Syntheses, Characterization, and Photo-Hydrogen-Evolving Properties of Tris(2,2'-bipyridine)ruthenium(II) Derivatives Tethered to an H2-Evolving (2-phenylpyridinato)platinum(II) Unit

Abstract
:1. Introduction
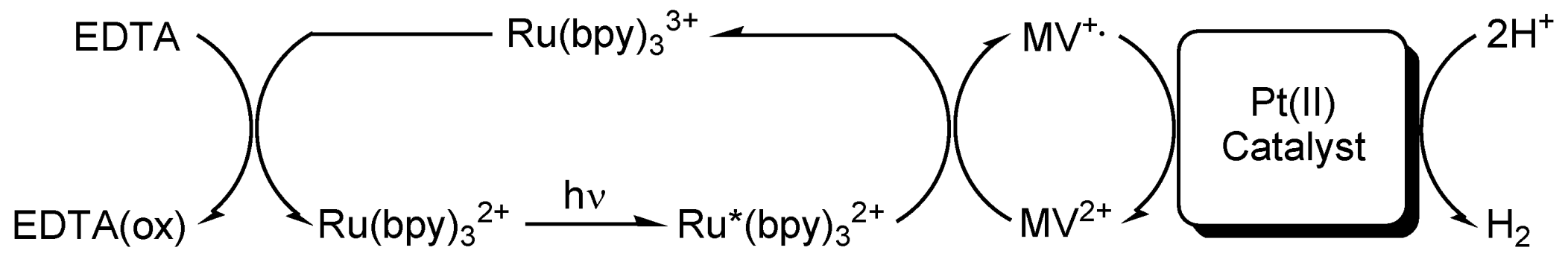
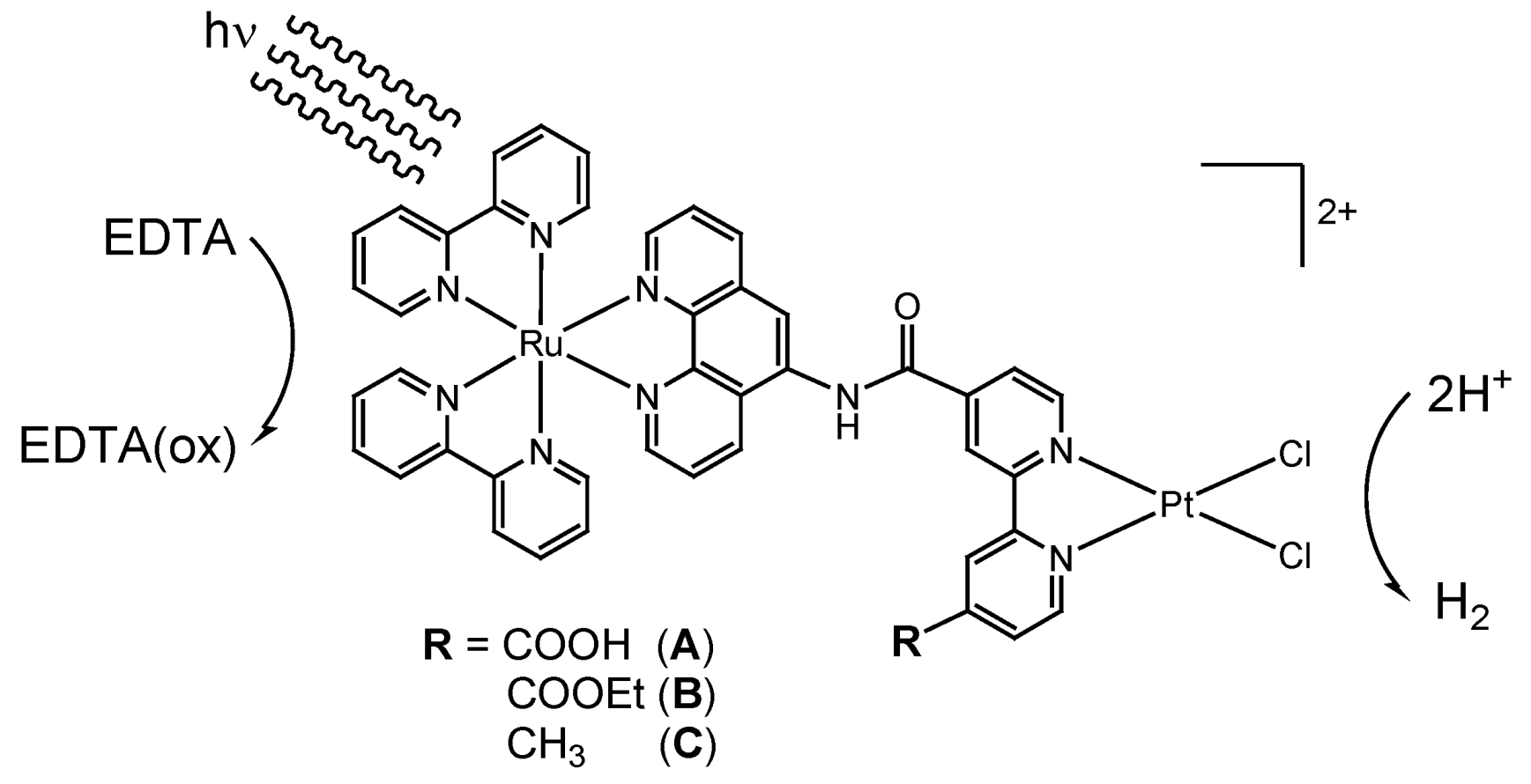
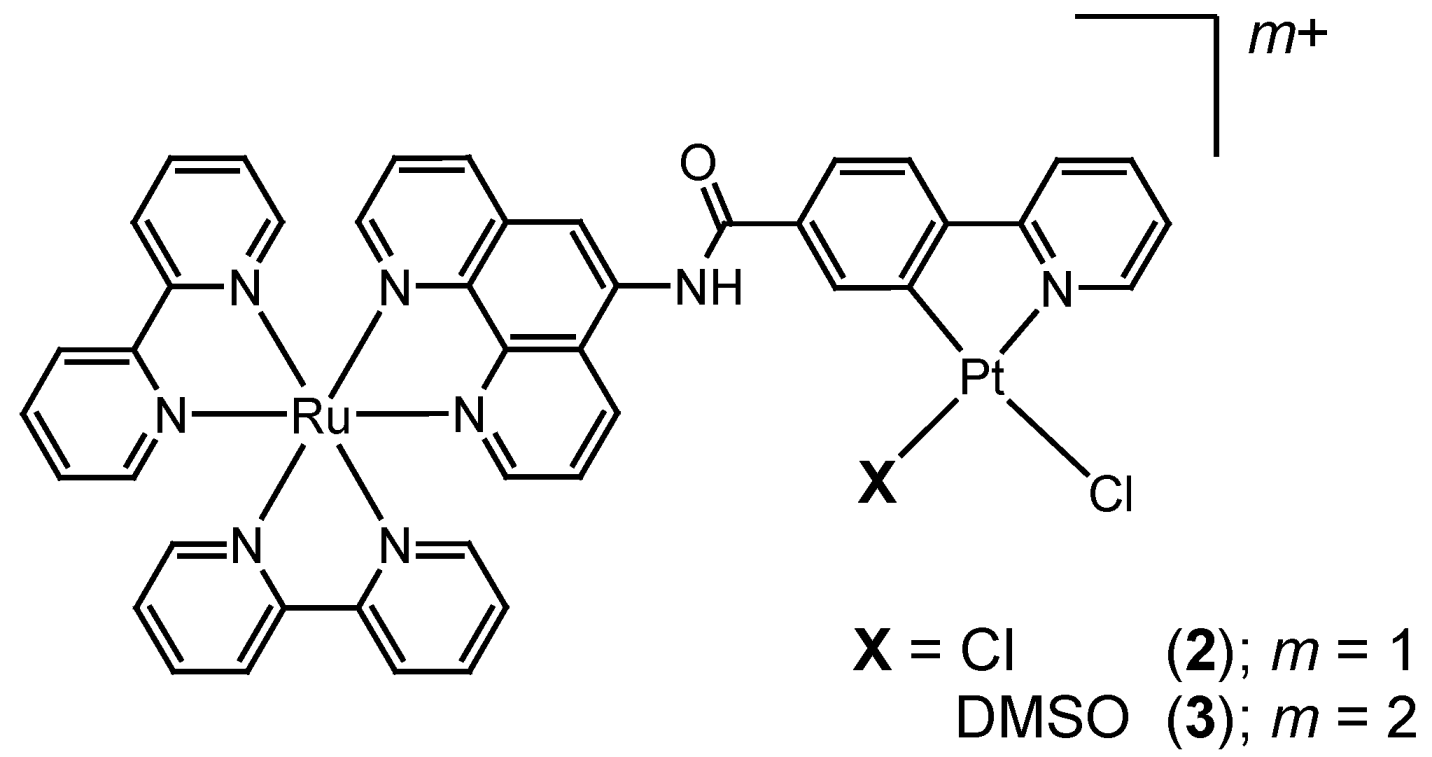
2. Results and Discussion
2.1. Syntheses

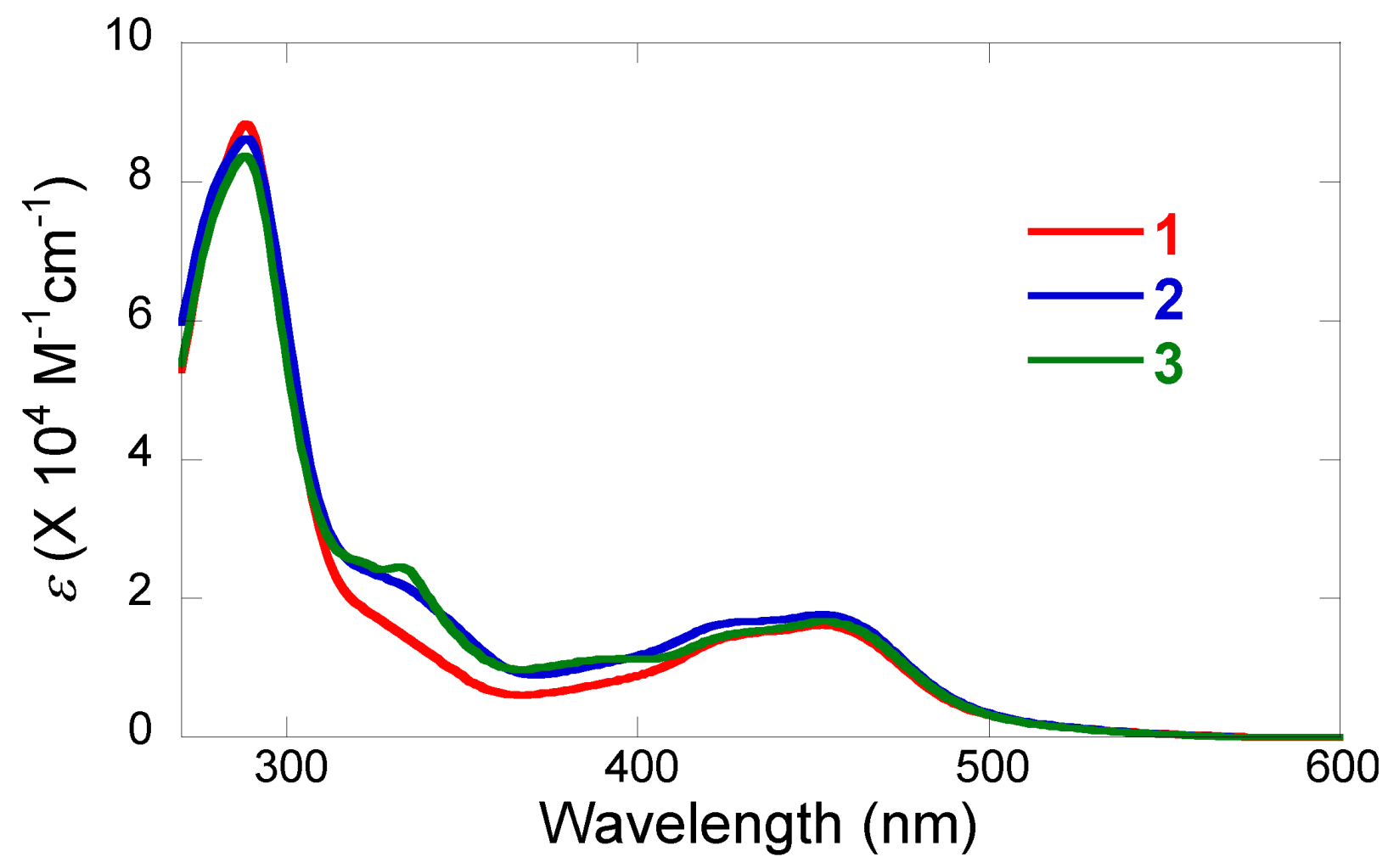
2.2. Spectroscopic study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |

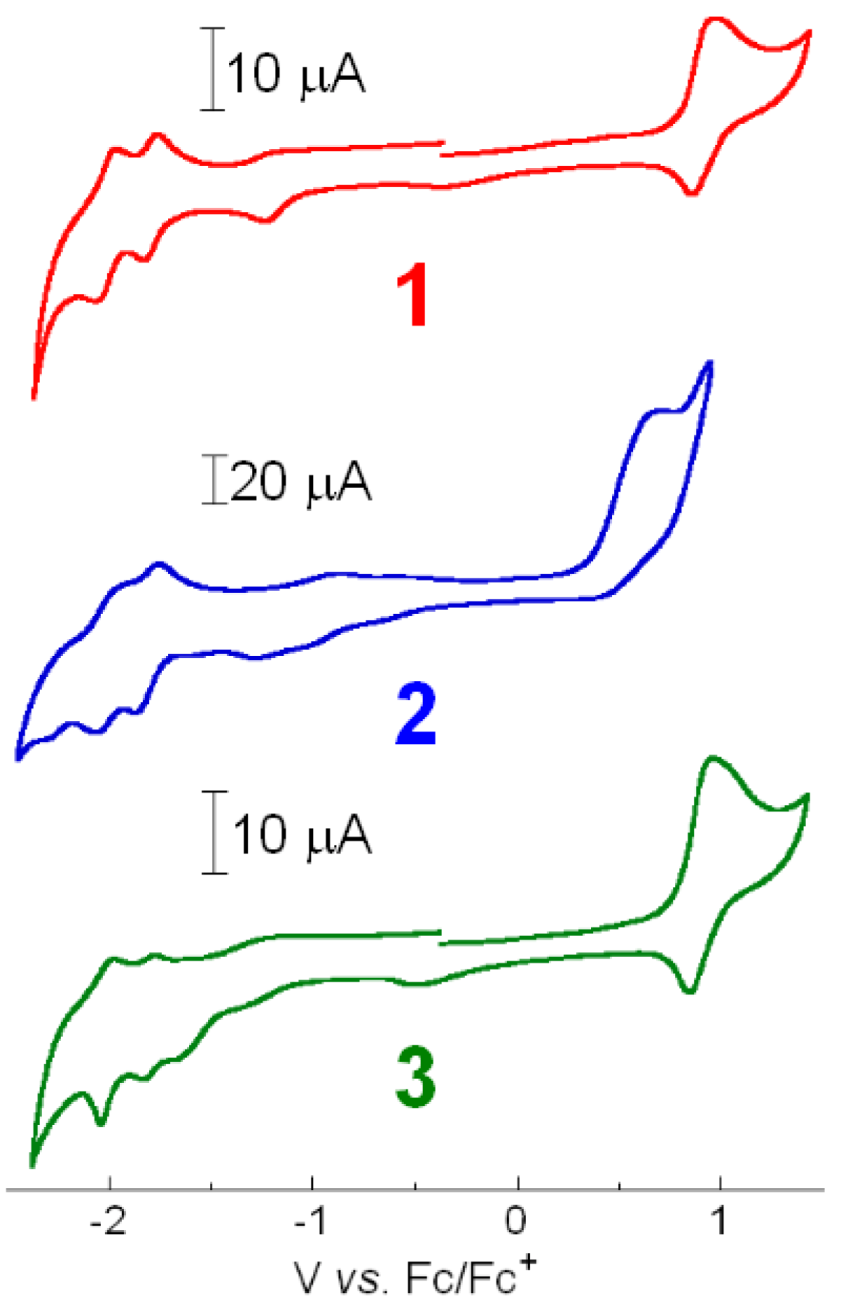
2.3. Electrochemistry
 |
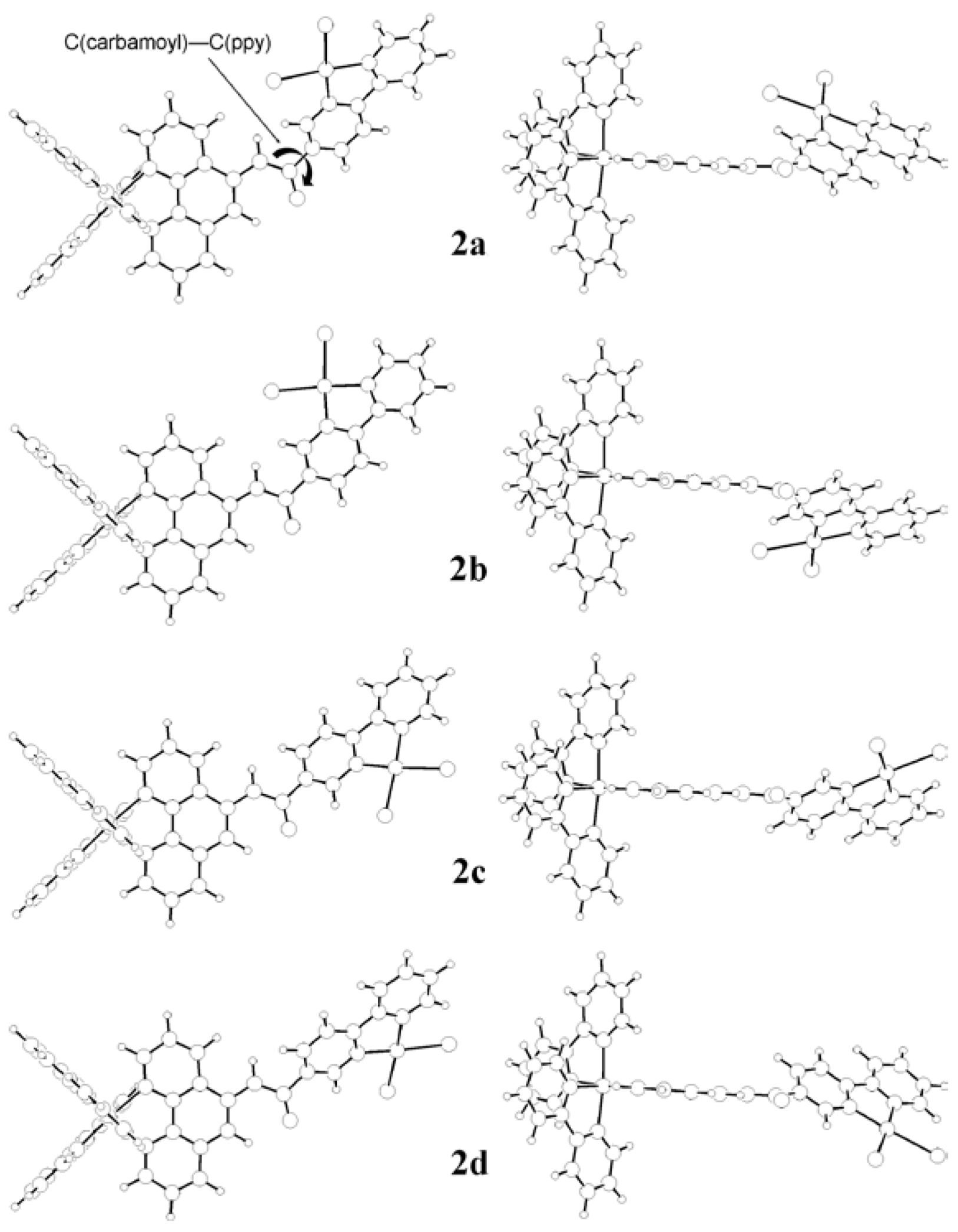
2.4. DFT studies
 |
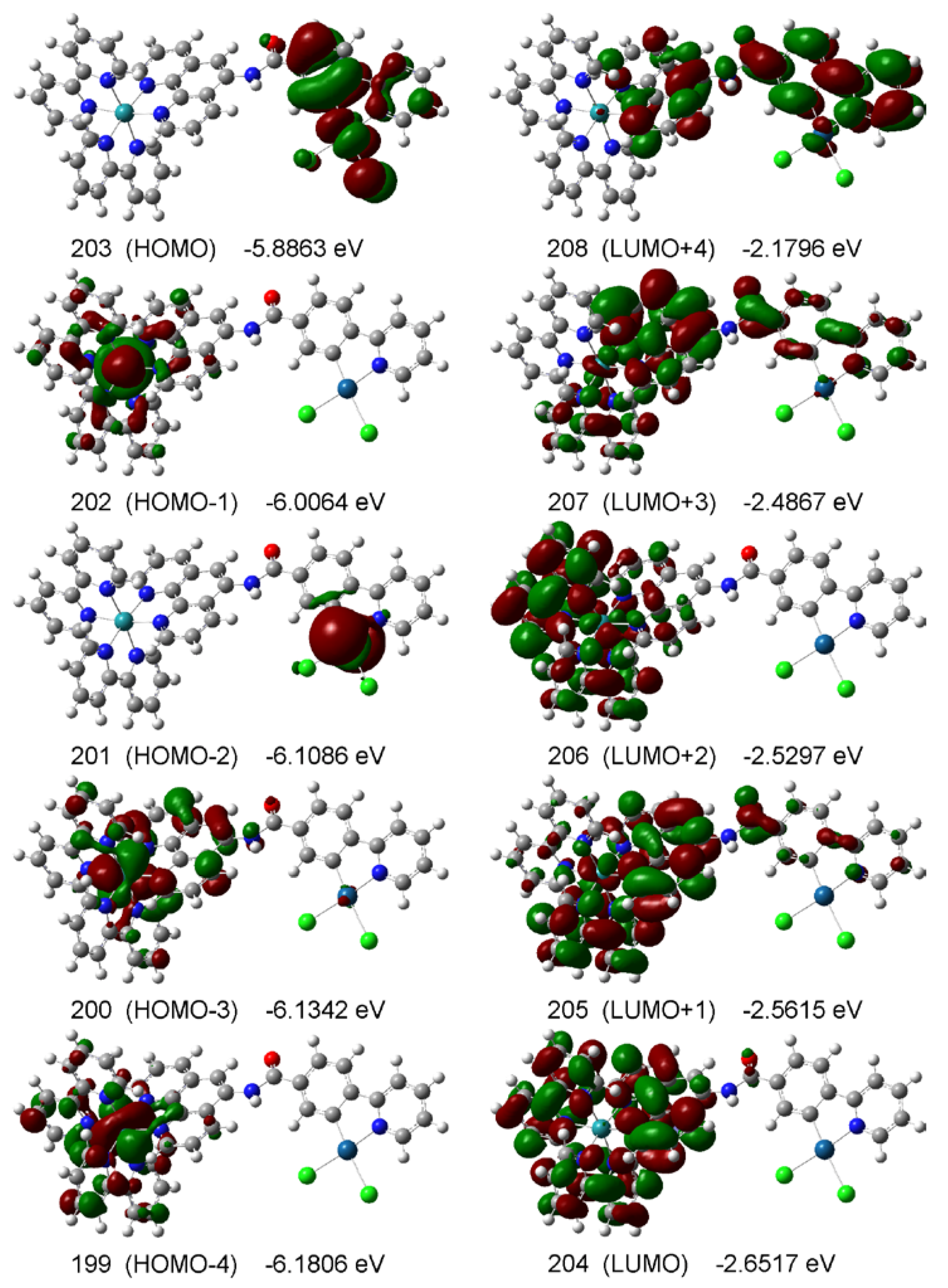
2.5. Photolysis experiments

3. Conclusions
4. Experimental
4.1. Materials and measurements
4.2. DFT calculations
4.3. Photolysis experiments
4.4. Syntheses
Acknowledgements
- Sample Availability: Samples of the compounds 1-3 are available from the authors.
References and Notes
- Lehn, J.-M.; Sauvage, J.-P. Chemical storage of light energy catalytic generation of hydrogen by visible light or sunlight. Irradiation of neutral aqueous solutions. Nouv. J. Chim. 1977, 1, 449–451. [Google Scholar]
- Kirch, M.; Lehn, J.-M.; Sauvage, J.-P. Hydrogen Generation by Visible Light Irradiation of Aqueous Solutions of Metal Complexes. An Approach to the Photochemical Conversion and Storage of Solar Energy. Helv. Chim. Acta 1979, 62, 1345–1384. [Google Scholar] [CrossRef]
- Kalyanasundaram, K.; Kiwi, J.; Grätzel, M. Hydrogen Evolution from Water by Visible Light, a Homogeneous Three Component Test System for Redox Catalysis. Helv. Chim. Acta 1978, 61, 2720–2730. [Google Scholar] [CrossRef]
- Grätzel, M. Artificial Photosynthesis: Water Cleavage into Hydrogen and Oxygen by Visible Light. Acc. Chem. Res. 1981, 14, 376–384. [Google Scholar] [CrossRef]
- Moradpour, A.; Amouyal, E.; Keller, P.; Kagan, H.B. Hydrogen production by visible light irradiation of aqueous solutions of tris(2,2'-bipyridine)ruthenium(2+). Nouv. J. Chim. 1978, 2, 547–549. [Google Scholar]
- Keller, P.; Moradpour, A.; Amouyal, E.; Kagan, H.B. Hydrogen production by visible-light using viologen-dye mediated redox cycles. Nouv. J. Chim. 1980, 4, 377–384. [Google Scholar]
- Sakai, K.; Matsumoto, K. Photochemical Reduction of Water to Hydrogen Catalyzed by Mixed-Valent Tetranuclear Platinum Complex. J. Coord. Chem. 1988, 18, 169–172. [Google Scholar] [CrossRef]
- Sakai, K.; Matsumoto, K. Homogeneous Catalysis of Platinum Blue Related Complexes in Photoreduction of Water into Hydrogen. J. Mol. Catal. 1990, 62, 1–14. [Google Scholar] [CrossRef]
- Sakai, K.; Kizaki, Y.; Tsubomura, T.; Matsumoto, K. Homogeneous Catalysis of Mixed-Valent Octanuclear Platinum Complexes in Photochemical Hydrogen Production from Water. J. Mol. Catal. 1993, 79, 141–152. [Google Scholar] [CrossRef]
- Ozawa, H.; Yokoyama, Y.; Haga, M.; Sakai, K. Syntheses, Characterization, and Photo-Hydrogen-Evolving Properties of Tris(2,2'-bipyridine)ruthenium(II) Derivatives Tethered to a cis-Pt(II)Cl2 Unit: Insights into the Structure-Activity Relationship. Dalton Trans. 2007, 1197–1206. [Google Scholar]
- Sakai, K.; Ozawa, H. Homogeneous catalysis of platinum(II) complexes in photochemical hydrogen production from water. Coord. Chem. Rev. 2007, 251, 2753–2766. [Google Scholar] [CrossRef]
- Kobayashi, M.; Masaoka, S.; Sakai, K. Synthesis, Crystal Structure, Solution and Spectroscopic Properties, and Hydrogen-Evolving Activity of [K(18-Crown-6)][Pt(II)(2-phenylpyridinato)Cl2. Photochem. Photobiol. Sci. 2009, 8, 196–203. [Google Scholar] [CrossRef]
- Ozawa, H.; Haga, M.; Sakai, K. A Photo-Hydrogen-Evolving Molecular Device Driving Visible-Light-Induced EDTA-Reduction of Water into Molecular Hydrogen. J. Am. Chem. Soc. 2006, 128, 4926–4927. [Google Scholar]
- Rau, S.; Schäfer, B.; Gleich, D.; Anders, E.; Rudolph, M.; Friedrich, M.; Görls, H.; Henry, W.; JVos, J.G. A Supramolecular Photocatalyst for the Production of Hydrogen and the Selective Hydrogenation of Tolane. Angew. Chem. Int. Ed. 2006, 45, 6215–6218. [Google Scholar]
- Rau, S.; Walther, D.; Vos, J.G. Inspired by nature: light driven organometallic catalysis by heterooligonuclear Ru(II) complexes. Dalton Trans. 2007, 915–919. [Google Scholar]
- Elvington, M.; Brewer, K.J. Photoinitiated Electron Collection at a Metal in a Rhodium-Centered Mixed-Metal Supramolecular Complex. Inorg. Chem. 2006, 45, 5242–5244. [Google Scholar] [CrossRef]
- Elvington, M.; Brown, J.; Arachchige, S.M.; Brewer, K.J. Photocatalytic Hydrogen Production from Water Employing A Ru, Rh, Ru Molecular Device for Photoinitiated Electron Collection. J. Am. Chem. Soc. 2007, 129, 10644–10645. [Google Scholar]
- Fihri, A.; Artero, V.; Razavet, M.; Baffert, C.; Leibl, W.; Fontecave, M. Cobaloxime-Based Photocatalytic Devices for Hydrogen Production. Angew. Chem. Int. Ed. 2008, 47, 564–567. [Google Scholar]
- Fihri, A.; Artero, V.; Pereira, A.; Fontecave, M. Efficient H2-producing photocatalytic systems based on cyclometalated iridium- and tricarbonylrhenium-diimine photosensitizers and cobaloxime catalysts. Dalton Trans. 2008, 5567–5569. [Google Scholar]
- Arachchige, S.M.; Brown, J.R.; Chang, E.; Jain, A.; Zigler, D.F.; Rangan, K.; Brewer, K.J. Design Considerations for a System for Photocatalytic Hydrogen Production from Water Employing Mixed-Metal Photochemical Molecular Devices for Photoinitiated Electron Collection. Inorg. Chem. 2009, 48, 1989–2000. [Google Scholar]
- Wang, M.; Na, Y.; Gorlov, M.; Sun, L. Light-driven hydrogen production catalysed by transition metal complexes in homogeneous systems. Dalton Trans. 2009, 6458–6467. [Google Scholar]
- Ozawa, H.; Kobayashi, M.; Balan, B.; Masaoka, S.; Sakai, K. Photo-Hydrogen-Evolving Molecular Catalysts Consisting of Polypyridyl Ruthenium(II) Photosensitizers and Platinum(II) Catalysts: Insights into the Reaction Mechanism. Chem. Asian J. 2010. [Google Scholar] [CrossRef]
- Ozawa, H.; Sakai, K. An Effect of Structural Modification in the Photo-hydrogen-evolving RuIIPtII Dimers. Chem. Lett. 2007, 36, 920–921. [Google Scholar] [CrossRef]
- Masaoka, S.; Mukawa, Y.; Sakai, K. Frontier Orbital Engineering of Photo-Hydrogen-Evolving Molecular Devices: A Clear Relationship Between the H2-Evolving Activity and the Energy Level of the LUMO. Dalton Trans. 2010, 39, 5868–5876. [Google Scholar] [CrossRef]
- Kobayashi, M.; Masaoka, S.; Sakai, K. N-(1,10-phenanthrolin-5-yl)-4-(pyridin-2-yl)benzamide monohydrate. Acta Cryst. 2008, E64, o1979. [Google Scholar]
- Kobayashi, M.; Masaoka, S.; Sakai, K. Chrolo(dimethylsulfoxide)(phenylpyridinato)platinum(II). Acta Cryst. 2008, E64, m1557. [Google Scholar]
- Craig, C.A.; Garces, F.O.; Watts, R.J.; Palmans, R.; Frank, A.J. Luminescence properties of two new Pt(II)-2-phenylpyridine complexes; the influence of metal-carbon bonds. Coord. Chem. Rev. 1990, 97, 193–208. [Google Scholar]
- Kvam, P.-I.; Puzyk, M.V.; Balashev, K.P.; Songstad, J. Spectroscopic and Electrochemical Properties of Some Mixed-Ligand Cyclometalated Platinum(II) Complexes Derived from 2-Phenylpyridine. Acta Chem. Scand. 1995, 49, 335–343. [Google Scholar] [CrossRef]
- Rehm, D.; Weller, A. Kinetik und Mechanismus der Elektronumbertragung bei der Fluoreszenzloschung in Acetonitril. Ber. Bunsen-Ges. Phys. Chem. 1969, 73, 834–839. [Google Scholar]
- Islam, A.; Sugihara, H.; Singh, L.P.; Hara, K.; Katoh, R.; Nagawa, Y.; Yanagida, M.; Takahashi, Y.; Murata, S.; Arakawa, H. Synthesis and photophysical properties of ruthenium(II) charge transfer sensitizers containing 4,4'-dicarboxy-2,2'-biquinoline and 5,8-dicarboxy-6,7-dihydro-dibenzo[1,10]-phenanthroline. Inorg. Chim. Acta 2001, 322, 7–16. [Google Scholar] [CrossRef]
- Huber, A.; Muller, L.; Elias, H.; Klement, R.; Valko, M. Cobalt(II) Complexes with Substituted Salen-Type Ligands and Their Dioxygen Affinity in N,N-Dimethylformamide at Various Temperatures. Eur. J. Inorg. Chem. 2005, 1459–1467. [Google Scholar]
- Yamauchi, K.; Masaoka, S.; Sakai, K. Evidence for Pt(II)-Based Molecular Catalysis in the Thermal Reduction of Water into Molecular Hydrogen. J. Am. Chem. Soc. 2009, 131, 8404–8406. [Google Scholar] [CrossRef]
- Sullivan, B.P.; Salmon, D.J.; Meyer, T.J. Mixed Phosphine 2,2'-Bipyridine Complexes of Ruthenium. Inorg. Chem. 1978, 17, 3334–3341. [Google Scholar] [CrossRef]
- Price, J.H.; Williamson, A.N.; Schramm, R.F.; Wayland, B.B. Palladium(II) and Platinum(II) Alkyl Sulfoxide Complexes. Examples of Sulfur-Bonded, Mixed Sulfur- and Oxygen-Bonded, and Totally Oxygen-Bonded Complexes. Inorg. Chem. 1972, 11, 1280–1284. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M. W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian 03; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Dunning, T.H.; Hay, P.J. In Modern Theoretical Chemistry; Schaefer, H.F., III, Ed.; Plenum: New York, NY, USA, 1976; pp. 1–28. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Cossi, M.; Scalmani, G.; Rega, N.; Barone, V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kobayashi, M.; Masaoka, S.; Sakai, K. Syntheses, Characterization, and Photo-Hydrogen-Evolving Properties of Tris(2,2'-bipyridine)ruthenium(II) Derivatives Tethered to an H2-Evolving (2-phenylpyridinato)platinum(II) Unit. Molecules 2010, 15, 4908-4923. https://doi.org/10.3390/molecules15074908
Kobayashi M, Masaoka S, Sakai K. Syntheses, Characterization, and Photo-Hydrogen-Evolving Properties of Tris(2,2'-bipyridine)ruthenium(II) Derivatives Tethered to an H2-Evolving (2-phenylpyridinato)platinum(II) Unit. Molecules. 2010; 15(7):4908-4923. https://doi.org/10.3390/molecules15074908
Chicago/Turabian StyleKobayashi, Masayuki, Shigeyuki Masaoka, and Ken Sakai. 2010. "Syntheses, Characterization, and Photo-Hydrogen-Evolving Properties of Tris(2,2'-bipyridine)ruthenium(II) Derivatives Tethered to an H2-Evolving (2-phenylpyridinato)platinum(II) Unit" Molecules 15, no. 7: 4908-4923. https://doi.org/10.3390/molecules15074908
APA StyleKobayashi, M., Masaoka, S., & Sakai, K. (2010). Syntheses, Characterization, and Photo-Hydrogen-Evolving Properties of Tris(2,2'-bipyridine)ruthenium(II) Derivatives Tethered to an H2-Evolving (2-phenylpyridinato)platinum(II) Unit. Molecules, 15(7), 4908-4923. https://doi.org/10.3390/molecules15074908