High Affinity, Developability and Functional Size: The Holy Grail of Combinatorial Antibody Library Generation

Abstract
:1. Introduction
2. Display Methods for Combinatorial Libraries
2.1. Ribosome Display
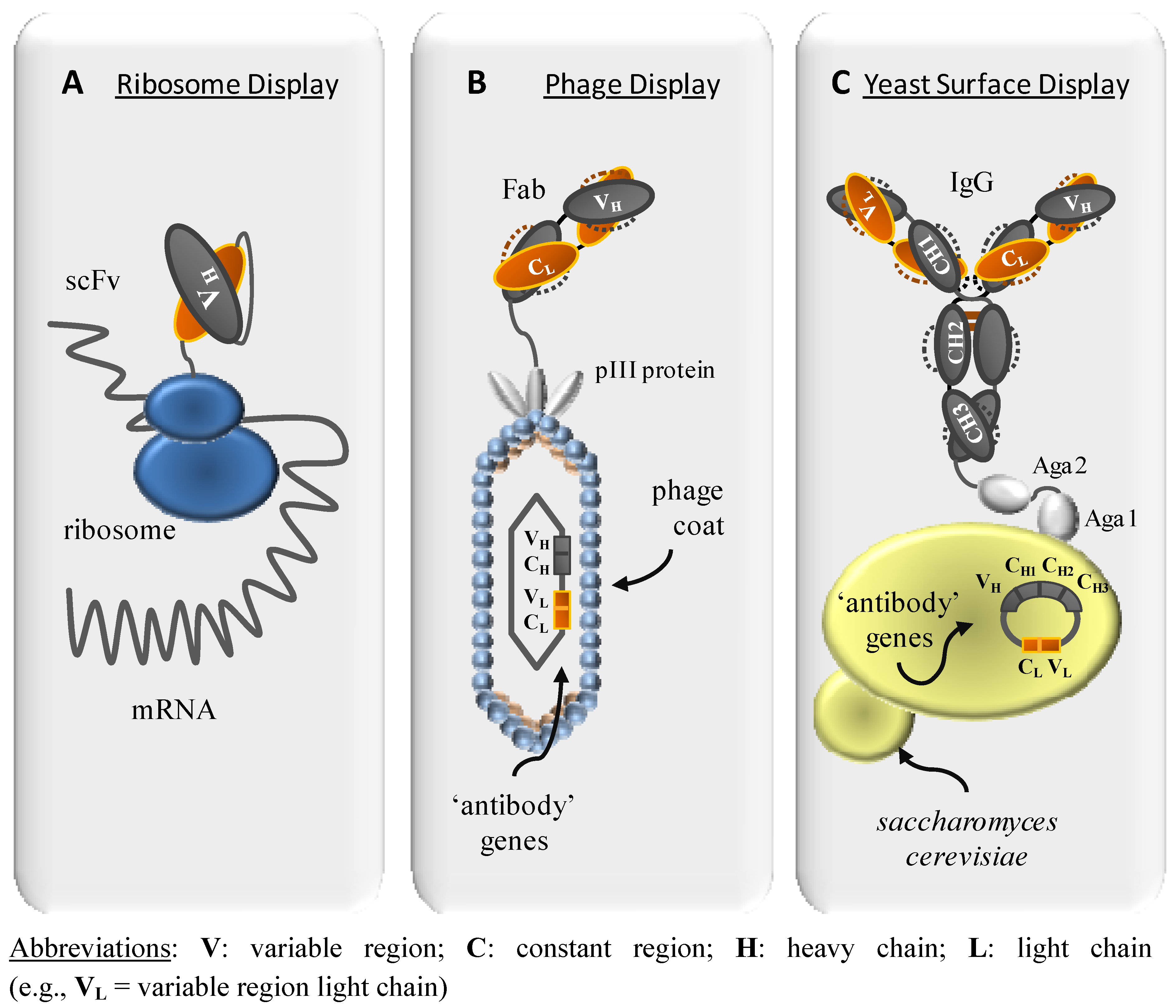
2.2. Yeast Surface Display
2.3. Phage display
3. Antibody-derived Fragments Used in Phage Display
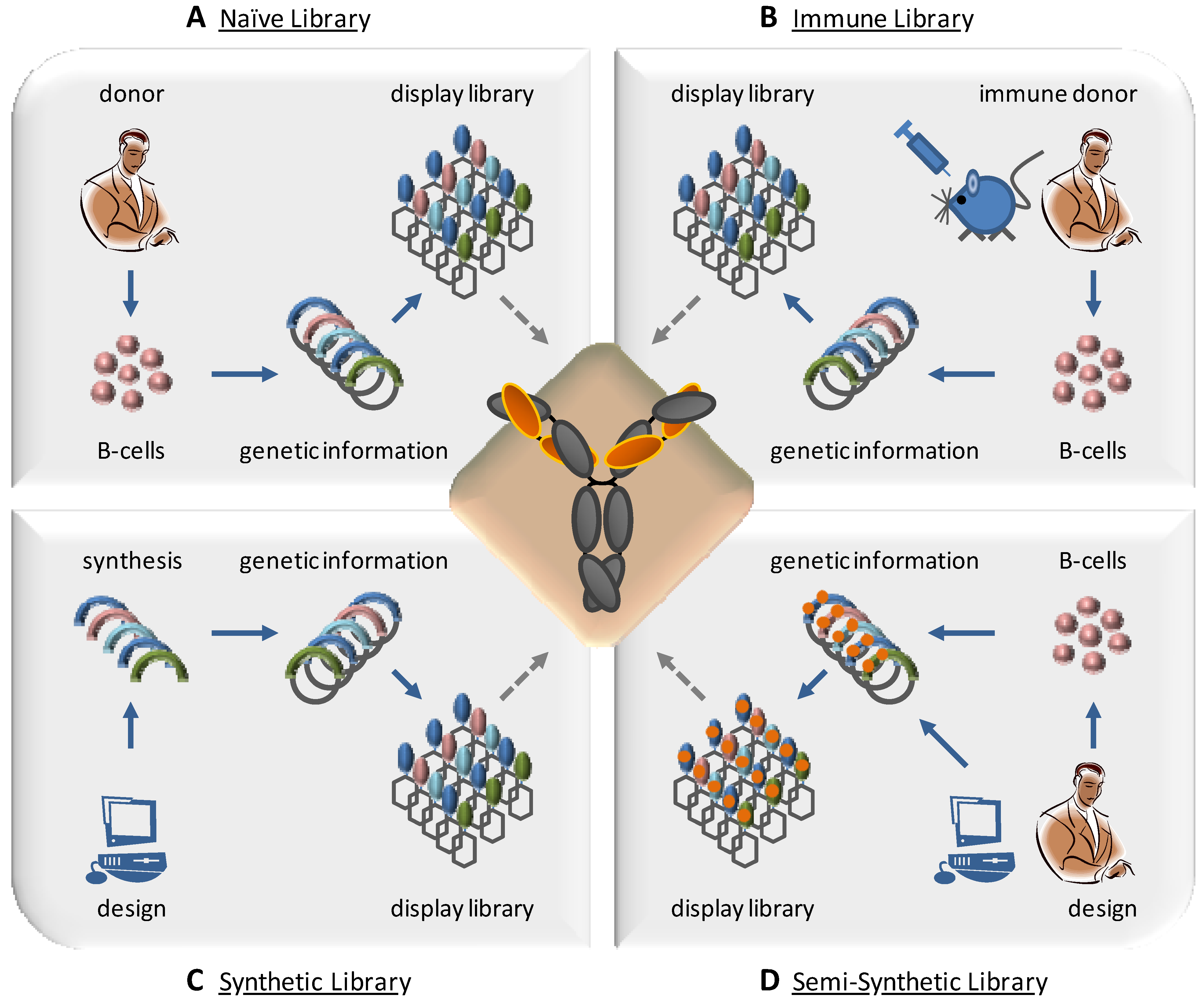
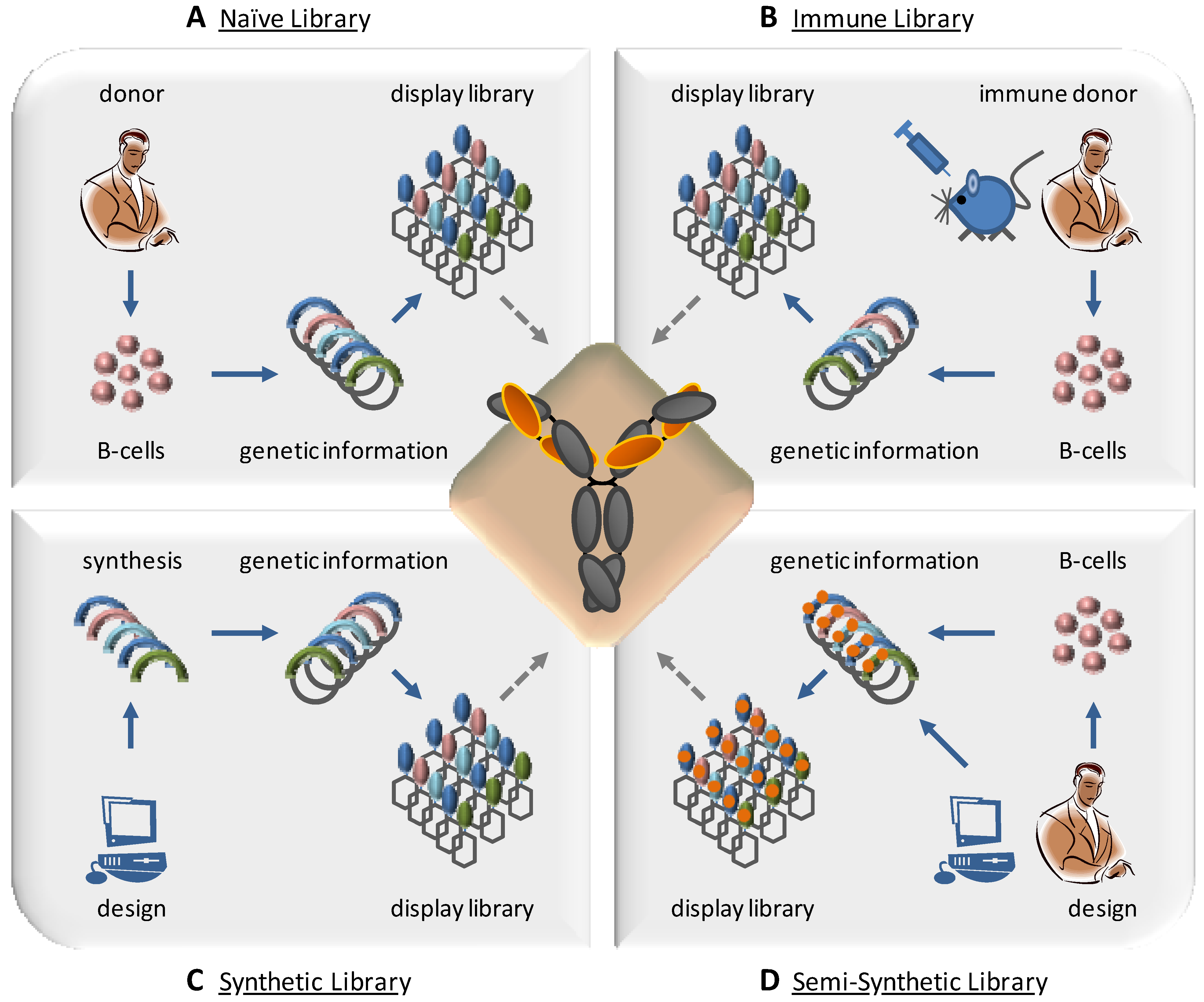
4. Design of Combinatorial Antibody Libraries

{kind=link}
{kind=link}
{kind=link}
| Library Name | Format | Framework | CDR Source | Library Size | Published Affinities | Reference |
|---|---|---|---|---|---|---|
| - | scFv | immune | immune | nM | [50] | |
| RAB-03/04-G01Crucell | immune | immune | 1 × 107 | [51] | ||
| - | naïve | naïve | >107 | low pM | [26,52] | |
| CAT1.0 MedImmune | naïve | naïve | 1.4 × 1010 | low to sub-nM | [27] | |
| CAT2.0 MedImmune | naïve | naïve | 1.3 × 1011 | [53] | ||
| - | • 49 VH frameworks • single VL framework | • synthetic CDR-H3 | 1 × 107 | µM | [54] | |
| - | • 49 VH frameworks • single VL framework | • synthetic CDR-H3 | 108 | [55] | ||
| - | • 49 VH frameworks • 7 VL frameworks | • synthetic CDR-H3 | 3.6 × 108 | 100nM to µM | [56] | |
| - | • single VH framework • single VL framework | • synthetic CDR-H3 and CDR-L3 | >3 × 108 | nM | [57] | |
| n-CoDeR® Bioinvent | scFv /Fab | • single VH framework • single VL framework | • natural | 109-1010 | sub-nM | [58,59] |
| Genentech | Fab | • single VH framework • single VL framework | • synthetic CDR-H1 -H2, -H3, (-L3) | up to 1010 | low nM | [60,61] |
| - | • 49 VH frameworks • 26 VL_k frameworks • 21 VL_l frameworks | • synthetic CDR-H3 | 6.5 × 1010 | up to single digit nM | [62] | |
| Dyax | • single VH framework • natural VL | • natural CDR-H3 • synthetic CDR-H1/-H2 | 1 × 1010 | sub-nM | [63] | |
| HuCAL GOLD®MorphoSys | • synthetic (consensus sequence) | synthetic | 1.6 × 1010 | pM | [64] | |
| HuCAL PLATINUM®MorphoSys | • synthetic (close to germline) | synthetic | 4.5 × 1010 |
4.1. Immune Libraries
4.3. Synthetic and Semi-Synthetic Libraries
4.4. Defined Frameworks: Maximal Diversity
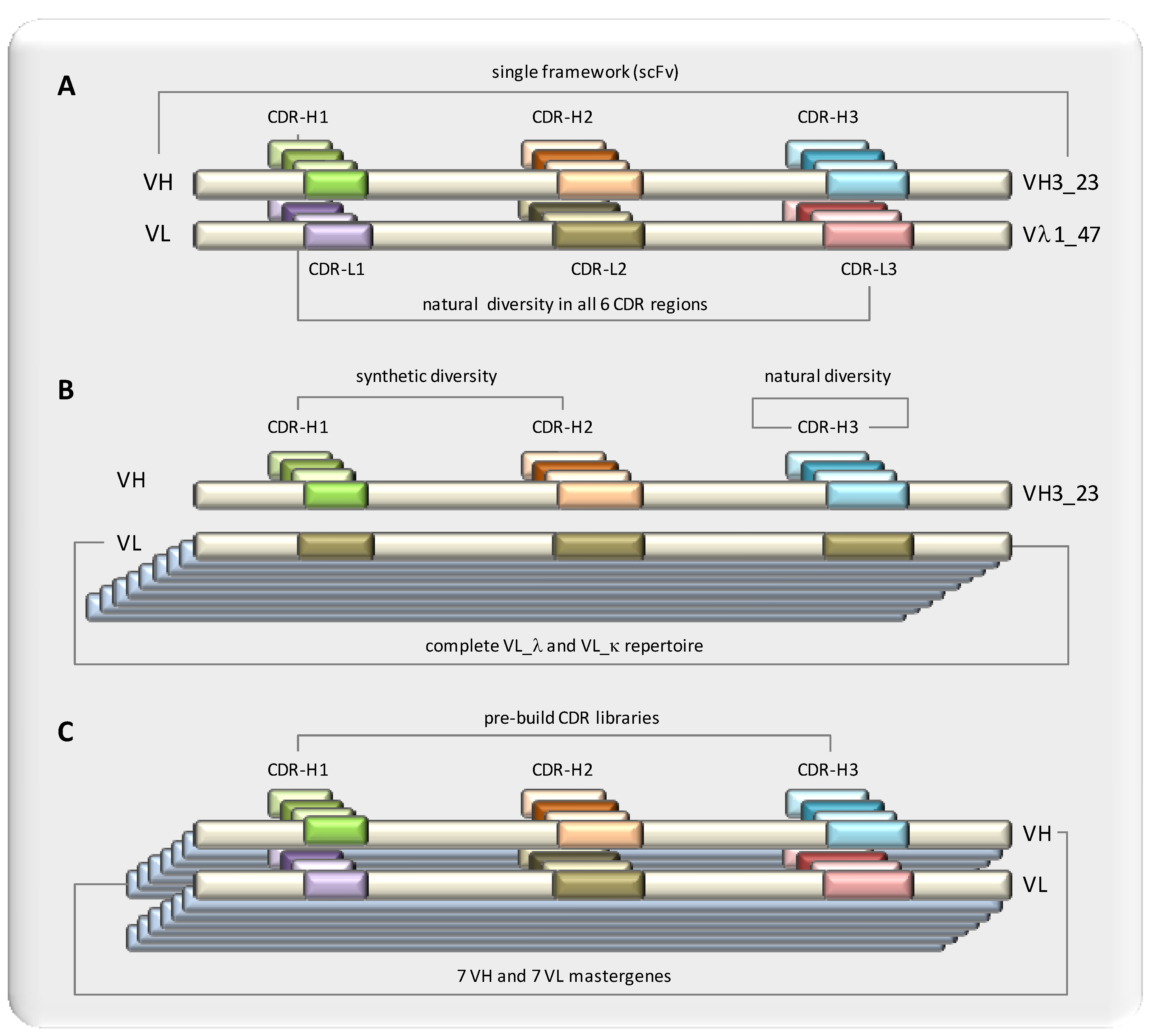
4.5. Elucidating Minimal Requirements: Restricted Diversity
4.6. Diversity Limited to Natural Design: Adimab Technology
4.7. Camelized Human VH Domains: Synthetic Libraries
5. Maturation Strategies
5.1. Non-Targeted Diversification
5.2. Targeted Diversification
6. Conclusions
References and Notes
- Perelson, A.S.; Oster, G.F. Theoretical studies of clonal selection: Minimal antibody repertoire size and reliability of self-non-self discrimination. J. Theor. Biol. 1979, 81, 645–670. [Google Scholar] [CrossRef]
- Nelson, A.L.; Dhimolea, E.; Reichert, J.M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 2010, 9, 767–774. [Google Scholar] [CrossRef]
- Brooks, S.A. Strategies for analysis of the glycosylation of proteins: Current status and future perspectives. Mol. Biotechnol. 2009, 43, 76–88. [Google Scholar] [CrossRef]
- Getts, D.R.; Getts, M.T.; McCarthy, D.P.; Chastain, E.M.; Miller, S.D. Have we overestimated the benefit of human(ized) antibodies? MAbs 2010, 2, 682–694. [Google Scholar] [CrossRef]
- Visintin, M.; Tse, E.; Axelson, H.; Rabbitts, T.H.; Cattaneo, A. Selection of antibodies for intracellular function using a two-hybrid in vivo system. Proc. Natl. Acad. Sci USA 1999, 96, 11723–11728. [Google Scholar] [CrossRef]
- Auf der Maur, A.; Tissot, K.; Barberis, A. Antigen-independent selection of intracellular stable antibody frameworks. Methods 2004, 34, 215–224. [Google Scholar] [CrossRef]
- Mossner, E.; Koch, H.; Plückthun, A. Fast selection of antibodies without antigen purification: adaptation of the protein fragment complementation assay to select antigen-antibody pairs. J. Mol. Biol. 2001, 308, 115–122. [Google Scholar] [CrossRef]
- Mattheakis, L.C.; Bhatt, R.R.; Dower, W.J. An in vitro polysome display system for identifying ligands from very large peptide libraries. Proc. Natl. Acad. Sci. USA 1994, 91, 9022–9026. [Google Scholar] [CrossRef]
- Hanes, J.; Plückthun, A. In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl. Acad. Sci. USA 1997, 94, 4937–4942. [Google Scholar] [CrossRef]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557. [Google Scholar] [CrossRef]
- Feldhaus, M.J.; Siegel, R.W.; Opresko, L.K.; Coleman, J.R.; Feldhaus, J.M.; Yeung, Y.A.; Cochran, J.R.; Heinzelman, P.; Colby, D.; Swers, J.; et al. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat. Biotechnol. 2003, 21, 163–170. [Google Scholar] [CrossRef]
- Weaver-Feldhaus, J.M.; Lou, J.; Coleman, J.R.; Siegel, R.W.; Marks, J.D.; Feldhaus, M.J. Yeast mating for combinatorial Fab library generation and surface display. FEBS Lett. 2004, 564, 24–34. [Google Scholar] [CrossRef]
- Feldhaus, M.J. Human Antibody Discovery and Optimization in Yeast. Presented at PEGS Protein Engineering Summit, 17-21 May 2010.
- Beerli, R.R.; Bauer, M.; Buser, R.B.; Gwerder, M.; Muntwiler, S.; Maurer, P.; Saudan, P.; Bachmann, M.F. Isolation of human monoclonal antibodies by mammalian cell display. Proc. Natl. Acad. Sci. USA 2008, 105, 14336–14341. [Google Scholar]
- Boder, E.T.; Midelfort, K.S.; Wittrup, K.D. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc. Natl. Acad. Sci. USA 2000, 97, 10701–10705. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar]
- Clackson, T.; Wells, J.A. In vitro selection from protein and peptide libraries. Trends Biotechnol. 1994, 12, 173–184. [Google Scholar] [CrossRef]
- Griffiths, A.D.; Malmqvist, M.; Marks, J.D.; Bye, J.M.; Embleton, M.J.; McCafferty, J.; Baier, M.; Holliger, K.P.; Gorick, B.D.; Hughes-Jones, N.C. Human anti-self antibodies with high specificity from phage display libraries. EMBO J. 1993, 12, 725–734. [Google Scholar]
- Duenas, M.; Borrebaeck, C.A. Novel helper phage design: intergenic region affects the assembly of bacteriophages and the size of antibody libraries. FEMS Microbiol. Lett. 1995, 125, 317–321. [Google Scholar] [Green Version]
- Rakonjac, J.; Jovanovic, G.; Model, P. Filamentous phage infection-mediated gene expression: Construction and propagation of the gIII deletion mutant helper phage R408d3. Gene 1997, 198, 99–103. [Google Scholar] [CrossRef]
- Rondot, S.; Koch, J.; Breitling, F.; Dübel, S. A helper phage to improve single-chain antibody presentation in phage display. Nat. Biotechnol. 2001, 19, 75–78. [Google Scholar] [CrossRef]
- Kramer, R.A.; Cox, F.; van der Horst, M.; van der Oudenrijn, S.; Res, P.C.; Bia, J.; Logtenberg, T.; de Kruif, J. A novel helper phage that improves phage display selection efficiency by preventing the amplification of phages without recombinant protein. Nucleic Acids Res. 2003, 31, e59. [Google Scholar] [CrossRef]
- Soltes, G.; Barker, H.; Marmai, K.; Pun, E.; Yuen, A.; Wiersma, E.J. A new helper phage and phagemid vector system improves viral display of antibody Fab fragments and avoids propagation of insert-less virions. J. Immunol. Methods 2003, 274, 233–244. [Google Scholar] [CrossRef]
- Baek, H.; Suk, K.H.; Kim, Y.H.; Cha, S. An improved helper phage system for efficient isolation of specific antibody molecules in phage display. Nucleic Acids Res. 2002, 30, e18. [Google Scholar] [CrossRef]
- Burton, D.R.; Barbas, C.F., III; Persson, M.A.; Koenig, S.; Chanock, R.M.; Lerner, R.A. A large array of human monoclonal antibodies to type 1 human immunodeficiency virus from combinatorial libraries of asymptomatic seropositive individuals. Proc. Natl. Acad. Sci. USA 1991, 88, 10134–10137. [Google Scholar]
- Marks, J.D.; Hoogenboom, H.R.; Bonnert, T.P.; McCafferty, J.; Griffiths, A.D.; Winter, G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J. Mol. Biol. 1991, 222, 581–597. [Google Scholar] [CrossRef]
- Vaughan, T.J.; Williams, A.J.; Pritchard, K.; Osbourn, J.K.; Pope, A.R.; Earnshaw, J.C.; McCafferty, J.; Hodits, R.A.; Wilton, J.; Johnson, K.S. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat. Biotechnol. 1996, 14, 309–314. [Google Scholar] [CrossRef]
- Bradbury, A.R.; Marks, J.D. Antibodies from phage antibody libraries. J. Immunol. Methods 2004, 290, 29–49. [Google Scholar] [CrossRef]
- Jung, S.; Honegger, A.; Plückthun, A. Selection for improved protein stability by phage display. J. Mol. Biol. 1999, 294, 163–180. [Google Scholar] [CrossRef]
- Persson, M.A.; Caothien, R.H.; Burton, D.R. Generation of diverse high-affinity human monoclonal antibodies by repertoire cloning. Proc. Natl. Acad. Sci. USA 1991, 88, 2432–2436. [Google Scholar] [CrossRef]
- Röthlisberger, D.; Honegger, A.; Plückthun, A. Domain interactions in the Fab fragment: A comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J. Mol. Biol. 2005, 347, 773–789. [Google Scholar] [CrossRef]
- Ward, E.S.; Gussow, D.; Griffiths, A.D.; Jones, P.T.; Winter, G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature 1989, 341, 544–546. [Google Scholar] [CrossRef]
- Ewert, S.; Cambillau, C.; Conrath, K.; Plückthun, A. Biophysical properties of camelid V(HH) domains compared to those of human V(H)3 domains. Biochemistry 2002, 41, 3628–3636. [Google Scholar] [CrossRef]
- Dumoulin, M.; Conrath, K.; Van, M.A.; Meersman, F.; Heremans, K.; Frenken, L.G.; Muyldermans, S.; Wyns, L.; Matagne, A. Single-domain antibody fragments with high conformational stability. Protein Sci. 2002, 11, 500–515. [Google Scholar]
- Davies, J.; Riechmann, L. Affinity improvement of single antibody VH domains: Residues in all three hypervariable regions affect antigen binding. Immunotechnology. 1996, 2, 169–179. [Google Scholar] [CrossRef]
- Davies, J.; Riechmann, L. Single antibody domains as small recognition units: Design and in vitro antigen selection of camelized, human VH domains with improved protein stability. Protein Eng. 1996, 9, 531–537. [Google Scholar] [CrossRef]
- Davies, J.; Riechmann, L. Antibody VH domains as small recognition units. Biotechnology (N.Y.) 1995, 13, 475–479. [Google Scholar] [CrossRef]
- Davies, J.; Riechmann, L. Camelising' human antibody fragments: NMR studies on VH domains. FEBS Lett. 1994, 339, 285–290. [Google Scholar] [CrossRef]
- Riechmann, L. Rearrangement of the former VL interface in the solution structure of a camelised, single antibody VH domain. J. Mol. Biol. 1996, 259, 957–969. [Google Scholar] [CrossRef]
- Riechmann, L.; Muyldermans, S. Single domain antibodies: comparison of camel VH and camelised human VH domains. J. Immunol. Methods 1999, 231, 25–38. [Google Scholar] [CrossRef]
- Muyldermans, S.; Cambillau, C.; Wyns, L. Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends Biochem. Sci. 2001, 26, 230–235. [Google Scholar] [CrossRef]
- Muyldermans, S. Single domain camel antibodies: current status. J. Biotechnol. 2001, 74, 277–302. [Google Scholar]
- Desmyter, A.; Transue, T.R.; Ghahroudi, M.A.; Thi, M.H.; Poortmans, F.; Hamers, R.; Muyldermans, S.; Wyns, L. Crystal structure of a camel single-domain VH antibody fragment in complex with lysozyme. Nat. Struct. Biol. 1996, 3, 803–811. [Google Scholar] [CrossRef]
- Desmyter, A.; Decanniere, K.; Muyldermans, S.; Wyns, L. Antigen specificity and high affinity binding provided by one single loop of a camel single-domain antibody. J. Biol. Chem. 2001, 276, 26285–26290. [Google Scholar] [CrossRef]
- Desmyter, A.; Spinelli, S.; Payan, F.; Lauwereys, M.; Wyns, L.; Muyldermans, S.; Cambillau, C. Three camelid VHH domains in complex with porcine pancreatic alpha-amylase. Inhibition and versatility of binding topology. J. Biol. Chem. 2002, 277, 23645–23650. [Google Scholar]
- Lauwereys, M.; Arbabi, G.M.; Desmyter, A.; Kinne, J.; Holzer, W.; De, G.E.; Wyns, L.; Muyldermans, S. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 1998, 17, 3512–3520. [Google Scholar] [CrossRef]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [Google Scholar]
- Chapman, A.P.; Antoniw, P.; Spitali, M.; West, S.; Stephens, S.; King, D.J. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat. Biotechnol. 1999, 17, 780–783. [Google Scholar] [CrossRef]
- Lee, L.S.; Conover, C.; Shi, C.; Whitlow, M.; Filpula, D. Prolonged circulating lives of single-chain Fv proteins conjugated with polyethylene glycol: a comparison of conjugation chemistries and compounds. Bioconjug.Chem. 1999, 10, 973–981. [Google Scholar] [CrossRef]
- Huse, W.D.; Sastry, L.; Iverson, S.A.; Kang, A.S.; Alting-Mees, M.; Burton, D.R.; Benkovic, S.J.; Lerner, R.A. Generation of a large combinatorial library of the immunoglobulin repertoire in phage lambda. Science 1989, 246, 1275–1281. [Google Scholar]
- Kramer, R.A.; Marissen, W.E.; Goudsmit, J.; Visser, T.J.; Clijsters-Van der Horst, M.; Bakker, A.Q.; de Jong, M.; Jongeneelen, M.; Thijsse, S.; Backus, H.H.; et al. The human antibody repertoire specific for rabies virus glycoprotein as selected from immune libraries. Eur. J. Immunol. 2005, 35, 2131–2145. [Google Scholar] [CrossRef]
- Marks, J.D.; Griffiths, A.D.; Malmqvist, M.; Clackson, T.P.; Bye, J.M.; Winter, G. By-passing immunization: building high affinity human antibodies by chain shuffling. Biotechnology (N.Y.) 1992, 10, 779–783. [Google Scholar] [CrossRef]
- Lloyd, C.; Lowe, D.; Edwards, B.; Welsh, F.; Dilks, T.; Hardman, C.; Vaughan, T. Modelling the human immune response: performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng. Des. Sel. 2009, 22, 159–168. [Google Scholar]
- Hoogenboom, H.R. Overview of antibody phage-display technology and its applications. Methods Mol. Biol. 2002, 178, 1–37. [Google Scholar]
- Nissim, A.; Hoogenboom, H.R.; Tomlinson, I.M.; Flynn, G.; Midgley, C.; Lane, D.; Winter, G. Antibody fragments from a 'single pot' phage display library as immunochemical reagents. EMBO J. 1994, 13, 692–698. [Google Scholar]
- de Kruif, J.; Boel, E.; Logtenberg, T. Selection and application of human single chain Fv antibody fragments from a semi-synthetic phage antibody display library with designed CDR3 regions. J. Mol. Biol. 1995, 248, 97–105. [Google Scholar] [CrossRef]
- Pini, A.; Viti, F.; Santucci, A.; Carnemolla, B.; Zardi, L.; Neri, P.; Neri, D. Design and use of a phage display library. Human antibodies with subnanomolar affinity against a marker of angiogenesis eluted from a two-dimensional gel. J. Biol. Chem. 1998, 273, 21769–21776. [Google Scholar]
- Carlsson, R.; Söderlind, E. n-CoDeR concept: unique types of antibodies for diagnostic use and therapy. Expert Rev. Mol. Diagn. 2001, 1, 102–108. [Google Scholar] [CrossRef]
- Söderlind, E.; Strandberg, L.; Jirholt, P.; Kobayashi, N.; Alexeiva, V.; Aberg, A.M.; Nilsson, A.; Jansson, B.; Ohlin, M.; Wingren, C.; Danielsson, L.; Carlsson, R.; Borrebaeck, C.A. Recombining germline-derived CDR sequences for creating diverse single-framework antibody libraries. Nat. Biotechnol. 2000, 18, 852–856. [Google Scholar] [CrossRef]
- Lee, C.V.; Liang, W.C.; Dennis, M.S.; Eigenbrot, C.; Sidhu, S.S.; Fuh, G. High-affinity human antibodies from phage-displayed synthetic Fab libraries with a single framework scaffold. J. Mol. Biol. 2004, 340, 1073–1093. [Google Scholar] [CrossRef]
- Liang, W.C.; Dennis, M.S.; Stawicki, S.; Chanthery, Y.; Pan, Q.; Chen, Y.; Eigenbrot, C.; Yin, J.; Koch, A.W.; Wu, X.; Ferrara, N.; Bagri, A.; Tessier-Lavigne, M.; Watts, R.J.; Wu, Y. Function blocking antibodies to neuropilin-1 generated from a designed human synthetic antibody phage library. J. Mol. Biol. 2007, 366, 815–829. [Google Scholar] [CrossRef]
- Griffiths, A.D.; Williams, S.C.; Hartley, O.; Tomlinson, I.M.; Waterhouse, P.; Crosby, W.L.; Kontermann, R.E.; Jones, P.T.; Low, N.M.; Allison, T.J. Isolation of high affinity human antibodies directly from large synthetic repertoires. EMBO J. 1994, 13, 3245–3260. [Google Scholar]
- Hoet, R.M.; Cohen, E.H.; Kent, R.B.; Rookey, K.; Schoonbroodt, S.; Hogan, S.; Rem, L.; Frans, N.; Daukandt, M.; Pieters, H.; et al. Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat. Biotechnol. 2005, 23, 344–348. [Google Scholar] [CrossRef]
- Rothe, C.; Urlinger, S.; Lohning, C.; Prassler, J.; Stark, Y.; Jager, U.; Hubner, B.; Bardroff, M.; Pradel, I.; Boss, M.; et al. The human combinatorial antibody library HuCAL GOLD combines diversification of all six CDRs according to the natural immune system with a novel display method for efficient selection of high-affinity antibodies. J. Mol. Biol. 2008, 376, 1182–1200. [Google Scholar] [CrossRef]
- Waterhouse, P.; Griffiths, A.D.; Johnson, K.S.; Winter, G. Combinatorial infection and in vivo recombination: a strategy for making large phage antibody repertoires. Nucleic Acids Res. 1993, 21, 2265–2266. [Google Scholar] [CrossRef]
- Sblattero, D.; Bradbury, A. Exploiting recombination in single bacteria to make large phage antibody libraries. Nat. Biotechnol. 2000, 18, 75–80. [Google Scholar] [CrossRef]
- Seehaus, T.; Breitling, F.; Dübel, S.; Klewinghaus, I.; Little, M. A vector for the removal of deletion mutants from antibody libraries. Gene 1992, 114, 235–237. [Google Scholar] [CrossRef]
- Azzazy, H.M.; Highsmith, W.E., Jr. Phage display technology: clinical applications and recent innovations. Clin. Biochem. 2002, 35, 425–445. [Google Scholar] [CrossRef]
- Benhar, I. Design of synthetic antibody libraries. Expert Opin. Biol. Ther. 2007, 7, 763–779. [Google Scholar] [CrossRef]
- de Carvalho, N.C.; Williamson, R.A.; Parren, P.W.; Lundkvist, A.; Burton, D.R.; Bjorling, E. Neutralizing human Fab fragments against measles virus recovered by phage display. J. Virol. 2002, 76, 251–258. [Google Scholar] [CrossRef]
- Zebedee, S.L.; Barbas, C.F., III; Hom, Y.L.; Caothien, R.H.; Graff, R.; DeGraw, J.; Pyati, J.; LaPolla, R.; Burton, D.R.; Lerner, R.A. Human combinatorial antibody libraries to hepatitis B surface antigen. Proc. Natl. Acad. Sci. USA 1992, 89, 3175–3179. [Google Scholar]
- Cai, X.; Garen, A. Anti-melanoma antibodies from melanoma patients immunized with genetically modified autologous tumor cells: selection of specific antibodies from single-chain Fv fusion phage libraries. Proc. Natl. Acad. Sci. USA 1995, 92, 6537–6541. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar]
- Muyldermans, S.; Atarhouch, T.; Saldanha, J.; Barbosa, J.A.; Hamers, R. Sequence and structure of VH domain from naturally occurring camel heavy chain immunoglobulins lacking light chains. Protein Eng. 1994, 7, 1129–1135. [Google Scholar] [CrossRef]
- Muyldermans, S.; Lauwereys, M. Unique single-domain antigen binding fragments derived from naturally occurring camel heavy-chain antibodies. J. Mol. Recognit. 1999, 12, 131–140. [Google Scholar] [CrossRef]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar]
- Sheets, M.D.; Amersdorfer, P.; Finnern, R.; Sargent, P.; Lindquist, E.; Schier, R.; Hemingsen, G.; Wong, C.; Gerhart, J.C.; Marks, J.D. Efficient construction of a large nonimmune phage antibody library: the production of high-affinity human single-chain antibodies to protein antigens. Proc. Natl. Acad. Sci. USA 1998, 95, 6157–6162. [Google Scholar]
- De Haard, H.J. Construction of large naive Fab libraries. Methods Mol. Biol. 2002, 178, 87–100. [Google Scholar]
- Hoogenboom, H.R.; Winter, G. By-passing immunisation. Human antibodies from synthetic repertoires of germline VH gene segments rearranged in vitro. J. Mol. Biol. 1992, 227, 381–388. [Google Scholar] [CrossRef]
- Schoonbroodt, S.; Steukers, M.; Viswanathan, M.; Frans, N.; Timmermans, M.; Wehnert, A.; Nguyen, M.; Ladner, R.C.; Hoet, R.M. Engineering antibody heavy chain CDR3 to create a phage display Fab library rich in antibodies that bind charged carbohydrates. J. Immunol. 2008, 181, 6213–6221. [Google Scholar]
- Knappik, A.; Ge, L.; Honegger, A.; Pack, P.; Fischer, M.; Wellnhofer, G.; Hoess, A.; Wolle, J.; Plückthun, A.; Virnekas, B. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J. Mol. Biol. 2000, 296, 57–86. [Google Scholar] [CrossRef]
- Rauchenberger, R.; Borges, E.; Thomassen-Wolf, E.; Rom, E.; Adar, R.; Yaniv, Y.; Malka, M.; Chumakov, I.; Kotzer, S.; Resnitzky, D.; Knappik, A.; Reiffert, S.; Prassler, J.; Jury, K.; Waldherr, D.; Bauer, S.; Kretzschmar, T.; Yayon, A.; Rothe, C. Human combinatorial Fab library yielding specific and functional antibodies against the human fibroblast growth factor receptor 3. J. Biol. Chem. 2003, 278, 38194–38205. [Google Scholar]
- Steipe, B.; Schiller, B.; Plückthun, A.; Steinbacher, S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J. Mol. Biol. 1994, 240, 188–192. [Google Scholar] [CrossRef]
- Krebs, B.; Rauchenberger, R.; Reiffert, S.; Rothe, C.; Tesar, M.; Thomassen, E.; Cao, M.; Dreier, T.; Fischer, D.; Hoss, A.; Inge, L.; Knappik, A.; Marget, M.; Pack, P.; Meng, X.Q.; Schier, R.; Sohlemann, P.; Winter, J.; Wolle, J.; Kretzschmar, T. High-throughput generation and engineering of recombinant human antibodies. J. Immunol. Methods 2001, 254, 67–84. [Google Scholar] [CrossRef]
- Virnekas, B.; Ge, L.; Plückthun, A.; Schneider, K.C.; Wellnhofer, G.; Moroney, S.E. Trinucleotide phosphoramidites: ideal reagents for the synthesis of mixed oligonucleotides for random mutagenesis. Nucleic Acids Res. 1994, 22, 5600–5607. [Google Scholar] [CrossRef]
- Sidhu, S.S.; Li, B.; Chen, Y.; Fellouse, F.A.; Eigenbrot, C.; Fuh, G. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J. Mol. Biol. 2004, 338, 299–310. [Google Scholar] [CrossRef]
- Fellouse, F.A.; Wiesmann, C.; Sidhu, S.S. Synthetic antibodies from a four-amino-acid code: a dominant role for tyrosine in antigen recognition. Proc. Natl. Acad. Sci. USA 2004, 101, 12467–12472. [Google Scholar]
- Fellouse, F.A.; Li, B.; Compaan, D.M.; Peden, A.A.; Hymowitz, S.G.; Sidhu, S.S. Molecular recognition by a binary code. J. Mol. Biol. 2005, 348, 1153–1162. [Google Scholar] [CrossRef]
- Fellouse, F.A.; Barthelemy, P.A.; Kelley, R.F.; Sidhu, S.S. Tyrosine plays a dominant functional role in the paratope of a synthetic antibody derived from a four amino acid code. J. Mol. Biol. 2006, 357, 100–114. [Google Scholar] [CrossRef]
- Tanha, J.; Xu, P.; Chen, Z.; Ni, F.; Kaplan, H.; Narang, S.A.; MacKenzie, C.R. Optimal design features of camelized human single-domain antibody libraries. J. Biol. Chem. 2001, 276, 24774–24780. [Google Scholar]
- Reiter, Y.; Schuck, P.; Boyd, L.F.; Plaksin, D. An antibody single-domain phage display library of a native heavy chain variable region: isolation of functional single-domain VH molecules with a unique interface. J. Mol. Biol. 1999, 290, 685–698. [Google Scholar] [CrossRef]
- Jespers, L.; Schon, O.; James, L.C.; Veprintsev, D.; Winter, G. Crystal structure of HEL4, a soluble, refoldable human V(H) single domain with a germ-line scaffold. J. Mol. Biol. 2004, 337, 893–903. [Google Scholar] [CrossRef]
- Jespers, L.; Schon, O.; Famm, K.; Winter, G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat. Biotechnol. 2004, 22, 1161–1165. [Google Scholar]
- Thie, H.; Voedisch, B.; Dübel, S.; Hust, M.; Schirrmann, T. Affinity maturation by phage display. Methods Mol. Biol. 2009, 525, 309–322. [Google Scholar] [CrossRef]
- Cadwell, R.C.; Joyce, G.F. Mutagenic PCR. PCR Methods Appl. 1994, 3, S136–S140. [Google Scholar] [CrossRef]
- Coia, G.; Hudson, P.J.; Irving, R.A. Protein affinity maturation in vivo using E. coli mutator cells. J. Immunol. Methods 2001, 251, 187–193. [Google Scholar] [CrossRef]
- Low, N.M.; Holliger, P.H.; Winter, G. Mimicking somatic hypermutation: affinity maturation of antibodies displayed on bacteriophage using a bacterial mutator strain. J. Mol. Biol. 1996, 260, 359–368. [Google Scholar] [CrossRef]
- Hawkins, R.E.; Russell, S.J.; Winter, G. Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J. Mol. Biol. 1992, 226, 889–896. [Google Scholar] [CrossRef]
- Chodorge, M.; Fourage, L.; Ravot, G.; Jermutus, L.; Minter, R. In vitro DNA recombination by L-Shuffling during ribosome display affinity maturation of an anti-Fas antibody increases the population of improved variants. Protein Eng. Des. Sel. 2008, 21, 343–351. [Google Scholar] [CrossRef]
- Dufner, P.; Jermutus, L.; Minter, R.R. Harnessing phage and ribosome display for antibody optimisation. Trends Biotechnol. 2006, 24, 523–529. [Google Scholar] [CrossRef]
- Schwesinger, F.; Ros, R.; Strunz, T.; Anselmetti, D.; Guntherodt, H.J.; Honegger, A.; Jermutus, L.; Tiefenauer, L.; Plückthun, A. Unbinding forces of single antibody-antigen complexes correlate with their thermal dissociation rates. Proc. Natl. Acad. Sci. USA 2000, 97, 9972–9977. [Google Scholar]
- Lu, D.; Shen, J.; Vil, M.D.; Zhang, H.; Jimenez, X.; Bohlen, P.; Witte, L.; Zhu, Z. Tailoring in vitro selection for a picomolar affinity human antibody directed against vascular endothelial growth factor receptor 2 for enhanced neutralizing activity. J. Biol. Chem. 2003, 278, 43496–43507. [Google Scholar]
- Schier, R.; Bye, J.; Apell, G.; McCall, A.; Adams, G.P.; Malmqvist, M.; Weiner, L.M.; Marks, J.D. Isolation of high-affinity monomeric human anti-c-erbB-2 single chain Fv using affinity-driven selection. J. Mol. Biol. 1996, 255, 28–43. [Google Scholar] [CrossRef]
- Stemmer, W.P. DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 10747–10751. [Google Scholar] [CrossRef]
- Barbas, C.F., III; Hu, D.; Dunlop, N.; Sawyer, L.; Cababa, D.; Hendry, R.M.; Nara, P.L.; Burton, D.R. In vitro evolution of a neutralizing human antibody to human immunodeficiency virus type 1 to enhance affinity and broaden strain cross-reactivity. Proc. Natl. Acad. Sci. USA 1994, 91, 3809–3813. [Google Scholar]
- Yang, W.P.; Green, K.; Pinz-Sweeney, S.; Briones, A.T.; Burton, D.R.; Barbas, C.F., III. CDR walking mutagenesis for the affinity maturation of a potent human anti-HIV-1 antibody into the picomolar range. J. Mol. Biol. 1995, 254, 392–403. [Google Scholar] [CrossRef]
- Prassler, J.; Steidl, S.; Urlinger, S. In vitro affinity maturation of HuCAL antibodies: complementarity determining region exchange and RapMAT technology. Immunotherapy 2009, 1, 571–583. [Google Scholar]
- Steidl, S.; Ratsch, O.; Brocks, B.; Durr, M.; Thomassen-Wolf, E. In vitro affinity maturation of human GM-CSF antibodies by targeted CDR-diversification. Mol. Immunol. 2008, 46, 135–144. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ponsel, D.; Neugebauer, J.; Ladetzki-Baehs, K.; Tissot, K. High Affinity, Developability and Functional Size: The Holy Grail of Combinatorial Antibody Library Generation. Molecules 2011, 16, 3675-3700. https://doi.org/10.3390/molecules16053675
Ponsel D, Neugebauer J, Ladetzki-Baehs K, Tissot K. High Affinity, Developability and Functional Size: The Holy Grail of Combinatorial Antibody Library Generation. Molecules. 2011; 16(5):3675-3700. https://doi.org/10.3390/molecules16053675
Chicago/Turabian StylePonsel, Dirk, Julia Neugebauer, Kathrin Ladetzki-Baehs, and Kathrin Tissot. 2011. "High Affinity, Developability and Functional Size: The Holy Grail of Combinatorial Antibody Library Generation" Molecules 16, no. 5: 3675-3700. https://doi.org/10.3390/molecules16053675
APA StylePonsel, D., Neugebauer, J., Ladetzki-Baehs, K., & Tissot, K. (2011). High Affinity, Developability and Functional Size: The Holy Grail of Combinatorial Antibody Library Generation. Molecules, 16(5), 3675-3700. https://doi.org/10.3390/molecules16053675