Oak (Quercus frainetto Ten.) Honeydew Honey—Approach to Screening of Volatile Organic Composition and Antioxidant Capacity (DPPH and FRAP Assay)


Abstract
:1. Introduction
2. Results and Discussion
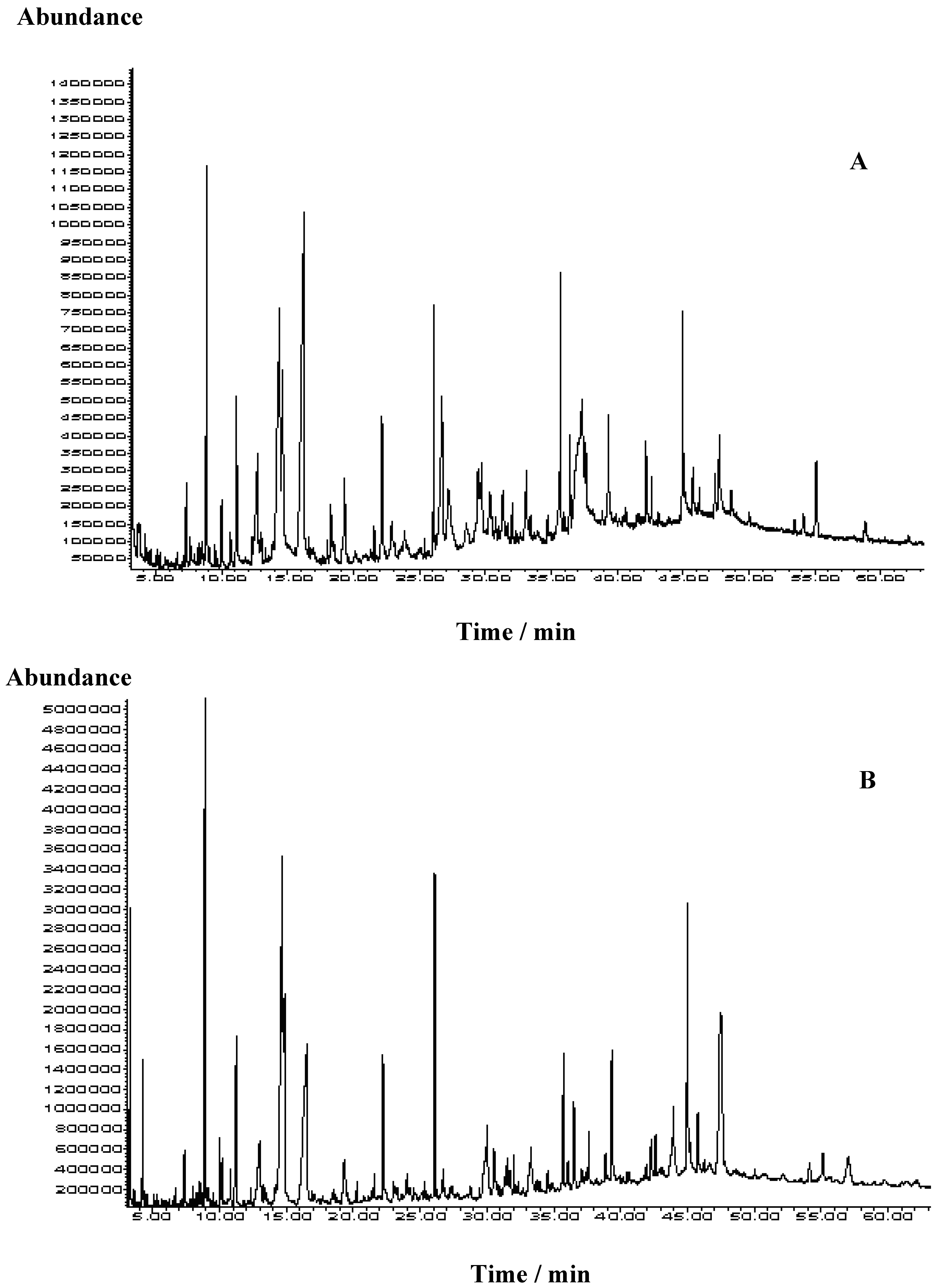
2.1. Volatiles Isolated by Headspace Solid-Phase Microextraction

{kind=link}
{kind=link}
| No. | Compound | RI | Area percentage (%) | |
|---|---|---|---|---|
| sample I | sample II | |||
| 1. | Pentan-1-ola | < 900 | 0.5 | 0.9 |
| 2. | 2-Methylbutan-1-ol | < 900 | 0.7 | - |
| 3. | 2-Furancarboxaldehyde | < 900 | 2.0 | - |
| 4. | 3-Methylbutanoic acid (Isovaleric acid) | < 900 | 0.8 | - |
| 5. | 2-Furanmethanol | < 900 | 1.3 | 5.2 |
| 6. | 1,3-Dimethylbenzene** | < 900 | 0.4 | - |
| 7. | 1-(2-Furanyl)-ethanone | 914 | 1.1 | - |
| 8. | 5-Methylfurfural | 969 | 8.4 | 3.5 |
| 9. | Hexanoic acida | 974 | 0.7 | 2.2 |
| 10. | 2-Ethyl-1,3-dimethylbenzene* | 1032 | 0.7 | - |
| 11. | 2-Ethylhexan-1-ol* | 1035 | 1.1 | - |
| 12. | Benzyl alcohola | 1037 | 2.4 | 4.8 |
| 13. | Phenylacetaldehydea | 1048 | 2.0 | 1.5 |
| 14. | trans-Linalool oxide (furan type) | 1076 | 18.1 | 13.8 |
| 15. | Methyl 2-furoate | 1084 | - | 2.8 |
| 16. | cis-Linalool oxide (furan type) | 1091 | 10.8 | 14.0 |
| 17. | Hotrienol | 1106 | 4.4 | 9.7 |
| 18. | 2-Phenylethanola | 1116 | 3.5 | 6.0 |
| 19. | 3,5,5-Trimethyl-cyclohex-2-en-1-one (α-Isophorone) | 1124 | 0.6 | - |
| 20. | 2-Ethylhexanoic acid | 1140 | 2.9 | - |
| 21. | Neroloxide | 1162 | 2.1 | - |
| 22. | Epoxylinalool | 1178 | 4.4 | 2.0 |
| 23. | Octanoic acida | 1190 | 8.5 | 9.4 |
| 24. | Lilac alcohol (isomer I) | 1208 | 0.8 | - |
| 25. | Lilac alcohol (isomer II) | 1237 | 1.3 | - |
| 26. | Ethyl phenylacetate | 1253 | 1.2 | - |
| 27. | Nonanoic acida | 1273 | 2.8 | 4.7 |
| 28. | Methyl cinnamate | 1312 | 1.1 | - |
| 29. | Decanoic acida | 1370 | 1.5 | 2.4 |
| 30. | Hexadecanoic acida | 1963 | 3.7 | 3.8 |
| Total identified | 89.8% | 86.7% | ||
2.2. Volatiles Isolated by Ultrasonic Solvent Extraction
| Area percentage (%) | ||||||
|---|---|---|---|---|---|---|
| No. | Compound | RI | sample I | sample II | ||
| A | B | A | B | |||
| 1. | 3-Methylbutanoic acid | < 900 | 0.1 | - | - | - |
| 2. | 2-Furanmethanol | < 900 | 1.7 | 0.3 | 3.0 | 3.7 |
| 3. | 1,3-Dimethylbenzene** | < 900 | 0.1 | - | - | 0.2 |
| 4. | 3-Hydroxy-pentan-2-one | < 900 | 0.2 | - | 0.5 | - |
| 5. | 1-(2-Furanyl)-ethanone | 914 | 0.3 | - | - | - |
| 6. | Hexane-2,5-dione | 931 | 0.2 | 0.1 | - | - |
| 7. | 5-Methylfurfural | 969 | 1.5 | 0.5 | 0.1 | 0.1 |
| 8. | Hexanoic acida | 974 | 0.1 | - | - | - |
| 9. | 2-Formylpyrrole | 1018 | 0.2 | - | 0.1 | 0.4 |
| 10. | p-Cymenea | 1031 | 0.1 | - | 0.2 | - |
| 11. | 2-Hydroxy-3-methyl-cyclopent-2-en-1-one | 1034 | 0.3 | 0.1 | - | 0.1 |
| 12. | Benzyl alcohola | 1037 | 1.2 | - | 1.4 | 0.6 |
| 13. | Pantoic lactone | 1046 | - | 0.5 | - | 0.8 |
| 14. | Phenylacetaldehydea | 1048 | 0.1 | - | - | - |
| 15. | 2-Acetylpyrrole | 1063 | 0.2 | 0.1 | 0.2 | 0.1 |
| 16. | trans-Linalool oxide | 1076 | 0.7 | - | 0.2 | - |
| 17. | 4,5-Dimethyl-2-formylfuran | 1078 | 0.4 | - | - | - |
| 18. | 6-Methyl-2-pyrazinylmethanol | 1084 | - | 0.3 | - | 0.2 |
| 19. | 1-(2-Furyl)-2-hydroxyethanone | 1087 | 5.5 | 4.6 | 5.1 | 8.8 |
| 20. | cis-Linalool oxide | 1091 | 0.4 | - | 0.2 | - |
| 21. | Linaloola | 1101 | 0.3 | - | 0.2 | - |
| 22. | 2-Phenylethanola | 1116 | 0.7 | - | 0.9 | - |
| 23. | 3-Hydroxy-2-methyl-4H-Pyran-4-one (Maltol) | 1119 | - | 0.5 | - | 1.3 |
| 24. | 2-Formyl-1-methylpyrrole | 1139 | 1.1 | 0.5 | 0.3 | 0.8 |
| 25. | 2,3-Dihydro-3,5-dihydroxy-6-methyl-4H-pyran-4-one | 1145 | 1.3 | 0.3 | 2.8 | 4.3 |
| 26. | Benzoic acida | 1162 | 4.5 | 1.8 | 4.3 | 3.5 |
| 27. | 3,5-Dimethylphenol** | 1181 | 0.3 | 0.1 | 0.4 | 0.2 |
| 28. | 3,7-Dimethyl-octa-1,5-dien-3,7-diol | 1191 | 0.4 | 0.1 | 0.5 | 0.8 |
| 29. | Dodecanea | 1200 | 0.2 | - | 0.1 | 0.5 |
| 30. | 1,2-Benzenediol | 1219 | 0.3 | - | 0.2 | - |
| 31. | 2,5-Di(hydroxymethyl)-furan* | 1229 | 2.2 | - | 10.9 | 17.2 |
| 32. | 5-Hydroxymethylfurfural | 1230 | 42.5 | 64.2 | 4.7 | 6.2 |
| 33. | Phenylacetic acida | 1269 | 8.1 | 4.7 | 16.4 | 11.6 |
| 34. | Nonanoic acida | 1273 | - | - | - | 0.1 |
| 35. | 2-Hydroxybenzoic acid | 1311 | 0.5 | 0.2 | 1.6 | - |
| 36. | 3-Methoxyacetophenone | 1321 | - | - | 0.3 | - |
| 37. | 3-Hydroxy-4-phenyl-butan-2-one | 1354 | - | 0.5 | 1.4 | 0.8 |
| 38. | Tetradecanea | 1400 | 0.3 | 0.1 | 0.4 | 0.4 |
| 39. | 4-Hydroxybenzyl alcohol | 1426 | 0.4 | 0.2 | 0.6 | 0.8 |
| 40. | Cinnamic acid | 1434 | 0.2 | 0.2 | - | 0.2 |
| 41. | 8-Hydroxyoctanoic acid | 1465 | 0.4 | 0.3 | 0.4 | 0.6 |
| 42. | Pentadecanea | 1500 | - | - | 0.2 | - |
| 43. | 4-Methyl-2,6-bis(1,1-dimethylethyl)-phenol | 1514 | 1.4 | - | 3.6 | 5.7 |
| 44. | 4-Hydroxybenzoic acid | 1558 | 1.7 | - | 2.9 | - |
| 45. | Vanillic acid | 1566 | 0.6 | 0.1 | 0.4 | 0.2 |
| 46. | Hexadecane | 1600 | 0.8 | - | 1.5 | 0.1 |
| 47. | 3-Oxo-α-ionol | 1656 | 0.3 | 0.2 | - | - |
| 48. | Homovanillic acid | 1659 | - | - | - | 0.5 |
| 49. | Syringaldehyde | 1662 | 0.1 | 0.1 | - | 0.4 |
| 50. | 8-Quinolinol | 1713 | 0.2 | 1.2 | - | - |
| 51. | Methyl syringatea | 1744 | 1.6 | 1.2 | 4.6 | 3.2 |
| 52. | Octadecanea | 1800 | 0.1 | - | 0.2 | - |
| 53. | Vomifoliol | 1802 | 1.2 | - | 1.8 | 2.4 |
| 54. | 4-Hydroxycinnamic acid | 1817 | 1.9 | - | 6.6 | - |
| 55. | Hexadecan-1-ola | 1882 | 1.2 | 0.8 | 2.0 | 2.9 |
| 56. | Hexadecanoic acida | 1963 | 0.3 | 0.6 | 1.7 | 0.9 |
| 57. | (Z)-Octadec-9-en-1-ol | 2060 | 2.7 | 2.8 | 3.9 | 8.1 |
| 58. | Octadecan-1-ola | 2084 | 0.4 | 0.6 | 0.8 | 1.5 |
| 59. | (Z)-Octadec-9-enoic acid | 2147 | - | 0.3 | 2.0 | - |
| 60. | Tricosanea | 2300 | 0.3 | 0.3 | 2.3 | 2.4 |
| Total identified | 91.8 | 88.4 | 91.9 | 92.6 | ||
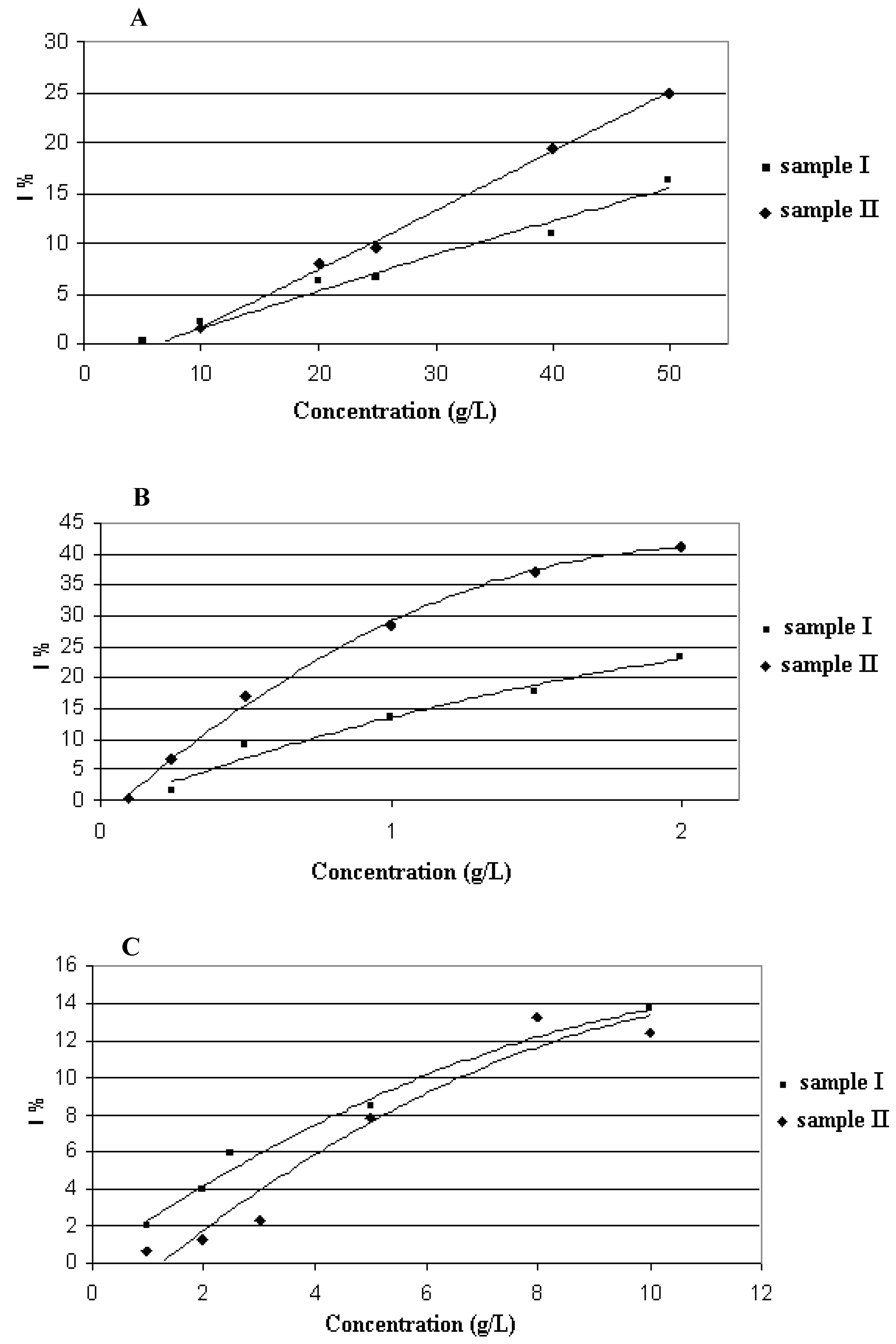
2.3. Antiradical and Antioxidant Capacity of the Honeydew and Extracts (DPPH and FRAP assay)
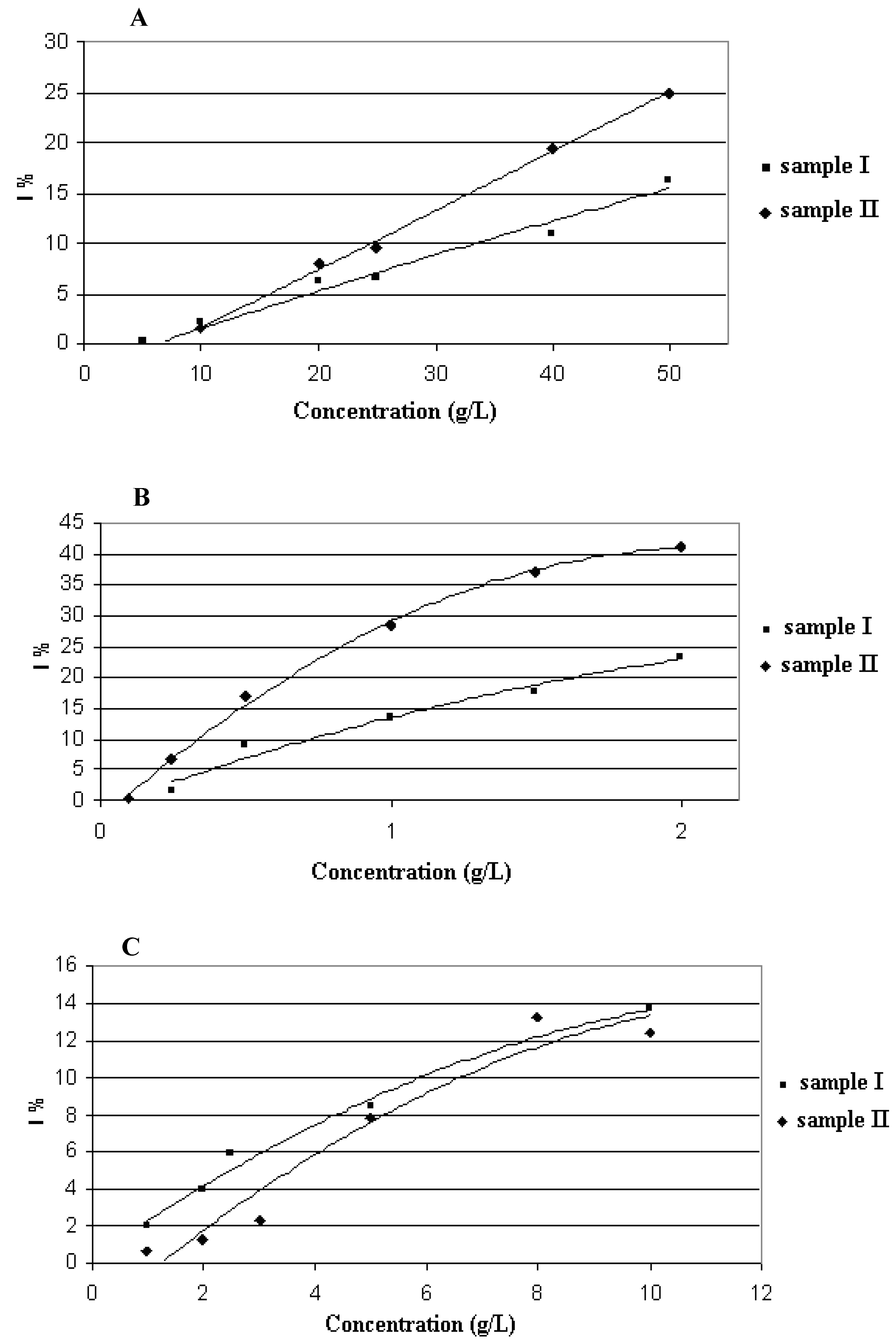
| sample I | sample II | |||
|---|---|---|---|---|
| DPPH | FRAP | DPPH | FRAP | |
| [mmol TEAC/kg] | [mmol Fe2+/kg] | [mmol TEAC/kg] | [mmol Fe2+/kg] | |
| oak honeydew | 4.5 | 4.8 | 5.1 | 16.1 |
| USE extract (solvent A; | ||||
| mIA = 0.0063 g; | 174.3 | 374.5 | 256.5 | 955.9 |
| mIIA = 0.0075 g) | ||||
| USE extract (solvent B; ; | 100.8 | 127.3 | 132.1 | 101.5 |
| mIB = 0.0102 g; | ||||
| mIIB = 0.0167 g)) | ||||
3. Experimental
3.1. Honey Samples
3.2. Headspace Solid-Phase Microextraction (HS-SPME)
3.3. Ultrasonic Solvent Extraction (USE)
3.4. Gas Chromatography and Mass Spectrometry (GC, GC/MS)
3.5. Data Analysis and Data Evaluation
3.6. Antiradical Activity (DPPH Assay)
3.7. Total Antioxidant Activity (FRAP Assay)
4. Conclusions
Acknowledgements
- Sample Availability: Contact the corresponding author.
References
- Soria, A.C.; Gonzales, M.; de Lorenzo, C.; Castro-Martinez, I.; Sanz, J. Characterization of artisanal honeys from Madrid (central Spain) on the basis of their melissopalynological, physicochemical and volatile composition data. Food Chem. 2004, 85, 121–130. [Google Scholar] [CrossRef]
- Castro-Vázquez, L.; Díaz-Maroto, M.C.; Pérez-Coello, M.S. Volatile composition and contribution to the aroma of Spanish honeydew honey. Identification of a new marker. J. Agric. Food Chem. 2006, 54, 4809–4813. [Google Scholar] [CrossRef]
- Frankel, S.; Robinson, G.E.; Berenbaum, M.R. Antioxidant capacity and correlated characteristics of 14 unifloral honeys. J. Apicultural Res. 1998, 37, 27–31. [Google Scholar]
- Wang, X.-H.; Gheldof, N.; Engeseth, N.J. Effect of processing and storageon antioxidant capacity of honey. J. Food Sci. 2004, 69, 96–101. [Google Scholar]
- Antony, S.M.; Han, I.Y.; Rieck, J.R.; Dawson, P.L. Antioxidative effect of Maillard reaction products formed fromhoney at different reaction times. J. Agric. Food Chem. 2000, 48, 3985–3989. [Google Scholar] [CrossRef]
- Gheldof, N.; Engeseth, N.J. Antioxidant capacity of honeys from various floral sources based on the determination of oxygen radical aborbance capacity and inhibition of in vitro lipoprotein oxidation in human serum samples. J. Agric. Food Chem. 2002, 50, 3050–3055. [Google Scholar] [CrossRef]
- Roginsky, V.; Lissi, E.A. Review of methods to determine chain-breaking antioxidant activity in food. Food Chem. 2005, 92, 235–254. [Google Scholar] [CrossRef]
- Jerković, I.; Marijanović, Z.; Kezić, J.; Gugić, M. Headspace, volatile and semi-volatile organic compoundsdiversity and radical scavenging activity of ultrasonic solventextracts from Amorpha fruticosa honey samples. Molecules 2009, 14, 2717–2728. [Google Scholar] [CrossRef]
- Chatonnet, P. Incidence du bois de chene sur la composition chimique et les quelités organoleptiques des vins. Aplications technologiques. Ph.D. Thesis, Université de Bordeaux II, France, 1991. [Google Scholar]
- Villamiel, M.; del Castillo, M.D.; Corzo, N.; Olano, A. Presence of furosine in honeys. J. Sci. Food Agric. 2001, 81, 790–793. [Google Scholar] [CrossRef]
- Jerković, I.; Tuberoso, C.I.G.; Marijanović, Z.; Jelić, M.; Kasum, A. Headspace, volatile and semi-volatile patterns of Paliurus spina-christi unifloral honey as markers of botanical origin. Food Chem. 2009, 112, 239–245. [Google Scholar] [CrossRef]
- Louveaux, J.; Maurizio, A.; Vorwohl, G. Methods of melissopalynology. Bee World 1978, 59, 39–153. [Google Scholar]
- Bogdanov, S.; Martin, P.; Lüllman, C. Harmonised methods of the European Honey Comission. Apidologie 1997, extra issue, 1–59. [Google Scholar]
- El-Sayed, A.M. The pherobase: database of insect pheromones and semiochemicals. Available online: http://www.pherobase.com/. Accessed on 25 May 2010.
- Tuberoso, C.I.G.; Bifulco, E.; Jerković, I.; Caboni, P.; Cabras, P.; Floris, I. Methyl syringate: a chemical marker of asphodel (Asphodelus microcarpus Salzm. et Viv.) monofloral honey. J. Agric. Food Chem. 2009, 57, 3895–3900. [Google Scholar] [CrossRef]
© 2010 by the authors;
Share and Cite
Jerković, I.; Marijanović, Z. Oak (Quercus frainetto Ten.) Honeydew Honey—Approach to Screening of Volatile Organic Composition and Antioxidant Capacity (DPPH and FRAP Assay). Molecules 2010, 15, 3744-3756. https://doi.org/10.3390/molecules15053744
Jerković I, Marijanović Z. Oak (Quercus frainetto Ten.) Honeydew Honey—Approach to Screening of Volatile Organic Composition and Antioxidant Capacity (DPPH and FRAP Assay). Molecules. 2010; 15(5):3744-3756. https://doi.org/10.3390/molecules15053744
Chicago/Turabian StyleJerković, Igor, and Zvonimir Marijanović. 2010. "Oak (Quercus frainetto Ten.) Honeydew Honey—Approach to Screening of Volatile Organic Composition and Antioxidant Capacity (DPPH and FRAP Assay)" Molecules 15, no. 5: 3744-3756. https://doi.org/10.3390/molecules15053744
APA StyleJerković, I., & Marijanović, Z. (2010). Oak (Quercus frainetto Ten.) Honeydew Honey—Approach to Screening of Volatile Organic Composition and Antioxidant Capacity (DPPH and FRAP Assay). Molecules, 15(5), 3744-3756. https://doi.org/10.3390/molecules15053744