Enantio and Diastereoselective Addition of Phenylacetylene to Racemic α-chloroketones

Abstract
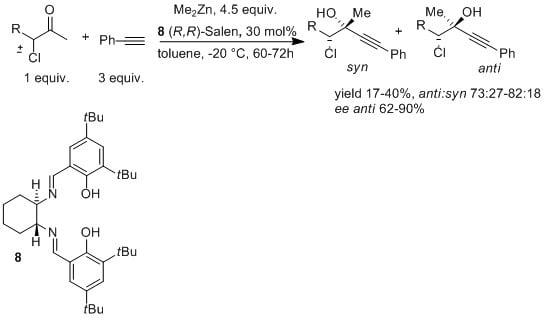
:
1. Introduction
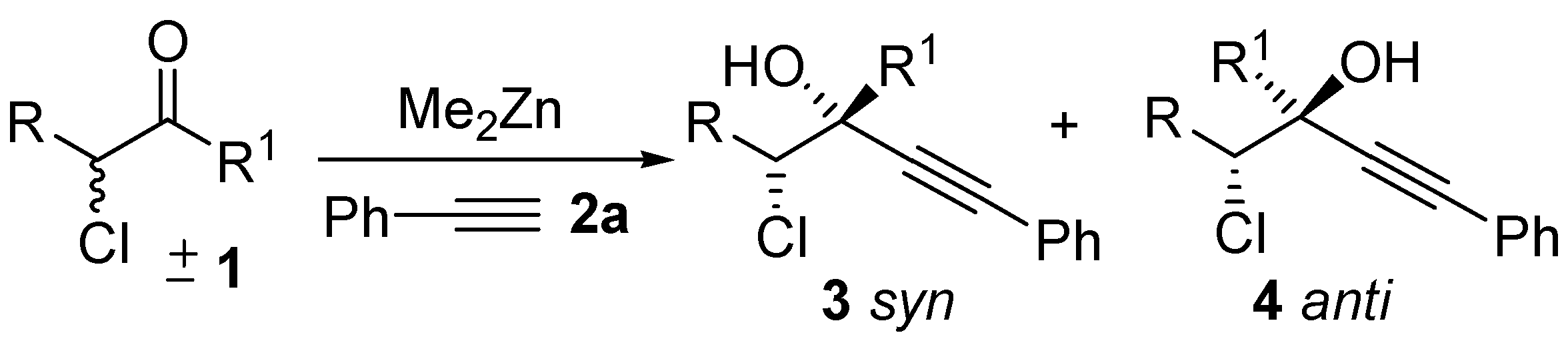
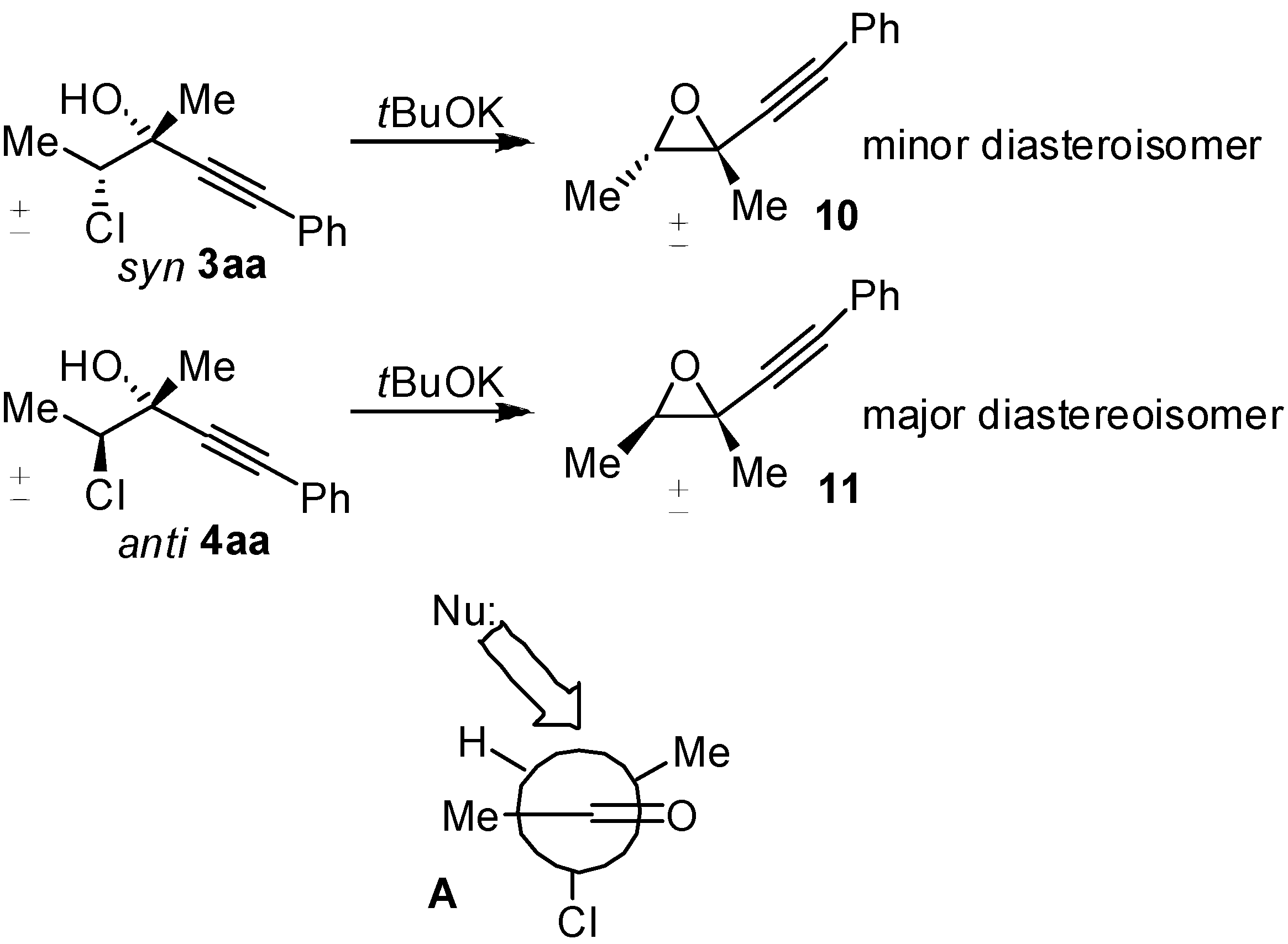
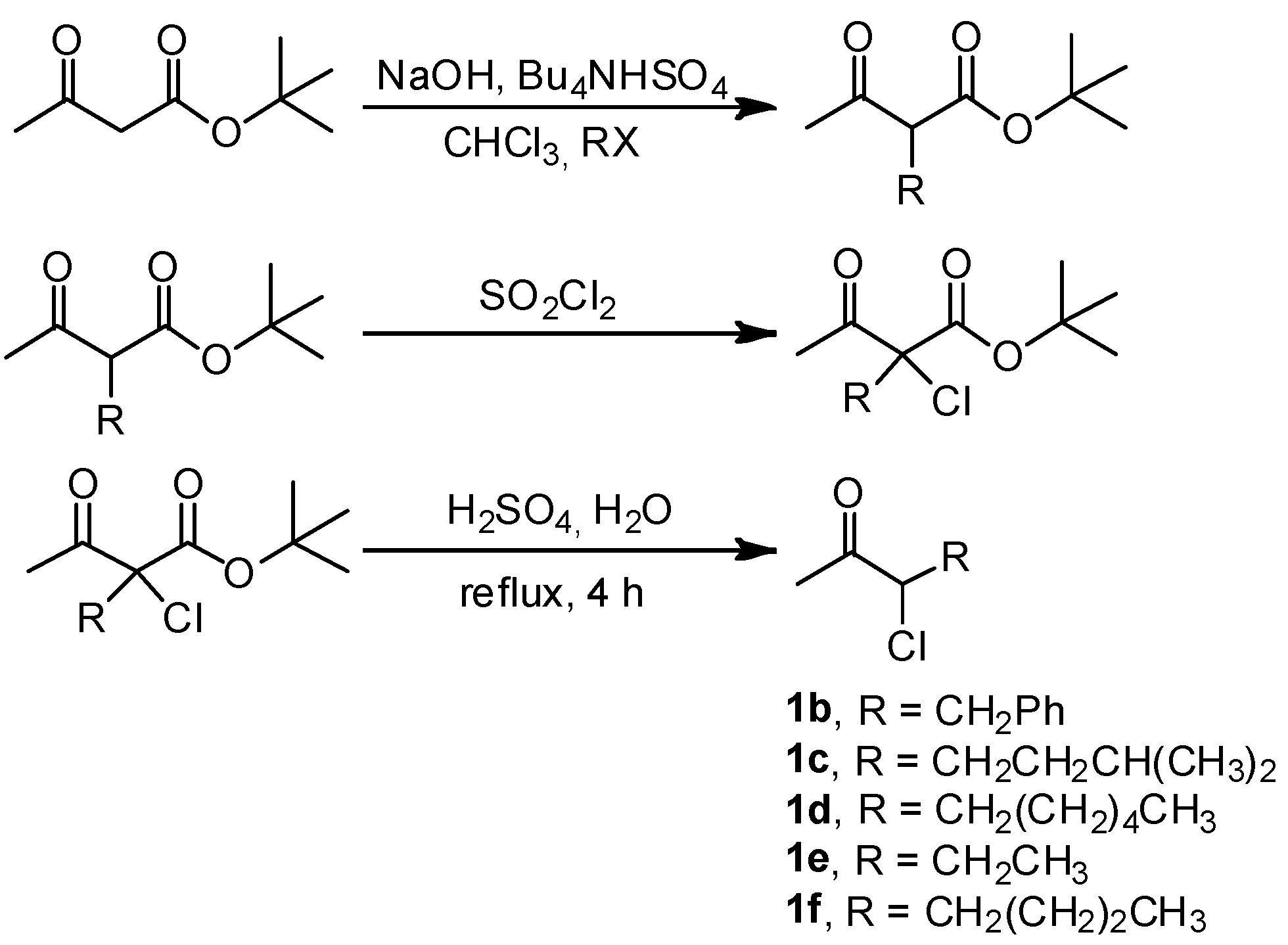
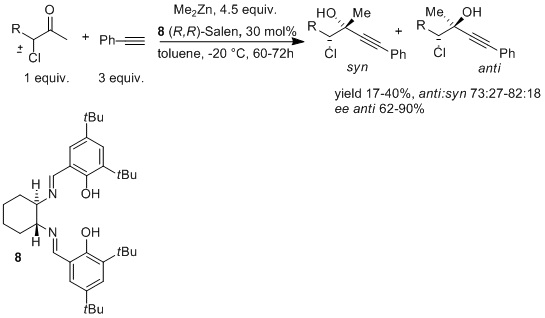
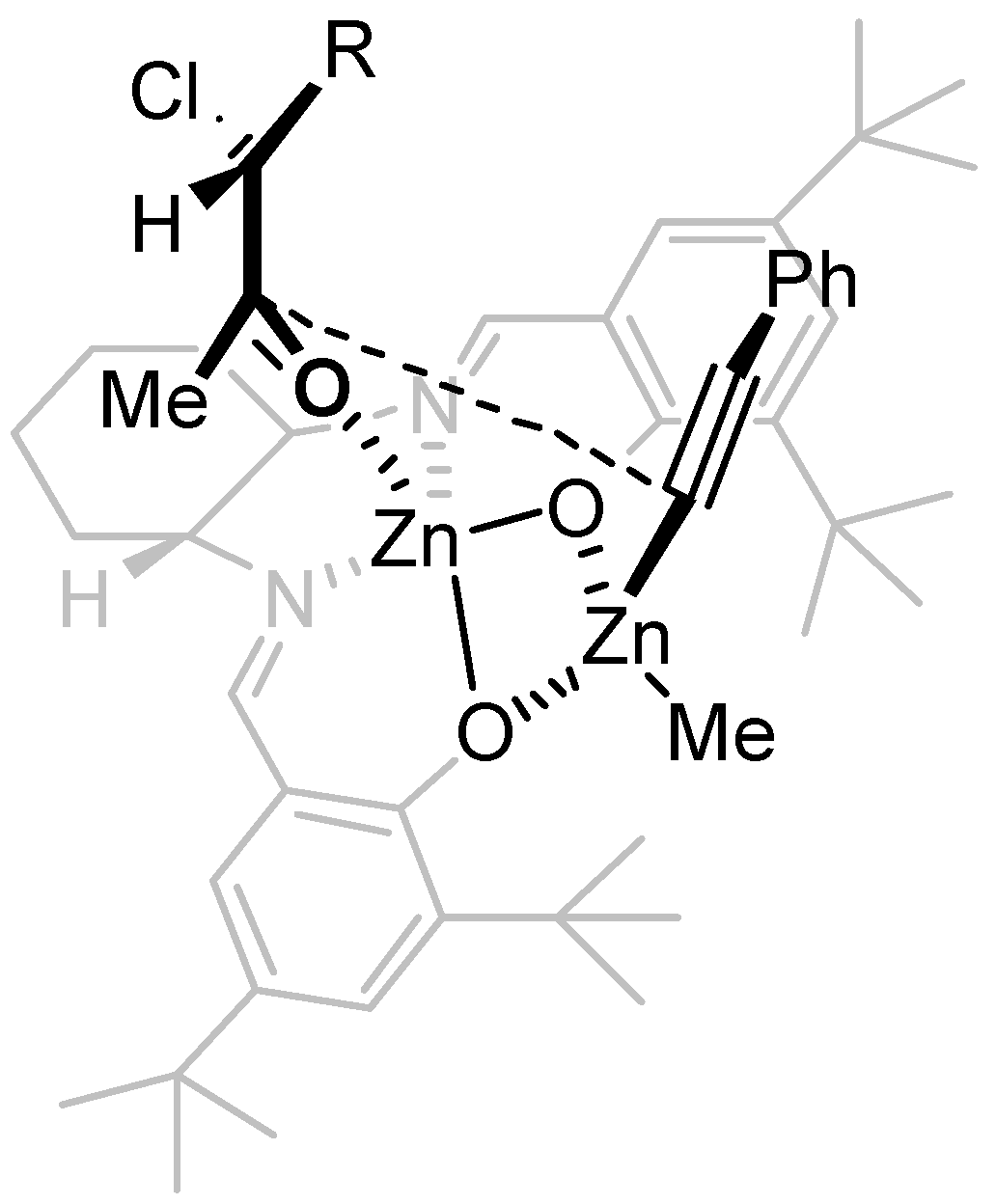
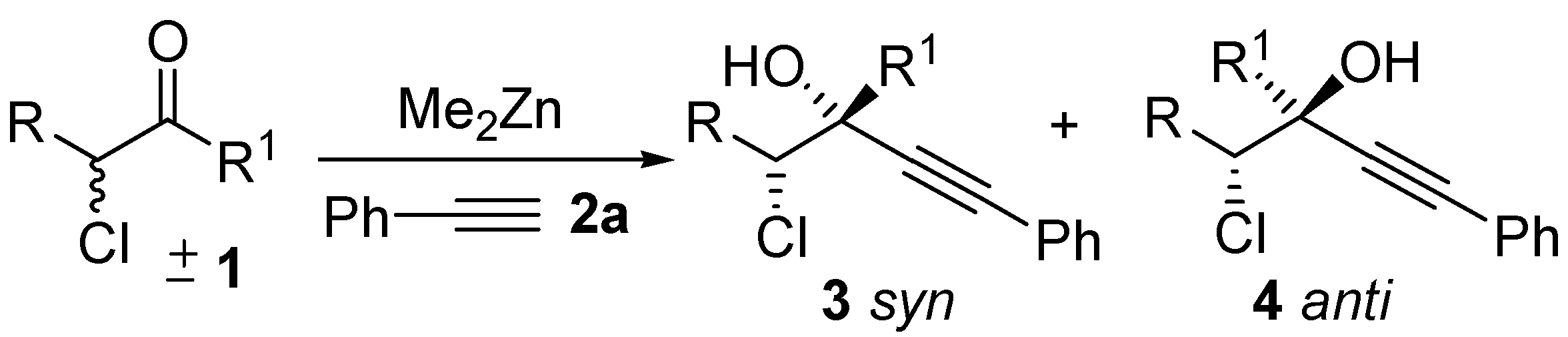
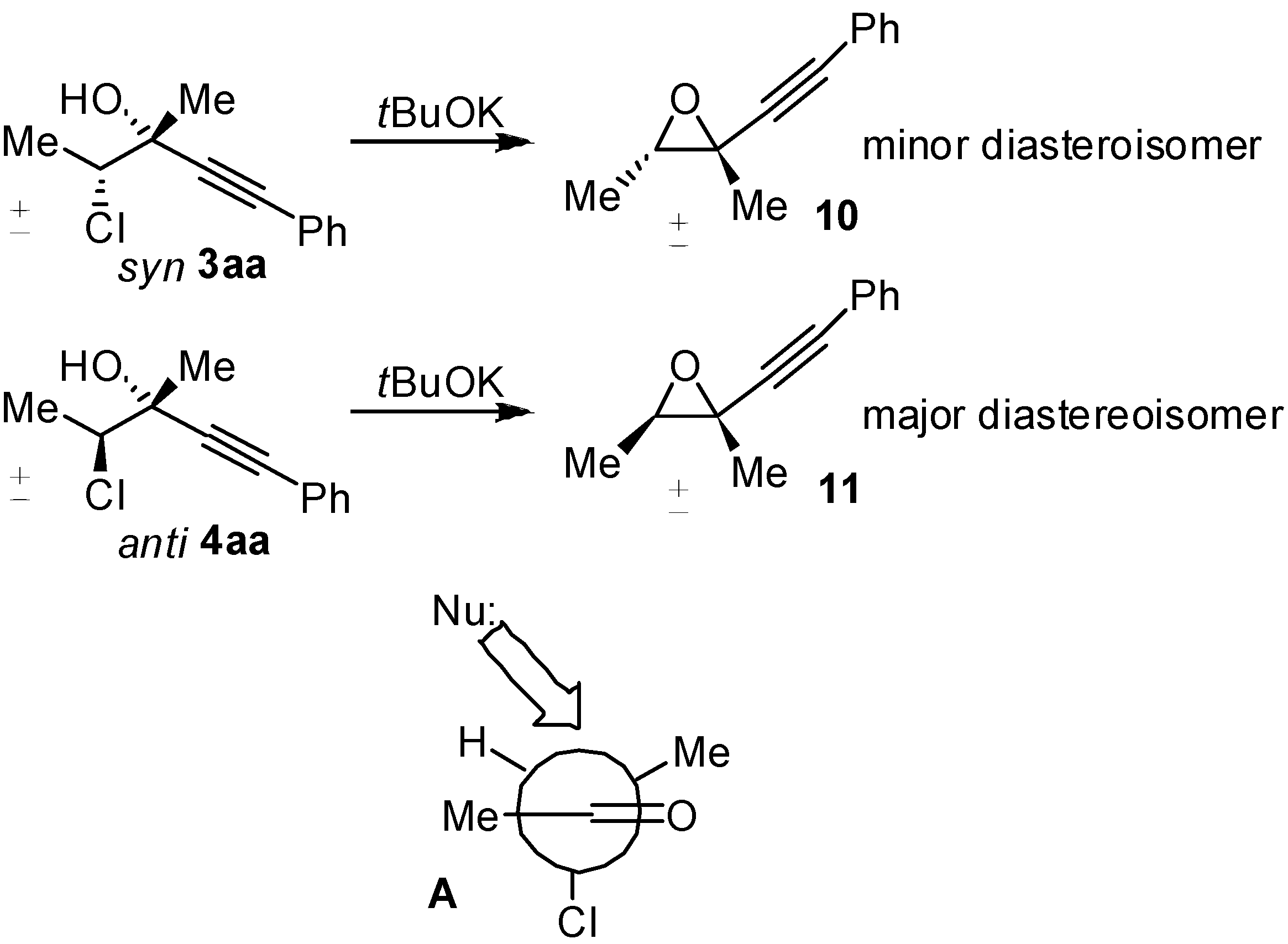
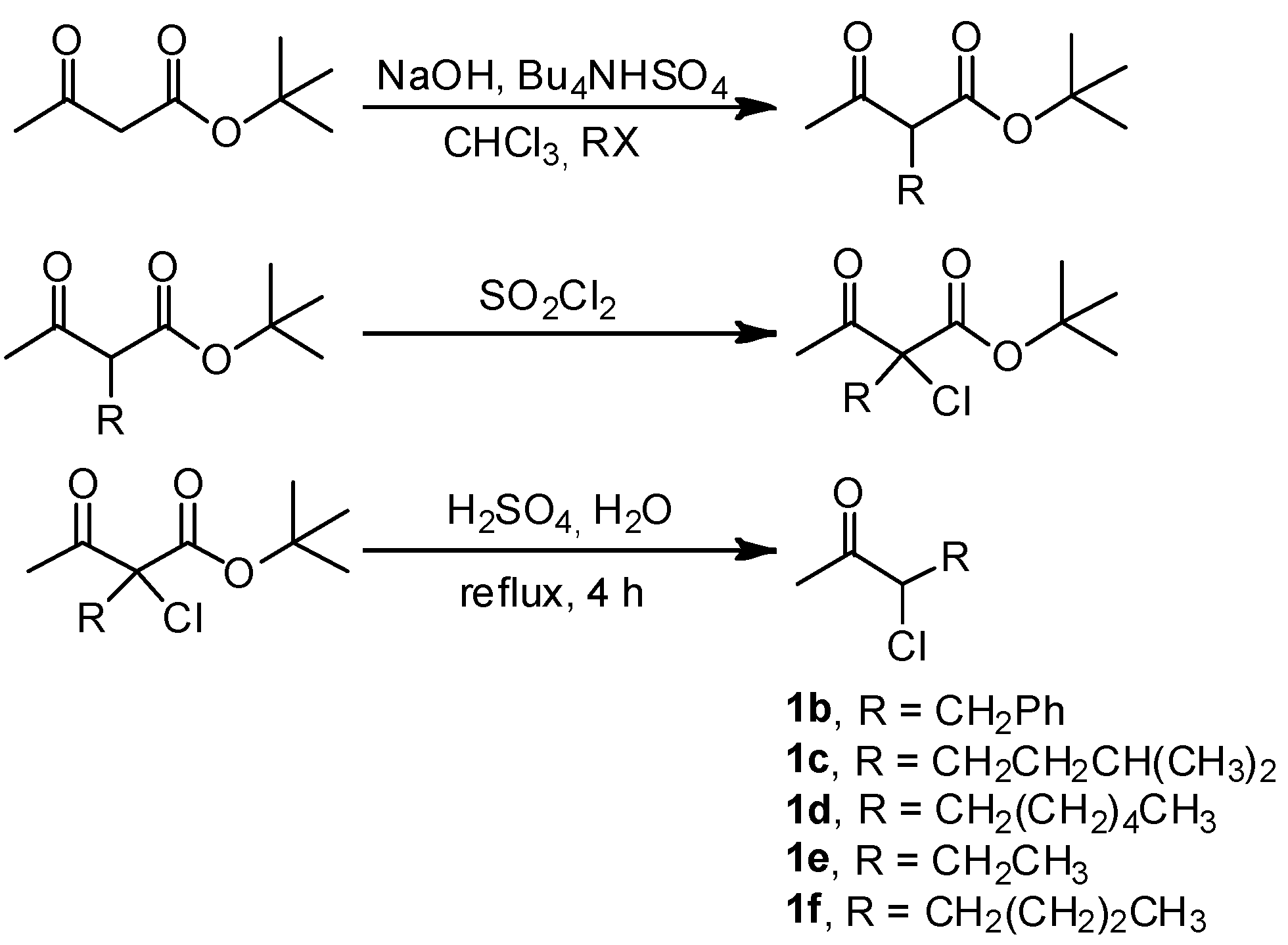
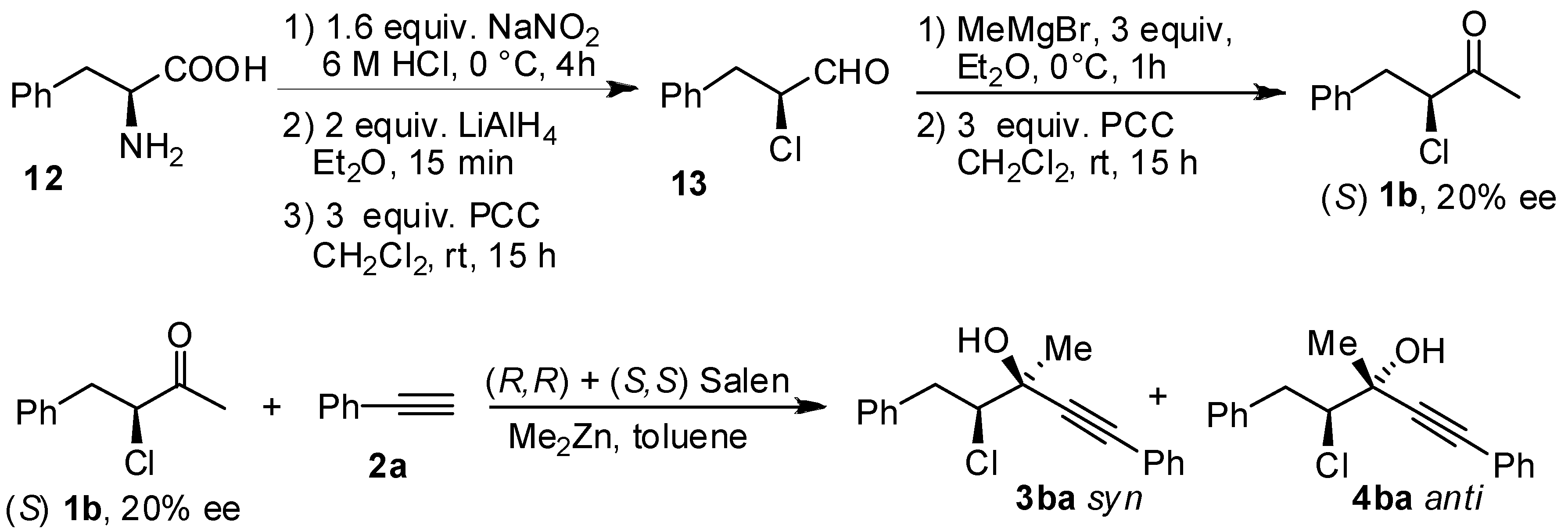
2. Results and Discussion

 | |||
|---|---|---|---|
| Entry a | ligand | syn% | anti% |
| 1 b | -- | 48 | 52 |
| 2 |  | 23 | 77 |
| 3 |  | 29 | 71 |
| 4 |  | 23 | 77 |
| 5 |  | 22 | 78 |
| 6 | 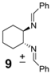 | 29 | 71 |
| 7 c | -- | 6 | 94 |
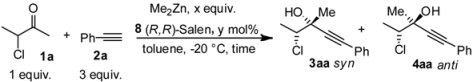 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry a | Me2Zn | Salen | solvent | dr b | T (h) | ee anti c | ee syn c | Yield d |
| (x equiv.) | (y mol %) | (mL) | ||||||
| 1 | 3 | 20 | 1 | 78:22 | 72 | 70 | 40 | 25 |
| 2 | 5 | 20 | 1 | 77:23 | 72 | 79 | 45 | 45 |
| 3 | 6 | 20 | 1 | 76:24 | 60 | 50 | 50 | 53 |
| 4 | 4.5 | 30 | 1.5 | 82:18 | 60 | 90 | 82 | 25 |
| 5 | 4.5 | 10 | 1 | 83:17 | 60 | 50 | 67 | 69 |
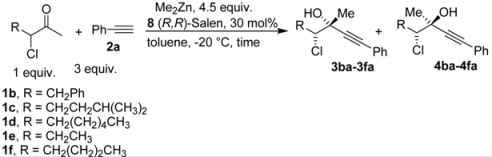 | ||||||
|---|---|---|---|---|---|---|
| Entry a | Ketone | dr b | T (h) | ee anti c | ee syn c | Yield d ( syn + anti) |
| 1 | 1a | 82:18 | 60 | 90 | 82 | 26 |
| 2 | 1b | 73:27 | 120 | 85 | 65 | 35 |
| 3 | 1c | 77:23 | 60 | 81 | 58 | 25 |
| 4 | 1d | 78:22 | 60 | 82 | 55 | 18 |
| 5 | 1e | 75:25 | 140 | 62 | 42 | 40 |
| 6 | 1f | 80:20 | 60 | 83 | 57 | 17 |
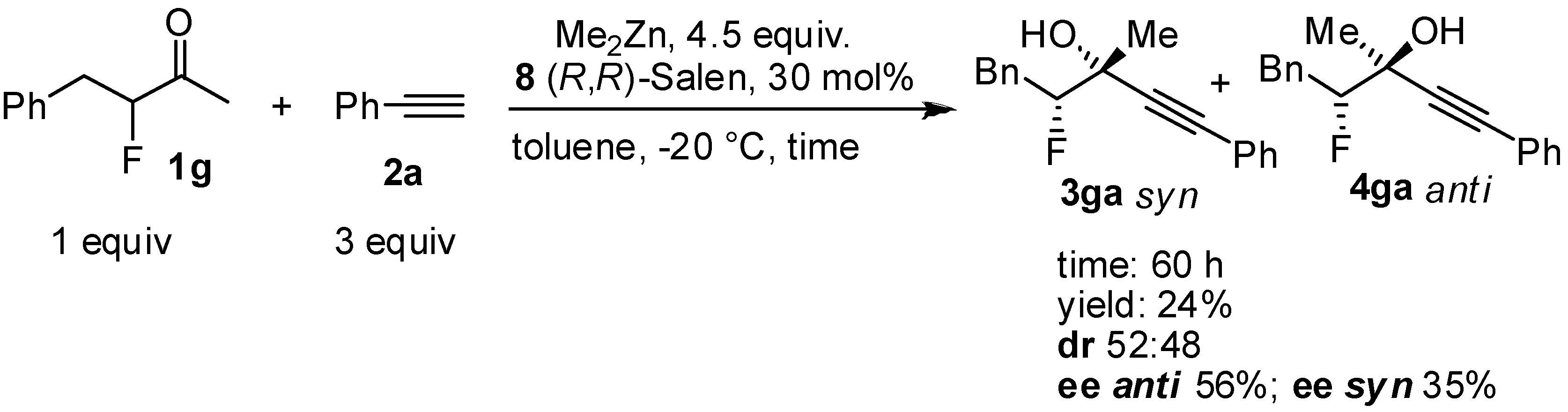
 | ||||||
|---|---|---|---|---|---|---|
| Entry a | Alkyne | dr b | ee anti c | ee syn c | Yield d ( syn + anti) | |
| 1 | HCCCH2Br, 2b | 64:36 | 66 | 53 | 25 | |
| 2 | HCCSiMe3,2c | 75:25 | 75 | 62 | 15 | |
| 3 | HCC(CH2)3CH3, 2d | 83:17 | 68 | 41 | 20 | |
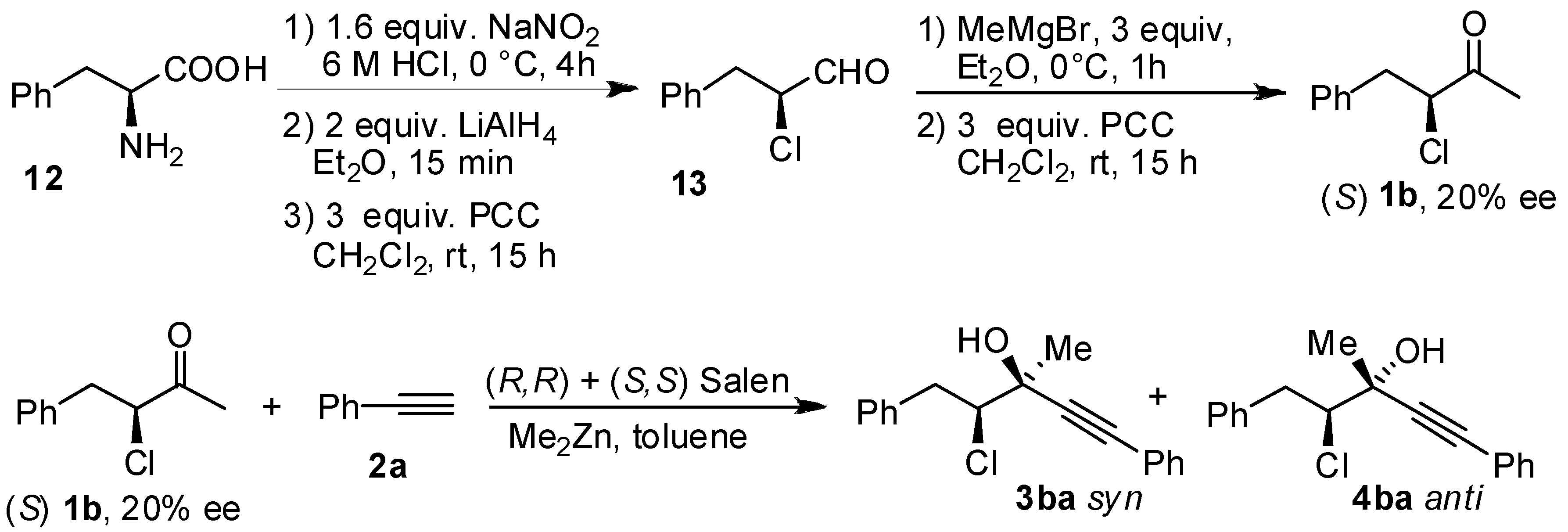
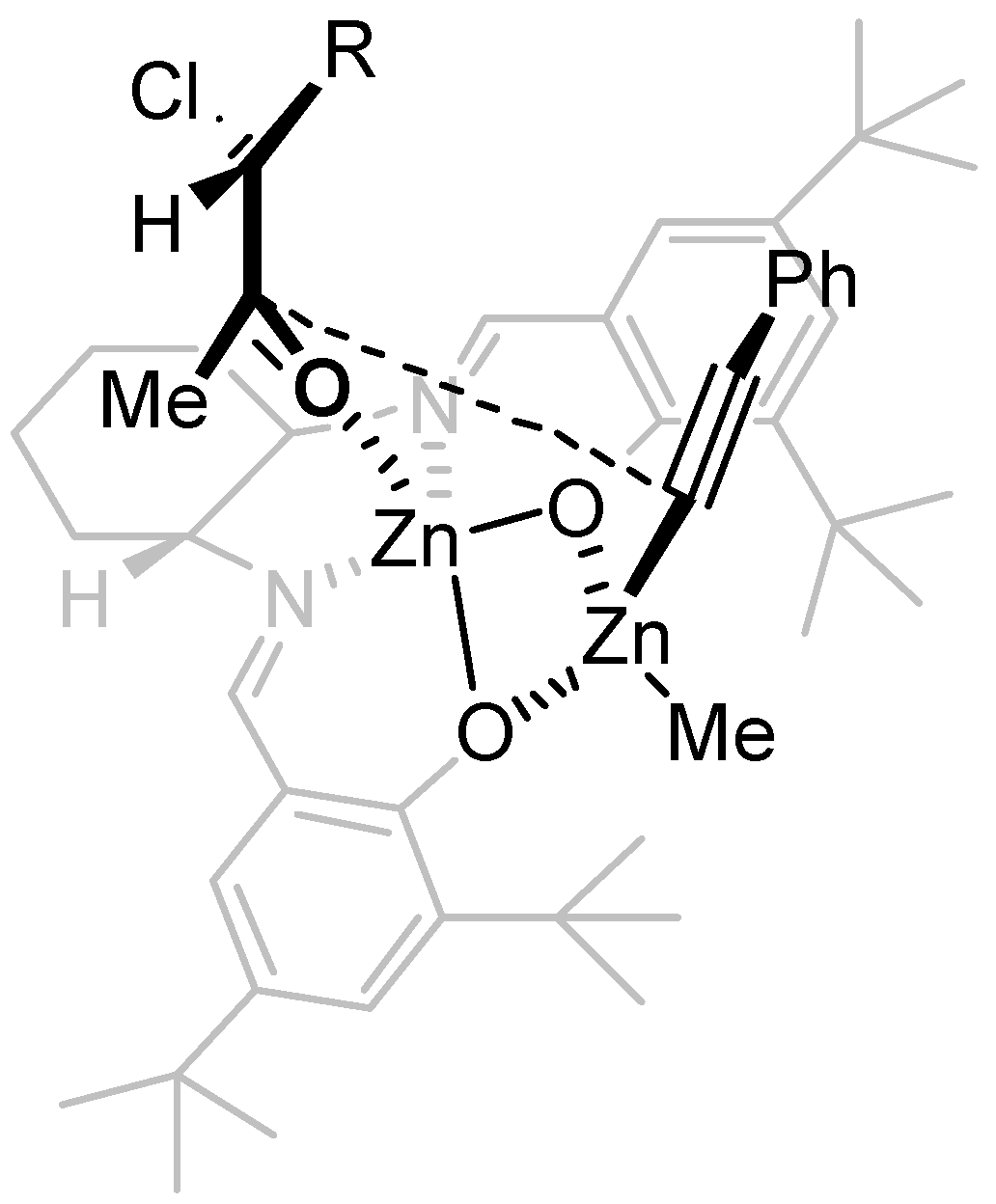
3. Experimental
3.1. General
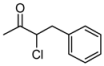
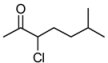
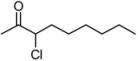
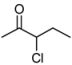
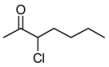
3.2. Addition of Alkynes to Chloroketones
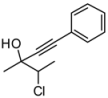
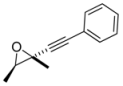
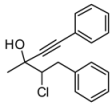
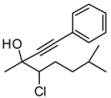
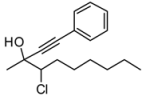
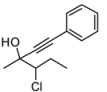
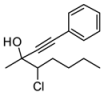
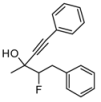
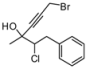
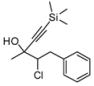
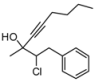
4. Conclusions
Acknowledgments
References
- Luderer, M.R.; Bailey, W.F.; Luderer, M.R.; Fair; J.D.; Dancer, R.J.; Sommer, M.B. Asymmetric addition of achiral organomagnesium reagents or organolithium to achiral aldehydes or ketones: A review. Tetrahedron: Asymmetry 2009, 20, 981–998. [Google Scholar] [CrossRef]
- Hatano, M.; Ishihara, K. Recent progress in the catalytic synthesis of tertiary alcohols from ketones with organometallic reagents. Synthesis 2008, 1647–1675. [Google Scholar]
- Cozzi, P.G.; Hilgraf, R.; Zimmerman, N. Acetylene in catalysis: enantioselective additions to carbonyl groups and imines and applications beyond. Eur. J. Org. Chem. 2004, 4095–4105. [Google Scholar]
- Blay, G.; Monleon, A.; Pedro, J.R. Recent development in asymmetric alkynylation of imines. Curr. Org. Chem. 2009, 13, 1498–1539. [Google Scholar] [CrossRef]
- Trost, B.M.; Weiss, A.H. The enantioselective alkyne nucleophiles to carbonyl groups. Adv. Synth. Catal. 2009, 351, 963–983. [Google Scholar] [CrossRef]
- Cozzi, P.G.; Rudolph, J.; Bolm, C.; Norrby, P.O.; Tomasini, C. Me2Zn-mediated addition of acetylene to aldehydes and ketones. J. Org. Chem. 2005, 70, 5733–5736. [Google Scholar]
- Cozzi, P.G. Enantioselective alkynylation of ketones catalyzed by Zn(Salen) complexes. Angew. Chem. Int. Ed. 2003, 42, 2895–2898. [Google Scholar] [CrossRef]
- Zhang, G.-W.; Meng, W.; Ma, H.; Nie, J.; Zhang, W.-Q.; Ma, J.-A. Catalytic enantioselective alkynylation of trifluoromethyl ketones: Pronounced metal fluoride effects and implications of zinc-to-titanium transmetallation. Angew. Chem. Int. Ed. 2011, 50, 3538–3542. [Google Scholar]
- Li, H.; Li, X.; Xue, F.; Huang, Y.; Jin, W.; Wan, B. Enantioselective alkynylzinc addition to carbonyl compounds by Tf-based sulfamide-amine alcohol catalysis. Chin. J. Chem. 2009, 27, 2013–2019. [Google Scholar] [CrossRef]
- Chen, C.; Hong, L.; Zhang, B.; Wang, R. Catalytic asymmetric addition of alkynylzinc reagents to ketones using polymer-supported chiral Schiff-base amino alcohols. Tetrahedron: Asymmetry 2008, 19, 191–196. [Google Scholar] [CrossRef]
- Lu, G.; Li, X.; Li, Y.-M.; Kwong, F.Y.; Chan, A.S.C. Highly enantioselective catalytic alkynylation of ketones - a convenient approach to optically active propargylic alcohols. Adv. Synth. Catal. 2006, 348, 1926–1933. [Google Scholar] [CrossRef]
- Ni, M.; Wang, R.; Han, Z.-J.; Mao, B.; Da, C.-S.; Liu, L.; Chen, C. Synthesis of new C2-symmetrical bissulfonamide ligands and application in the enantioselective addition of alkynylzinc to aldehydes and ketones. Adv. Synth. Catal. 2005, 347, 1659–1665. [Google Scholar] [CrossRef]
- Lu, G.; Li, X.; Jia, X.; Chan, W.L.; Chan, A.S.C. Enantioselective alkynylation of aromatic ketones catalyzed by chiral camphorsulfonamide ligands. Angew. Chem., Int. Ed. 2003, 42, 5057–5058. [Google Scholar] [CrossRef]
- Roman, B.I.; De Kimpe, N. Stevens, C.V. Synthesis of β -, γ -, δ -
![Molecules 16 05298 i026]() - Halogenated Ketones and Aldehydes. Chem. Rev. 2010, 110, 5914–5988. [Google Scholar] [CrossRef]
- Halogenated Ketones and Aldehydes. Chem. Rev. 2010, 110, 5914–5988. [Google Scholar] [CrossRef] - Brun, E.M.; Gil, S.; Mestres, R.; Parra, M.; Villar, F. Enediolates and dienediolates of carboxylic acids in synthesis. Synthesis of β,γ-epoxyacids from α- chloroketones. Tetrahedron Lett. 1998, 39, 1055–1058. [Google Scholar]
- Li, F.Q.; Zhong, S.; Lu, G.; Chan, A.S.C. Zn-Salen catalyzed asymmetric alkynylation of alkyl acylsilanes. Adv. Synth. Catal. 2009, 351, 1956–1960. [Google Scholar]
- Bernard, D.; Doutheau, A.; Gore, J.; Moulinoux, J.; Quemener, V.; Chantepie, J.; Quash, G. γ-Amino-α-acetylinic epoxides. Preparation and biological activity due to an aldehyde riductase inhibition. Tetrahedron 1989, 45, 1429–1439. [Google Scholar] [CrossRef]
- Chérest, M.; Felkin, H.; Prudent, N. Torsional strain involving partial bonds. The stereochemistry of the lithium aluminium hydride reduction of some simple open-chain ketones. Tetrahedron Lett. 1968, 2199–2204. [Google Scholar]
- Anh, N.T. Regio and stereoselectivities in some nucleophilic reactions. Top. Curr. Chem. 1980, 88, 145–162. [Google Scholar] [CrossRef]
- Anh, N.T.; Eistenstein, O. Theoretical interpretation of 1-2 asymmetric induction - Importance of antiperiplanarity. Nouv. J. Chim. 1977, 1, 61–70. [Google Scholar]
- Wong, S.S.; Paddon-Row, M.N. Theoretical evidence in support of the Anh-Eisenstein electronic model in controlling π-facial stereoselectivity in nucleophilic additions to carbonyl compounds. J. Chem. Soc. Chem. Commun. 1990, 456–458. [Google Scholar]
- Smith, R.J.; Trzoss, M.; Böhl, M.; Bienz, S. The Cram rule revisited once more - Revision of the Felkin-Anh model. Eur. J. Org. Chem. 2002, 2770–2775. [Google Scholar]
- De Kimpe, N.; Brunet, P. A convenient synthesis of 3-chloro-2-alkanones. Synthesis 1990, 7, 595–596. [Google Scholar] [CrossRef]
- Dekeukeleire, S.; D’hooghe, M.; Törnroos, K.W.; De Kimpe, N. Stereoselective synthesis of chiral 4-(1-chloroalkyl)-β-lactams starting from amino acids and their transformation into functionalized chiral azetidines and pyrrolidines. J. Org. Chem. 2010, 75, 5934–5940. [Google Scholar]
- Cornforth, J.W.; Cornforth, R.H.; Mathew, K.K. A general stereoselective synthesis of olefins. J. Chem. Soc. 1959, 112–127. [Google Scholar]
- Brinkmann, H.; Hoffmann, R.W. Stereoselective synthesis of alcohols. XXXV Addition of E and Z crotylboronate to chiral α substituted aldehydes. Chem. Ber. 1990, 123, 2395–2401. [Google Scholar] [CrossRef]
- Frenking, G.; Köhler, K.F.; Reetz, M.T. On the origin of π-facial diastereoselectivity in nucleophilic additions to chiral carbonyl compounds. 2. Calculated transition state structures for the addition of nucleophiles to propionaldehyde 1, chloroacetyldehyde 2, and 2-chloropropionaldehyde 3. Tetrahedron 1991, 47, 9005–9018. [Google Scholar] [CrossRef]
- Mengel, A.; Reiser, O. Around and beyond the Cram rule. Chem. Rev. 1999, 99, 1191–1223. [Google Scholar] [CrossRef]
- Concellòn, J.M.; Rodrìguez-Solla, H.; Simal, C.; Gòmez, C. A convenicent syntehsis of Z allylsilanes with good stereoselectivity promoted by SmI2. Synlett 2007, 75–78. [Google Scholar]
- Kang, B.; Britton, R. A general method for the synthesis of non-racemic trans epoxide containing insect sex pheromones. Org. Lett. 2007; 9, 5083–5086. [Google Scholar]
- The sense of the absolute configuration is reversed. However, also in the case of choroketone, (S) configurated stereogenic centers are formeddue to priority of the halogenated chain.
- Hutchinson, J.; Sandford, G.; Vaughan, J.F.S. Alkylation and decarboxylation of ethyl 2-fluoro-3 oxobutanoate as a route to functionalized α-fluoro ketones. Tetrahedron 1998, 54, 2867–2876. [Google Scholar]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
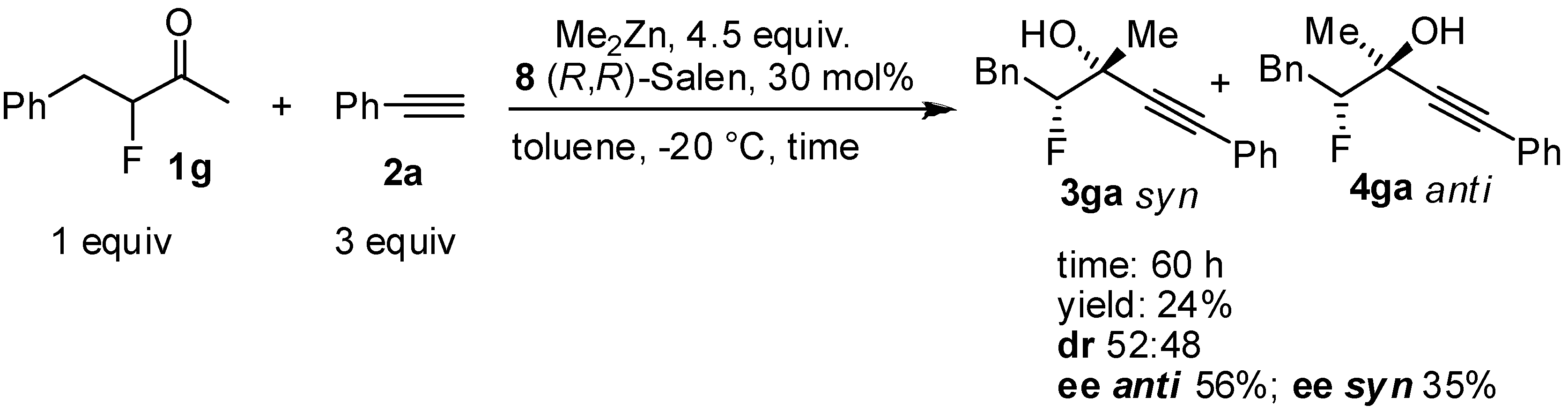{kind=link}
{kind=link}
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Alesi, S.; Emer, E.; Capdevila, M.G.; Petruzziello, D.; Gualandi, A.; Cozzi, P.G. Enantio and Diastereoselective Addition of Phenylacetylene to Racemic α-chloroketones. Molecules 2011, 16, 5298-5314. https://doi.org/10.3390/molecules16065298
Alesi S, Emer E, Capdevila MG, Petruzziello D, Gualandi A, Cozzi PG. Enantio and Diastereoselective Addition of Phenylacetylene to Racemic α-chloroketones. Molecules. 2011; 16(6):5298-5314. https://doi.org/10.3390/molecules16065298
Chicago/Turabian StyleAlesi, Silvia, Enrico Emer, Montse Guiteras Capdevila, Diego Petruzziello, Andrea Gualandi, and Pier Giorgio Cozzi. 2011. "Enantio and Diastereoselective Addition of Phenylacetylene to Racemic α-chloroketones" Molecules 16, no. 6: 5298-5314. https://doi.org/10.3390/molecules16065298
APA StyleAlesi, S., Emer, E., Capdevila, M. G., Petruzziello, D., Gualandi, A., & Cozzi, P. G. (2011). Enantio and Diastereoselective Addition of Phenylacetylene to Racemic α-chloroketones. Molecules, 16(6), 5298-5314. https://doi.org/10.3390/molecules16065298