H.; Chun, H.S.; Lee, S.Y.; Endou, H.; Kanai, Y.; Kim, D.K. BCH, an inhibitor system L amino acid transporters, induces apoptosis in cancer cells. BiolPharm. Bull. 2008, 31, 1096–1100. [Google Scholar] [CrossRef]
- Sebastianelli, L.; Ledonne, A.; Marrone, M.C.; Bernadi, G.; Mercuri, N.B. The L-amino acid carrier inhibitor 2-aminobicyclo[2.2.1]heptane-2-carboxylic acid (BCH) reduces L-dopa-elicited responses in dopaminergic neurons of the substantia nigra pars compata. Exp. Neurol. 2008, 212, 230–233. [Google Scholar]
- Smith, P.W.; Trivedi, N.; Howes, P.D.; Sollis, S.I.; Rahim, G.; Bethell, R.C.; Lynn, S. Synthesis of tetrasubstitued bicyclo[2.2.2]octane as a potencial inhibitor of influenza virus sialidase. Bioorg. Med. Chem. Lett. 1999, 9, 611–614. [Google Scholar] [CrossRef]
- Babu, Y.S.; Chand, P.; Kotian, P.L. Influenza Neuraminidase Inhibitors as Antiviral Agents; Academic Press: Oxford, UK, 2006; pp. 287–297, Annual reports in medicinal chemistry Elsevier book Series on SciencesDirect. [Google Scholar]
- Zand, R.; Sellinger, O.Z.; Water, R.; Harris, R. α-Aminocyclic and bicyclic alkane carboxylic acids: Differential effects on selected amino acids of rat brain cortex. J. Neurochem. 1974, 23, 1201–1206. [Google Scholar] [CrossRef]
- Abellán, T.; Chinchilla, R.; Galindo, N. New oxazinone and pyrazinone derivatives as chiral reagents for the asymmetric synthesis of a-amino acids. J. Heterocyclic Chem. 2000, 37, 467–479. [Google Scholar] [CrossRef]
- Chang, L.W.; Hagmann, W.K.; MacCoss, M. Cyclic amino acid derivateves as cell adhesion inhibitors. PCT Int. Appl. WO1999/26615 CA, 1999. [Google Scholar]
- Chang, L.L.; Troung, Q.; Doss, G.A.; MacCoss, M.; Lyons, K.; McCauley, E.; Mumford, R.; Forrest, G.; Vincent, S.; Schmidt, J.A.; Hagmann, K.W. Highly constrained bicyclic VLA-4 antagonists. Bioorg. Med. Chem. Lett. 2007, 17, 597–601. [Google Scholar]
- Otani, Y.; Futaki, T.; Kiwada, T.; Sugiura, Y.; Muranaka, A.; Kobayashi, N.; Uchiyama, M.; Yamagichi, K.; Ohwada, T. Oligomers of β-amino acid bearing non-planar amides form ordered structures. Tetrahedron 2006, 62, 11635–11644. [Google Scholar]
- Chang, L.L.; Truong, Q.; Doss, G.A.; MacCoss, M.; Lyons, K.; McCauley, E.; Mumford, R.; Forrest, G.; Vincent, S.; Schmidt, J.A.; Hagmann, W.K. Highly constrained bicyclic VLA-4 antagonists. Bioorg. Med. Chem. Lett. 2007, 17, 597–601. [Google Scholar]
- Fülöp, F.; Bernáth, G.; Pihlaja, K. Synthesis, stereochemistry and transformations of cyclopentane-, cyclohexane-, cycloheptane-, and cyclooctane-fused 1,3-oxazines, 1,3-thiazines and pyrimidines. Adv. Heterocycl. Chem. 1998, 69, 349–477. [Google Scholar]
- Duan, J.; Jiang, B.; Sheppeck, J.; Gilmore, J. Modulators of the glucocorticoid receptor AP-1, and/or NF-κB activity and use thereof. PCT Int Appl WO 2005/070207 CA, 2005. [Google Scholar]
- Mehta, G.; Le Droumaguet, C.; Islam, K.; Anoop, A.; Jemmis, E.D. Face-selectivity in [4+2]-cycloadditions to novel polycyclic benzoquinones. Remarkable stereodirecting effects of a remote cyclopropane ring and an olefinic bond. Tetrahedron Lett. 2003, 44, 3109–3113. [Google Scholar] [CrossRef]
- Fairbust, J.; Horwell, D.C.; Timms, G.H. Novel bicyclo[2.2.2]octanyl-1,4-benzodiazepinones. Their syntheses and rearrangement to bicyclooct-2-enylbenzimidazoles. J. Heterocyclic Chem. 1977, 14, 1199–1201. [Google Scholar] [CrossRef]
- Alibert, S.; Santelli-Rouvier, C.; Pradines, B.; Houdoin, C.; Parzy, D.; Karolak-Wojciechowska, J.; Barbe, J. Synthesis and effects on Chloroquine Susceptibility in Plasmodium falciparum of a series of new dihydroanthracene derivatives. J. Med. Chem. 2002, 45, 3195–3209. [Google Scholar] [CrossRef]
- Saito, K.; Ono, K.; Sano, M.; Kiso, S.; Takoda, T. Palladium catalyzed double substitution reactions of iodophenol and iodoaniline derivatives with homo-conjugated compounds to form cyclic ether and cyclic amines through homo-conjugated interaction. Heterocycles 2002, 57, 1781–1786. [Google Scholar] [CrossRef]
- Moriconi, E.J.; Crawford, W.C. The reaction of chlorosulfonyl isocyanate with bridged bi- and tricyclic olefins. J. Org. Chem. 1968, 33, 370–378. [Google Scholar] [CrossRef]
- Winn, C.M.; Vaughen, W.R. Stereochemistry of pyrrolidine addition to bicycle[2.2.2]oct-2-ene-carbonitrile. J. Org. Chem. 1968, 33, 2371–2374. [Google Scholar] [CrossRef]
- Calmes, M.; Escale, F.; Didierjean, C.; Martinez, J. Synthesis of enantiopure trans N-Boc-3-aminobicyclo[2.2.2]octane-2-carboxylic acids and their bicyclic 1,3-amino alcohol derivatives via the [4+2] cycloaddition of 1,3-cyclohexadiene to a chiral β-nitroacrylate. Chirality 2011, 23, 245–249. [Google Scholar] [CrossRef]
- Palkó, M.; Kiss, L.; Fülöp, F. Syntheses of hydroxylated cyclic β-amino acid derivatives. Curr. Med. Chem. 2005, 12, 3063–3083. [Google Scholar] [CrossRef]
- Fülöp, F.; Palkó, M.; Forró, E.; Dervarics, M.; Martinek, T.A.; Sillanpää, R. Facile regio- and diastereoselective syntheses of hydroxylated β–aminocyclohexanecarboxylic acids. Eur. J. Org. Chem. 2005, 3214–3220. [Google Scholar]
- Palkó, M.; Sándor, E.; Sohár, P.; Fülöp, F. Synthesis and stereostructure of 2-amino 5- and 6-hydroxybicyclo[2.2.1]heptane-2-carboxylic acids. Monats. Chem. 2005, 136, 2051–2058. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Gyónfalvi, S.; Forró, E.; Hetényi, A.; De Kimpe, N.; Fülöp, F. Synthesis of 3- and 4-hydroxy-2-aminocyclohexanecarboxyl acids by idolactonisation. Eur. J. Org. Chem. 2005, 4017–4023. [Google Scholar]
- Benedek, G.; Palkó, M.; Wéber, E.; Martinek, T.A.; Forró, E.; Fülöp, F. Efficient synthesis of hydroxy-substituted cispentacin derivatives. Eur. J. Org. Chem. 2008, 3724–3730. [Google Scholar]
- Palkó, M.; Benedek, G.; Forró, E.; Wéber, E.; Hänninen, M.; Sillanpää, R.; Fülöp, F. Synthesis of mono- and dihydroxy-substituted 2-amino-cyclooctanecarboxylic acid enantiomers. Tetrahedron Asymmetry 2010, 21, 957–961. [Google Scholar] [CrossRef]
- Songis, O.; Didierjean, C.; Martinez, J.; Calmes, M. The iodolactone approach to enantiopure oxiranes of constrained chiral cyclic β-amino acids. Tetrahedron Asymmetry 2008, 19, 2135–2139. [Google Scholar] [CrossRef]
- Kiss, L.; Fülöp, F. Selective syntheses of functionalized cyclic β-amino acids via transformation of the ring C-C double bonds. Synlett 2010, 1302–1314, and references therein. [Google Scholar]
- Yokota, Y.; Cortez, G.S.; Romo, D. Nucleophilic opening of bicyclic b-lactones via acyl C-O and alkyl C-O cleavage: Catalytic, asymmetric synthesis of a versatile, carbocyclic nucleoside precursor and protected transpentacin. Tetrahedron 2002, 58, 7075–7080. [Google Scholar] [CrossRef]
- Gomez-Vidal, J.A.; Silverman, R.B. Short, highly efficient syntheses of protected 3-azido- and 4-azidoproline and their precursors. Org. Lett. 2001, 3, 2481–2484. [Google Scholar] [CrossRef]
- Nelson, S.G.; Spences, K.L.; Cheung, W.S.; Mamie, S.J. Divergent reaction pathways in amine additions to β-lactone electrophiles. An application to β-peptide synthesis. Tetrahedron 2002, 58, 7081–7091. [Google Scholar] [CrossRef]
- Yan, Z.; Weaving, R.; Dauban, P.; Dodd, R.H. Enantiospecific synthesis of 3,4-disubstituted glutamic acids via controlled stepwise ring-opening of 2,3-aziridino-γ-lactone. Tetrahedron Lett. 2002, 43, 7593–7595. [Google Scholar] [CrossRef]
- Stájer, G.; Szabó, A.E.; Pintye, J.; Bernáth, G. Stereochemical studies. Part 86. Saturated heterocycles part 81. Preparation of New thiouracils via retrodiene decomposition of methylene-bridged quinazolone thiones. J. Chem. Soc. Perkin Trans. I 1985, 2483–2487. [Google Scholar]
- Stájer, G.; Csende, F.; Fülöp, F. The retro Diels-Alder reaction as a valuable tool for the synthesis of heterocycles. Curr. Org. Chem. 2003, 7, 1423–1432. [Google Scholar] [CrossRef]
- Miklós, F.; Stájer, G.; Fülöp, F. Preparation of heterocycles by microwave-induced retro Diels-Alder reaction. Lett. Org. Chem. 2006, 3, 915–916. [Google Scholar] [CrossRef]
- Stájer, G.; Szabó, A.E.; Bernáth, G. A facile method for the preparation of 2-substituted pyrimidin-4(3H)-ones by a retro-Diels-Alder reaction. Synthesis 1987, 290–292. [Google Scholar]
- Holly, S.; Sohár, P. Theoretical and Technical Introduction to the Series Absorption Spectra in the Infrared Region; Láng, L., Prichard, W.H., Eds.; Akadémiai Kiadó: Budapest, Hungary, 1975; pp. 83,84,95. [Google Scholar]
- Sohár, P. Nuclear Magnetic Resonance Spectroscopy; CRC Press: Boca Raton, FL, USA, 1984; Volume 1-2, a) 2, p. 153; b) 2, pp. 180, 182; c) 2, pp. 2, 30, 32; d) 1, 32 & 2, pp. 154, 155; e) 1, pp. 34, 35 and 2, p. 4. [Google Scholar]
- Pretsch, E.; Clerc, T.; Seibl, J.; Simon, N. Tabellen zur Strukturaufklärung organischer Verbindungen mit spektroskopischen Methoden; Springer Verlag: Berlin, Germany, 1976; p. C70. [Google Scholar]
- Karplus, M. Contact electron-spin coupling of nuclear magnetic moments. J. Chem. Phys. 1959, 30, 11–16. [Google Scholar] [CrossRef]
- Karplus, M. Proton spin coupling by pi-electrons. J. Chem. Phys. 1960, 33, 1842–1849. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Palkó, M.; Sohár, P.; Fülöp, F. Synthesis and Transformations of di-endo-3-Aminobicyclo-[2.2.2]oct-5-ene-2-carboxylic Acid Derivatives. Molecules 2011, 16, 7691-7705. https://doi.org/10.3390/molecules16097691
Palkó M, Sohár P, Fülöp F. Synthesis and Transformations of di-endo-3-Aminobicyclo-[2.2.2]oct-5-ene-2-carboxylic Acid Derivatives. Molecules. 2011; 16(9):7691-7705. https://doi.org/10.3390/molecules16097691
Chicago/Turabian StylePalkó, Márta, Pál Sohár, and Ferenc Fülöp. 2011. "Synthesis and Transformations of di-endo-3-Aminobicyclo-[2.2.2]oct-5-ene-2-carboxylic Acid Derivatives" Molecules 16, no. 9: 7691-7705. https://doi.org/10.3390/molecules16097691
APA StylePalkó, M., Sohár, P., & Fülöp, F. (2011). Synthesis and Transformations of di-endo-3-Aminobicyclo-[2.2.2]oct-5-ene-2-carboxylic Acid Derivatives. Molecules, 16(9), 7691-7705. https://doi.org/10.3390/molecules16097691