Synthesis and Spectrosopic Identification of Hybrid 3-(Triethoxysilyl)propylamine Phosphine Ruthenium(II) Complexes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
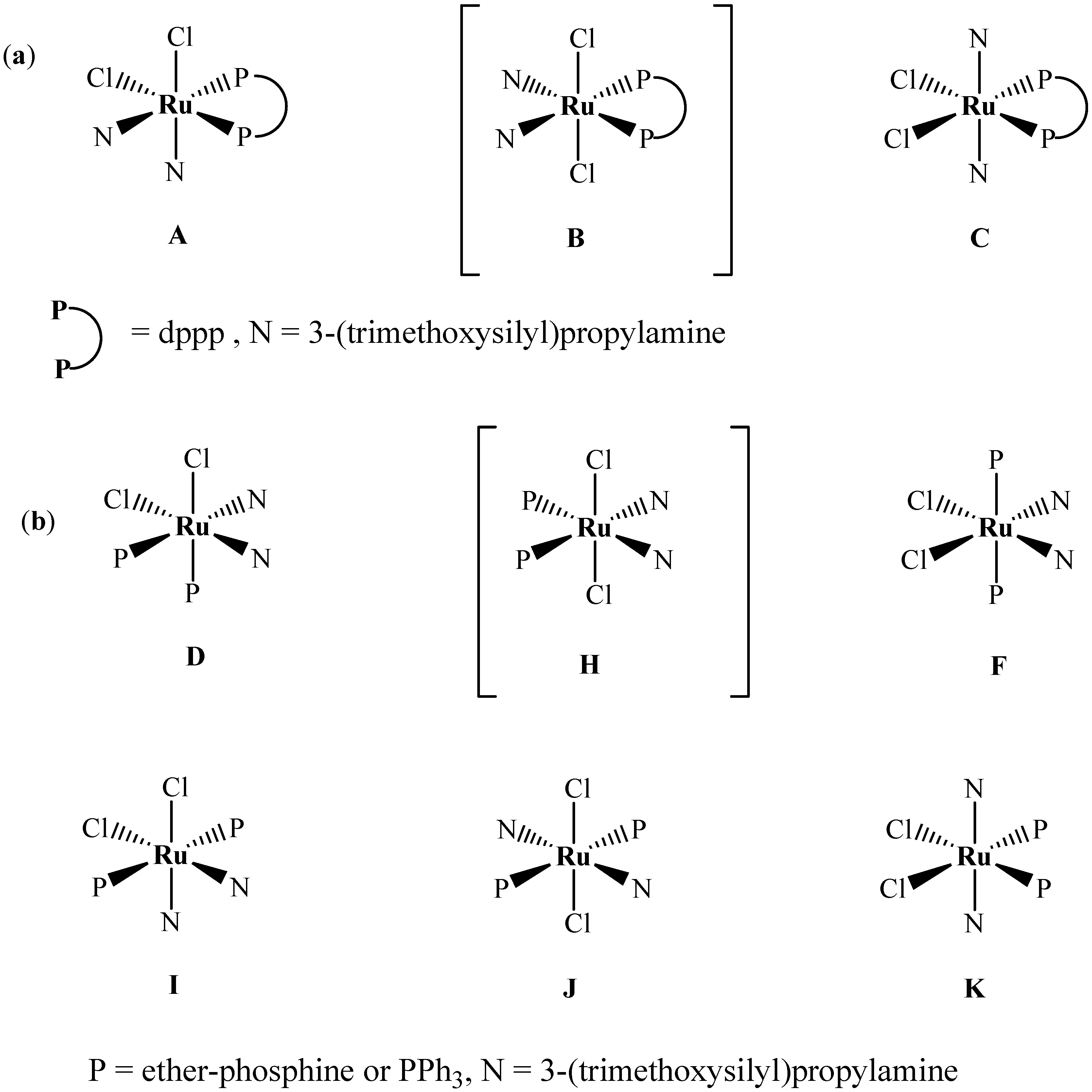{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
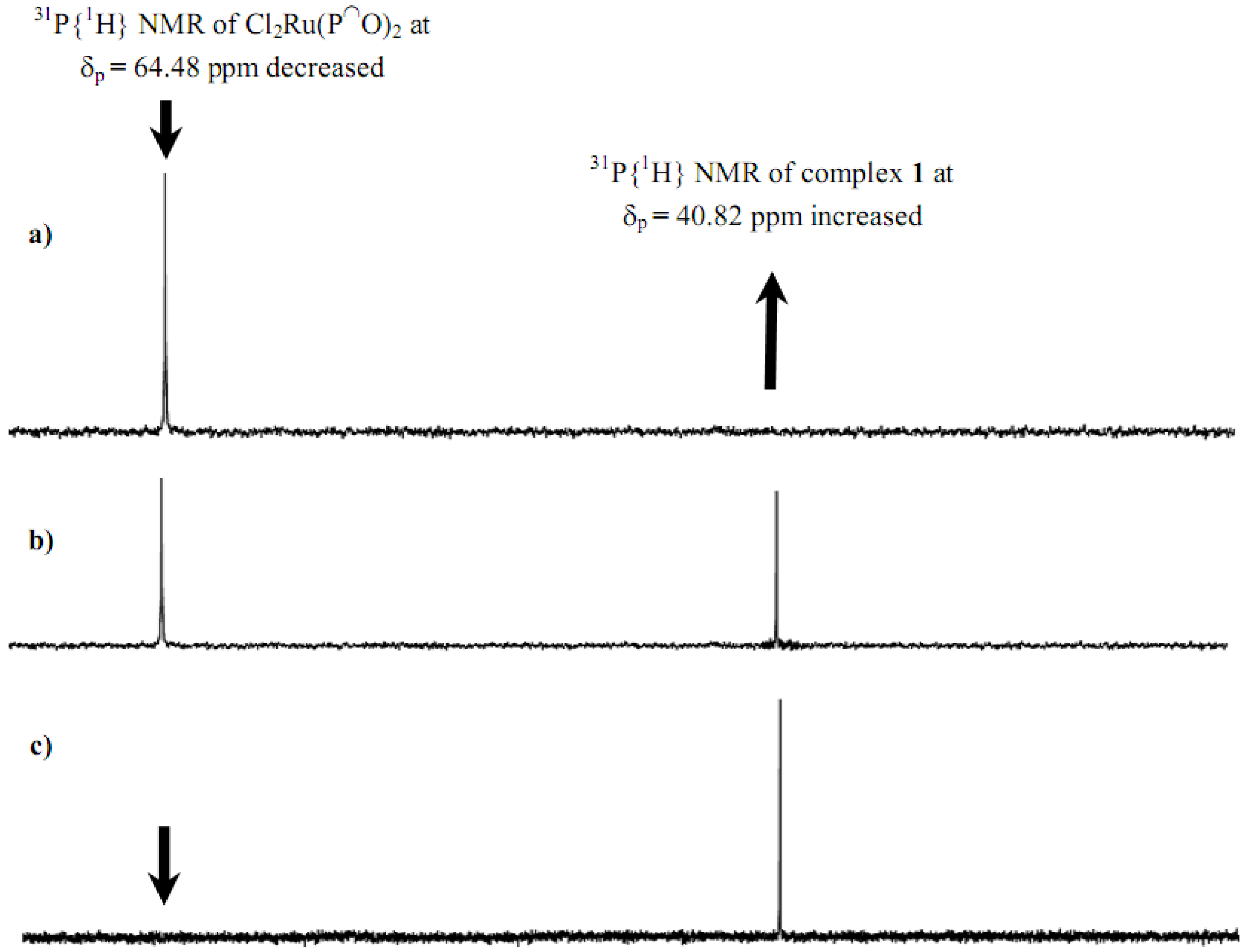
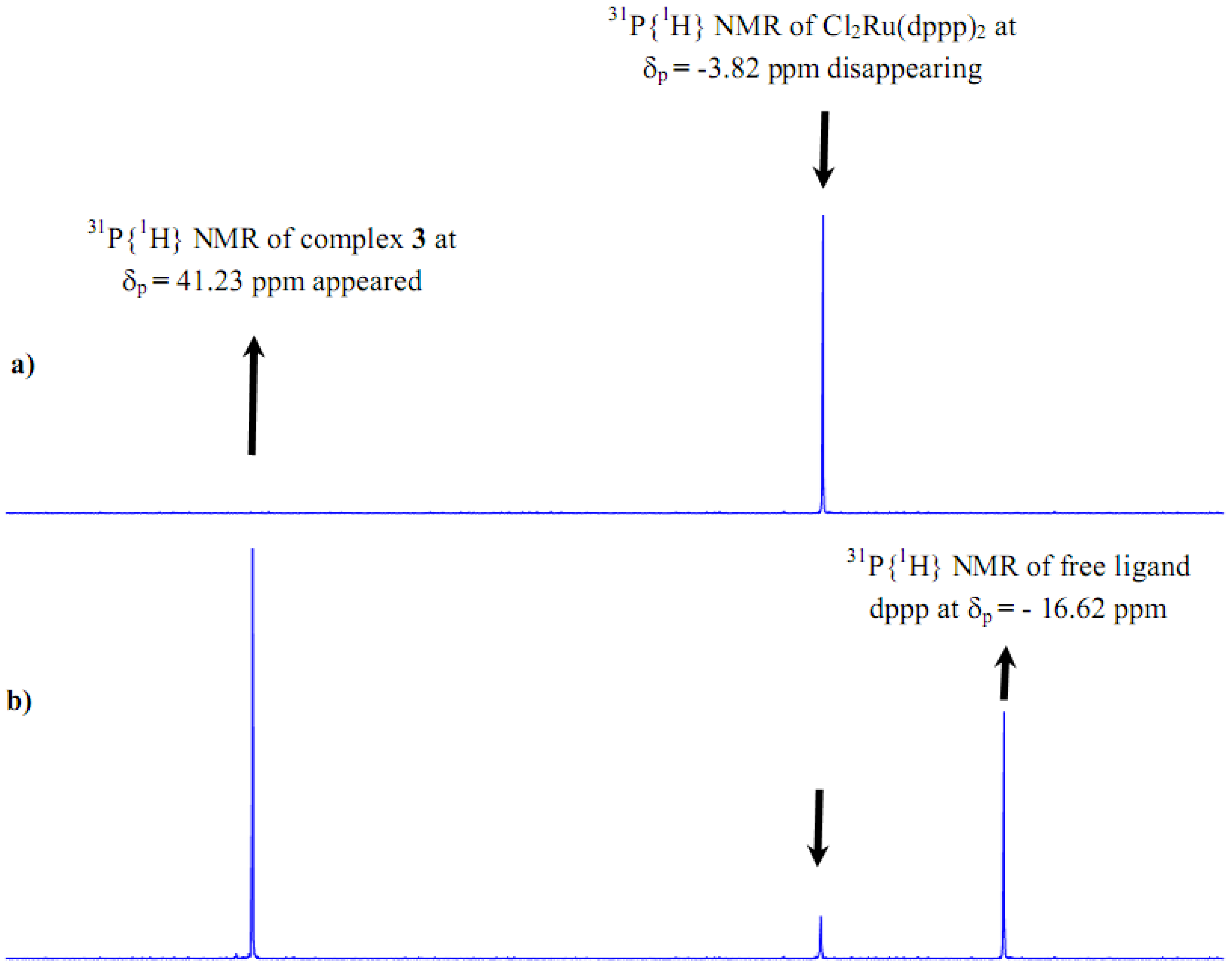
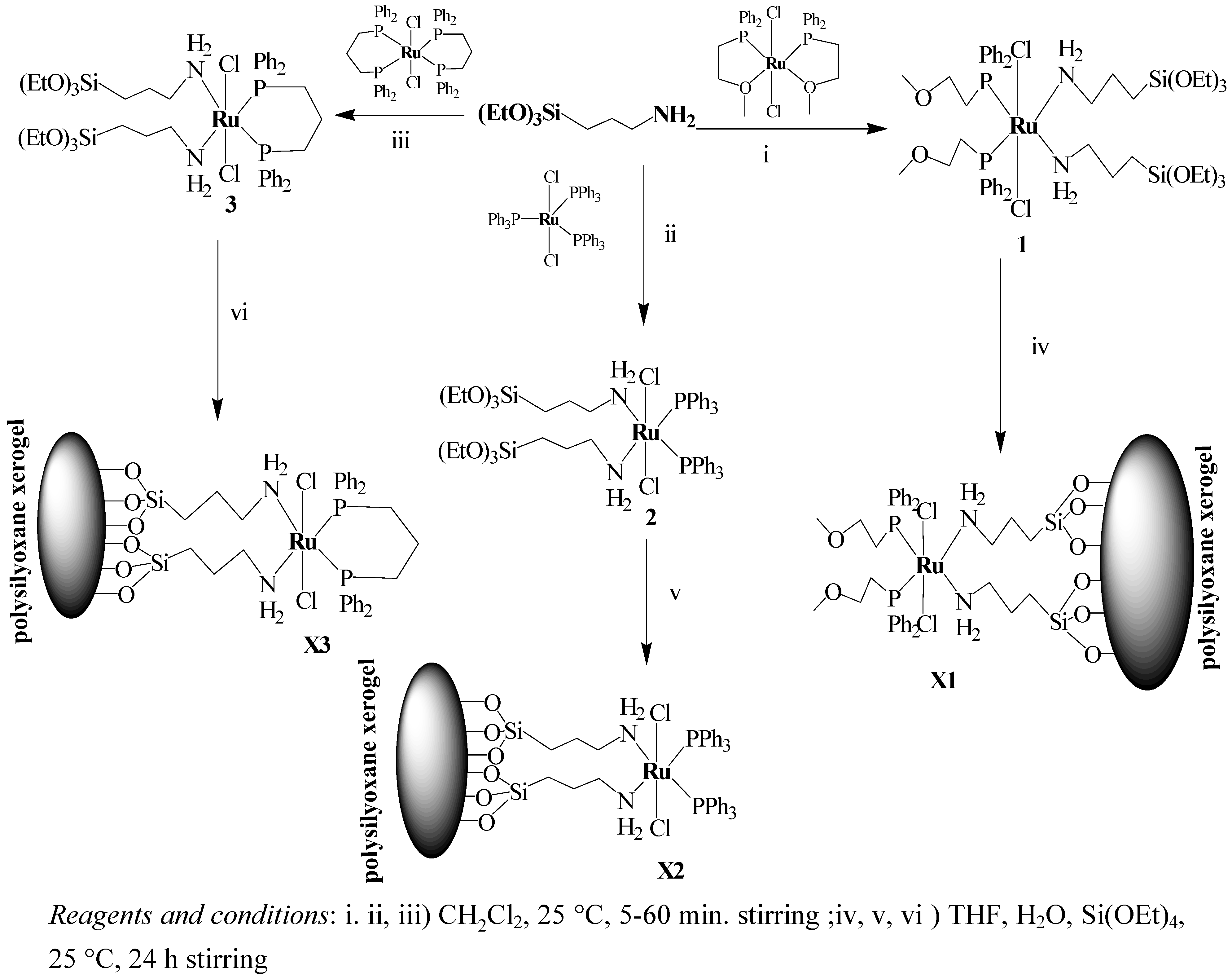
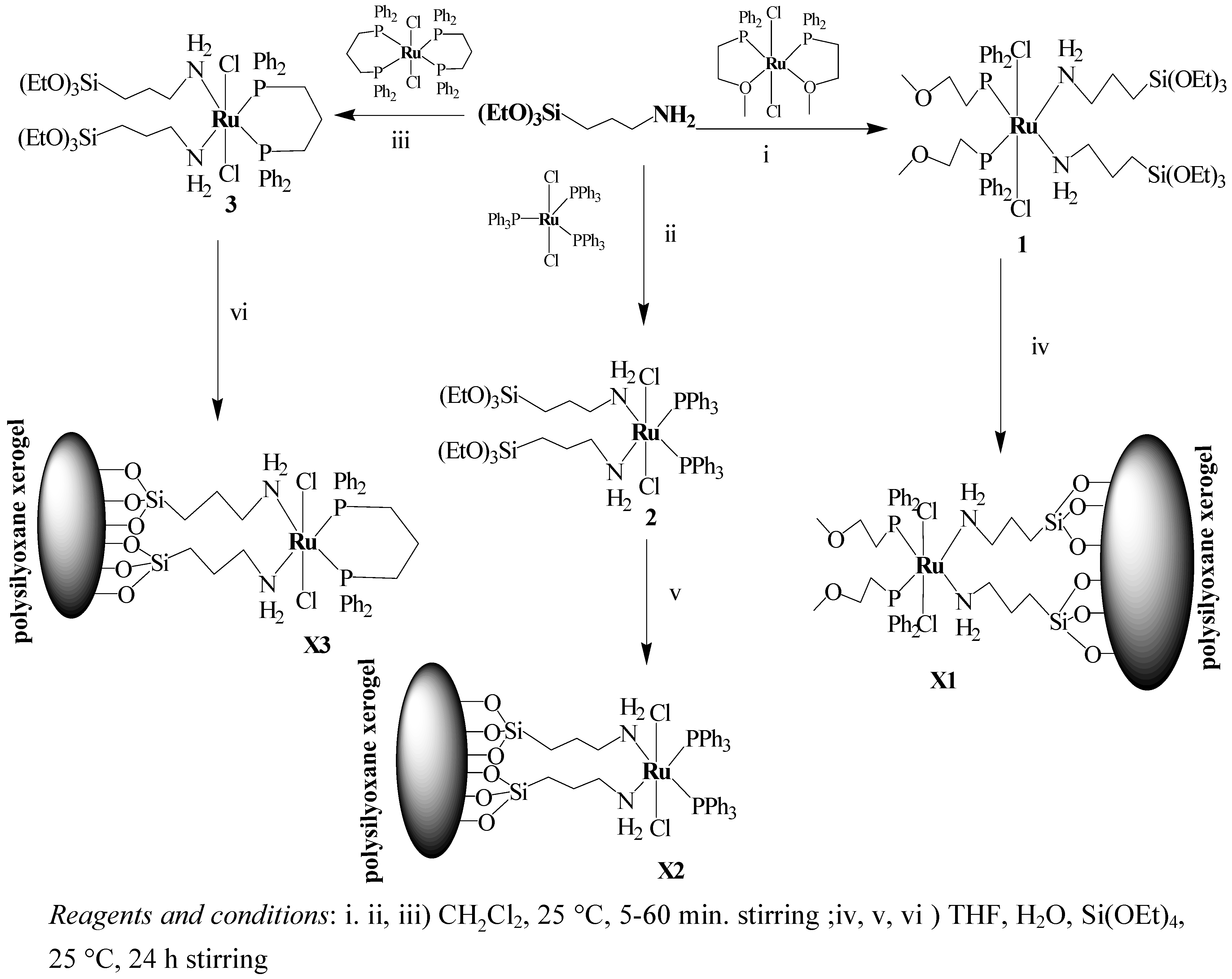
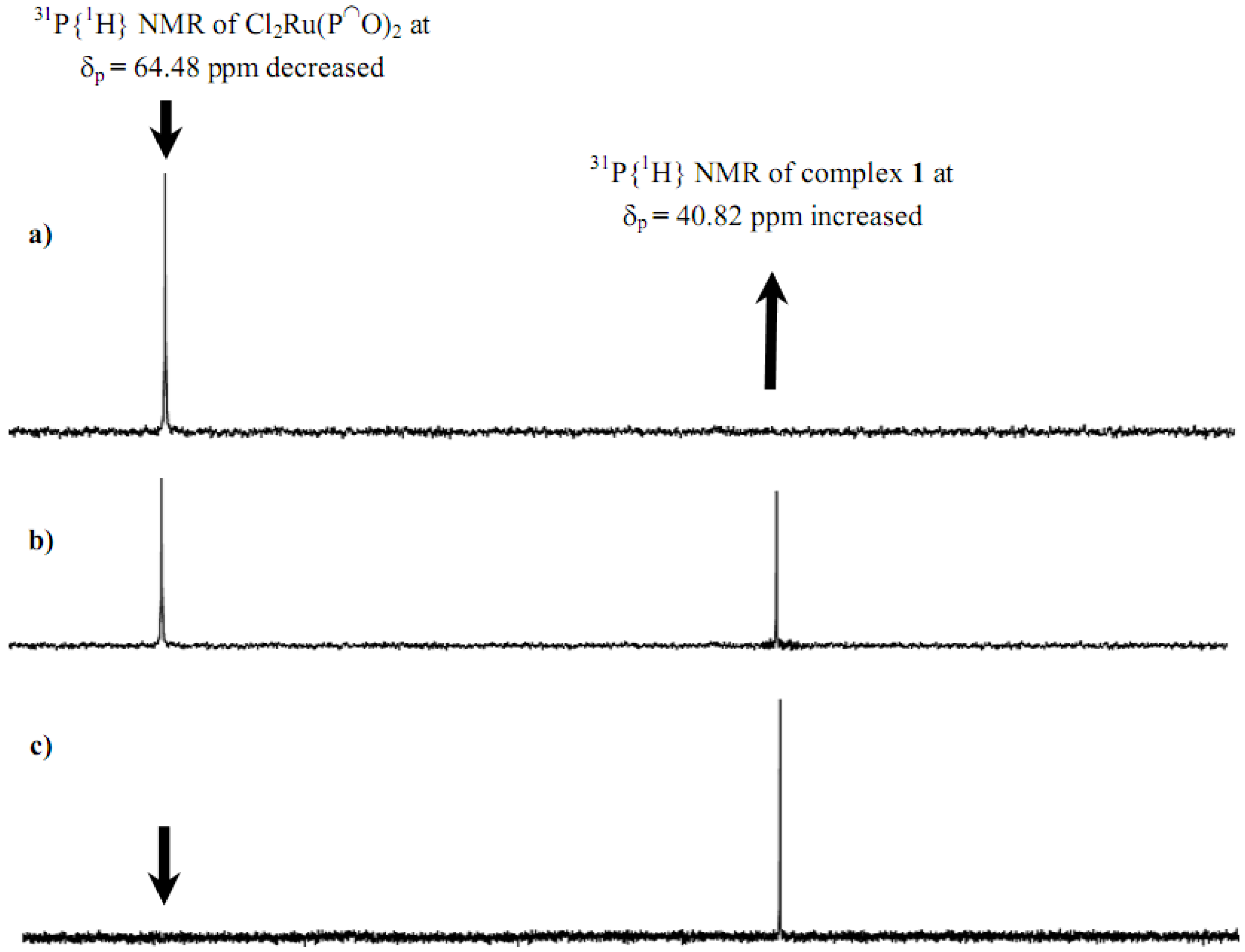
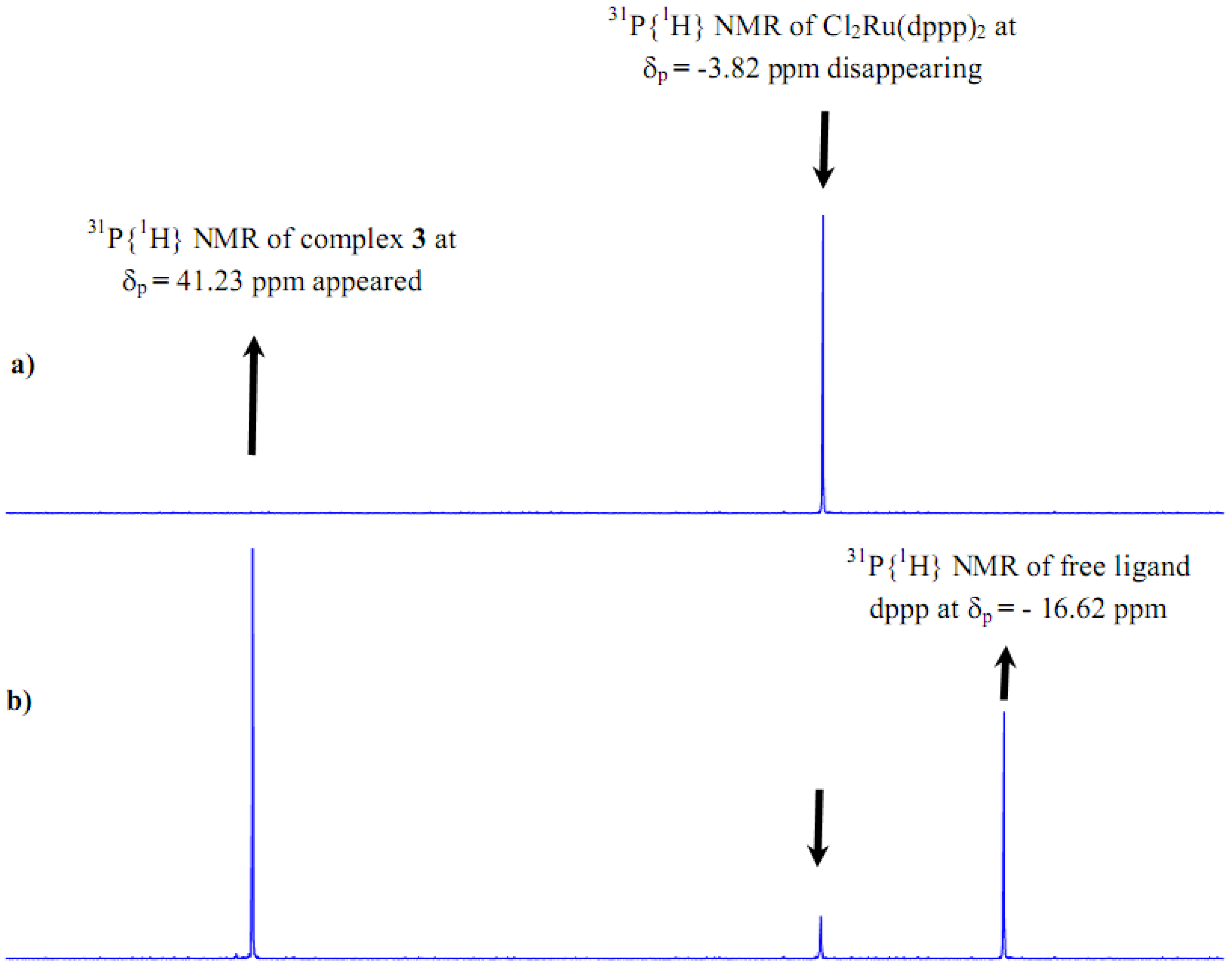
2.1. Synthesis and 31P-NMR investigation of ruthenium(II) complexes 1-3 and xerogels X1-X3
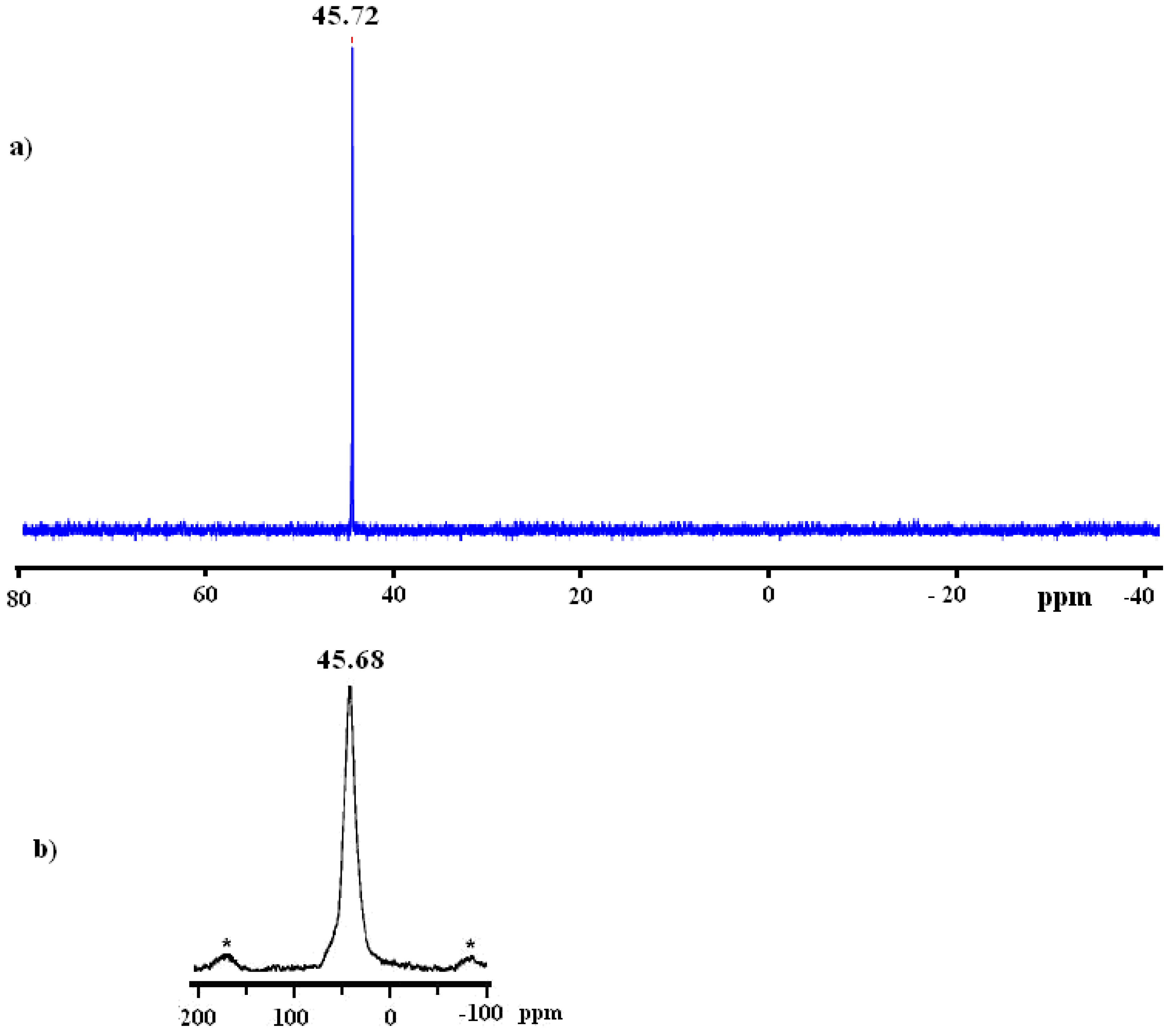
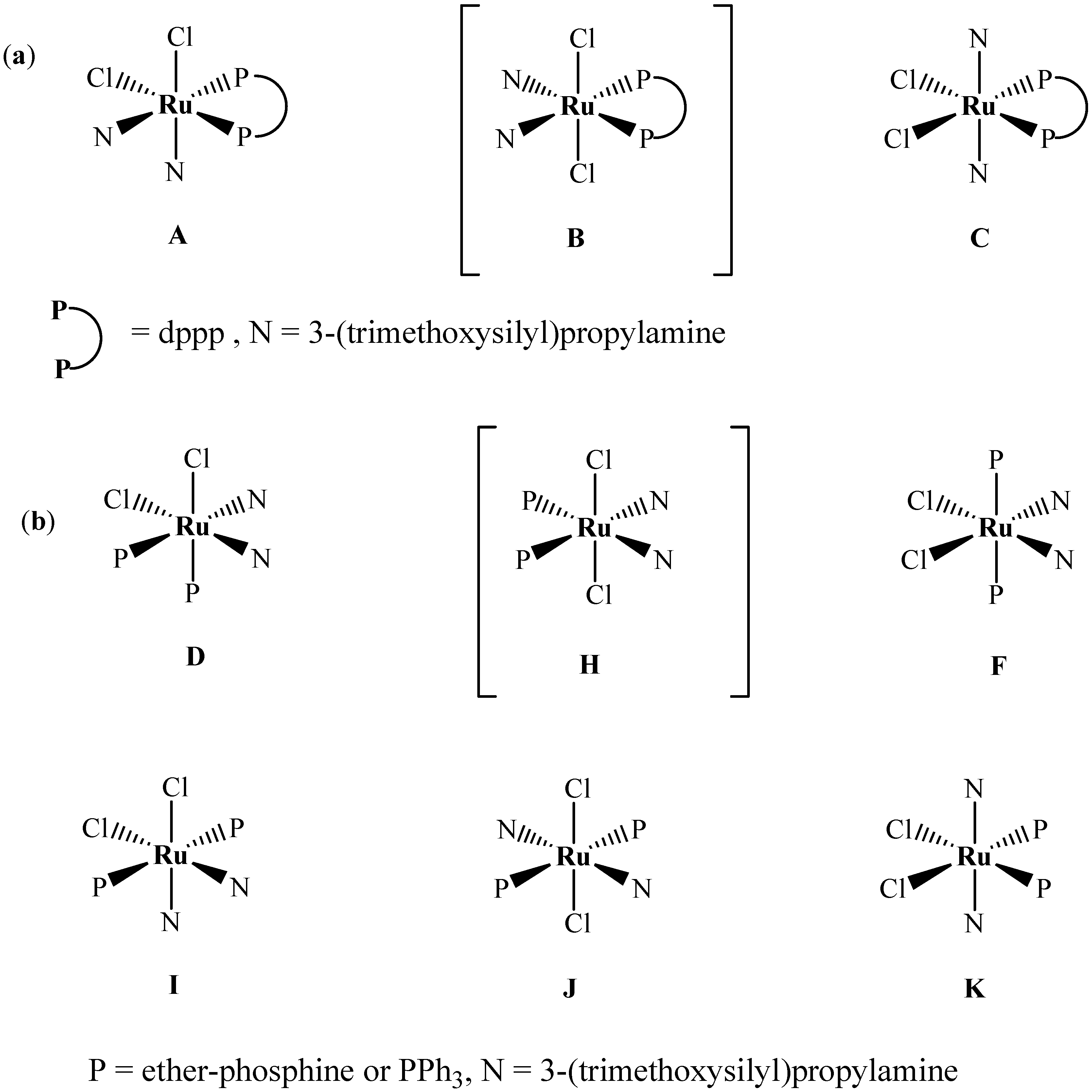
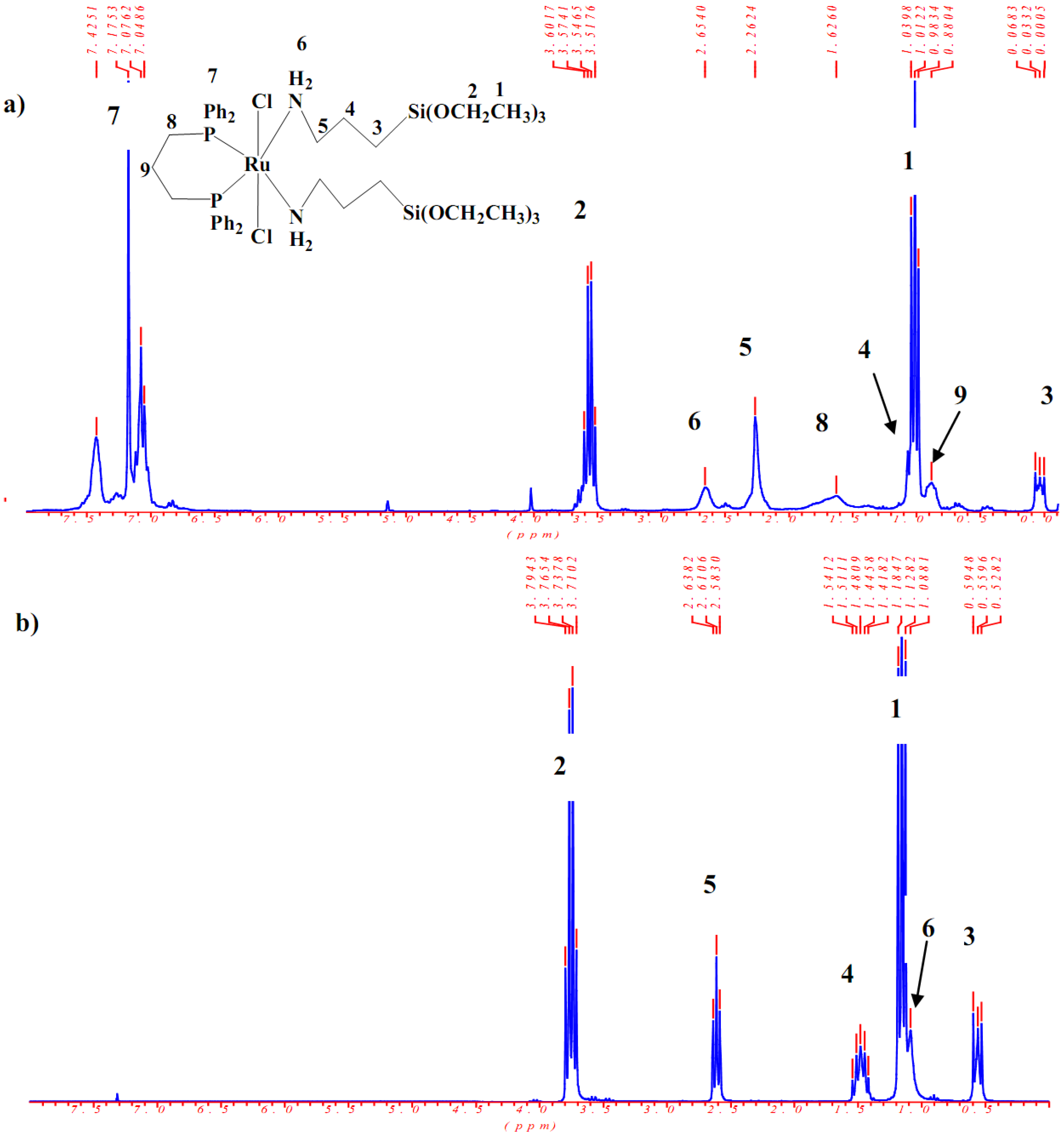
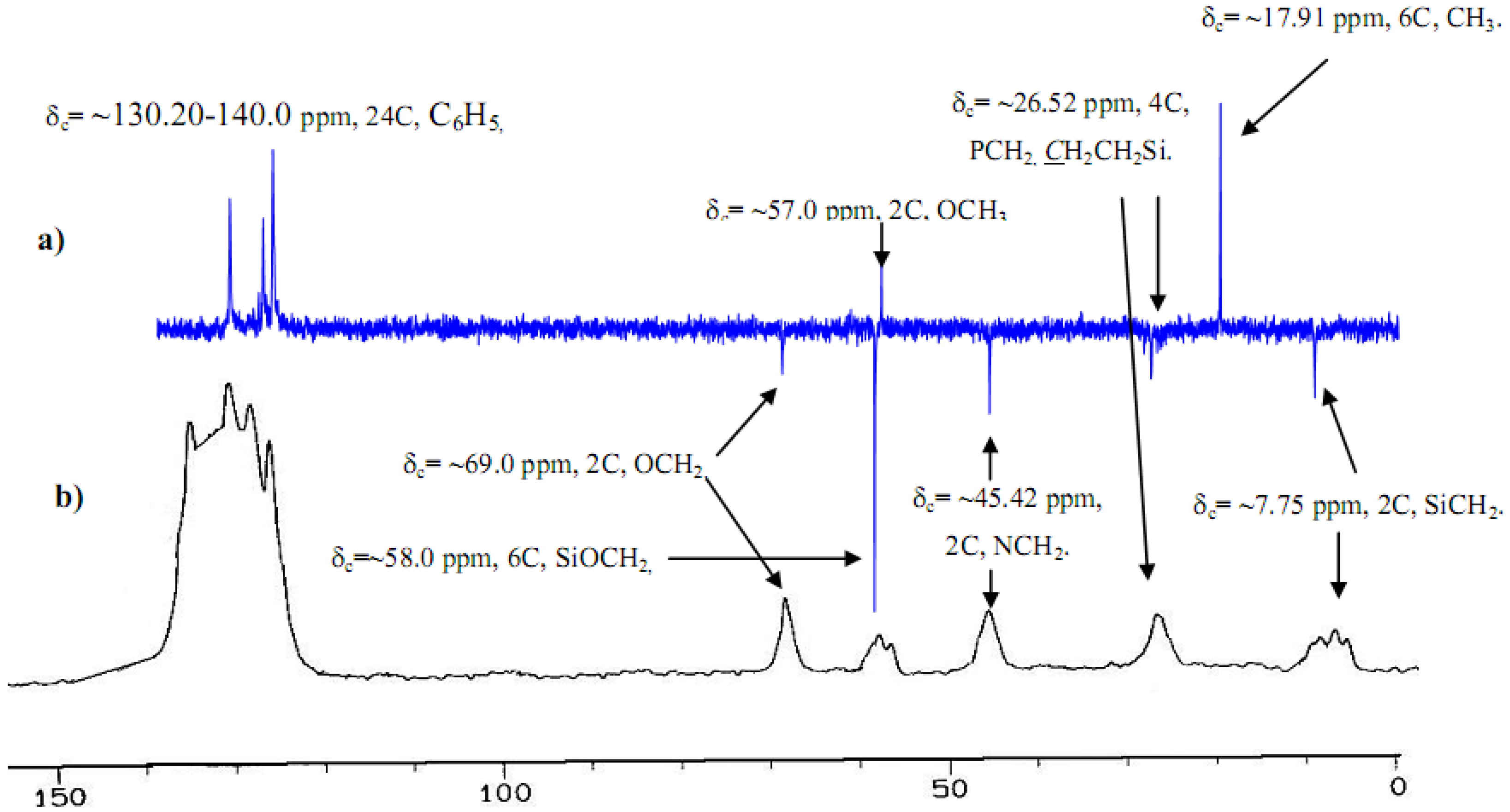
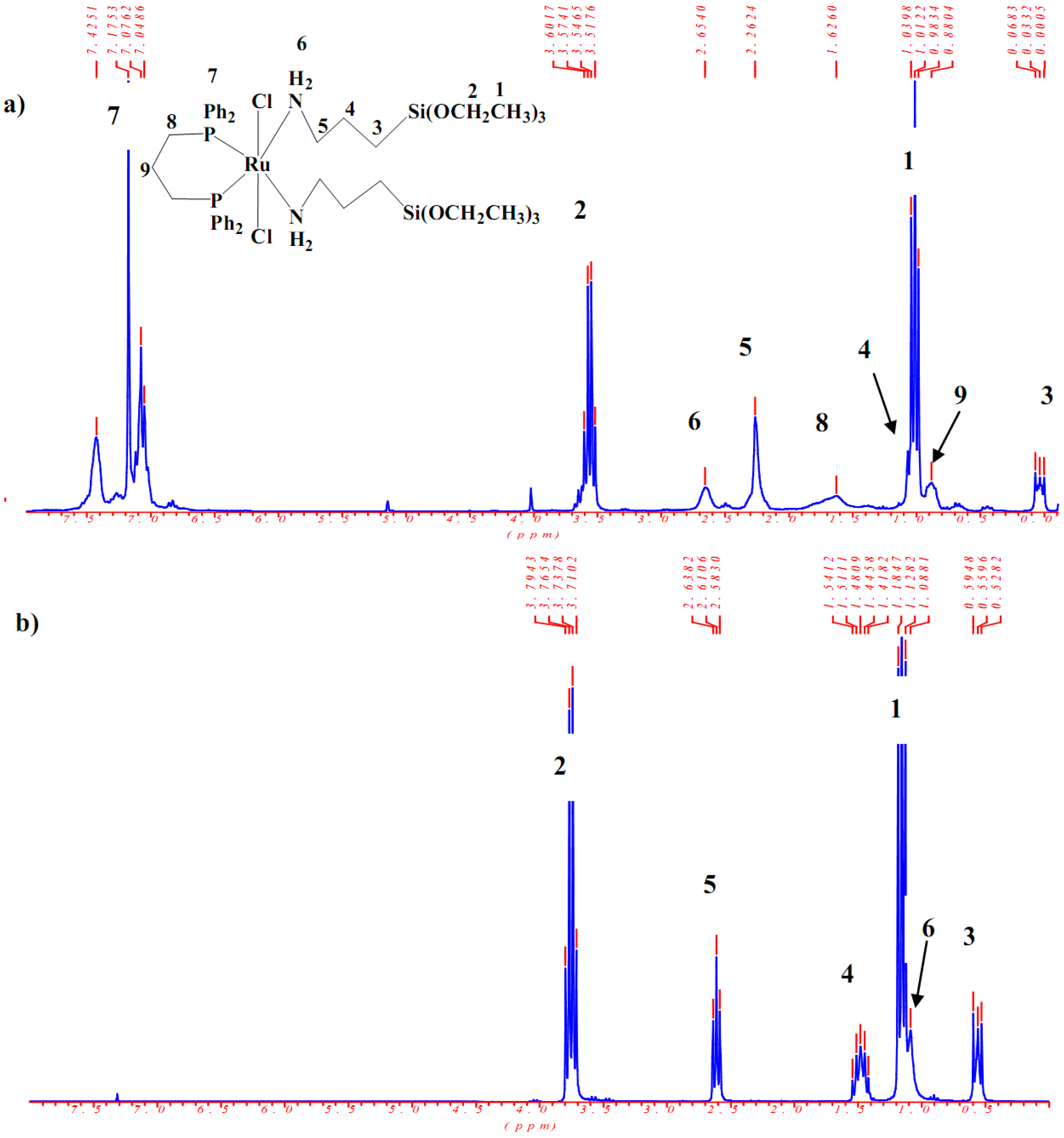
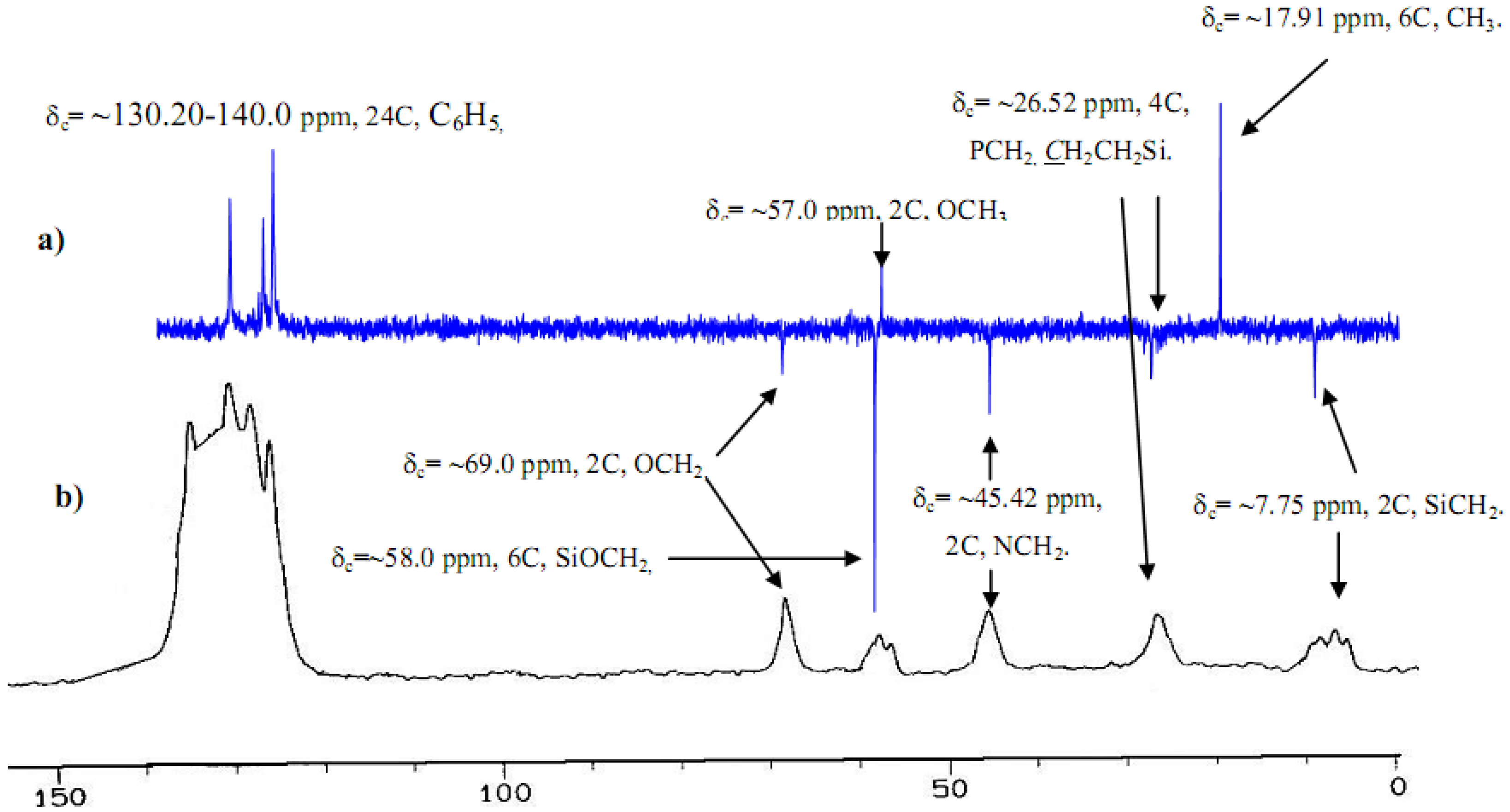
2.2. H and C NMR investigations
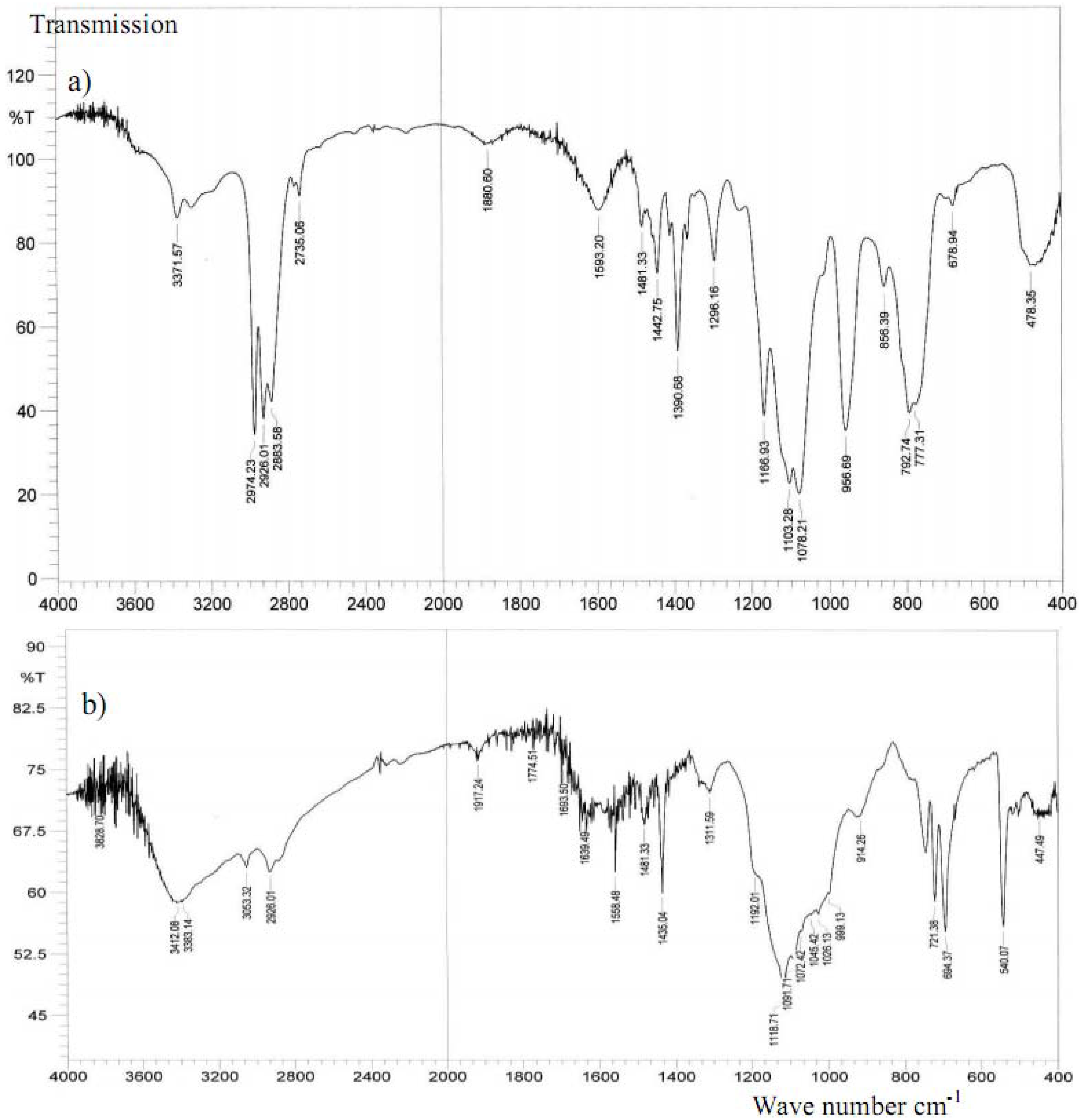
2.3. IR investigations
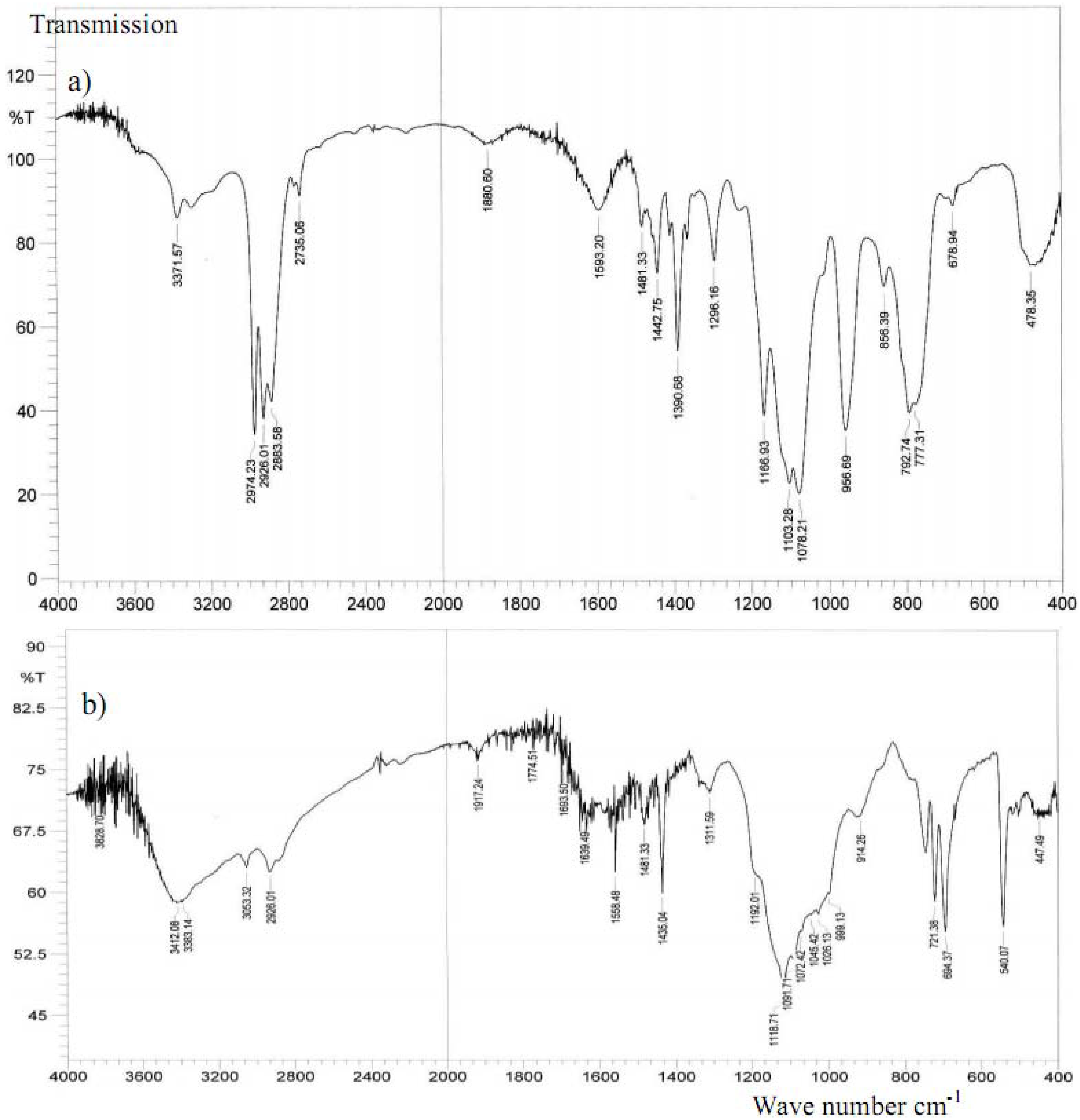
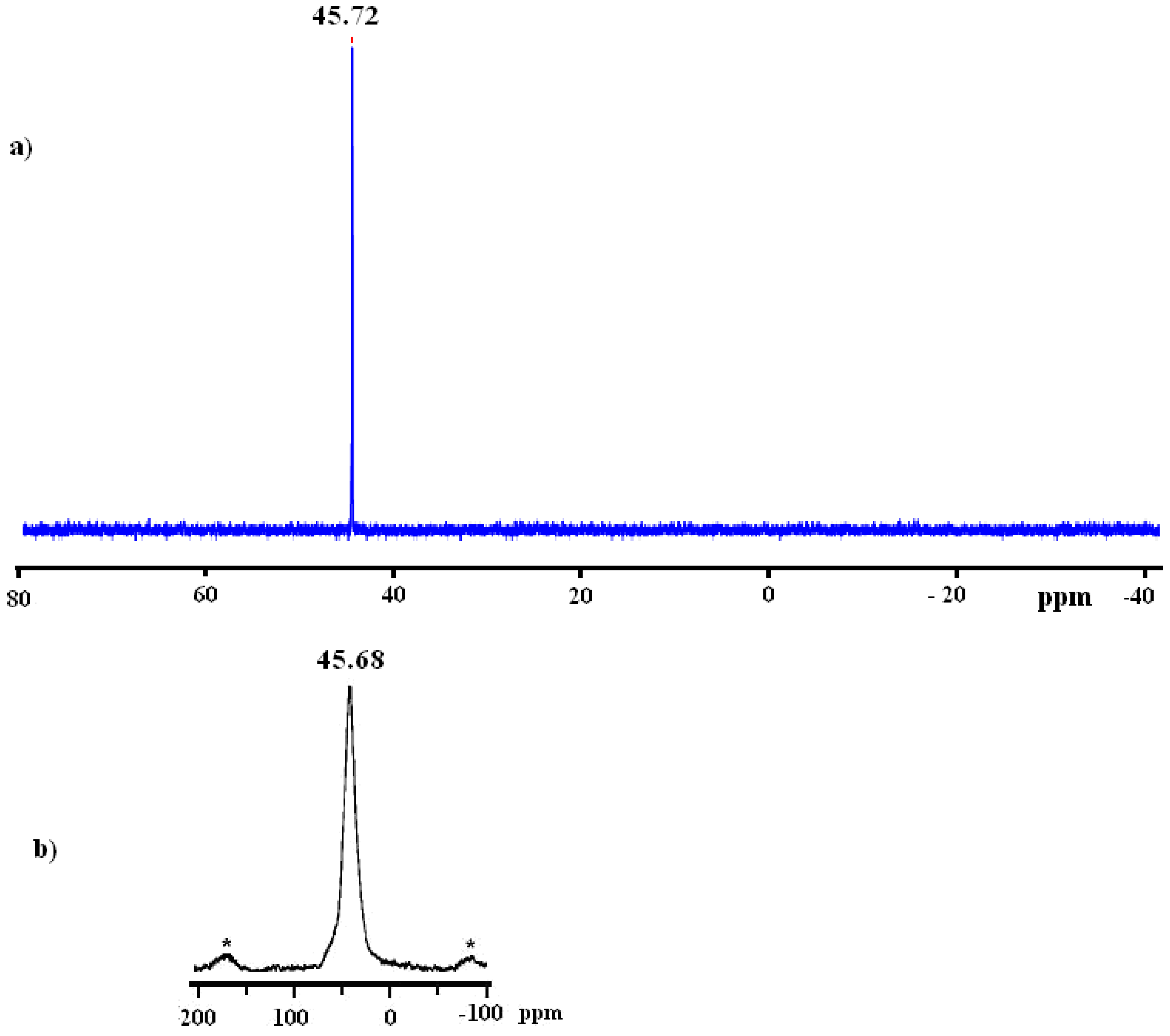
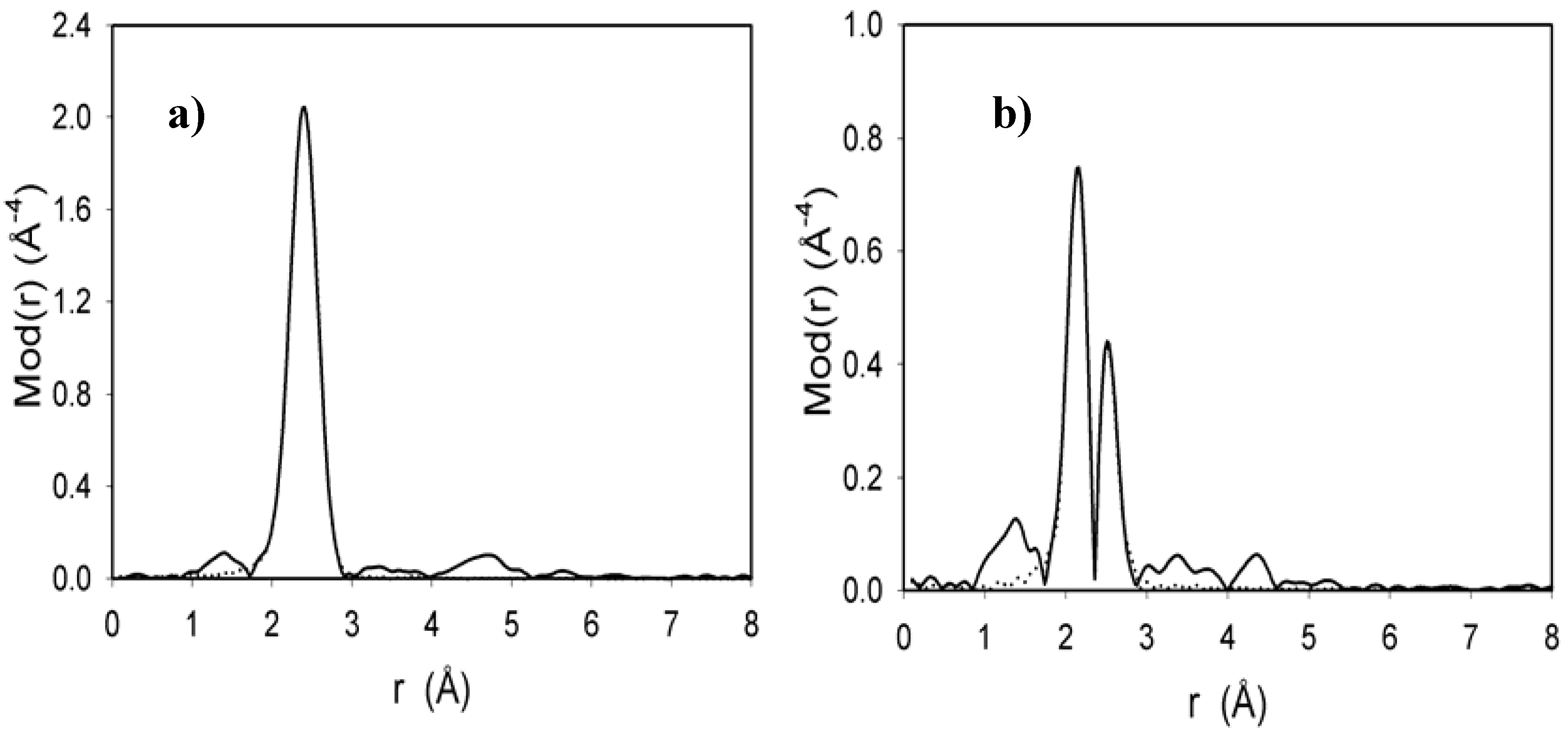
2.4. EXAFS measurement of Cl2Ru(dppp)2 complex and xerogel X3
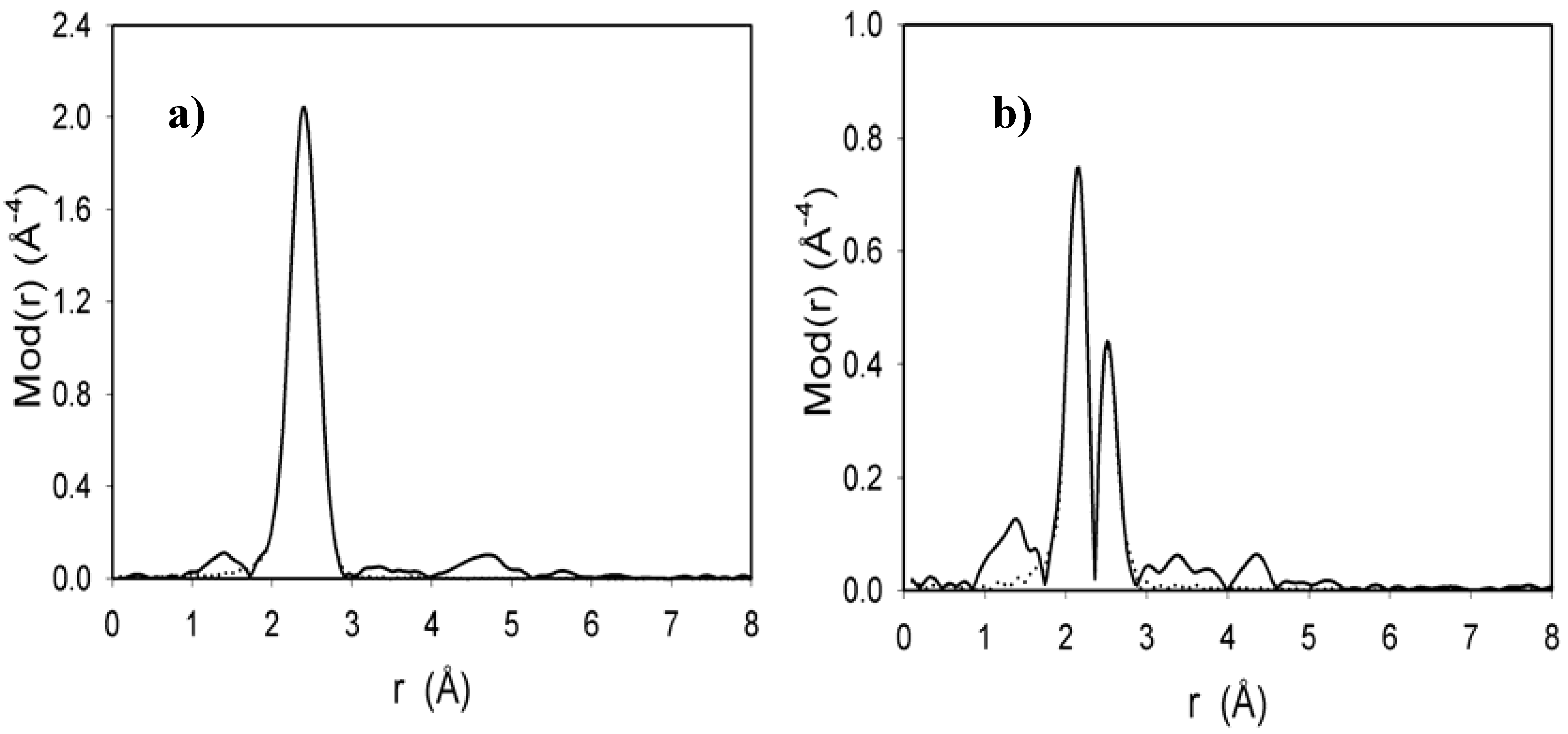
3. Experimental
3.1. General remarks, materials, and instrumentation
3.2. General procedure for the preparation of the complex 1-3
3.3. General procedure for sol–gel processing of xerogel X1-X3
4. Conclusions
Acknowledgements
References and Notes
- Warad, I.; Al-Othman, Z.; Al-Resayes, S.; Al-Deyab, S.; Kenawy, E. Synthesis and characterization of novel inorganic-organic hybrid Ru(II) complexes and their application in selective hydrogenation. Molecules 2010, 15, 1028–1040. [Google Scholar]
- Jakob, A.; Ecorchard, P.; Linseis, M; Winter, R. Synthesis, solid state structure and spectro-electrochemistry of ferrocene-ethynyl phosphine and phosphine oxide transition metal complexes. J. Organomet. Chem. 2010, 694, 655–666. [Google Scholar]
- Warad, I.; Siddiqui, M.; Al-Resayes, S.; Al-Warthan, A.; Mahfouz, R. Synthesis, characterization, crystal structure and chemical behavior of [1,1-bis(diphenylphosphinomethyl)ethene]ruthenium-(II) complex toward primary alkylamine addition. Trans. Met. Chem. 2009, 34, 347–354. [Google Scholar] [CrossRef]
- Chang, C.-P.; Weng, C.-M.; Hong, F.-E. Preparation of cobalt sandwich diphosphine ligand [(η5-C5H4iPr)Co(η4-C4(PPh2)2Ph2)] and its chelated Palladium complex: Application of diphosphine ligand in the preparation of mono-substituted ferrocenylarenes. Inorg. Chim. Acta 2010, 363, 412–417. [Google Scholar] [CrossRef]
- Michelin, R.; Sgarbossa, P.; Scarso, A.; Strukul, G. The Baeyer–Villiger oxidation of ketones: A paradigm for the role of soft Lewis acidity in homogeneous catalysis. Coord. Chem. Rev. 2010, 254, 646–660. [Google Scholar]
- Wang, Z.-W.; Cao, Q.-Y.; Huang, X.; Lin, S.; Gao, X.-C. Synthesis, structure and electronic spectra of new three-coordinated Copper(I) complexes with tricyclohexylphosphine and diimine ligands. Inorg. Chim. Acta 2010, 363, 15–19. [Google Scholar]
- James, B.; Lorenzini, F. Developments in the chemistry of tris(hydroxymethyl)phosphine. Coord. Chem. Rev. 2010, 254, 420–430. [Google Scholar]
- Drozdzak, R.; Allaert, B.; Ledoux, N.; Dragutan, I.; Dragutan, V.; Verpoort, F. Ruthenium complexes bearing bidentate Schiff base ligands as efficient catalysts for organic and polymer syntheses. Coord. Chem. Rev. 2005, 249, 3055–3074. [Google Scholar]
- Tfouni, E.; Doro, F.; Gomes, A.; Silva, R.; Metzker, G.; Grac, P.; Benini, Z.; Franco, D. Immobilized ruthenium complexes and aspects of their reactivity. Coord. Chem. Rev. 2010, 254, 355–371. [Google Scholar]
- Lindner, E.; Mayer, H. A.; Warad, I.; Eichele, K. Synthesis, characterization, and catalytic application of a new family of diamine(diphosphine)ruthenium(II) complexes. J. Organomet. Chem. 2003, 665, 176–185. [Google Scholar]
- Xi, Z.; Hao, W.; Wang, P.; Cai, M. Ruthenium(III) chloride catalyzed acylation of alcohols, phenols, and thiols in room temperature ionic liquids. Molecules 2009, 14, 3528–3537. [Google Scholar] [CrossRef]
- Duraczynska, D.; Serwicka, E.M.; Drelinkiewicz, A.; Olejniczak, Z. Ruthenium(II) phosphine/ mesoporous silica catalysts: The impact of active phase loading and active site density on catalytic activity in hydrogenation of phenylacetylene. Appl. Catal. A. Gen. 2009, 371, 166–172. [Google Scholar] [CrossRef]
- Premkumar, J.; Khoo, S. Immobilization of ruthenium(II)bipyridyl complex at highly oxidized glassy carbon electrodes. Electrochem. Commun. 2004, 6, 984–989. [Google Scholar] [CrossRef]
- Noyori, R. Asymmetric Catalysis in Organic Synthesis; Wiley and Sons: New York, NY, USA, 1994; pp. 16–46. [Google Scholar]
- Noyori, R. Asymmetric Catalysis: Science and Opportunities. Adv. Synth. Catal. 2003, 345, 15–32, (Nobel Lecture). [Google Scholar] [CrossRef]
- Lindner, E.; Lu, Z.-L.; Mayer, A.H.; Speiser, B.; Tittel, C.; Warad, I. Cyclic voltammetric redox screening of homogeneous ruthenium(II) hydrogenation catalysts. Electrochem. Commun. 2005, 7, 1013–1020. [Google Scholar] [CrossRef]
- Lindner, E.; Warad, I.; Eichele, K.; Mayer, H.A. Synthesis and structures of an array of diamine(ether-phosphine)ruthenium(II) complexes and their application in the catalytic hydrogenation of trans-4-phenyl-3-butene-2-one. Inorg. Chim. Acta 2003, 350, 49–56. [Google Scholar] [CrossRef]
- Lu, Z.-L.; Eichele, K.; Warad, I.; Mayer, H.A.; Lindner, E.; Jiang, Z.; Schurig, V. Bis(methoxyethyldimethylphosphine)ruthenium(II) complexes as transfer hydrogenation catalysts. Z. Anorg. Allg. Chem. 2003, 629, 1308–1315. [Google Scholar] [CrossRef]
- Warad, I.; Lindner, E.; Eichele, K.; Mayer, A.H. Cationic Diamine(ether–phosphine)ruthenium(II) complexes as precursors for the hydrogenation of trans-4-phenyl-3-butene-2-one. Inorg. Chim. Acta 2004, 357, 1847–1853. [Google Scholar] [CrossRef]
- Lindner, E.; Ghanem, A.; Warad, I.; Eichele, K.; Mayer, H.A.; Schurig, V. Asymmetric hydrogenation of an unsaturated ketone by diamine(ether–phosphine)ruthenium(II) complexes and lipase-catalyzed kinetic resolution: A consecutive approach. Tetrahedron Asymmetry 2003, 14, 1045–1050. [Google Scholar] [CrossRef]
- Lindner, E.; Al-Gharabli, S.; Warad, I.; Mayer, H.A.; Steinbrecher, S.; Plies, E.; Seiler, M.; Bertagnolli, H. Diaminediphosphineruthenium(II) interphase catalysts for the hydrogenation of α,ß-unsaturated ketones. Z. Anorg. Allg. Chem. 2003, 629, 161–171. [Google Scholar] [CrossRef]
- Lu, Z.-L.; Eichele, K.; Warad, I.; Mayer, H.A.E.; Lindner; Jiang, Z.; Schurig, Schurig. Bis(methoxyethyldimethylphosphine)ruthenium(II) complexes as transfer hydrogenation catalysts. Z. Anorg. Allg. Chem. 2003, 629, 1308–1315. [Google Scholar] [CrossRef]
- Warad, I.; Al-Resayes, S.; Eichele, E. Crystal structure of trans-dichloro-1,3-propanediamine-bis[(2-methoxyethyl)diphenylphosphine]ruthenium(II), RuCl2-(C3H10N2)(C15H17OP)2. Z. Kristallogr. NCS 2006, 221, 275–277. [Google Scholar]
- Warad, I. Synthesis and crystal structure of cis-dichloro-1,2-ethylenediamine-bis[1,4-(diphenylphosphino)butane]ruthenium(II) dichloromethane disolvate, RuCl2(C2H8N2) (C28H28P2)-2CH2Cl2. Z. Kristallogr. NCS 2007, 222, 415–417. [Google Scholar]
- Ohkuma, T.; Koizumi, M.; Muniz, K.; Hilt, G.; Kabuta, C.; Noyori, R. Trans-RuH(η1- BH4)(binap)(1,2-diamine): A Catalyst for asymmetric hydrogenation of simple ketones under base-Free conditions". J. Am. Chem. Soc. 2002, 124, 6508–6509 and reference there in. [Google Scholar]
- Hashiguchi, S.; Fujii, A.; Haack, K.-J.; Matsumura, K.; Ikariya, T.; Noyori, R. Kinetic resolution of racemis secondary alcohols by Ru(II)-catalyzed hydrogen transfer. Angew. Chem. Int. Ed. Engl. 1997, 36, 288–290. [Google Scholar] [CrossRef]
- Haack, K.-J.; Hashiguchi, S.; Fujii, A.; Ikariya, T.; Noyori, R. The catalyst precursor, catalyst, and intermediate in the Ru-II-promoted asymmetric hydrogen transfer between alcohols and ketones. Angew. Chem. Int. Ed. Engl. 1997, 36, 285–288. [Google Scholar] [CrossRef]
- Abdur-Rashid, K.; Faatz, M.; Lough, A.J.; Morris, H.R. Catalytic Cycle for the asymmetric hydrogenation of prochiral ketones to chiral alcohols: Direct hydride and proton transfer from chiral catalysts trans-Ru(H)2(diphosphine)(diamine) to ketones and direct Addition of Dihydrogen to the Resulting Hydridoamido Complexes. J. Am. Chem. Soc. 2001, 123, 7473–7474 and reference their in. [Google Scholar]
- Brunel, D.; Bellocq, N.; Sutra, P.; Cauvel, A.; Laspearas, M.; Moreau, P.; Renzo, F.; Galarneau, A.; Fajula, F. Transition-metal ligands bound onto the micelle-templated silica surface. Coord. Chem. Rev. 1998, 178–180, 1085–1108. [Google Scholar]
- Lindner, E.; Salesch, T.; Brugger, S.; Steinbrecher, S.; Plies, E.; Seiler, M.; Bertagnolli, H.; Mayer, A.M. Accessibility studies of sol-gel processed phosphane-substituted iridium(I) complexes in the interphase. Eur. J. Inorg. Chem. 2002, 1998–2006. [Google Scholar]
- Sayah, R.; Flochc, M.; Framery, E.; Dufaud, V. Immobilization of chiral cationic diphosphine rhodium complexes in nanopores of mesoporous silica and application in asymmetric hydrogenation. J. Mol. Cat. A. Chem. 2010, 315, 51–59. [Google Scholar] [CrossRef]
- Ohkuma, T.; Takeno, H.; Honda, Y.; Noyori, R. Asymmetric hydrogenation of ketones with polymer-bound BINAP diamine ruthenium catalysts. Adv. Synth. Catal. 2001, 343, 369–375. [Google Scholar] [CrossRef]
- Lu, Z.-L.; Lindner, E.; Mayer, H.A. Applications of sol-gel-processed interphase Catalysts. Chem. Rev. 2002, 102, 3543–3578. [Google Scholar] [CrossRef]
- Chai, L.T.; Wang, W.W.; Wang, Q.R; Tao, Q.R. Asymmetric hydrogenation of aromatic ketones with MeO-PEG supported BIOHEP/DPEN ruthenium catalysts. J. Mol. Cat. A 2007, 270, 83–88. [Google Scholar] [CrossRef]
- Kang, C.; Huang, J.; He, W.; Zhang, F. Periodic mesoporous silica-immobilized palladium(II) complex as an effective and reusable catalyst for water-medium carbon–carbon coupling reactions. J. Organomet. Chem. 2010, 695, 120–127. [Google Scholar]
- Bergbreiter, D. Using soluble polymers to recover catalysts and Ligands. Chem. Rev. 2002, 102, 3345–3384. [Google Scholar] [CrossRef]
- Song, C.; Lee, S. Supported chiral catalysts on inorganic materials. Chem. Rev. 2002, 102, 3495–3524. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2010 by the authors;
Share and Cite
Warad, I.; Al-Resayes, S.; Al-Othman, Z.; Al-Deyab, S.S.; Kenawy, E.-R. Synthesis and Spectrosopic Identification of Hybrid 3-(Triethoxysilyl)propylamine Phosphine Ruthenium(II) Complexes. Molecules 2010, 15, 3618-3633. https://doi.org/10.3390/molecules15053618
Warad I, Al-Resayes S, Al-Othman Z, Al-Deyab SS, Kenawy E-R. Synthesis and Spectrosopic Identification of Hybrid 3-(Triethoxysilyl)propylamine Phosphine Ruthenium(II) Complexes. Molecules. 2010; 15(5):3618-3633. https://doi.org/10.3390/molecules15053618
Chicago/Turabian StyleWarad, Ismail, Saud Al-Resayes, Zeid Al-Othman, Salem S. Al-Deyab, and El-Refaie Kenawy. 2010. "Synthesis and Spectrosopic Identification of Hybrid 3-(Triethoxysilyl)propylamine Phosphine Ruthenium(II) Complexes" Molecules 15, no. 5: 3618-3633. https://doi.org/10.3390/molecules15053618
APA StyleWarad, I., Al-Resayes, S., Al-Othman, Z., Al-Deyab, S. S., & Kenawy, E.-R. (2010). Synthesis and Spectrosopic Identification of Hybrid 3-(Triethoxysilyl)propylamine Phosphine Ruthenium(II) Complexes. Molecules, 15(5), 3618-3633. https://doi.org/10.3390/molecules15053618