Synthesis of ent-Kaurane Diterpene Monoglycosides

Abstract
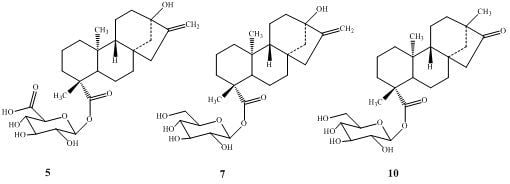
:
1. Introduction
2. Results and Discussion
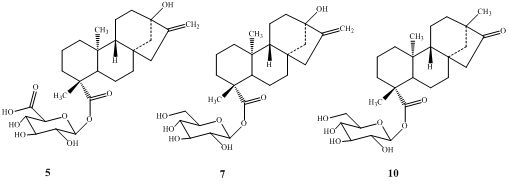
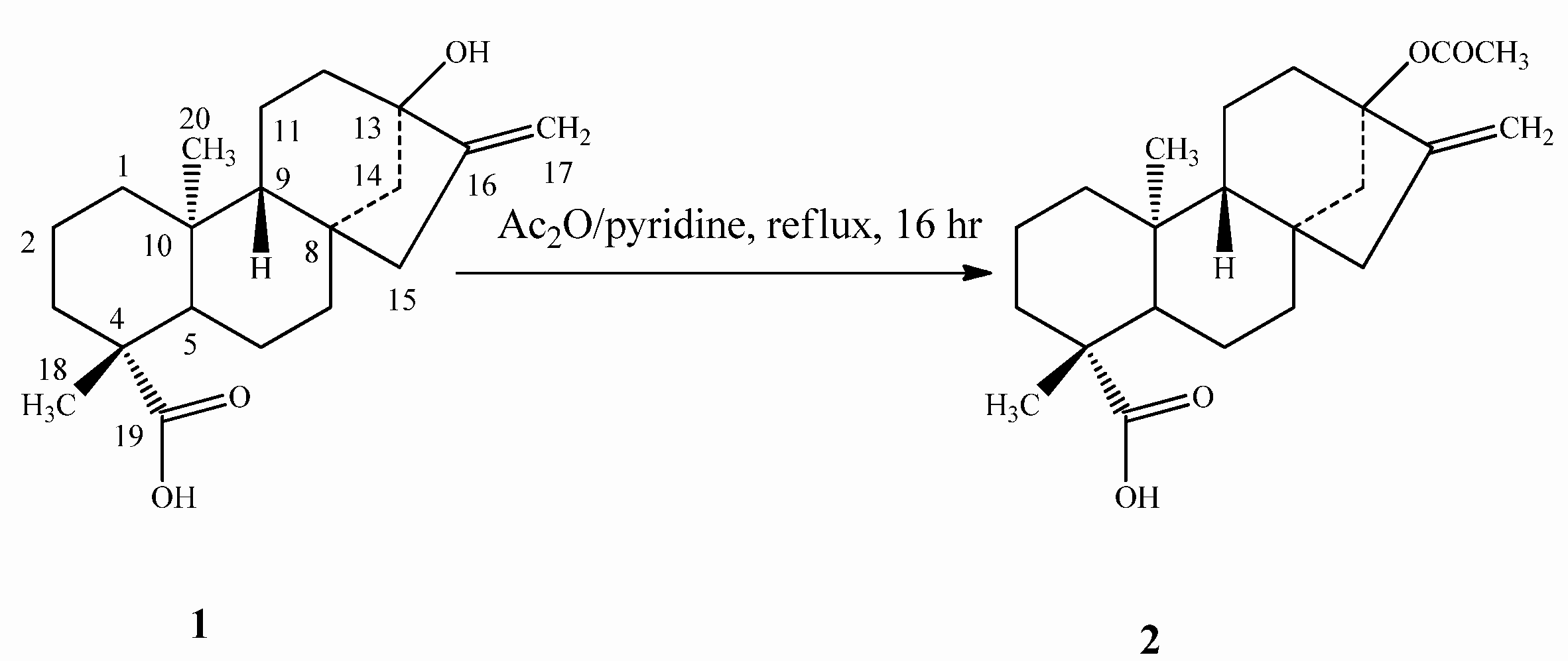
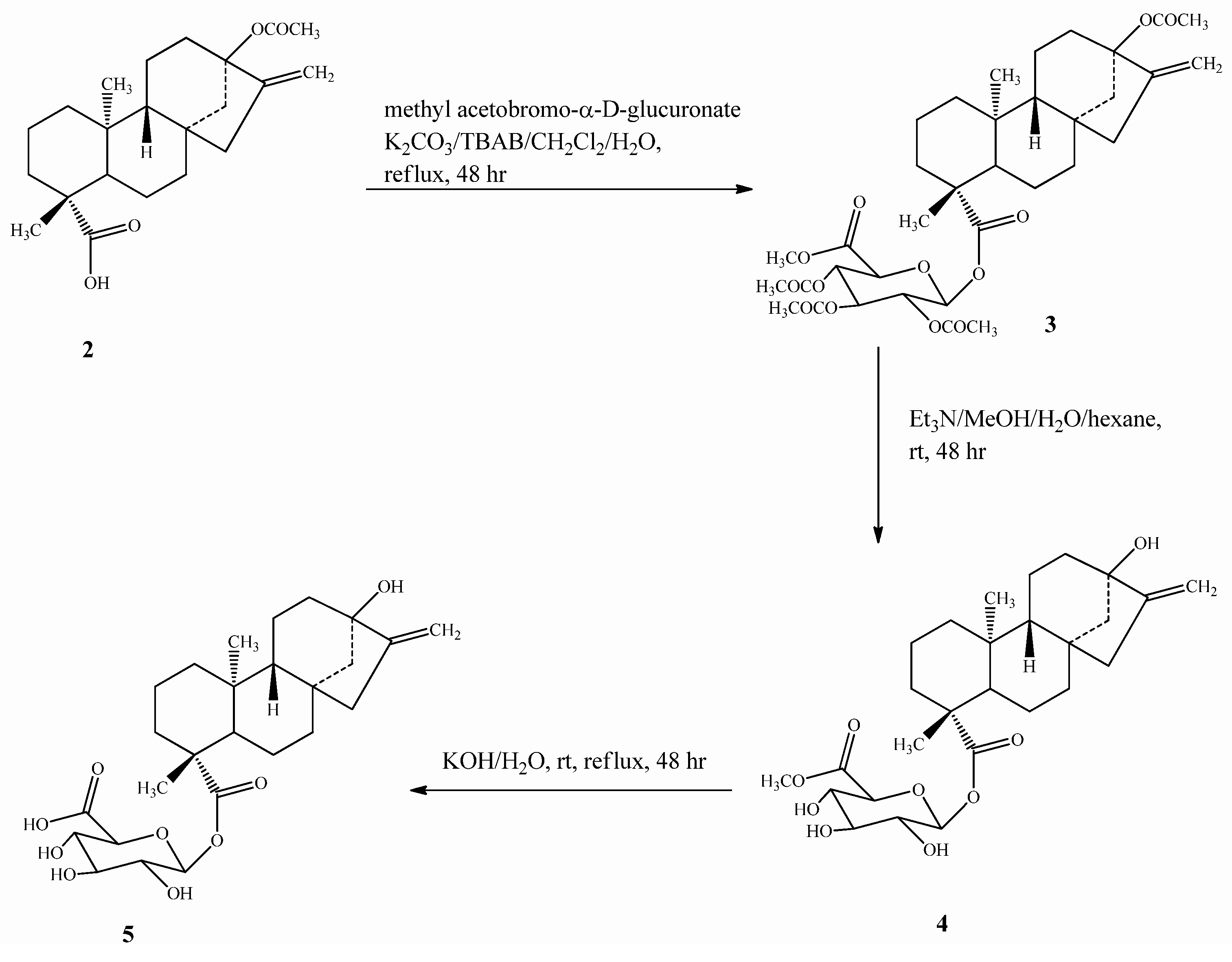
2.1. Chemistry
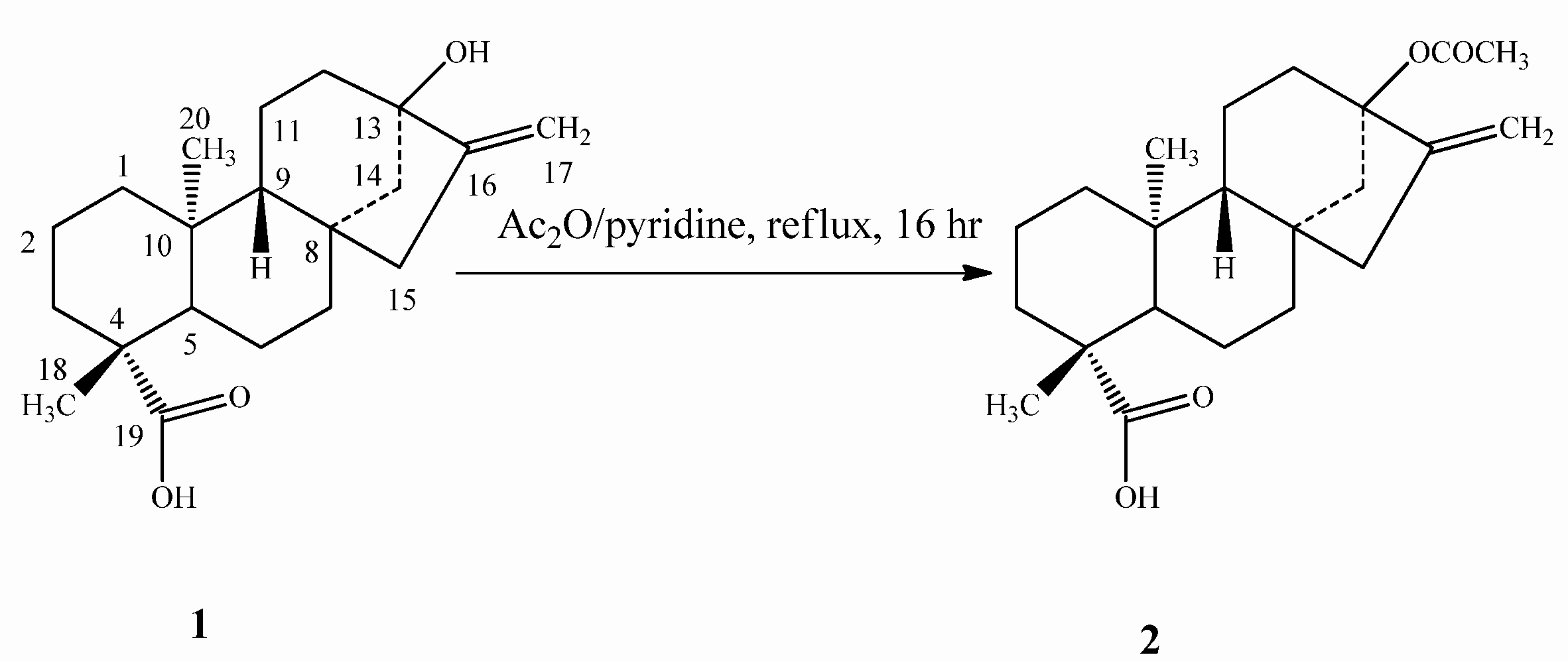
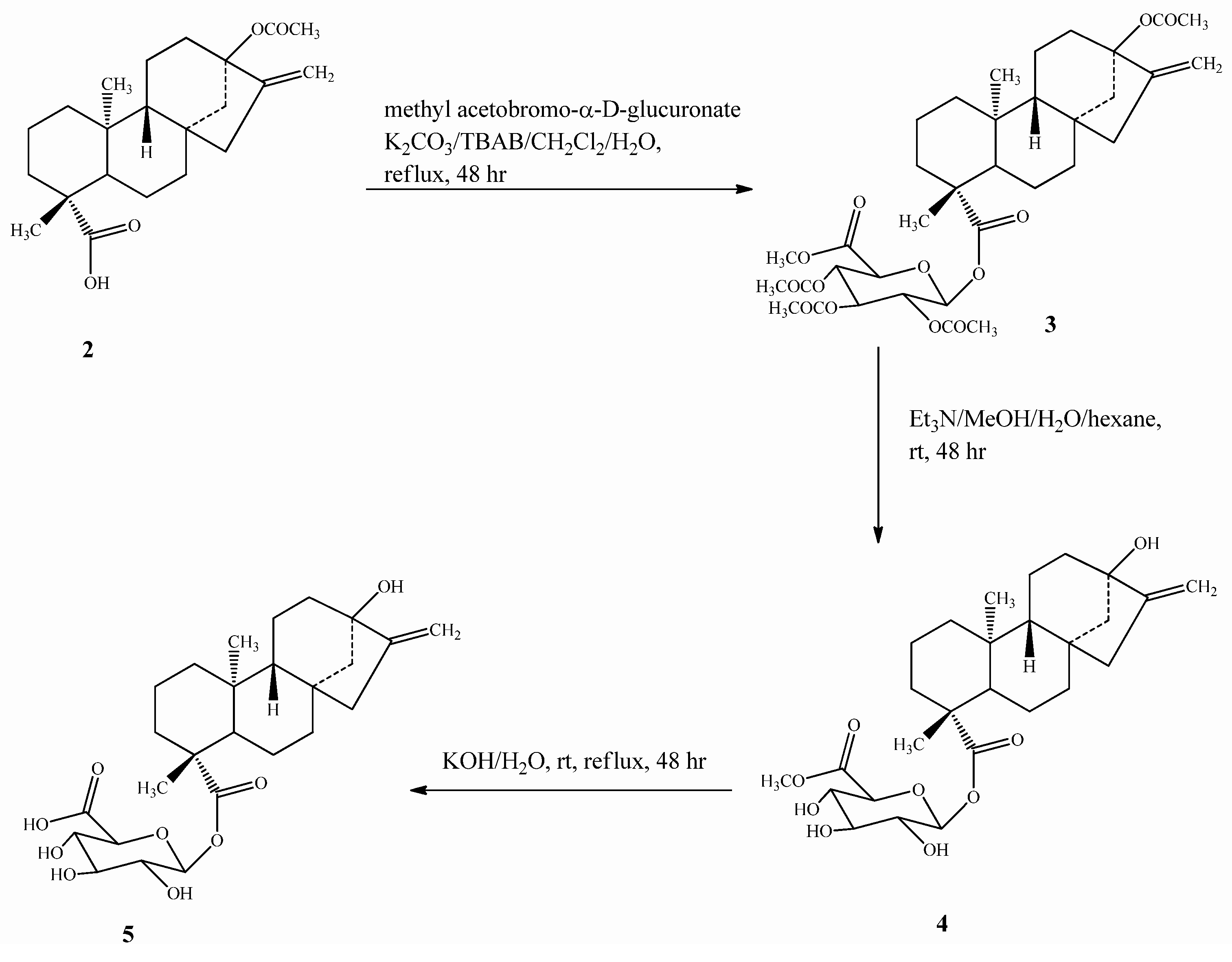
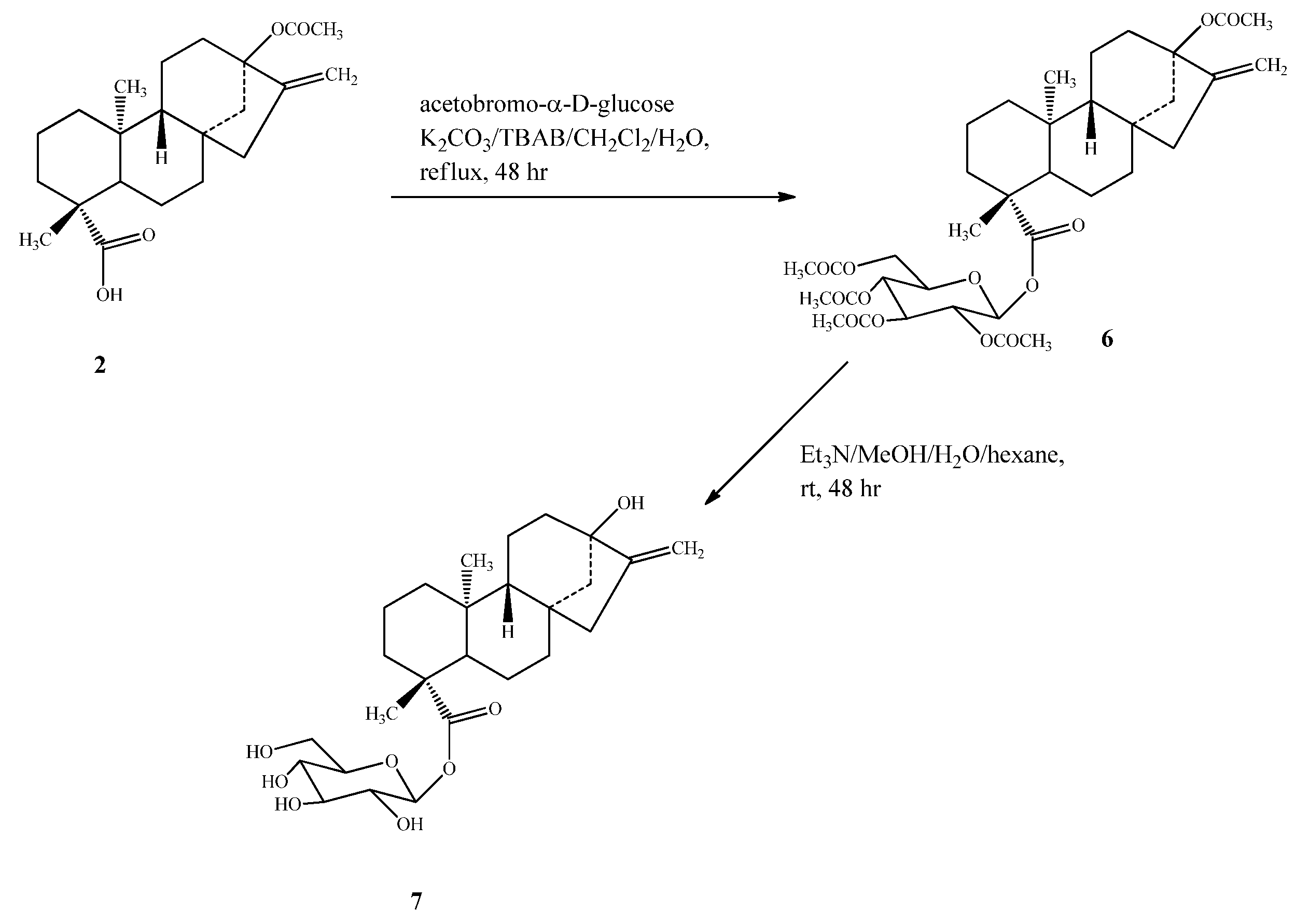
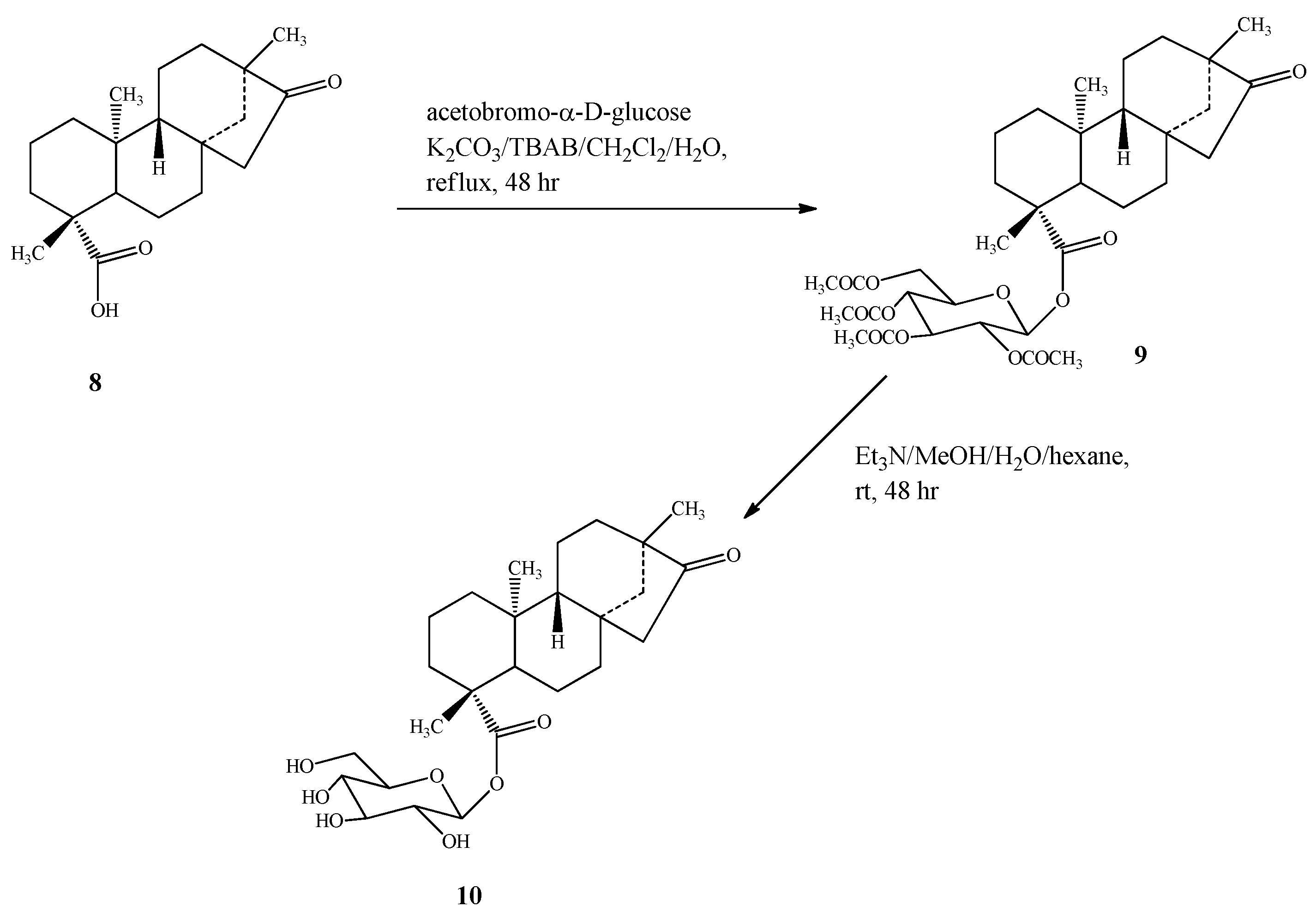
2.2. Spectroscopy

{kind=link}
{kind=link}
{kind=link}
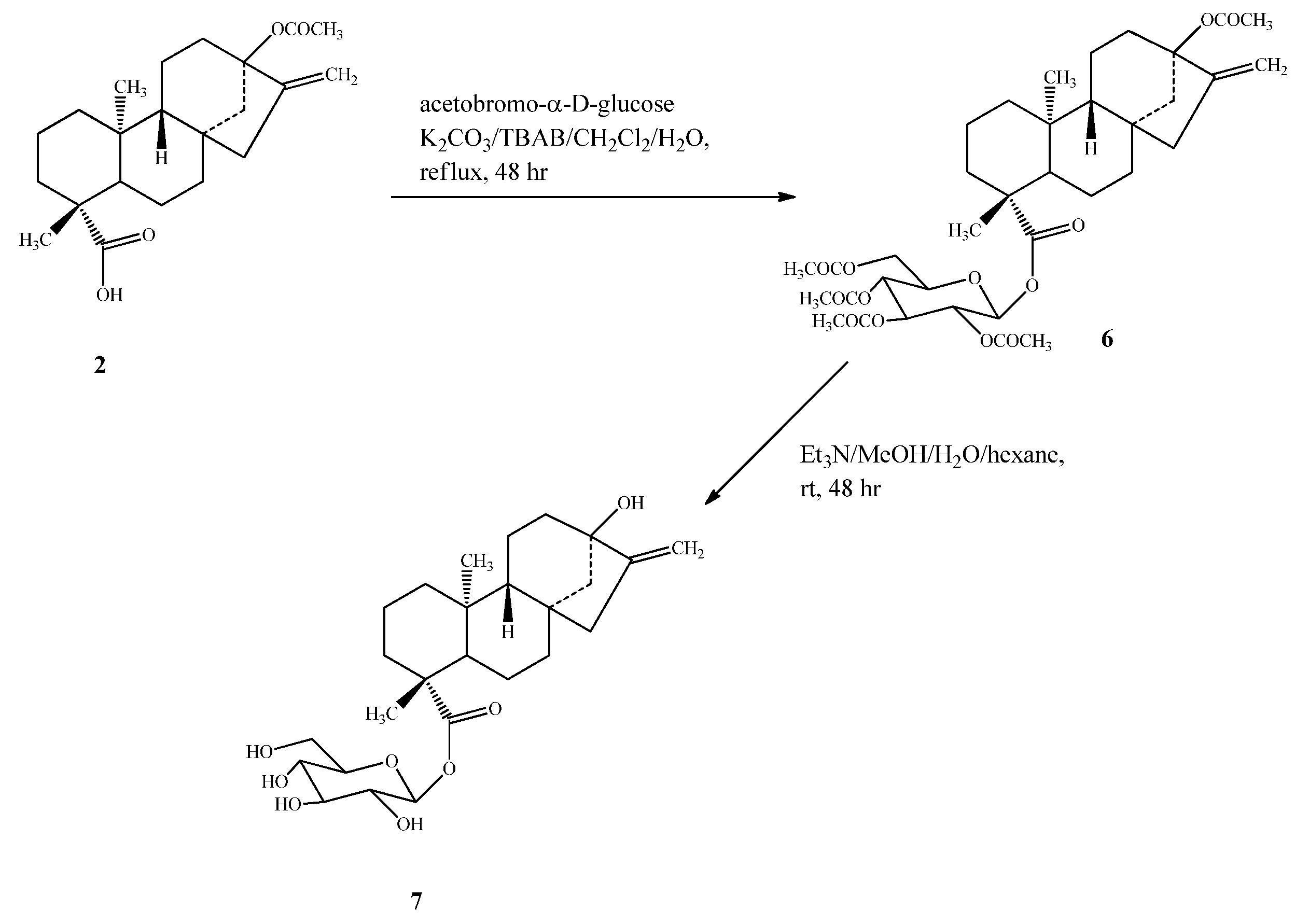{kind=link}
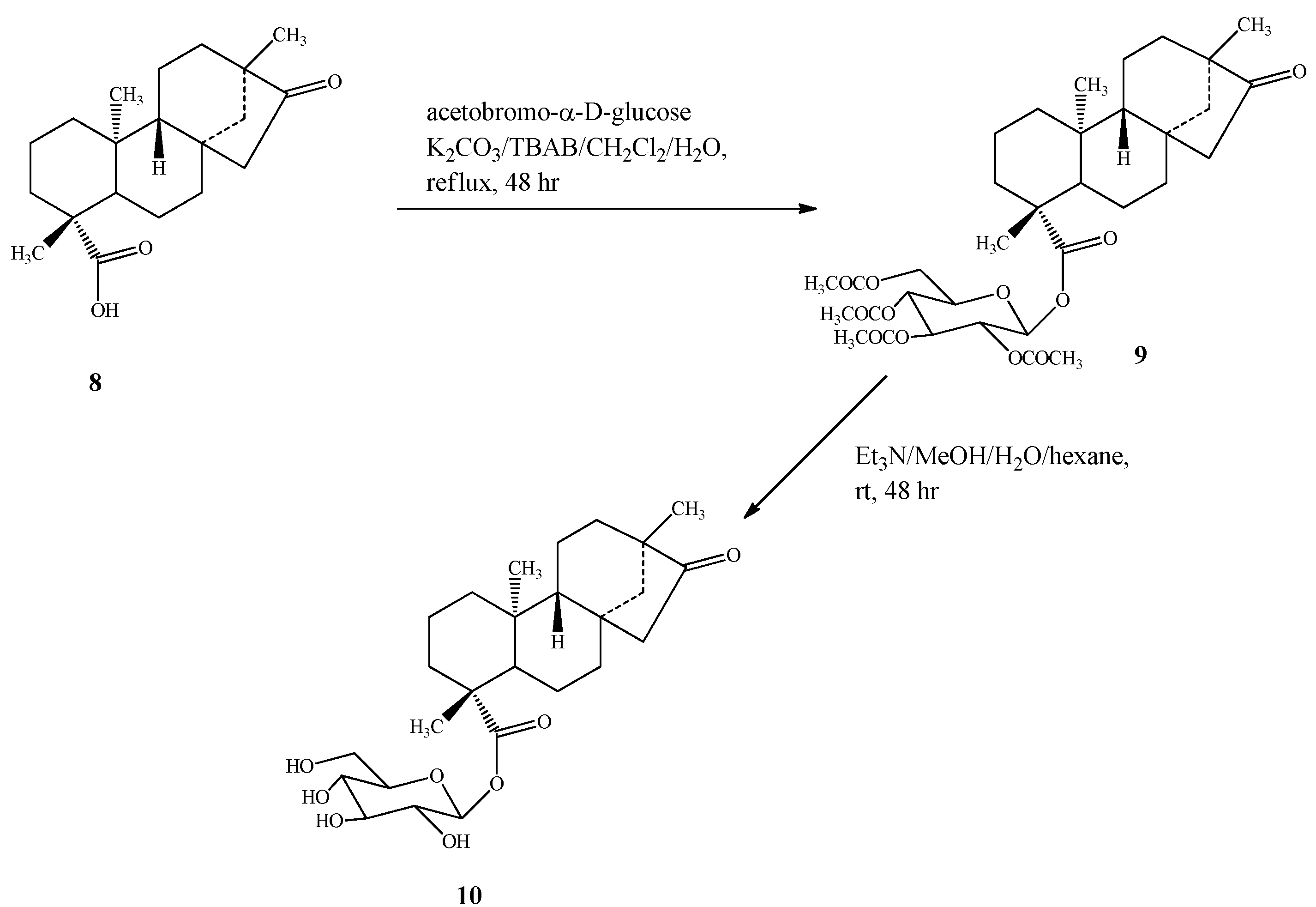{kind=link}
| Position | 5 | 7 | 10 |
|---|---|---|---|
| 1 | 0.86 (m, 1H), 1.86 (m, 1H) | 0.86 (m, 1H), 1.88 (m, 1H) | 0.96 (m, 1H), 1.68 (m, 1H) |
| 2 | 1.39 (m, 1H), 1.90 (m, 1H) | 1.43 (m, 1H),1.93 (m, 1H) | 1.36 (m, 1H), 1.92 (m, 1H) |
| 3 | 1.02 (m, 1H), 2.26 (d, 11.9, 1H) | 1.06 (m, 1H), 2.19 (d, 12.4, 1H) | 1.05 (m, 1H), 2.16 (d, 13.2, 1H) |
| 5 | 1.08 (m, 1H) | 1.12 (m, 1H) | 1.20 (m, 1H) |
| 6 | 1.82 (m, 1H), 1.93 (m, 1H) | 1.85 (m, 1H), 1.96 (m, 1H) | 1.86 (m, 2H) |
| 7 | 1.45 (m, 1H), 1.56 (m, 1H) | 1.44 (m, 1H), 1.54 (m, 1H) | 1.47 (m, 1H), 1.66 (m, 1H) |
| 9 | 0.88 (m, 1H) | 0.96 (m, 1H) | 1.24 (m, 1H) |
| 11 | 1.60 (m, 1H), 1.79 (m, 1H) | 1.61 (m, 1H), 1.77 (m, 1H) | 1.25 (m, 1H), 1.70 (m, 1H) |
| 12 | 1.53 (m, 1H), 1.87 (m, 1H) | 1.46 (m, 1H), 1.74 (m, 1H) | 1.42 (m, 1H), 1.56 (m, 1H) |
| 14 | 1.58 (m, 1H), 2.23 (d, 12.1, 1H) | 1.28 (m, 1H), 2.11 (m, 1H) | 1.44 (m, 1H), 1.58 (m, 1H) |
| 15 | 2.02 (m, 1H), 2.16 (d, J = 17.4, 1H) | 2.06 (m, 1H), 2.17 (m, 1H) | 1.80 (m, 1H), 2.65 (dd, 3.1, 17.2, 1H) |
| 17 | 4.59 (s, 1H), 4.78 (br s, 1H) | 4.77 (br s, 1H), 4.93 (s, 1H) | |
| 18 | 1.23 (s, 3H) | 1.20 (s, 3H) | 1.24 (s, 3H) |
| 20 | 0.97 (s, 3H) | 0.98 (s, 3H) | 0.93 (s, 3H) |
| 1′ | 5.44 (d, 7.8, 1H) | 5.41 (d, J = 8.2 Hz, 1H) | 5.38 (d, 8.2, 1H) |
| 2′ | 3.60 (dd, 8.2, 9.1, 1H) | 3.35 (dd, 7.1, 7.6, 1H) | 3.32 (dd, 7.2, 7.8, 1H) |
| 3′ | 3.42 (dd, 8.1, 8.9, 1H) | 3.44 (dd, 8.3, 9.1, 1H) | 3.43 (dd, 8.1, 9.1, 1H) |
| 4′ | 3.49 (dd, 8.1, 9.2, 1H) | 3.34 (dd, 8.2, 9.4, 1H) | 3.34 (dd, 8.1, 9.1, 1H) |
| 5′ | 3.72 (d, 8.2, 1H) | 3.39 (ddd, 8.1, 2.1, 7.4, 1H) | 3.37 (ddd, 8.2, 1.9, 7.2, 1H) |
| 6′ | 3.66 (dd, 2.1, 12.1, 1H), 3.81 (dd, 4.2, 12.1, 1H) | 3.68 (dd, 1.9, 12.1, 1H), 3.82 (dd, 3.9, 12.1, 1H) |
| Position | 1 | 2 | 3 |
|---|---|---|---|
| 1 | 42.0 | 41.5 | 39.5 |
| 2 | 20.3 | 20.2 | 19.1 |
| 3 | 39.2 | 38.6 | 38.0 |
| 4 | 45.2 | 45.1 | 43.9 |
| 5 | 58.7 | 58.5 | 57.6 |
| 6 | 23.1 | 22.8 | 20.7 |
| 7 | 42.8 | 42.4 | 41.4 |
| 8 | 42.9 | 43.0 | 48.4 |
| 9 | 55.5 | 55.0 | 53.9 |
| 10 | 40.9 | 40.5 | 37.6 |
| 11 | 21.5 | 21.3 | 21.1 |
| 12 | 40.7 | 40.4 | 38.6 |
| 13 | 81.0 | 88.5 | 39.1 |
| 14 | 47.4 | 47.2 | 54.6 |
| 15 | 48.8 | 48.6 | 48.3 |
| 16 | 157.2 | 157.0 | 223.8 |
| 17 | 103.5 | 103.4 | |
| 18 | 29.1 | 29.1 | 27.6 |
| 19 | 178.2 | 178.0 | 176.8 |
| 20 | 16.5 | 16.3 | 19.1 |
| 1′ | 95.6 | 95.6 | 95.5 |
| 2′ | 78.7 | 74.0 | 73.8 |
| 3′ | 74.0 | 77.7 | 77.3 |
| 4′ | 73.6 | 71.0 | 70.8 |
| 5′ | 77.7 | 77.8 | 77.6 |
| 6′ | 177.5 | 61.2 | 61.1 |
3. Experimental
3.1. General
3.2. Isolation
4. Conclusions
Acknowledgements
- Samples Availability: Samples of the three synthesized steviol glycosides 5 , 7 , and 10 are available from the authors.
References
- Brandle, J.E.; Starrratt, A.N.; Gijen, M. Stevia rebaudiana: Its agricultural, biological and chemical properties. Can. J. Plant Sci. 1998, 78, 527–536. [Google Scholar] [CrossRef]
- Chaturvedula, V.S.P.; Rhea, J.; Milanowski, D.; Mocek, U.; Prakash, I. Two minor diterpene glycosides from the leaves of Stevia rebaudiana. Nat. Prod. Commun. 2011, 6, 175–178. [Google Scholar]
- Chaturvedula, V.S.P.; Mani, U.; Prakash, I. Diterpene glycosides from Stevia rebaudiana. Molecules 2011, 16, 3552–3562. [Google Scholar] [CrossRef]
- Chaturvedula, V.S.P.; Prakash, I. A new diterpenoid glycoside from Stevia rebaudiana. Molecules 2011, 16, 2937–2943. [Google Scholar] [CrossRef]
- Chaturvedula, V.S.P.; Prakash, I. Structures of the novel diterpene glycosides from Stevia rebaudiana. Carbohydr. Res. 2011, 346, 1057–1060. [Google Scholar] [CrossRef]
- Chaturvedula, V.S.P.; Prakash, I. Additional minor diterpene glycosides from Stevia rebaudiana. Nat. Prod. Commun. 2011, 6, 1059–1062. [Google Scholar]
- Chaturvedula, V.S.P.; Clos, J.F.; Rhea, J.; Milanowski, D.; Mocek, U.; DuBois, G.E.; Prakash, I. Minor diterpene glycosides from the leaves of Stevia rebaudiana. Phytochemistry Lett. 2011, 4, 209–212. [Google Scholar]
- Chaturvedula, V.S.P.; Mani, U.; Prakash, I. Structures of the novel α-glucosyl linked diterpene glycosides from Stevia rebaudiana. Carbohydr. Res. 2011, 346, 2034–2038. [Google Scholar] [CrossRef]
- Chaturvedula, V.S.P.; Prakash, I. Stability study of steviol glycosides in mock beverages using fluorescent light exposure under ICH guidelines. Int. J. Pharm. Pharm. Sci. 2011, 3, 316–323. [Google Scholar]
- Geuns, J.M.C.; Buyse, J.; Vankeirsblick, A.; Temme, E.H.M.; Compernolle, F.; Toppet, S. Identification of steviol glucuronide in human urine. J. Agric. Food Chem. 2008, 56, 8507–8513. [Google Scholar] [CrossRef]
- Bliard, C.; Massiot, G.; Nazabadioko, S. Glycosylation of acids under phase transfer conditions; Partial synthesis of saponins. Tetrahedron Lett. 1994, 35, 6107–6108. [Google Scholar] [CrossRef]
- Gianfagna, T.; Zeevaart, J.A.D.; Lusk, W.J. Synthesis of [2H] gibberellins from steviol using the fungus Gibberella fujikuroi. Phytochemistry 1983, 22, 427–430. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chaturvedula, V.S.P.; Klucik, J.; Upreti, M.; Prakash, I. Synthesis of ent-Kaurane Diterpene Monoglycosides. Molecules 2011, 16, 8402-8409. https://doi.org/10.3390/molecules16108402
Chaturvedula VSP, Klucik J, Upreti M, Prakash I. Synthesis of ent-Kaurane Diterpene Monoglycosides. Molecules. 2011; 16(10):8402-8409. https://doi.org/10.3390/molecules16108402
Chicago/Turabian StyleChaturvedula, Venkata Sai Prakash, Josef Klucik, Mani Upreti, and Indra Prakash. 2011. "Synthesis of ent-Kaurane Diterpene Monoglycosides" Molecules 16, no. 10: 8402-8409. https://doi.org/10.3390/molecules16108402
APA StyleChaturvedula, V. S. P., Klucik, J., Upreti, M., & Prakash, I. (2011). Synthesis of ent-Kaurane Diterpene Monoglycosides. Molecules, 16(10), 8402-8409. https://doi.org/10.3390/molecules16108402